
Abstract
Spatial transcriptomics and epigenomics have enabled mapping gene regulation in the tissue context. However, it remains poorly understood how spatial gene expression patterns are orchestrated by enhancers. Here we build eSpatial, a computational framework that deciphers spatially resolved enhancer regulation of gene expression by integrating spatial profiles of gene expression and chromatin accessibility. Applying eSpatial to diverse spatial datasets, including mouse embryo and brain, as well as human melanoma and breast cancer, we reveal a “spatial enhancer code”, in which divergent combinations of enhancers regulate the same gene in spatially segregated domains. We validate the spatial enhancer code using public spatial datasets such as VISTA, Allen in situ hybridization (ISH), and H3K27ac MERFISH. Moreover, we conduct transgenic reporter assays and in vivo CRISPR/Cas9-mediated perturbation experiments to confirm the Atoh1 spatial enhancer code in determining Atoh1 spatial expression in mouse embryonic spinal cord and brain. Our study establishes the spatial enhancer code concept, revealing how combinations of enhancers dynamically shape gene expression across diverse biological contexts, providing insights into tissue-specific regulatory mechanisms and tumor heterogeneity.
Similar content being viewed by others


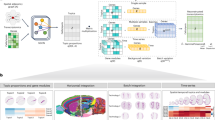
Introduction
Spatiotemporal regulation of gene expression is crucial for the development and function of multicellular organisms. This process relies on the selective activation or repression of gene expression, predominantly governed by cis-regulatory elements like enhancers1. Accurate identification and understanding of active enhancers hold paramount importance in this context. In a heroic effort, millions of epigenomic elements have been identified through sequencing-based approaches that profile chromatin accessibility and histone modifications at both ensemble and single-cell levels2,3,4. Typically, enhancers undergo classification based on chromatin accessibility, transcription factor occupancy, and the enrichment of specific histone modifications, notably including H3K4me1 and H3K27ac. Generally, a single gene can be regulated by multiple enhancers existing as a cluster in the genome. Some of these dispersed enhancer clusters have been labeled as “super-enhancers”5,6, “enhancer clusters”7, “shadow enhancers”8, or “redundant enhancers”9, providing redundancy to ensure robust transcriptional activity during development. However, deciphering how these enhancers precisely govern gene transcription remains challenging10,11. Furthermore, the current methods result in the loss of spatial activity distribution of these enhancers, which constrains our understanding of how the enhancers shape cell identity in the native context of complex tissues.
The emergence of spatial omics technologies has enabled mapping of spatially resolved epigenome and transcriptome. Notably, spatial-ATAC sequencing technologies allow the precise mapping of spatially resolved chromatin accessibility within tissue sections12,13. Furthermore, novel approaches have emerged that enable joint profiling of the epigenome and transcriptome by simultaneously sequencing chromatin accessibility and gene expression within the same tissue section. Examples include spatial epigenome-transcriptome co-profiling14, microfluidic indexing-based spatial-ATAC and RNA-seq (MISAR-seq)15, and multimodal Slide-tags16. These technologies offer new insights into spatial epigenetic regulation within tissues during development and diseases. However, the complex regulatory logic in the context of tissue organization remains largely unknown17,18. For instance, how combinations of enhancers coordinate to regulate spatial gene expression within the same cell type across different regions has yet to be systematically investigated.
Here, we developed eSpatial, a computational framework that deciphers spatially resolved enhancer regulation by integrating spatial epigenomics and transcriptomics data. By applying eSpatial across diverse biological contexts and conducting extensive validations, we demonstrate that distinct combinations of enhancers are utilized within spatial domains. This phenomenon, which we term the “spatial enhancer code,” highlights how divergent enhancer combinations orchestrate spatially distinct gene expression patterns across tissues. Our findings provide a perspective on enhancer regulation that goes beyond traditional gene-centric approaches, uncovering a previously uncharacterized layer of spatial regulatory complexity. Notably, the spatial enhancer activities discussed throughout this manuscript refer to their locations within a tissue, rather than the three-dimensional positioning of cis-regulatory elements within a nucleus19.
Results
eSpatial, a computational framework to decipher spatial enhancer code
To decipher spatially resolved enhancers regulation, we first collected published spatial epigenome profiling from mouse embryonic development and human disease, including embryonic day 11 (e11) and 13 (e13) mouse embryos, postnatal day 22 (P22) mouse brain, human melanoma and breast cancer (Fig. 1a). Then we built a bioinformatical framework eSpatial to decipher spatially resolved enhancer regulation of gene expression patterns based on spatial epigenome profiling (Fig. 1a), including spatial gene expression and chromatin accessibility co-profiling14,15 or spatial chromatin accessibility profiling12,13. Briefly, eSpatial identifies enhancer units within enhancer clusters based on spatial patterns of enhancer activity and evaluates how different combinations of these enhancer units contribute to gene expression across distinct spatial domains within tissues. Through eSpatial, we uncover a phenomenon where divergent combinations of enhancers regulate the same gene, but in spatially distinct domains, which we termed “spatial enhancer code” (Fig. 1a). Next, we validated spatial enhancer code using public datasets such as VISTA, Allen in situ hybridization (ISH), and H3K27ac MERFISH data. Finally, we utilized enhancer transgenic reporter assays and CRISPR/Cas9-mediated perturbation to in vivo validate the role of the spatial enhancer code of Atoh1 in controlling its expression in mouse embryonic spinal cord and brain (Fig. 1a). In the following sections, we will detail the discovery and validation of the spatial enhancer code.

a Schematic representation of the workflow in this study, including data collection, spatial enhancer code, and in vivo validation. b Spatial distribution of major cell types in spatial-ATAC-RNA-seq of the P22 mouse brain by integration of RNA pixels and scRNA-seq data. Oligo, oligodendrocyte; OPC, Oligodendrocyte precursor cell. Pixel size, 20 µm; scale bars, 1 mm. c Spatial distribution of RNA (c) and ATAC (d) clusters in spatial-ATAC-RNA-seq of the P22 mouse brain. Pixel size, 20 µm; scale bars, 1 mm. d Spatial distribution of identified spatial domains based on combined spatial profiles of chromatin accessibility and gene expression. Anatomical annotation defined from Nissl staining was obtained from ref. 14. Pixel size, 20 µm; scale bars, 1 mm. e Spatial mapping of denoised gene expression for selected marker genes across different spatial domains. f Heatmap showing gene expression (left) and enhancers chromatin accessibility (right) for 59,125 gene-enhancer pairs, clustered into 9 modules (K1–K9) based on gene expression. The relevant markers and representative enhancers were highlighted. The major cell types across spatial domains are shown in Supplementary Fig. 2b. g Spatial mapping of denoised chromatin accessibility for representative enhancers across different spatial domains. Source data are provided as a Source Data file.
Spatial domains in P22 mouse brain
To evaluate the performance of eSpatial, we first applied it to investigate enhancer regulation in postnatal day 22 (P22) mouse brain coronal sections (at bregma 1). The spatial epigenome-transcriptome co-profiling was captured by Zhang et al.14, containing 9215 spots with 260,032 accessible peaks and 22,914 expressed genes. We initially annotated the cell types of spots by integrating spatial-RNA data with the juvenile central nervous system (CNS) scRNA-seq atlas20. We observed distinct spatial distributions of major cell types and their marker genes (Fig. 1b and Supplementary Fig. 1b). Then we performed spatial clustering analysis on gene expression and chromatin accessibility profiling were performed independently, and 11 RNA clusters (R0-R10) and 15 ATAC clusters were identified (A0-A14) (Fig. 1c and Supplementary Fig. 1c). Notably, the ATAC clusters exhibited clearer spatial shape than RNA clusters. For example, ATAC clusters A1, A4, and A6 presented layer distribution in the excitatory neurons (Fig. 1c, left), which cannot be observed in RNA clusters R2, R3, R5, and R9 (Fig. 1c, right). This observation suggests the potential role of chromatin accessibility, alongside gene expression, in specifying the spatial distribution of excitatory neurons. Motivated by these findings, we combined spatial profiles of chromatin accessibility and gene expression, together with the spatial coordinates, unveiling 14 spatial domains (D0–D13, Fig. 1d and “Methods”). Strikingly, chromatin accessibility together with gene expression identified clearer distinctions among excitatory neurons compared to those solely based on chromatin accessibility or gene expression, as evidenced by domains D0, D1, and D2 (Fig. 1d). This integration highlighted distinct spatial distributions of major cell types.
To delineate the gene regulatory intricacies underlying each spatial domain, we first quantified the spatial specificity of cis-regulatory elements (CREs) and genes. Interestingly, consistent with the superior spatial specificity observed in spatial-ATAC data, we noted that the chromatin accessibility of CREs showed significantly higher spatial specificity than gene expression in each spatial domain (Supplementary Fig. 2a, p-value < 2.2e-16, two-sided Student’s t-test). These results suggest the role of CREs in shaping spatial patterns. Next, we identified spatial-specific cis-regulatory elements (sCREs) for individual spatial domains based on the spatial specificity of CREs (Methods). Most of the sCREs displayed highly localized chromatin accessibility across diverse brain spatial domains (Supplementary Fig. 2c). For example, sCREs associated with excitatory neurons showed spatial specificity across distinct layers, such as sCRE1 in D0, sCRE2 in D1, and sCRE3 in D3 (Supplementary Fig. 2c–e). Furthermore, the GREAT analysis21 corroborated a robust alignment between gene regulatory pathways and anatomical annotations (Supplementary Data 1 and “Methods”). Motif enrichment analysis identified specific motifs enriched in each spatial domain. Notably, motifs like Egr1 and Egr2 were enriched in sCREs exhibiting spatial specificity in D0, while Neurog1 and Olig2 motifs were enriched in D1, Tbr1, and Eomes motifs in D4 (Supplementary Fig. 2c, right and Supplementary Data 2). These results suggest that the specification of spatial domains might involve different regulatory networks composed of spatial-domain-specific transcription factors (TFs).
Taken together, eSpatial identifies spatial domains within cell types and spatial-specific cis-regulatory elements, laying a foundation for investigating the gene regulatory programs driving spatial patterns of cell types.
Combinations of divergent spatial enhancer units encode gene expression
Enhancer clusters regulate gene expression patterns during development; however, it remains largely unexplored to what extent and how multiple enhancers regulate spatial gene expression precisely. We used eSpatial to identify putative enhancer clusters for each gene by linking enhancers to the potential target gene based on the correlation between gene expression and enhancer accessibility22 (Methods). In total, we identified 59,125 putative enhancer-gene pairs, involving 59,125 enhancers linked to 11,538 genes (Fig. 1e–g). We compared the peak-to-gene links identified by eSpatial with those from publicly available single-cell multiomic data of the same tissue and developmental stage (e.g., mouse cortex)22. The results showed substantial overlap, validating eSpatial’s ability to capture core regulatory relationships (Supplementary Fig. 2f). Additionally, eSpatial identified several links absent in single-cell data, suggesting that spatial information enables the discovery of regulatory interactions specific to tissue architecture and microenvironmental context. Moreover, we benchmarked peak-to-gene links identified by eSpatial and other single-cell methods, such as ArchR23 and Signac24 (Supplementary Fig. 2f). We found the peak-to-gene links identified by eSpatial exhibited slightly higher enrichment of the reference gene-enhancer links compared with those from ArchR and Signac, underscoring its superior performance in uncovering regulatory relationships from spatial data (Supplementary Fig. 2g).
To explore the spatial gene expression patterns across diverse spatial domains, we utilized the k-means clustering algorithm and identified 9 gene expression modules (K1–K9, Fig. 1f, left). Gene modules exhibited cell-type-specific expression, such as K8 in oligodendroglia and K9 in Neuroblast. Genes within each module were enriched for cell-type-specific Gene Ontology (GO) terms, including “cerebral cortex development” (K1), “oligodendrocyte development” (K8), and “cerebral cortex radially oriented cell migration” (K9) (Supplementary Data 1). To explore the phenomenon of different combinations of enhancers regulating the same gene across spatial domains within the same cell type (Fig. 2a and Supplementary Fig. 1a), we focused on regulatory elements of excitatory neurons across cortical layers (Fig. 1b, d–g). We found this phenomenon occurs not only across tissues or cell types but also within spatially distinct domains of the same cell type. For instance, Sox10, a marker of oligodendroglia25, resided in the K8 module expressed in D11 (Fig. 1e). Similarly, Nrn1, a marker of excitatory neurons, was in the K1 module expressed in excitatory neurons26. (Fig. 1e, f and Supplementary Fig. 2h, i). Intriguingly, for these cell-type-specific genes, enhancers within their enhancer clusters displayed domain-specific activity within cell types (Fig. 1e–g), indicating that the spatial specificity of enhancer activity extends beyond gene expression and reflects distinct regulatory mechanisms across spatial domains.

a Bar plot illustrating the number of spatial domains in which genes are expressed. Pie chart showing the proportion of genes expressed across multiple spatial domains (≥2) versus those specific to a single domain. b Heatmap displaying the spatial enhancer units regulating K1 genes in Fig. 1f. These enhancers were clustered into 17 patterns (M1–M17, columns) based on their spatial specificity across K1 genes expressed domains (D0, D1, D2, D3, D13, and D4). c Schematic representation of divergent regulation and coherent regulation by enhancer clusters. d Bar plot showing the number of enhancer clusters (y-axis) containing enhancers from various numbers of enhancer units (x-axis). Pie chart showing the proportion of divergent regulation (≥2 enhancer units within a cluster) versus coherent regulation (only 1 enhancer pattern). e The combination of enhancers encodes Nrn1 spatial expression. Genome tracks presenting chromatin accessibility, peak site (marked by open regions, enhancer clusters, and enhancer units), gene tracks (left), and gene expression (right) for the Nrn1 locus in cortex regions (D0, D1, D2, and D3). Spatial mapping of denoised chromatin accessibility of representative enhancers from three distinct enhancer units (M12, M3, and M1) and gene expression (top). f Spatial distribution of cell types in spatial-ATAC-RNA-seq of the P22 mouse brain by integration of ATAC pixels and scATAC-seq data22. Arrows indicate the dorsal-ventral (D-V) and deep-superficial (D-S) axes in the coronal section. Pixel size, 20 µm; scale bars, 1 mm. g Directionality analysis of chromatin accessibility along D-V or D-S axes for the top 1500 cell-type-specific accessible peaks (ranked by log2[fold change]) across individual cell types. Each point represents a putative enhancer, positioned by Spearman’s correlation coefficient (R) between peak accessibility and spatial coordinates (n = cell-type-specific counts, coordinates normalized to 0–1 scale), with color indicating statistical significance (−log10 P-value) of correlation. Positive or negative R values denote ventral/superficial or dorsal/deep directional biases, respectively. The top 5 enhancers with the highest absolute R values were explicitly labeled. h Spatial mapping of denoised Hlf gene expression and denoised chromatin accessibility of representative directional enhancers (from g) associated with Hlf showing ITL23GL-specific chromatin accessibility and directionality preference. Schematic representation of the location of Hlf gene and Hlf putative enhancers. i Spatial mapping of denoised Bean1 gene expression and denoised chromatin accessibility of representative directional enhancers (from g) associated with Bean1 showing CTGL-specific chromatin accessibility and directionality preference. Schematic representation of the location of Bean1 gene and Bean1 putative enhancers. Source data are provided as a Source Data file.
To explore regulatory mechanisms across spatial domains, we defined spatial enhancer units based on the spatial patterns of constitute enhancers within enhancer clusters. This approach aimed to characterize their activity patterns in the spatial domains where their target genes were expressed (Methods). For example, for the enhancers linked to genes in the K1 module, we identified 17 spatial enhancer units (M1–M17). Several enhancer units (e.g., M1, M2) exhibited activity across multiple domains, while others displayed specific activity in individual spatial domains (e.g., M15, M16, M17) (Fig. 2b). These observations promoted us to explore whether different enhancer units collectively encode gene expression patterns across different spatial domains. To test this hypothesis, we quantified the diversity of spatial enhancer units within individual enhancer clusters. The enhancer clusters were categorized into two groups based on the number of enhancer units: spatially divergent and coherent regulation scenarios. Specifically, genes expressed across multiple spatial domains with combinations of different spatial enhancer units were classified as divergent regulation, whereas those regulated by the same enhancer unit were termed coherent regulation hereafter (Fig. 2c). Intriguingly, a majority of enhancer clusters (6940, 60%) contained at least two enhancer units (Fig. 2d). For example, the expression of Nrn1 in distinct spatial domains (D0, D1, D2, D3) was regulated by enhancer combinations showcasing three different spatial patterns. Enhancer unit M1 was accessible across all Nrn1-expressing spatial domains, while Enhancer unit M3 exhibited relatively higher accessibility in D0 and D1. In contrast, enhancer unit M12 exhibited higher accessibility in D2 and D3 (Fig. 2e). These findings suggest that Nrn1 expression in D0 and D1 may be encoded by a combination of enhancer units M1 and M3, while its expression in D2 and D3 might be driven by enhancer units M1 and M12.
To further elucidate the mechanisms underlying divergent enhancer units combinations, we investigated the potential role of TFs (Methods). By focusing on enhancers regulating genes expressed in excitatory neurons, we observed differential enrichment of several TF motifs across specific spatial domains (Supplementary Fig. 3a, b). For instance, the Fra1 motif was enriched across all enhancers in excitatory neurons. However, enhancers associated with specific spatial domains exhibited unique TF enrichment patterns. Specifically, enhancers in domains D1 and D3 showed significant enrichment for Mef2c and Mef2a motifs, while enhancers in domains D4 exhibited notable enrichment for Rfx5 motif. Moreover, enhancers with similar spatial patterns tend to share a significantly higher number of TF binding sites compared to enhancers with divergent spatial patterns (Supplementary Fig. 3c). These findings might underscore the pivotal role of distinct TFs binding in coordinating the combinatorial regulation of enhancers driving excitatory neuron-specific gene expression. To explore the relationship between super-enhancers and divergent enhancer combinations, we obtained super-enhancer data from publicly available databases27 for three brain-related tissues: cortex, brain (E14.5), and cerebellum. We then performed an enrichment analysis to assess the overlap between divergent enhancers, coherent enhancers, and super-enhancers across these tissues. Our analysis revealed a significant enrichment of divergent enhancers within super-enhancer regions (Supplementary Fig. 3d). This finding suggests that enhancers within the same super-enhancer tend to exhibit coordinated, yet not completely uniform, activity.
To better distinguish subtle spatial differences from cell-type variations, we annotated the spatial distribution of cell types by integrating spatial-ATAC-seq data from P22 mouse brain with a previously established single-cell ATAC-seq atlas of the adult mouse cerebrum22 (Fig. 2f, Supplementary Fig. 3e). We then investigated the spatial accessibility of putative enhancers within individual cell types, following the approach in a recent study28. Specifically, we quantified the spatial distribution of enhancers accessibility by correlating chromatin accessibility along either dorsal-ventral (D-V) or deep-superficial (D-S) spatial axes (Fig. 2f, g and “Methods”). Strikingly, we found ~20% of putative enhancers showed directional bias in chromatin accessibility along these spatial axes (Supplementary Fig. 3f, top). Of note, putative enhancers within the same enhancer cluster targeting the same gene can exhibit distinct spatial accessibility patterns with different directionality preference (Supplementary Fig. 3f, bottom). For example, Hlf expressed within the ITL23GL excitatory neuron subtype, contains putative enhancers located at +83 kb, +58 kb, and −29 kb relative to the transcription start site. These enhancers exhibited distinct spatially accessibility along the D-V axis: the +83 kb and −29 kb enhancers showed dorsal- and ventral-specific accessibility, respectively (Fig. 2h). Similarly, in the CTGL subtype, putative enhancers of Bean1 displayed spatially variable accessibility along the D-S axis (Fig. 2i). These results indicate the spatially divergent regulation of enhancer cluster exists not only across different neuron subtypes located in various cortical layers, but also within individual neuron subtypes.
Combinations of divergent enhancer units encode gene expression in human tumors
After demonstrating the spatial enhancer code in normal tissues, we wondered whether it also encoded gene expression in the disease samples. Therefore, we employed eSpatial to investigate epigenetic regulation in the establishment of microenvironments in human tumors. We first applied eSpatial to a multimodal Slide-tags dataset of a metastatic melanoma sample16 (Fig. 3a). The original study analyzed the spatial data and reported that the metastatic sample contained two distinct tumor compartments, denoted as Compartment 1 (C1) and Compartment 2 (C2), where compartment 1 representing a mesenchymal-like cell state and compartment 2 comprising a melanocytic-like population16 (Fig. 3b). We asked the role of enhancers in regulating gene expression in these two distinct spatial compartments, with a specific focus on the well-annotated tumor cells and T cells from the original study. We identified 15,641 enhancers associated with 7457 genes and clustered them into gene modules based on their gene expression patterns (Fig. 3c, left). Notably, nearly half of the genes (49.8%) exhibited expression in both compartments (Fig. 3d), including genes in module K1 expressed in tumor cells, and genes in K4 expressed in T cells. As tumor cells are intrinsically very different from T cells, we investigated the spatial enhancer regulation for tumor cells and T cells separately. Intriguingly, enhancers associated with those genes expressed in tumor cells in both compartments showed distinct spatial activity (Fig. 3c, right), which were categorized into three spatial enhancer units (M1–M3). Enhancer unit M1, active in both compartments; enhancer unit M2, specifically active in compartment 1; and enhancer unit M3, specifically active in compartment 2 (Fig. 3f). Further exploration into the potential mechanism underlying compartment-specific enhancers revealed enriched accessible motifs within these enhancer units (Fig. 3g and Supplementary Fig. 4a). Notably, motifs corresponding to FOS/JUN-family members were enriched in M2 enhancer unit, specifically accessible in Compartment 1 (Fig. 3g, left). This enrichment aligns with findings that the FOS/JUN-family plays a key role in the mesenchymal-like melanoma state29,30. Conversely, the motif corresponding to MITF was enriched in M3 enhancer unit specifically accessible in Compartment 2 (Fig. 3g, right), consistent with MITF’s role in maintaining the melanocytic lineage29,30. These findings suggest that the spatial-specific transcription factors regulate tumor development through their spatial enhancer activation, further confirming the power of our eSpatial analysis in dissecting the epigenetic regulatory mechanisms underlying spatial-specific regulation of tumors.

a Schematic representation (top) and spatial distribution (bottom) of two distinct spatial compartments defined by Slide-tags multiome which was adapted from the original literature16. b Spatial distribution of tumor cells and T cells in the human melanoma sample. c Heatmap showing gene expression (left) and enhancer chromatin accessibility (right) for 15,641 gene-enhancer pairs, clustered into 6 modules (K1–K6) based on gene expression in tumor cells and T cells from two compartments. d Bar plot illustrating the number of compartments in which genes are expressed. Pie chart showing the proportion of genes expressed in two compartments versus those specific to one compartment. e Bar plot showing the number of enhancer clusters (y-axis) composed of enhancers displaying various numbers of enhancer units (x-axis). Pie chart depicting the proportion of divergent regulation (≥2 enhancer units within a cluster) versus coherent regulation (only 1 enhancer pattern). f Heatmap displaying the spatial enhancer units regulating module K1 genes in (c). These enhancers were clustered into 3 enhancer units (M1–M3, rows) based on their binarized spatial specificity in two compartments. g Motif enrichment rank of enhancers with spatial patterns (M2 and M3) from (f). Top 10 motifs are highlighted. P values calculated by a one-sided hypergeometric test. h Spatial mapping of denoised DNMT3A gene expression and denoised chromatin accessibility of three DNMT3A enhancers from different enhancer units (M1, M2, and M3). Schematic representation of the location of DNMT3A gene and DNMT3A enhancers. i Spatial distribution of tumor, immune-rich, myeloid, and normal tissue regions in a human breast cancer sample. Schematic representation of spatial-ATAC was from a published study13. j A zoom-in window of spatial distribution of tumor, immune-rich, myeloid, and normal tissue regions in the human breast cancer sample from (i). k Left: Heatmap showing gene scores clustered into 9 modules (K1–K9). Right: Heatmap displaying the spatial enhancer units regulating K1 genes in (k). These enhancers were clustered into 3 enhancer units (M1–M3, rows) based on their binarized spatial specificity. l Spatial mapping of denoised NOTCH1 gene expression and denoised chromatin accessibility of three NOTCH1 enhancers from different enhancer units (M1, M2, and M3) in the tumor and myeloid regions. Schematic representation of the location of NOTCH1 gene and NOTCH1 enhancers. Source data are provided as a Source Data file.
Moreover, we observed a significant prevalence (26.3%) of divergent combinations of enhancer units in controlling gene expression in human melanomas (Fig. 3e). For example, a cluster of enhancers displaying three distinct patterns (C1-specific, C2-specific, and active in both) regulated the spatial expression of DNMT3A in tumor cells across the two compartments (Fig. 3h). Similarly, the expression of CD8A in T cells across the two compartments was regulated by a combination of two enhancer units (C1-specific and active in both) (Supplementary Fig. 4b, c).
To further evaluate the versatility of eSpatial across diverse disease contexts, we applied eSpatial analysis to a spatial-ATAC dataset of human breast cancer13. This allowed us to delineate the enhancer regulation of spatial regions of tumor, myeloid, immune-rich, and normal tissue, as defined by the original study (Fig. 3i, j). Once again, we identified genes with shared gene expression across the different regions, while their associated enhancers showed distinct spatial patterns (Fig. 3k and Supplementary Fig. 4d, f). Moreover, we noted a significant prevalence (22.6%) of divergent combinations of enhancer units, controlling spatial gene expression (Supplementary Fig. 4e). For example, the gene NOTCH1 was expressed in both tumor and myeloid regions, yet its associated enhancer clusters exhibited distinct combinations of three enhancer units (Fig. 3l). Similarly, RUNX3 was expressed in both myeloid and normal/immune-rich regions, while its enhancer clusters contained three different enhancer units (Supplementary Fig. 4g).
In summary, these results confirm that the divergent combinatorial use of enhancer units within enhancer cluster is a widespread mechanism in tumors, mirroring the patterns initially observed in normal tissues.
Functional validations of spatial-specific enhancers in mouse embryos at e11 using VISTA enhancer activity data
Next, we sought to assess the functional relevance of spatial-specific enhancers. Given the fact that VISTA houses abundant enhancer activity experimental data from transgenic report assays in mouse embryos at embryonic day 11.5 (e11.5), we applied eSpatial on the spatial-ATAC profile of mouse embryos at e1112. Of note, this dataset provided spatial chromatin accessibility data without matched spatial transcriptome information. Given this absence, we derived gene activity scores based on chromatin accessibility within their gene bodies, aligning with the approach commonly employed in scATAC-seq and spatial-ATAC studies12,13,14,15,23,24 (Methods). Subsequently, we conducted eSpatial analysis utilizing the gene activity score matrix, chromatin accessibility matrix, and spot coordinate matrix. It led to 7 spatial domains (D0–D6), each spatial domain exhibiting strong agreement with anatomical regions, such as D0 in the brain, D1 in the spinal cord, D2 in the notochord, D5 and D6 in the heart/limb regions (Fig. 4a and Supplementary Fig. 5a, b). As anticipated, well-known markers of the central neural systems, such as Sox2 and Pax631, displayed high gene activity scores in D0 and D1 (Fig. 4b and Supplementary Fig. 5c, d). Conversely, markers associated with the heart, like Tcf21 and Ror232 exhibited the highest gene activity scores in D5 and D6. (Fig. 4b and Supplementary Fig. 5c, d). Importantly, these gene activity scores showed consistent spatial expression with in situ hybridization (ISH) images at e11.5 mouse embryos obtained from the Allen Brain Atlas (Fig. 4b).

a Spatial distribution of spatial domains in e11 mouse embryos. Pixel size, 20 µm; spatial-ATAC-seq data was from ref. 12. b Left: Allen RNA ISH images in e11.5 mouse embryos. Right: spatial mapping of gene scores in e11 mouse embryos for selected marker genes across different spatial domains. c Heatmap showing gene expression (left) and chromatin accessibility (right) for 29,485 gene-enhancer pairs. Gene expression was clustered using k-means clustering (k = 7). The relevant markers and representative enhancers were highlighted. d Spatial mapping of denoised chromatin accessibility (left) and the corresponding VISTA enhancer reporter activity for e11.5 mouse embryos (right) for representative enhancers. e Enrichment of VISTA-validated enhancers among enhancers in (c) and all CREs. *p < 0.05, **p < 0.01, ***p < 0.001; n.s. not significant (binomial test; exact p-value < 2.2e-16). f The percentage of VISTA-validated enhancers in brain, spinal cord, and heart regions overlapped with sCREs from spatial-ATAC data. g Pie chart displaying the proportion of gene expression patterns (top: multiple domains and single domain) and enhancer cluster regulation patterns (bottom: divergent regulation and coherent regulation). h Heatmap showing enhancer units for enhancers regulating genes in K1 as depicted in (c). i The combination of enhancer units encodes Cmtm8 spatial expression. Genome tracks showing the chromatin accessibility, peak site (marked by open region, enhancer cluster, and enhancer unit), gene tracks (left), and gene expression (right) for Cmtm8 in D0 and D1. Spatial mapping of denoised chromatin accessibility of three representative enhancers from different enhancer units (M2, M4, and M6) and gene expression (top). Source data are provided as a Source Data file.
Next, to understand the spatial gene expression patterns across diverse tissues, we utilized eSpatial to cluster spatial gene activity and found the majority of genes (7039, 82%) exhibited expression across multiple spatial domains (Fig. 4c, left, 4g, top, and Supplementary Fig. 5h, left). Notably, several neuron-related markers, including Rbfox3, Notch1, Pax6, and Sox2, demonstrated expression in the central nervous system (CNS) regions (D0, D1, and D2) (Fig. 4c, left). To delineate the underlying gene regulatory intricacies, we identified 29,485 putative enhancer-gene pairs, involving 29,485 putative enhancers linked to 8584 genes (Fig. 4c). Impressively, the enhancers controlling those genes expressed in CNS regions showed distinct spatial activity. For instance, the enhancers in the brain and spinal cord (E1 in D0, E2 in D0 and D1) displayed significant agreement between the observed chromatin accessibility in spatial-ATAC data and the LacZ signals in VISTA enhancer reporter data (Fig. 4c, right, 4d and Supplementary Fig. 5e–g).
We next evaluated the enhancer activity of all constituted enhancers within enhancer clusters by leveraging the enhancer activity data sourced from VISTA33. Overall, our observations indicated a notably higher VISTA enrichment (3.6-fold) among the constitute enhancers in comparison to CREs (Fig. 4e), which suggested that the enhancers identified above may be indeed active enhancers. In addition, we further distinguished the experimentally validated enhancer activity in different tissues. We observed that 79.7% (295/370) of VISTA elements in brain regions, 79.4% (170/214) in the spinal cord region, and 79.2% (163/206) in the heart region exhibited spatial chromatin accessibility corresponding to their respective domains, as observed in the spatial-ATAC data of mouse embryos at e11 (Fig. 4f).
Similar to the observations in the P22 mouse brain (Fig. 2), we noted a prevalent presence of divergent combinations of enhancer units within enhancer clusters in controlling spatial gene expression in e11 mouse embryos (Fig. 4g, bottom and Supplementary Fig. 5h, right). For example, a cluster of enhancers comprising three different enhancer units regulated the expression of Cmtm8. Enhancer unit M2 was accessible in both D0 and D1, while enhancer units M4 and M6 patterns exhibited accessibility exclusively in D0 and D1, respectively. It suggested that Cmtm8 expression in D0 was encoded by the combination of enhancer units M2 and M4, while in D1, it was governed by the combination of enhancer units M2 and M6 (Fig. 4h, i).
Taken together, these data demonstrate the capability of eSpatial in identifying functionally relevant, spatial-specific enhancers, further substantiating the concept of the spatial enhancer code. Notably, eSpatial showcases adaptability even in datasets with only spatial chromatin accessibility, extending its utility beyond datasets composed of spatial epigenomic-transcriptomic co-profiling.
Validations of enhancer spatial activity in the mouse brain at e13.5 by ISH and H3K27ac MERFISH data
To further validate the efficacy of eSpatial analysis, we applied it to the joint profiling of chromatin accessibility and transcriptome in the e13 mouse embryo14. This analysis identified 12 spatial domains (D0–D11, Fig. 5a and Supplementary Fig. 6a, b), aligning well with the anatomical annotation in the mouse embryo. For example, D0 and D1 corresponded to the forebrain region, D2 and D3 to the hindbrain region, and D4 specifically to the eye region (Fig. 5a, b). Spatial domains exhibited distinct patterns of spatial-specific gene expression (Supplementary Fig. 6c). For instance, Rbfox3, a CNS maker playing a prominent role in neural tissue development34, showed specific gene expression in the forebrain and hindbrain regions (D0 and D2) (Supplementary Fig. 6d). Conversely, Six6, an essential gene involved in eye development35, exhibited specific gene expression in the eye region (D4, Supplementary Fig. 6d).

a Spatial distribution of spatial domains in e13 mouse embryos. Pixel size, 50 µm; scale bars, 1 mm. Spatial data was from ref. 14. b Schematic highlighting different brain regions (forebrain, midbrain, and hindbrain in solid color shades, while the cortex, diencephalon, and prosomere in dotted color lines) of an imaged sagittal slice of an e13.5 mouse brain from the H3K27ac MERFISH data. The background shows the DAPI signal. Scale bars: 1 mm. The picture was adapted from ref. 37. c Spatial mapping of denoised gene expression (left) and Allen RNA ISH images (right) for Neurog2, Tbr1, and Foxg1. d Spatial mapping of denoised chromatin accessibility (left) and epigenomic MERFISH images of H3K27ac signals (right) for the enhancers controlling Neurog2, Tbr1, and Foxg1. e Heatmap showing gene expression (left) and chromatin accessibility (right) for 14,504 gene-enhancer pairs. Gene expression clustered via k-means clustering (k = 7). The relevant markers and representative enhancers are highlighted. f Pie chart demonstrating the proportion of gene expression patterns (left: multiple domains and single domain) and enhancer cluster regulation patterns (right: divergent regulation and coherent regulation). g Heatmap showing the enhancer units for enhancers regulating K1 genes as depicted in (e). h The combination of enhancer units encodes Bcl11a spatial expression. Genome tracks showing the chromatin accessibility, peak site (labeled by open region, enhancer cluster, and enhancer unit), gene tracks (left), and gene expression (right) for Bcl11a in D0 and D2. Spatial mapping of denoised chromatin accessibility of three representative enhancers in distinct enhancer units (M2, M4, and M6) and gene expression (top). Source data are provided as a Source Data file.
H3K27ac peaks distal from transcription start sites often denote active enhancers36. A recent study reported a spatially resolved H3K27ac MERFISH (multiplexed error-robust fluorescence in situ hybridization) and mapped a high-resolution spatial atlas of hundreds of putative enhancers in the e13.5 embryonic mouse brain37. Additionally, the Allen database hosts a collection of brain in situ hybridization (ISH) data (http://developingmouse.brain-map.org/static/atlas). Leveraging these two datasets, we sought to validate the spatial enhancer regulation of gene expression identified by eSpatial. Indeed, the spatial enhancer activity and gene expression identified by eSpatial agreed with H3K27ac MERFISH and the ISH data. For example, Neurog2, Tbr1, and Foxg1 displayed expression in the forebrain region according to the spatial transcriptome and Allen brain ISH atlas (Fig. 5c). Consistently, their enhancers showed specific accessibility in the corresponding domains in e13.5 embryonic mouse forebrain region, which is consistent with the signals detected in H3K27ac MERFISH data (Fig. 5d).
Furthermore, we delineated the spatial enhancer code in the e13 mouse embryo and uncovered how divergent combinations of enhancer units encode spatial gene expression. Our analysis identified 14,504 putative gene-enhancer pairs for 7017 genes. We observed genes in the K1 module were expressed in both forebrain and hindbrain regions (D0 and D2), while their linked enhancers showed spatial-specific activity (Fig. 5e). Moreover, a portion of genes expressed across multiple spatial domains and exhibited regulation by divergent combinations of enhancers (Fig. 5f). For example, Bcl11a, expressed in both the forebrain and hindbrain, was regulated by three enhancer units, where M4 in the forebrain, M6 in the hindbrain and M2 in both domains (Fig. 5g, h).
As a parallel validation, we applied eSpatial to the mouse fetal brain data (e13.5) generated through MISAR-seq (microfluidic indexing-based spatial assay for transposase-accessible chromatin and RNA-sequencing)15 (Supplementary Fig. 7a and “Methods”). Consistent with the results of joint spatial profiling of chromatin accessibility and transcriptome in e13 mouse embryo14 (Fig. 5), ISH and H3K27ac MERFISH data validated the enhancer activity and gene expression in space, such as Neurog2, Tbr1, and Foxg1 (Supplementary Fig. 7b, c). Additionally, the expression of Bcl11a in the forebrain and hindbrain was regulated by combinations of distinct enhancer units (Supplementary Fig. 7e).
Together, the agreement between H3K27ac MERFISH and ISH data with the spatial enhancer activity identified by eSpatial in two independent datasets reinforces its efficacy in deciphering the spatial enhancer code.
Transgenic reporter assays validate spatially divergent activity of Atoh1 enhancers in mouse embryonic spinal cord and hindbrain
To experimentally validate the spatial enhancer code, we focused on four enhancers (E0, E1, E2, and E3) located downstream of Atoh1 we previously identified38,39,40. Our recent study revealed the activity of these enhancers in the neurons during mouse spinal cord development39. Additionally, Atoh1 is known to be expressed in neurons of the hindbrain and is essential for cerebellum development41. However, it remains unexplored how the Atoh1 enhancer cluster orchestrates Atoh1 expression in both neurons of the spinal cord and hindbrain.
To this end, we applied eSpatial to investigate the Atoh1 enhancer cluster using the spatial-ATAC data of e11 mouse embryos (Fig. 6a). We first calculated Atoh1 gene activity scores based on its chromatin accessibility, and found that Atoh1 showed high gene activity scores in the spinal cord and hindbrain of mouse e11 (Fig. 6b). Consistently, our fluorescence in situ hybridization (FISH) analysis further confirmed Atoh1 expression in the neurons of both the hindbrain and spinal cord of e11 mice (Fig. 6c).

Spatial mapping of Atoh1 gene activity scores (a) in the spinal cord and hindbrain in e11 mouse embryos (b), where spatial-ATAC-seq data were from a published study12. c Fluorescence in situ hybridization (FISH) images depicting Atoh1 expression at e11 mouse embryos. The experiment was repeated independently 3 times with similar results. Scale bars, 500 µm. d Heatmap showing the accessibility levels of Atoh1 enhancers. The spatial specificity scores of Atoh1 enhancers in the e11 spinal cord and hindbrain were labeled on the heatmap. e Genome tracks showing the chromatin accessibility, peak coordinates, gene activity sores (left), and gene tracks (right) for the Atoh1 locus in the spinal cord and hindbrain of e11 mouse. Spatial mapping of denoised chromatin accessibility of four Atoh1 enhancers (E0, E1, E2, and E3) in the e11 mouse spinal cord and hindbrain. f Whole-mount X-gal staining of E0 (top), E2 (middle), and E3 (bottom) reporter embryos at e11. The blue signal displays the β-gal activity of the LacZ reporter driven by the indicated enhancers, representing the activities of the Atoh1 enhancers. The experiment was repeated independently 3 times with similar results. Scale bars, 500 µm. g, h Schematic diagram represented sampled embryonic tissue (left), imaging perspectives (middle), and eSpatial prediction (right). The blue dot line indicates the section direction. The pink box denotes the RNAscope imaged region in the spinal cord (g). The pale green box denotes the RNAscope imaged region of the hindbrain (h). i–l RNAscope detection of Atoh1 mRNA in E0/E2/E3/E0 + 3 KO or WT mice in e11 spinal cord. Left: Representative images show staining for Atoh1 RNAscope probes (red) and DAPI (blue). Right: Quantification of Atoh1 RNAscope probe signal. Values shown are the mean ± standard error of the mean; n = 5 independent experiments. Statistical significance was determined by two-sided Student’s t-test: E0-WT vs. E0-KO (p < 0.0001), E2-WT vs. E2-KO (p = 0.8792), E3-WT vs. E3-KO (p = 0.0296), and E0 + 3-WT vs. E0 + 3-DKO (p < 0.0001). *p < 0.05, **p < 0.01, ***p < 0.001; n.s., not significant. Scale bar, 50 μm. WT wild type, KO knockout. m–p RNAscope detection of Atoh1 mRNA in E0/E2/E3/E0 + 3 KO or WT mice in e11 hindbrain. Left: Representative images show staining for Atoh1 RNAscope probes (red) and DAPI (blue). Right: Quantification of Atoh1 RNAscope probe signal. Values shown are the mean ± standard error of the mean; n = 5 independent experiments. Statistical significance was determined by two-sided Student’s t-test: E0-WT vs. E0-KO (p < 0.0001), E2-WT vs. E2-KO (p = 0.9910), E3-WT vs. E3-KO (p = 0.8914), and E0 + 3-WT vs. E0 + 3-DKO (p < 0.0001). *p < 0.05, **p < 0.01, ***p < 0.001; n.s., not significant. Scale bar, 50 μm. WT wild type, KO knockout. Source data are provided as a Source Data file.
Next, we depicted the spatial patterns of Atoh1 enhancers in the hindbrain and spinal cord of e11 (Methods), and observed the phenomenon similar to those in the P22 mouse brain, as well as e11 and e13 mouse embryos (Figs. 2–5). Atoh1 exhibited consistent gene activity in the spinal cord and hindbrain at e11. However, the associated enhancers (E0, E1, E2, and E3) exhibited distinct chromatin accessibility and acted as four enhancer units (Fig. 6d). Specifically, E0 showed high accessibility in both the spinal cord and hindbrain, while E1 displayed no accessibility in either region. E2 showed weak accessibility in the spinal cord but lacked accessibility in the hindbrain. Conversely, E3 mirrored the high accessibility in the spinal cord as E0, yet displayed weaker accessibility in the hindbrain (Fig. 6d, e). Due to its lack of accessibility in both the spinal cord and hindbrain, E1 was excluded in the subsequent analysis. Therefore, we focus on testing the activity of the E0, E2, and E3 enhancers by using transgenic reporter mice for these enhancers.
To validate the activity of the E0, E2, and E3 enhancers of Atoh1, we conducted enhancer transgenic reporter assays to experimentally assess the spatiotemporal activity of these enhancers in vivo (Fig. 6f). Briefly, E0, E2, and E3 were used as putative enhancers to drive the expression of the β-galactosidase gene (LacZ) downstream of a minimal promoter39 (Supplementary Fig. 8a), then the enhancer activity can be visualized through X-gal staining42,43,44. E0 showcased β-gal activity in both the spinal cord and hindbrain, with obviously stronger activity in the hindbrain than in the spinal cord (Fig. 6f, top). E2 displayed modest β-gal activity in the e11 spinal cord but lacked activity in the e11 hindbrain (Fig. 6f, middle). On the other hand, E3 exhibited β-gal activity in both the spinal cord and hindbrain, with noticeably stronger activity in the spinal cord than in the hindbrain (Fig. 6f, bottom). These observations aligned with the results from the eSpatial analysis (Fig. 6d), confirming the spatial-specific activity of Atoh1 enhancers. These observations suggest that the Atoh1 expression in the spinal cord may be encoded by the combination of E0 and E3, whereas in the hindbrain, it primarily relies on E0 (Fig. 6g, h).
In vivo CRISPR/Cas9-mediated perturbation validates spatial enhancer code of Atoh1 in mouse embryos at e11
To further explore the spatial enhancer code encoding Atoh1 spatial expression, we employed enhancer knock-out mice (E0, E2, and E3) generated by CRISPR/Cas9-mediated genome editing and assessed Atoh1 expression in the e11 spinal cord and hindbrain (Supplementary Fig. 8b–d). Deletion of either E0 or E3 led to a significant decrease in Atoh1 expression in the spinal cord compared with their wild-type siblings (Fig. 6i, k). Notably, deletion of E0 resulted in almost complete depletion of Atoh1 expression in the hindbrain at e11, while deletion of either E2 or E3 did not (Fig. 6m–o). Moreover, we assessed the impact of deleting the E0 and E3 simultaneously by generating double knock-out mice (E0 + 3 DKO) thorough targeting E3 in E0 heterozygotes using CRISPR/Cas9 genome editing39 (Supplementary Fig. 8e). The E0 + 3 DKO mice exhibited a more pronounced reduction in Atoh1 mRNA levels compared to the deletion of E0 or E3 alone in the e11 spinal cord (Fig. 6i, k, l). In the e11 hindbrain, the Atoh1 mRNA levels in E0 + 3 DKO mice were decreased to a level resembling that in E0 KO mice (Fig. 6m, p). These findings indicate a collaborative role of E0 and E3 in regulating the Atoh1 expression in the spinal cord, while E0 alone regulates Atoh1 expression in the hindbrain at e11. In addition, minimal changes in Atoh1 expression were observed in mice with E2 deletion in both the spinal cord and the hindbrain at e11 (Fig. 6j, n). Collectively, our results provide compelling genetic evidence that the combinatorial use of divergent enhancer units within the Atoh1 enhancer cluster orchestrates Atoh1 expression, supporting the concept of a spatial enhancer code that spatially regulates gene expression.
Discussion
The regulation of gene expression by enhancer clusters is a widespread phenomenon. Models like shadow enhancers, super-enhancers5, stretch enhancers, enhancer clusters, and domains of regulatory chromatin (DORC)45 have been proposed to decipher the underlying cis-regulatory principles11. However, these models lack precise spatial information and the divergence between enhancers within a cluster to fully characterize spatiotemporal gene regulation. In this study, we developed eSpatial, a comprehensive framework for spatial enhancer-gene analysis that allows users to systematically decipher spatial enhancer-gene relationships in complex tissues at the resolution of individual enhancers. eSpatial encompasses seven key steps (Supplementary Fig. 1a): integration of spatial transcriptomics and epigenomics data (Steps 1–2), cell types and spatial domain detection (Steps 3–4), and decoding spatial patterns of enhancer-gene regulation (Steps 5–7). This capability cannot be achieved by existing spatial tools, which are typically designed for specific tasks. In addition, unlike existing single-cell methods23,24, eSpatial incorporates spatial information with multiomics features at multiple steps of the workflow, including spatial-aware cell clustering and peak-gene linkage in spatial context. This refinement is crucial for linking chromatin accessibility with gene expression in complex tissues. By incorporating these spatial-aware adjustments, eSpatial provides a deeper understanding of gene regulation in the context of tissue organization, offering insights that cannot be achieved by existing single-cell methods.
By leveraging spatial epigenomic data in both developmental and disease contexts, we revealed that the phenomenon of different combination of enhancers regulating the same gene not only exists across tissue/cell types, which has been previously reported7, but also within spatial domains of the same cell type. This later aspect, which has not been previously addressed in existing studies, led us to propose the concept of a “spatial enhancer code.” This phenomenon describes how divergent combinations of enhancers regulate the same gene in spatially segregated domains. For example, we demonstrated that Nrn1, expressed in different spatial domains of excitatory neurons, and DNMT3A, expressed in distinct compartments of T cells in melanoma, are controlled by distinct enhancer combinations. These findings provide a critical understanding of tissue microenvironments and tumor heterogeneity, highlighting the complexity of spatial regulatory dynamics.
To provide experimental validation of the spatial enhancer code governing the spatial expression of Atoh1, we conducted in vivo transgenic reporter assays and CRISPR/Cas9-mediated perturbation. We extended previous insights into the Atoh1 enhancer cluster, unveiling the spatial enhancer code model that explains the distinct regulation of the Atoh1 expression of neurons across different tissues. Specifically, our validations highlight that the functional role of E3 is in the e11 spinal cord but not in the hindbrain, contrasting the involvement of E0 in both the spinal cord and hindbrain. These experiments provide proof-of-concept, affirming the spatial enhancer code concept, in which divergent combinations of enhancers control spatial gene expression, as revealed by eSpatial analysis.
While this study has achieved promising insights into spatial gene regulation, it does encounter some limitations. A major constraint is its reliance on spatial epigenome profiling, which remains largely unavailable for various human and mouse tissues and low resolution and potential aggregation of multiple cell types into a single spatial spot. The potential of spatial information to resolve subtle regulatory differences within a single cell type, such as interneurons or astrocytes, has yet to be fully explored. Future studies focusing on these cell types, combined with advancements in spatial profiling technologies, will be crucial for achieving a comprehensive understanding of spatial gene expression dynamics. Last, beyond the Atoh1 locus, functional studies of more spatial enhancer codes are imperative to better understand spatial gene expression in the future.
Methods
Overview of eSpatial
eSpatial is a framework to decipher the spatial regulation of enhancer clusters controlling the same gene based on spatial chromatin accessibility profiling. Briefly, eSpatial analysis comprises the following seven key steps.
Step 1. Prepare input matrix (Input)
The input matrices in eSpatial analysis consist of a spatial chromatin accessibility matrix, a spatial gene expression matrix (optional), and a spot coordinate matrix. We downloaded spatial chromatin accessibility profiling data from public literature and generated distinct spatial matrices for each dataset independently.
For the spatial-ATAC-RNA-seq data of the P22 mouse brain (data from ref. 14) in Figs. 1 and 2, we obtained the preprocessed fragments files, a gene-by-spot matrix (with 22,914 genes in 9215 spots), and spot coordinate matrix from the Gene Expression Omnibus with accession number GSE205055. To generate the spatial peak-by-spot matrix of the P22 mouse brain, we retrieved the cis-regulatory elements from the single-cell ATAC-seq mouse brain atlas data22. Using the fragments files and cis-regulatory elements, we employed the function FeatureMatrix() of Signac v.1.11.024 to generate the peak-by-spot matrix, encompassing 260,032 peaks in 9215 spots.
For the spatial-ATAC-RNA-seq data relating to e13 mouse embryos (data from ref. 14) and spatial-ATAC data of e11 mouse embryos (data from ref. 12) in Figs. 4 and 5, we first downloaded the preprocessed fragments files, gene-by-spot matrix (with 20,900 genes in 2187 spots), and spots coordinates matrix from the Gene Expression Omnibus (GEO) with accession number GSE205055 and GSE171943. Then we downloaded the ATAC peaks of all mouse tissues at e11.5 and e13.5 from ENCODE2 and e13.5 epigenomic MERFISH loci from NCBI GEO data repository (GSE191069). Then we merged them using reduce() function in GenomicRanges package. Finally, we created a peak-by-spot matrix within a common set of 181,633 peaks.
For MISAR-seq data of the e13.5 mouse fetal brain (data from ref. 15) in Supplementary Fig. 7, we downloaded the preprocessed fragments files, gene-by-spot matrix (with 32,285 genes in 1777 spots) and spots coordinates matrix from the National Genomics Data Center with accession number OEP003285. Then we used the fragments files and the common peak set of e11.5 and e13.5 mouse embryos to generate the peak-by-spot matrix (with the function FeatureMatrix() of Signac v.1.11.024) with 181,633 peaks in 1777 spots.
The Slide-tags data of a human melanoma sample (data from ref. 16) in Fig. 3 was downloaded from the Broad Institute Single Cell Portal under the following accession numbers SCP2176. The dataset includes a peak-by-cell matrix (with 53,451 peaks in 2535 cells), gene-by-cell matrix (with 36,601 genes in 2535 cells), cell coordinate matrix, and cell type annotation information.
For the spatial-ATAC data of a human breast cancer sample (data from ref. 13) in Fig. 3, the fragments files, spots coordinate matrix, and regions annotation information were kindly shared by the authors of the original study. Then we processed the fragments file using the standard pipeline of ArchR v.1.0.223 to generate the peak-by-spot matrix with 51,571 peaks in 3538 spots.
Step 2. Reduce dimension and construct spatial neighbor network (Preprocessing)
eSpatial conducted dimension reduction for the gene-by-spot matrix and peak-by-spot matrix separately. For the gene-by-spot matrix, it employed principal component analysis (PCA) for dimensionality reduction and returned the top 20 principal components (PCs) matrix to generate a PC-by-spot matrix. For the peak-by-spot matrix, eSpatial adopted an iterative latent semantic indexing (LSI) approach to reduce dimensions and returned the top 20 LSI matrix to generate an LSI-by-spot matrix, excluding the foremost LSI corresponding to sequencing depth variation. To integrate the similarity among adjacent spots of a given spot, eSpatial converted the spatial coordinates into an undirected neighbor network based on Euclidean distance between two spots by using the function STAGATE.Cal_Spatial_Net() of STAGATE v.1.0.146. When the distance between two spots is below a predetermined radius, these spots are connected within the spatial neighbor network.
Step 3. Define cell types
Seurat v.4.4.047 was used for RNA data integration and major cell type identification. The “SCTransform()” function normalized both spatial-RNA and scRNA-seq data. The “SelectIntegrationFeatures()” function identified common features between the datasets, while “FindIntegrationAnchors()” and “IntegrateData()” were used to integrate the data. Clustering of the integrated dataset confirmed a strong match between the spatial and scRNA-seq data. The “FindTransferAnchors()” function identified transfer anchors, which were applied for label transfer using the “TransferData()” function.
Step 4. Detect spatial domains
To identify the spatial domains, eSpatial performed spatial clustering by STAGATE v.1.0.1, which was originally designed to identify spatial domains from spatially resolved transcriptomics data. STAGATE utilized a graph attention autoencoder to acquire low-dimensional latent embeddings with both spatial information and gene expressions. To adapt the application of STAGATE in spatial-ATAC-RNA co-profiling or spatial-ATAC profiling, eSpatial provided three options to identify spatial domains: (1) “spatial-RNA”: eSpatial followed the STAGATE pipeline with default parameters to detect spatial domain by only using PCA dimension matrix from spatial-RNA profiling and spatial neighbor network. (2) “spatial-ATAC”: eSpatial followed the STAGATE pipeline except replacing PCA dimension matrix with LSI dimension matrix which is from spatial-ATAC profiling, then combined spatial neighbor network. (3) “spatial-ATAC-RNA”: eSpatial first combined the two reduced dimension matrices (PCA dimension matrix from spatial-RNA profiling and LSI dimension matrix from spatial-ATAC profiling) and took the combined dimension matrix and spatial neighbor network as input of STAGATE. Finally, the identified spatial domains by STAGATE underwent a smoothing process based on the spatial neighbor network.
Step 5. Define enhancer clusters of genes
Regulatory elements with chromatin accessibility were defined as putative enhancers or simply enhancers. However, it is important to note that while these putative regulatory elements are referred to as enhancers here, further functional characterization might be required for a more detailed study of individual elements. Based on this, eSpatial identified the enhancer clusters of genes by adapting the method that we developed previously48 with some modifications. Briefly, eSpatial first aggregated the gene expression and chromatin accessibility profiles for cells in each spatial neighbor network. This aggregation was inspired by the pseudobulk approach that has been developed for single-cell RNA/ATAC data49, which produced counts that enable adjustments to mitigate the impact of sparse spatial data. Next, given a gene, eSpatial selected the CREs located within a 100 kb window, excluding those located within ±2 kb around each annotated transcriptional start site (TSS), as its candidate enhancers. For each gene-enhancer pair, eSpatial then calculated the Spearman correlation between enhancer chromatin accessibility and gene expression. Lastly, the enhancers exhibiting a significant correlation (p-value < 0.01, one-tailed Student’s t-test) were defined as a putative enhancer cluster regulating the specific target gene.
Step 6. Identify spatial enhancer units based on spatial patterns of enhancers
eSpatial identified spatial enhancer units by mapping enhancer activity patterns to gene expression modules across spatial domains. Initially, gene expression modules were defined using k-means clustering to group genes with similar spatial expression profiles. For each module, eSpatial mapped the regulatory enhancers based on their spatial specificity scores. These scores were binarized, with “1” indicating active spatial activity within a given domain. By clustering the binary enhancer activity profiles, eSpatial captured distinct spatial activity patterns of enhancers corresponding to each gene module. These distinct patterns of spatial enhancer activity across domains were defined as spatial enhancer units. To keep the robustness of these patterns, eSpatial filtered out enhancer units that involve fewer than 10 enhancers. Thus, eSpatial deciphered spatial patterns of their linked enhancers for genes expressed in similar spatial domains.
Step 7. Decode the combinations of divergent enhancer units
eSpatial decodes the combinations of divergent enhancer units by quantifying the diversity of spatial enhancer units within each enhancer cluster. It categorizes enhancer regulation into two distinct modes: divergent regulation and coherent regulation. Divergent regulation occurs when an enhancer cluster comprises multiple spatial enhancer units, indicating that the target genes are regulated by enhancers with differing spatial activities across tissue domains. In contrast, coherent regulation is characterized by a single spatial enhancer unit within an enhancer cluster, suggesting that the target genes are regulated by enhancers with similar spatial activity across spatial domains.
Identify spatial-specific genes and cis-regulatory elements (sGEs/sCREs)
To identify the spatial-specific cis-regulatory elements, eSpatial first quantified the spatial specificity of each gene and cis-regulatory element (each peak of peak-by-spot matrix) in each spatial domain. Briefly, for each gene/cis-regulatory element in a specific spatial domain, eSpatial calculated its fold change of the average log-normalized counts in a specific spatial domain versus the rest spatial domains. This computation generated a matrix indicating the spatial specificity of genes/CREs in each spatial domain. Next, eSpatial selected candidate sGEs/sCREs based on the maximum spatial specificity of the CREs. Any gene or CREs surpassing a threshold (0.25 for genes, 0.1 for CREs) and exhibiting a significantly higher spatial specificity compared to others (one-tailed Student’s t-test, p-value < 0.01) were considered as candidate spatial-specific genes (sGEs)/cis-regulatory elements (sCREs). Finally, eSpatial mapped these candidate sGEs/sCREs to the corresponding spatial domains with the maximum spatial specificity. Thus, eSpatial identified candidate spatial-specific genes/cis-regulatory elements for each spatial domain.
Benchmark of peak-to-gene links from spatially resolved eSpatial and conventional single-cell multiomics approaches
To evaluate eSpatial’s performance, we compared its identified peak-to-gene links with those derived from conventional single-cell multiomics approaches (ArchR v.1.0.223 and Signac v.1.11.024) on the same spatial epigenomic data. The comparison was based on the gene-enhancer links from the mouse cortex (same tissue)22 as the reference. For each approach, the peak-to-gene links were converted to BED format, and overlaps with the reference were determined using bedtools v.2.31.1 intersect50. Enrichment of overlaps was then calculated by constructing contingency tables and applying Fisher’s exact test. Venn diagrams were used to visualize the overlap between approaches, and a bar plot of normalized odds ratios was generated to compare the relative enrichment of peak-to-gene links across datasets.
Visualize the spatial pattern of enhancers within enhancer clusters
eSpatial first visualized gene expression and chromatin accessibility in each spot by applying a denoising deep count autoencoder (DCA v.0.3.1) to unsupervised denoise spatial chromatin accessibility and transcriptome51. It separated the peak-by-spot matrix and gene-by-spot based on the spatial domains and denoised the separated matrix independently using DCA. For the separated peak-by-spot matrix, eSpatial specified the following parameters: –nosizefactors –nonorminput –nologinput, whereas DCA was run with default settings on the separated gene-by-spot matrix. Then eSpatial prepared the genome tracks for each spatial domain to visualize the chromatin landscape by applying the function plotBrowserTrack() of ArchR v.1.0.2 package23.
Characterization of sCREs with GREAT and motif analysis
We used the R implementation of GREAT v.4.0.421 to identify enriched gene ontologies to associate sCREs with the biological processes (BP) of their putative target genes. Then we performed a hypergeometric test to determine the probability of observing the motif at the given frequency by chance using Signac v.1.11.024,52, comparing it with a background peak set matched for GC content or the rest of the genome.
Motif enrichment analysis
We performed motif enrichment analysis using Homer v.5.153 with the mm10 reference genome. Known motifs were identified using the findMotifsGenome.pl command, with the option -size given to scan the entire peak region. Randomly selected background regions were used for motif discovery.
Super-enhancers enrichment analysis
Super-enhancers (SEs) were identified from publicly available databases27 (dbSUPER, https://asntech.org/dbsuper/) and integrated with spatial link data to map enhancer-gene pairs associated with SEs. SE regions were intersected with enhancer clusters using the bedtools v.2.31.150 to identify overlapping enhancer clusters. The proportion of SEs relative to total enhancer clusters was calculated for each tissue and normalized against a shuffled background to generate enrichment scores. Statistical significance was assessed using the binomial test.
Transcription factor similarity analysis
We analyzed transcription factor (TF) similarity between enhancer pairs associated with the same gene. For each gene, enhancer pairs were categorized as either belonging to the same module type (referred to as “same spatial pattern”) or different module types (“different spatial pattern”). TF binding profiles were retrieved from CistromeDB54, and overlaps between TF binding regions and enhancers were identified. TF similarity between enhancer pairs was quantified by counting the number of shared TFs bound to both enhancers.
Directionality analysis of chromatin accessibility
We performed a directionality analysis to identify putative enhancers exhibiting spatial biases in chromatin accessibility along the Dorsal-Ventral (D-V) or Deep-Superficial (D-S) axes within interneurons, following the approach in a recent study28. For each cell type, we selected the top 1500 cell-type-specific accessible peaks ranked by log2[fold change] compared to other cell types. The spatial bias of each enhancer was assessed by calculating the Spearman correlation coefficient (R) between denoised chromatin accessibility and spatial coordinates (normalized to 0–1 range), where positive R values indicate ventral/superficial preference and negative values indicate dorsal/deep preference. The statistical significance of each correlation was determined by its associated p-value, with the absolute magnitude of R reflecting the strength of spatial preference.
Functional validations of spatial enhancers using public data
We performed functional validations of spatial enhancers using publicly available datasets, including VISTA, MERFISH, and Allen RNA ISH data. We downloaded the VISTA catalog from the VISTA database (https://enhancer.lbl.gov/) and selected the elements activated in mice. In addition, we further distinguished the reported enhancer activity in different tissues. The loci of VISTA elements were transferred from the mm9 genome to the mm10 genome using rtracklayer. In total, we collected 697 elements activated in at least one mouse tissue. Among these, 370 VISTA elements exhibited enhancer activity in brain regions (including forebrain, midbrain, and hindbrain), 214 were from neural tube regions, and 206 were from heart regions in mouse embryos. Then, we detected the overlapping elements between the VISTA elements and sCREs identified in this study by using the rtracklayer package v.1.58.055. MERFISH images37 were downloaded from Zenodo (https://doi.org/10.5281/zenodo.7075964). ISH data were downloaded from the Allen database (http://developingmouse.brain-map.org/static/atlas).
Depict spatial patterns of Atoh1 enhancers
To quantitatively assess the spatial patterns of Atoh1 enhancers (E0, E1, E2, and E3), we applied eSpatial to evaluate the spatial specificity degree of these enhancers with some optimization. Briefly, enhancers showing high spatial specificity (spatial specificity > cutoff, where the cutoff equals the average spatial specificity score plus one standard deviation of all enhancers in e11 mouse embryos) were categorized as “highly accessible enhancers.” Enhancers showing positive spatial specificity (spatial specificity >0) were categorized as “accessible enhancers.” Enhancers showing a negative spatial specificity score (spatial specificity ≤0) were categorized as “closed enhancers.”
Experimental model and subject details
The care and use of mice were followed by the United States National Institutes of Health Guidelines and the Chinese Ministry of Public Health Guidelines. All the mouse experiments were performed according to the ethical guidelines established by the Animal Care and Use Committee of the Institute of Genetics and Development Biology, Chinese Academy of Sciences. Mice were housed in a standard ventilated cage with a 12 h light/12 h dark cycle. 3–4 mice were housed in a cage. The ambient temperature is 25 °C with relative humidity of 50%. All mice have ad libitum access to water and chow. E0-KO, E2-KO, E3-KO, and E0 + 3-DKO mice are from our lab, which have been reported39.
LacZ transgenic mice generation
The strains of E0-lacZ, E2-lacZ, and E3-lacZ transgenic mice are from our lab, which have been reported39. Briefly, E0/E2/E3 fragments were PCR amplified and cloned into the Hsp68-LacZ vector to create the pHsp68-E0/E2/E3-lacZ plasmid. The plasmid and PiggyBac vector were co-injected into CD1 one-cell–stage zygotes. PCR was performed to screen for founder mice 0 (F0) harboring an insertion of E0/E2/E3-lacZ in their genomic DNA. Plasmids, primers, and transgenic mouse strains are listed in Supplementary Table 2.
X-Gal staining
Embryos were dissected at e11. Removed a small piece of tissue from each embryo for genotyping by PCR examination of the lacZ gene. The embryos were fixed by cold 0.125% glutaraldehyde (e11 for 30 min). Then, we washed it three times in PBS for 5 min. Embryos were transferred to a freshly made X-Gal staining solution containing 100 mM phosphate buffer (pH 7.2), 2 mM MgCl2, 5 mM EGTA, 0.02% NP40, 0.01% sodium-deoxycholate, 50 mM K3Fe(CN)6, 50 mM K4Fe(CN)6, and 1 mg/ml X-Gal. Embryos were incubated in X-Gal staining solution at 37 °C. Incubation time varied from several hours to overnight, depending on the strength of β-Gal expression. After staining, embryos were washed three times with PBS and stored in freshly made 4% formaldehyde. The embryos were again washed with PBS three times before photographing. Primers and transgenic mouse strains are listed in the key resources table (Supplementary Table 2).
RNAscope and preparation of spinal cord sections
Fresh mouse embryos at e11 were embedded in an Optimal Cutting Temperature (O.C.T) compound. The sagittal frozen sections were taken at 10-μm thickness and then stored at −80 °C. RNAscope was performed as instructed by the manufacturer (ACD, CA, USA). Images were acquired on a Leica SP8 microscope. Primers and mouse strains are listed in the key resources table (Supplementary Table 2).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The spatial-ATAC-RNA-seq data of the P22 mouse brain, e13 mouse embryo, and e11 mouse embryo used in this study are available in the Gene Expression Omnibus (GEO) under accession codes GSE205055 and GSE171943. The MISAR-seq data of the e13.5 mouse fetal brain used in this study are available in the National Genomics Data Center under accession number OEP003285. The H3K27ac MERFISH data of the e13.5 mouse fetal brain used in this study are available in Zenodo under the https://doi.org/10.5281/zenodo.7075964. The Slide-tags snRNA+snATAC data of human melanoma used in this study are available in the Broad Institute Single Cell Portal under the accession number SCP2176. The spatial-ATAC data of human breast cancer used in this study were kindly shared by the authors of the original study. The experimentally validated VISTA enhancers in human and mouse noncoding fragments with gene-enhancer activity assessed in transgenic mice are accessible through the VISTA Enhancer Browser at https://enhancer.lbl.gov/. The Allen Brain Reference Atlases ISH data used in this study are accessible through the Allen Brain Atlas at https://atlas.brain-map.org/. All datasets’ details were published previously (Supplementary Table 1). Source data are provided with this paper.
Code availability
The source code for eSpatial is available on GitHub (https://github.com/xmuhuanglab/eSpatial), Zenodo (https://doi.org/10.5281/zenodo.15383127)56 and CodeOcean (https://codeocean.com/capsule/2493559/tree/v1).
References
Allis, C. D. & Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17, 487–500 (2016).
Gorkin, D. U. et al. An atlas of dynamic chromatin landscapes in mouse fetal development. Nature 583, 744–751 (2020).
Gray, L. T. et al. Layer-specific chromatin accessibility landscapes reveal regulatory networks in adult mouse visual cortex. eLife 6, e21883 (2017).
Visel, A. et al. A high-resolution enhancer atlas of the developing telencephalon. Cell 152, 895–908 (2013).
Hnisz, D. et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013).
Blobel, G. A., Higgs, D. R., Mitchell, J. A., Notani, D. & Young, R. A. Testing the super-enhancer concept. Nat. Rev. Genet. 22, 749–755 (2021).
Bahr, C. et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 553, 515–520 (2018).
Hong, J. W., Hendrix, D. A. & Levine, M. S. Shadow enhancers as a source of evolutionary novelty. Science 321, 1314 (2008).
Osterwalder, M. et al. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 554, 239–243 (2018).
Smith, G. D., Ching, W. H., Cornejo-Paramo, P. & Wong, E. S. Decoding enhancer complexity with machine learning and high-throughput discovery. Genome Biol. 24, 116 (2023).
Kim, S. & Wysocka, J. Deciphering the multi-scale, quantitative cis-regulatory code. Mol. Cell 83, 373–392 (2023).
Deng, Y. et al. Spatial profiling of chromatin accessibility in mouse and human tissues. Nature 609, 375–383 (2022).
Llorens-Bobadilla, E. et al. Solid-phase capture and profiling of open chromatin by spatial ATAC. Nat. Biotechnol. 41, 1085–1088 (2023).
Zhang, D. et al. Spatial epigenome-transcriptome co-profiling of mammalian tissues. Nature 616, 113–122 (2023).
Jiang, F. et al. Simultaneous profiling of spatial gene expression and chromatin accessibility during mouse brain development. Nat. Methods 20, 1048–1057 (2023).
Russell, A. J. C. et al. Slide-tags enables single-nucleus barcoding for multimodal spatial genomics. Nature 625, 101–109 (2024).
Qin, T. et al. Comprehensive enhancer-target gene assignments improve gene set level interpretation of genome-wide regulatory data. Genome Biol. 23, 105 (2022).
Bravo Gonzalez-Blas, C. et al. Identification of genomic enhancers through spatial integration of single-cell transcriptomics and epigenomics. Mol. Syst. Biol. 16, e9438 (2020).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Zeisel, A. et al. Molecular architecture of the mouse nervous system. Cell 174, 999–1014.e22 (2018).
McLean, C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010).
Li, Y. E. et al. An atlas of gene regulatory elements in adult mouse cerebrum. Nature 598, 129–136 (2021).
Granja, J. M. et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411 (2021).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019).
Sock, E. & Wegner, M. Using the lineage determinants Olig2 and Sox10 to explore transcriptional regulation of oligodendrocyte development. Dev. Neurobiol. 81, 892–901 (2021).
Utsunomiya, S. et al. Ezh1 regulates expression of Cpg15/Neuritin in mouse cortical neurons. Drug Discov. Ther. 15, 55–65 (2021).
Khan, A. & Zhang, X. dbSUPER: a database of super-enhancers in mouse and human genome. Nucleic Acids Res. 44, D164–D171 (2016).
Song, L., Chen, W., Hou, J., Guo, M. & Yang J. Spatially resolved mapping of cells associated with human complex traits. Nature 641, 932–941 (2025).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Pedri, D., Karras, P., Landeloos, E., Marine, J. C. & Rambow, F. Epithelial-to-mesenchymal-like transition events in melanoma. FEBS J. 289, 1352–1368 (2022).
Gomez-Lopez, S. et al. Sox2 and Pax6 maintain the proliferative and developmental potential of gliogenic neural stem cells In vitro. Glia 59, 1588–1599 (2011).
Tani, H. et al. Direct reprogramming improves cardiac function and reverses fibrosis in chronic myocardial infarction. Circulation 147, 223–238 (2023).
Visel, A., Minovitsky, S., Dubchak, I. & Pennacchio, L. A. VISTA Enhancer Browser—a database of tissue-specific human enhancers. Nucleic Acids Res. 35, D88–D92 (2007).
Huang, D. F. et al. Neuronal splicing regulator RBFOX3 mediates seizures via regulating Vamp1 expression preferentially in NPY-expressing GABAergic neurons. Proc. Natl. Acad. Sci. USA 119, e2203632119 (2022).
Diacou, R., Zhao, Y., Zheng, D., Cvekl, A. & Liu, W. Six3 and Six6 are jointly required for the maintenance of multipotent retinal progenitors through both positive and negative regulation. Cell Rep. 25, 2510–2523.e4 (2018).
Rada-Iglesias, A. et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283 (2011).
Lu, T., Ang, C. E. & Zhuang, X. Spatially resolved epigenomic profiling of single cells in complex tissues. Cell 185, 4448–4464.e4417 (2022).
Mulvaney, J. & Dabdoub, A. Atoh1, an essential transcription factor in neurogenesis and intestinal and inner ear development: function, regulation, and context dependency. J. Assoc. Res. Otolaryngol. 13, 281–293 (2012).
Shu, M. et al. Single-cell chromatin accessibility identifies enhancer networks driving gene expression during spinal cord development in mouse. Dev. Cell 57, 2761–2775.e6 (2022).
Luo, Z. et al. Three distinct Atoh1 enhancers cooperate for sound receptor hair cell development. Proc. Natl. Acad. Sci. USA 119, e2119850119 (2022).
Ben-Arie, N. et al. Math1 is essential for genesis of cerebellar granule neurons. Nature 390, 169–172 (1997).
Rossant, J., Zirngibl, R., Cado, D., Shago, M. & Giguere, V. Expression of a retinoic acid response element-hsplacZ transgene defines specific domains of transcriptional activity during mouse embryogenesis. Genes Dev. 5, 1333–1344 (1991).
Kothary, R. et al. Inducible expression of an hsp68-lacZ hybrid gene in transgenic mice. Development 105, 707–714 (1989).
Pennacchio, L. A. et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature 444, 499–502 (2006).
Ma, S. et al. Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell 183, 1103–1116.e20 (2020).
Dong, K. & Zhang, S. Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder. Nat. Commun. 13, 1739 (2022).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Hong, D. et al. Complexity of enhancer networks predicts cell identity and disease genes revealed by single-cell multi-omics analysis. Brief. Bioinform. 24, bbac508 (2023).
Granja, J. M. et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat. Biotechnol. 37, 1458–1465 (2019).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Eraslan, G., Simon, L. M., Mircea, M., Mueller, N. S. & Theis, F. J. Single-cell RNA-seq denoising using a deep count autoencoder. Nat. Commun. 10, 390 (2019).
Hao, Y. et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 42, 293–304 (2024).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Taing, L. et al. Cistrome Data Browser: integrated search, analysis and visualization of chromatin data. Nucleic Acids Res. 52, D61–D66 (2024).
Lawrence, M., Gentleman, R. & Carey, V. rtracklayer: an R package for interfacing with genome browsers. Bioinformatics 25, 1841–1842 (2009).
Hong, D. et al. Divergent combinations of enhancers encode spatial gene expression. Zenodo (2025).
Acknowledgements
We thank Dr Guo-Cheng Yuan for critical reading of the manuscript. This work was supported by the National Natural Science Foundation of China (92474104, 32370586 to J.H., 32330059 to J.D., 32300695 to M.S.), the National Key Research and Development Program of China (2023YFC2508100 to J.H., 2020YFA0804000 to F.L., 2023YFC2412501 to J.D.), the Fundamental Research Funds for the Central Universities (20720230068 to J.H.), and the CAS Project for Young Scientists in Basic Research (YSBR-012 to F.L., YSBR-073 to Y.Z.). We also thank the support from the Youth Innovation Promotion Association CAS (Grant No. Y202031 to Y.Z.), CAMS Innovation Fund for Medical Sciences (2022-I2M-3-002 to J.D., 2024-I2M-3-008 to M.S.) and the Wang Deyao Outstanding Graduate Scholarship Program of Xiamen University.
Author information
Authors and Affiliations
Contributions
J.H., F.L., J.D., D. Hong, and M.S. conceived and designed the experiments. D. Hong, J.L., L.L., and M.Z. performed the computational analysis. M.S., H.C., Y.D., D. Hu, and Z.L. generated transgenic and knock-out mice. M.S., H.C., Y.Z., and B.X. performed staining experiments. D. Hong, M.S., J.L., J.H., and F.L. wrote the manuscript with the input from the other authors. J.H., F.L., and J.D. supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hong, D., Shu, M., Liu, J. et al. Divergent combinations of enhancers encode spatial gene expression. Nat Commun 16, 5091 (2025). https://doi.org/10.1038/s41467-025-60482-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60482-1
