
Abstract
The Jahn-Teller and cooperative Jahn-Teller effects are phenomena that induce asymmetry in individual ions and solid-state lattices and are commonly observed in structures containing specific transition metals, such as copper and manganese. Although the Jahn-Teller effect causes lattice distortions that stress electrode materials in rechargeable batteries, strategically utilising the strain generated by cooperative Jahn-Teller distortions can enhance structural stability. Here we introduce the cooperative Jahn-Teller effect on MnO2 by constructing a two-dimensional superlattice structure with graphene crated in the bulk MnO2/graphene composite material. The strong interaction between MnO2 and graphene increases the concentration of high-spin Mn3+ ions, creating orderly long-range biaxial strains that are compressive in the out-of-plane direction and tensile in the in-plane direction. These strains mitigate Zn2+ intercalation stress and proton corrosion, enabling over 5000 cycles with 165 mAh g−1 capacity retention at 5 C (1 C = 308 mA g−1) in aqueous zinc-ion batteries. Our approach offers an effective strategy to significantly enhance the lifetime of rechargeable batteries by introducing the cooperative Jahn-Teller effect that overcomes the stress of ion insertion in electrode materials.
Similar content being viewed by others

Introduction
The structural integrity of electrode materials plays a critical role in determining the cycle life and overall performance of rechargeable batteries, such as lithium-ion batteries, sodium-ion batteries, and aqueous zinc-ion batteries1,2,3,4. A significant challenge lies in the mechanical degradation of electrode materials caused by volumetric changes and stress resulting from the insertion and extraction of ions during the charge/discharge cycling process5,6,7. Strain engineering has been employed to solve this conundrum8: this includes inducing lattice mismatch using heterostructures9,10,11, introducing low-strain/zero-strain pillar structures12, or applying specific substrates with different properties, notably thermal expansion coefficients13,14. While these methods focus on surface or interface area, there is an opportunity to improve effectiveness by mitigating diffusion-induced lattice stress within localised regions. Introducing long-range strain offers a more effective approach to stabilize crystal structures by uniformly distributing strain to prevent the concentration of stress that typically leads to cracking and deformation. Current strategies for achieving long-range strain engineering primarily focus on mechanical transfer techniques or epitaxial growth utilizing substrates8. While these methods have shown promise in enhancing the performance of various functional materials, such as superconductivity, electrical, magnetic, and optical properties15, engineering of the long-range strain within the bulk materials, that are critical for electrode materials, remains a significant challenge.
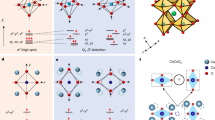
The cooperative Jahn-Teller effect (CJTE), commonly observed in Mn-based oxides like typical perovskite-type LaMnO3, could be utilized to generate a long-range strain field within crystals16,17,18,19. The Birnessite δ-MnO2 phase (illustrated in Fig. 1a) is considered a promising cathode material for aqueous zinc-ion batteries but suffers from structural degradation during cycling20. The ideal crystal structure of layered MnO2 comprises edge-sharing [MnO6] octahedra with exclusively MnIV centres, which should theoretically prevent the Jahn-Teller effect (JTE) and maintain strain-free flat MnO2 planes. However, the intercalation of ions, such as Li+, Na+, or Zn2+ can result in an increase in high-spin d4(t2g3eg1) Mn3+ ions, as observed in a typical Na-exchanged Birnessite with the formula Na0.31(MnIV0.69MnIII0.31)O2·0.4H2O21. The high-spin Mn3+ ions prompt the JTE due to the occupation of one electron in the doubly degenerated eg orbital, resulting in the elongation of two longitudinal Mn−O bonds in the [MnIIIO6] octahedra (as depicted in Fig. 1b)22,23. The occurrence of CJTE not only distorts individual [MnIIIO6] octahedra but also affects the orbital ordering of the neighboring [MnIVO6] octahedra through the shared edges, inducing strain in the surrounding matrix and lowering the overall energy of the system19, thus CJTE has been applied to overcome the charge disproportionation of MnO2 for aqueous zinc-ion batteries24. However, in partially CJTE, the distribution of JTE-active centres is localized, limiting its ability to effectively propagate long-range structural coherence. If the MnIII:MnIV ratio reaches 1:1, the MnO2 lattice could adopt an alternating arrangement of [MnIIIO6] (JTE-active) and [MnIVO6] (CJTE-affected) octahedra, defined as fully CJTE (f-CJTE). This structural alignment ensures a more continuous and extended interaction between JTE-active and non-active centres, reinforcing long-range orbital and lattice distortions across the MnO2 plane, as illustrated in Fig. 1a. The introduction of f-CJTE is crucial for maximizing the effectiveness of CJTE, as it eliminates isolated, short-range distortions and instead creates a globally interconnected strain network. This long-range ordered distortion acts as a framework to link separate local strain areas, culminating in a continuous biaxial strain field.

a Schematic illustration depicting the distinct structural scenarios in layered MnO2, including no Jahn-Teller effect (JTE), partially cooperative Jahn-Teller effect (CJTE), and fully CJTE. The corresponding strain distributions are also highlighted: an unstressed lattice for MnO2 without JTE, localized strains regions for partially CJTE, and a continuous long-range biaxial strain field for fully CJTE. No JTE-affected MnIV atoms, JTE centre MnIII atoms, CJTE-affected MnIV atoms, and O atoms are marked in purple, gold, green, and red, respectively. b Schematic of formed JTE in [MnO6] octahedra (where Mn and O atoms are marked in purple and red, respectively), where a, b and c are crystal axes and the distortion vector ωr (r represents x, y, or z axis in [MnO6] octahedra) can be projected to the c axis and ab plane as ωrc and ωrab. c Statistics of log(|ωrc|/|ωrab|) values for each Mn sites in simulated bilayer MnO2 and MnO2/graphene superlattice.
Herein, we successfully introduce long-range biaxial strain via f-CJTE by constructing two-dimensional MnO2/graphene superlattices using monolayer-stacked δ-MnO2 and graphene nanosheets. Both theoretical calculations and experimental investigations confirm the existence of f-CJTE of MnO2 in the superlattices. The derived long-range biaxial strains, consisting of compressive out-of-plane and tensile in-plane strains on the MnO2 layers, were further studied by transmission electron microscopy (TEM) and geometric phase analysis (GPA) technique in nanoscale and Raman spectroscopy in macroscale. This superlattice structure exhibits good cycling stability over 5000 cycles (lasting more than 1200 h) in aqueous zinc-ion batteries. This outstanding performance is attributed to the biaxial strains counteracting the stresses induced by ion intercalation, as evidenced by in situ static ultraviolet-visible (UV-vis) spectroscopy and in situ synchrotron radiation X-ray powder diffraction (SR-XRPD) characterizations.
Results
Existence of CJTE in MnO2/graphene superlattice
Density functional theory (DFT) calculations were first employed to predict the existence of the CTJE in the MnO2/graphene superlattice. The bilayer MnO2 derived from Na-exchanged Birnessite, and a MnO2/graphene superlattice unit, consisting of MnO2 and graphene monolayers, were adopted for the calculations (Supplementary Fig. 1 and Supplementary Data 1). Compared with bare MnO2, the Mn 3d states in the MnO2/graphene superlattice exhibit a significant downward shift in energy, as shown in the projected density of states (PDOS) (Supplementary Fig. 2). This shift could be caused by charge redistribution and electronic hybridization between MnO2 and graphene, leading to a partial reduction of Mn species. The introduction of Mn3+ induces additional electronic states near the conduction band, leading to a reduced band gap in the MnO2/graphene superlattice. This suggests that introducing graphene causes a stronger JTE due to the lower energy from completely reduced degeneracy25. This JTE distortion is further verified by the calculated shape variations within [MnO6] octahedra (Supplementary Fig. 3). As shown in Fig. 1b, [MnO6] octahedra in Birnessite δ-MnO2 normally experiences Q3 Jahn-Teller distortion with a predominant elongation of dz2-orbital ordering17. Every orbital axis (aligned with Mn−O bonds) has the chance for stretching, accompanied by the shrinkage of a corresponding perpendicular plane, where the increment is indicated as the distortion vector ωr. To elucidate the relationship with strain within the crystalline configuration, the distortion vector should be projected on the crystal axis as ωcr and ωabr, considering the 2D structure of δ-MnO2, as shown in an example of [Mn(34)O6] octahedra (Supplementary Fig. 3). The type of strain can be identified from the comparison between magnitudes |ωcr| and |ωabr|. When |ωcr|>|ωabr|, it suggests a local tensile strain on [MnO6] octahedra along the c axis, while conversely, the ab plane experiences tensile strain when |ωcr|<|ωabr|. The calculation results are shown in Fig. 1c and Supplementary Tables 1 & 2. A long-range biaxial strain may exist in MnO2/graphene superlattice for the consistency of [MnO6] orbital ordering, while only localized strains occur in bilayer MnO2 since nearly one of third [MnO6] octahedra possess strains with the opposite direction.
The MnO2/graphene superlattice materials were prepared by a flocculation approach via a typical electrostatic self-assembly26,27,28 between negative-charged monolayer-stacked δ-MnO2 (Supplementary Fig. 4 and Supplementary Note 1) and positive-charged graphene (p-Gr) nanosheets, obtained by modifying reduced graphene oxide with cationic polymer poly(diallyldimethylammonium chloride (PDDA) (Supplementary Fig. 5 and Supplementary Note 2). The thicknesses of monolayer-stacked δ-MnO2 and p-Gr nanosheets were determined to be 0.95 nm and 1.61 nm, respectively, by atomic force microscopy (AFM in Supplementary Fig. 4 & 5). The colloidal solutions of these two species were mixed with a mass ratio of 2.8:1, determined by the theoretical crystal lattice matching26. The actual weight percentage of MnO2 was calculated to be 74.7% from thermogravimetric analysis (TGA) results (Supplementary Fig. 6), close to the theoretical value of 73.7%. The flocculation-like sediment (Supplementary Fig. 7 and Supplementary Note 3) was separated from the clear supernatant via centrifugation. X-ray diffraction (XRD) and TEM characterizations indicated the successful synthesis of MnO2/graphene superlattices. The powder XRD in Supplementary Fig. 8 shows that the obtained MnO2/graphene superlattice has a (002) peak located at 6.88° with a d spacing of 1.28 nm, corresponding to a secondary order of reflection for infinite (00 l) planes. Accordingly, the thickness for one repeat unit in the MnO2/graphene superlattice structure, for example the d spacing for (001) facet, is estimated to be 2.57 nm, which is consistent with the combined thicknesses of monolayer-stacked δ-MnO2 and p-Gr nanosheets. The cross-section TEM images and the associated line profiles of bare MnO2 nanosheets (Fig. 2a) and MnO2/graphene superlattice (Fig. 2b) are consistent with the XRD result. The strong and weak peaks in the intensity profile of MnO2/graphene superlattice correspond to δ-MnO2 and p-Gr nanosheets, respectively.

Cross-section TEM images (scale bar 5 nm) and line profiles (scanning at the arrow areas) with a corresponding schematic illustration of a restacked MnO2 nanosheets and b MnO2/graphene superlattice (where p-Gr is positively charged graphene). The purple, red, and brown spheres represent Mn, O, and C atoms, respectively. c Magnetization (M) versus temperature (T) curves with dM/dT versus T curves (inset) and d 1/χ (where χ is susceptibility) versus T curves with Curie-Weiss fitting of bare MnO2 and MnO2/graphene superlattice in an applied magnetic field of 10 Oe.
X-ray photoelectron spectroscopy (XPS) characterizations (Supplementary Fig. 9) were conducted to quantify the valence states of Mn in bare MnO2 and MnO2/graphene superlattice. In the high-resolution Mn 2p spectra, the Mn 2p1/2 and 2p3/2 peaks of bare MnO2 are located at 654.1 and 642.4 eV, respectively. The corresponding peaks in the MnO2/graphene superlattice are blue-shifted by 0.2 eV, suggesting a slight decrease in overall Mn valence. This phenomenon is also found in the Mn 3s spectra, with a higher magnitude blue-shift of 0.4 eV. Analysis of simulated peaks shows an increased content of MnIII species in MnO2/graphene superlattice, resulting in a lower Mn valence state of +3.52 compared to +3.84 for bare MnO2.
Zero field cooling (ZFC) and field cooling (FC) magnetization curves in Fig. 2c were employed to measure the magnetic properties of MnO2/graphene superlattice and bare MnO2. There is an anomaly broad peak at 33.4 K before TN temperature (the transition temperature of “spin glass status” to antiferromagnetic/ferromagnetic interaction) in the ZFC/FC curve, which is more apparent in dM/dT curves. According to previous reports, a specific spin ordering in superlattice structure may exist, related to the magnetoelastic coupling29,30,31. The magnetoelastic effect, namely the inverse magnetostrictive effect, is the change in the magnetic susceptibility of a material when subjected to mechanical stress. The anomaly in ZFC and FC magnetization curves confirms the existence of CJTE in the MnO2/graphene superlattice30.
The Curie-Weiss fitting in Fig. 2d was applied to measure the average oxidation state of mixed-valent Mn in both samples. The 1/χ versus T curve was fitted via Curie-Weiss law as shown in the following Eq. 1:
where θ is the Curie-Weiss temperature, χ0 corresponds to the temperature-independent factors to the susceptibility, and C is the Curie-Weiss constant that is associated with the effective magnetic moment in Eq. 2:
where n is the number of magnetic atoms per cell unit, kB is the Boltzmann constant, and μeff and μB are the effective magnetic moment and Bohr magneton, respectively. The Curie constant C can be calculated from fitted slope 1/C in Fig. 2d with values of 1.765 and 1.400 emu·K·Oe−1·mol−1 for bare MnO2 and MnO2/graphene, respectively. Thus, the effective magnetic moments μeff of bare MnO2 and MnO2/graphene are determined as 3.76μB and 3.35μB, respectively. Both samples have mixed-valent Mn, including Mn4+ (3.87μB) and low-spin Mn3+ (2.83μB), compared with standard effective moments. The presence of low-spin Mn3+ rather than high-spin Mn3+ (4.90μB) suggests the existence of JTE under octahedral fields32,33,34. The proportions of Mn4+ and Mn3+ in two samples can be estimated based on their contributions. The ratio of Mn3+/Mn4+ in MnO2/graphene is calculated to be 1:1, which matches the theoretical threshold of f-CJTE.
DFT-based charge density difference analysis (Supplementary Fig. 10) was conducted to investigate the electronic interaction between MnO2 and graphene monolayers. In the MnO2/graphene superlattice, yellow regions represent areas of electron accumulation, whereas blue regions denote electron depletion. Notably, pronounced electron accumulation appears near the MnO2 surface at the interface, while the graphene surface exhibits a depletion zone, indicative of significant hole localization. In comparison, no significant charge redistribution is observed in a typical MnO2 bilayer structure. This distinct interfacial behaviour is attributed to substantial charge transfer via a Faradaic process (Supplementary Fig. 11 and Supplementary Note 4), which alters the occupancy of Mn 3d orbitals.
CJTE-induced long-range biaxial strains in MnO2/graphene superlattice
The CJTE-induced lattice distortion of MnO2 in the superlattice structure has been directly observed from the aberration-corrected high-angle annular dark field scanning TEM (HAADF-STEM) and inverse fast Fourier transformed (iFFT) image (marked in yellow square) in Fig. 3a, where the bright spots indicate the positions of Mn atoms in the lattices. The arrangement of Mn atoms exhibits deviation with periodic lattice twisting, which is indicative of a standard CJTE18. Figure 3b shows the corresponding FFT pattern, revealing asymmetric hexagonal diffraction spots that match a single crystal MnO2. The angle between (100) and (−110) facets is measured to be 113°, which is much lower than 120° in a typical hexagonal Birnessite phase35. The deformations of Mn-centred [MnO6] octahedra confirm the existence of severe lattice distortion for MnO2 in the superlattice structure.

a HAADF-STEM image (scale bar, 1 nm) recorded along the [002] direction with iFFT image (in the yellow square) showing the existence of CJTE in MnO2/graphene superlattice along with b its corresponding FFT pattern (scale bar, 5 nm−1). The calculated c εxx, d εyy, and e εxy strain components via GPA. f Distribution and g statistics analysis of ratios of orthometric strain εc versus εab.
The local strain field relaxation distribution in the MnO2/graphene superlattice was characterized by the GPA technique36 using the HAADF-STEM image, with the x- and y-axes aligned parallel to the [100] and [−110] zone axes, respectively. The simulated strain field maps, presented in Fig. 3c-e, illustrate the εxx, εyy, and εxy strain components, where εxy reflects shear strain within the xy-plane. The overall strain values range from −1.5 to 1.5%, with negative values indicating compressive strains and positive values indicating tensile strains. The strain distribution appears relatively uniform, with dominant tensile strain along the [−110] directions, while both compressive and tensile strains coexist along the [100] direction. The combination of εxx and εyy represents in-plane strain distribution within the hexagonal lattice. According to Poisson’s ratio (υ) for layered structures37, defined as:
where εin-plane and εout-of-plane represent strains within the layered [MnO6] plane and perpendicular to the plane, which can be defined in terms of crystallographic axes as εab and εc, respectively. The εab component is given by the sum of εxx and εyy, while εc corresponds to εzz. Therefore, εzz was calculated based on a Poisson’s ratio of 0.14, as reported for δ-MnO238. The mapping results of εzz, shown as a contour plot in Supplementary Fig. 12, indicate a strain field significantly dominated by compressive strains.
Similar to the orthometric vector components of ωrab and ωrc within [MnO6] octahedra in the aforementioned theoretical calculations, the comparison of |εc| and |εab| values was conducted and presented as mapping results in Fig. 3f and statistics distribution in Fig. 3g. In the bar chart, red and blue indicate locations where |εc|>|εab| and |εc|<|εab|, respectively. The statistical data demonstrate a Gaussian distribution of |εc|/|εab| ratios, suggesting a random and even arrangement within MnO2 nanosheets. The map is dominated (97.0%) by blue regions, confirming that |εc|<|εab|. As CJTE is widely accepted to be the origin of the strain, especially in materials containing Mn16,39,40, the overall TEM and GPA results confirmed the hypothesis that the f-CJTE generates long-range tensile strains within the ab plane and the perpendicular compressive strains along the c direction in the crystal lattice.
Raman spectroscopy was employed to analyze the Mn−O bonding in bare MnO2 and MnO2/graphene superlattice. Two dominant Mn−O stretching modes are identified in [MnO6] octahedra. These include: the in-plane stretching vibration (νpar), associated with the Ag symmetric stretching vibration of [MnO6] octahedra, which occurs parallel to the linked [MnO6] sheets and is originated from the shear motions of O atoms within the layers, and the out-of-plane mode (νperp) perpendicular to the sheets and corresponds to expansion and compression motions of [MnO6] sheets along the layer stacking direction (Fig. 4a, b)41,42. Previous investigations revealed that both νpar and νperp stretching vibrations are very sensitive to the intercalated species among interlayers42,43. In Birnessite-type MnO2, the larger cation insertion or substitution can weaken interlayer interaction, resulting in a redshift of νperp stretching vibrations44. In the case of MnO2/graphene superlattice, as shown in Fig. 4c, the remarkable blue-shift of the νperp band from 631 to 653 nm−1 is attributed to the intercalation of p-Gr nanosheets. Meanwhile, the νpar counterpart is redshifted.

The schematic illustrating νpar and νperp stretching modes (corresponding to the in-plane stretching vibration parallel to [MnO6] sheets and out-of-plane stretching vibration perpendicular to the sheets, respectively) of Mn−O bonds in Raman spectra for a bilayer MnO2 and b MnO2/graphene superlattice structures. The purple, red, and brown spheres represent Mn, O, and C atoms, respectively. c Raman spectra of bare MnO2 and MnO2/graphene superlattice with statistics distribution of νpar/νperp ratios of bare MnO2 and MnO2/graphene superlattice in d contour mapping and e bar chart.
There are significant intensity changes of νpar and νperp stretching vibrations when comparing Raman spectra between bare MnO2 and MnO2/graphene superlattice. To validate the intensity variation of these two stretching modes, the intensity ratios, νpar/νperp, were statistically calculated based on areal-scanned Raman spectra for both bare MnO2 and MnO2/graphene superlattice, presented in contour graphs (Fig. 4d) and a statistical bar chart (Fig. 4e). Bare MnO2 has significantly higher overall νpar/νperp ratios than MnO2/graphene superlattice, which is attributed to the greater MnIV occupying rate42 shown by the XPS (Supplementary Fig. 9) and the Curie-Weiss fitting (Fig. 2d) results. Another contributing factor could be related to the superlattice architecture, in which the sizeable lateral-sized graphene nanosheets cap every monolayer-stacked MnO2 nanosheet via a strong interaction, including electrostatic force and possible hydrogen bonding to surface hydroxyl groups of MnO243. Furthermore, the relatively lower νpar/νperp ratios are irregularly distributed on aggregated MnO2 nanosheets, while the distribution of νpar/νperp ratios on the MnO2/graphene superlattice is in a narrow range, as observed from the contour graph and statistical bar chart that statistically obeys a Gaussian distribution. Moreover, based on Badger’s rule45,46,47 (Supplementary Note 5), which describes the relationship between force constant and bond length, the Raman spectra analyses elucidate that the overwhelmingly increased out-of-plane and decreased in-plane stretching vibrations in MnO2 after coupling with graphene are ascribed to the overall elongated and compressed Mn−O bonds along the same orientations. These experimental results also align with the category of |ωrc|<|ωrab| observed in the theoretically calculated results, providing strong evidence for the existence of long-range biaxial strain in the superlattice.
Electrochemical performance of MnO2/graphene superlattice as cathodes in aqueous zinc-ion batteries
The structural stability of bare MnO2 and MnO2/graphene superlattices in aqueous electrolyte (2 M ZnSO4 with 0.1 M MnSO4 additive) was evaluated using in situ UV-vis characterizations, with schematic illustrations of the configuration and setup provided in Supplementary Fig. 13. The measurements were conducted both in static conditions (Supplementary Fig. 14) and under electrochemical control (Supplementary Fig. 15). To assess static structural stability (Supplementary Fig. 14 and Supplementary Note 6), both electrodes were immersed in the aqueous electrolyte following the specific ratio described in Methods section. The Mn dissolution status was reflected in the UV-vis spectra by the variation of peak position, corresponding to the emergence of soluble Mn2+ ions, δ-MnO2 or p-Gr fragments. Notably, the MnO2/graphene electrode remained stable over 20 h with slight dissolution of Mn into Mn2+ ions, while bare MnO2 experienced a severe erosion in aqueous electrolyte, resulting in structural collapse evidenced by the appearance of δ-MnO2 fragments. Coupled with Zn metal anodes, home-made two-electrode cells were employed for in situ UV-vis tests under electrochemical control at a specific current of 1 C (308 mA g−1) for both electrodes, as shown in Supplementary Fig. 15. While Mn dissolution is inherently limited by the oxidation of Mn2+ ions back to MnO2 at certain voltages during the charge process48, the overall spectral shifts in the UV-vis spectra for the MnO2/graphene superlattice remain significantly smaller than those of bare MnO2 over 20 cycles, indicating its enhanced ability to suppress Mn dissolution.
The electrochemical performance of bare MnO2 and MnO2/graphene superlattice cathodes was evaluated in full aqueous zinc-ion batteries using zinc foil as the anode. Supplementary Fig. 16 presents cycling behaviour at a current rate of 1 C. The MnO2/graphene superlattice electrode delivers an initial specific capacity of 270 mAh g−1 with an initial Coulombic efficiency of 93.4%. After a short activation stage, the capacity stabilizes with Coulombic efficiencies approaching 100% and retains 91.2% of its capacity after 700 cycles. In contrast, the bare MnO2 electrode exhibits lower initial capacity, a prolonged activation phase, and a rapid capacity decay, retaining only ~50 mAh g−1 after 500 cycles. These differences are further reflected in the galvanostatic discharge curves. The MnO2/graphene superlattice electrode shows highly consistent discharge profiles throughout cycling (Fig. 5a), with a notable inflection point at ~1.25 V, separating two distinct discharge plateaus. This transition marks a shift in the dominant charge storage mechanism: proton insertion above 1.25 V and Zn2+ insertion below it49. For the bare MnO2 electrode (Supplementary Fig. 17), the separation between these processes becomes increasingly blurred with cycling, as the faster proton insertion reaction begins to dominate50. The decline in Zn2+-insertion contribution and overall capacity is attributed to progressive MnO2 inactivation. In situ UV-vis spectroscopy further confirms that Mn2+ dissolution and fragmentation of the MnO2 structure are the main causes of its limited cycling stability.

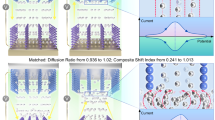
a Galvanostatic discharge profile of MnO2/graphene superlattice electrodes within various cycles. b Long-term cycling life after a short activation (Supplementary Fig. 18) at 1 C (308 mA g−1) for 10 cycles of bare MnO2 and MnO2/graphene superlattice electrodes tested at a specific current of 5 C. c In situ SR-XRPD patterns of MnO2/graphene superlattice within a full cycle at a specific current of 1 C under 25°C±5°C in the ambient condition. Schematic illustration of d the structural degradation of bare MnO2 electrodes within two steps and e good structural stability of MnO2/graphene superlattice electrodes.
When tested at a high rate of 5 C (Fig. 5b) after an activation at 1 C for ten cycles (Supplementary Fig. 18), the specific capacity of bare MnO2 electrodes quickly dropped to 100 mAh g−1 within 400 cycles, and only 60 mAh g−1 was maintained after 5000 cycles. Conversely, the MnO2/graphene superlattice electrodes presented a similar initial specific capacity as bare MnO2 electrode with a value of 175 mAh g−1, but much superior long-term cycling stability over 5000 cycles with a high reversible capacity of 165 mAh g−1. It should be noted that the operating time of full cells using MnO2/graphene superlattice electrodes is over 1200 h here. Comparative data for previously reported manganese oxide cathodes are provided in Supplementary Table 3. The long lifespan of the superlattice electrode firmly proves the good suppression effect on Mn dissolution and stabilization of MnO2 in aqueous zinc-ion batteries.
In situ SR-XRPD was employed to investigate the Zn storage mechanism of the MnO2/graphene superlattice (Fig. 5c and Supplementary Fig. 19). The SR-XRPD patterns can be divided into three regions in terms of H+ insertion, Zn2+ insertion, and H+/Zn2+ extraction. The conventional XRD pattern (Supplementary Fig. 8) indicates the (002) peak of MnO2/graphene superlattice is located at 6.88°, while the synchrotron powder diffraction (PD) patterns reflect the (003) peak of superlattices at 10.34°, associated with the initial d spacing of 2.57 nm as well. During the galvanostatic discharge/charge processes, the interspacing of the superlattice almost remained within the H+ insertion stage due to the ultrasmall proton radius, but the intensities of H+-intercalated (003) peaks increased. The ionic radius of Zn2+ ions (~0.75 Å)51 is much larger and thus causes a dramatic peak shift to 10.13°, accompanied by continuous Zn2+ ion insertion. Interestingly, the (003) peak vanishes and is then replaced by the (006) peak at 20.82° when the concentration of Zn2+ in the superlattice is high enough. Another finding is the emergence and disappearance of the Groutite-phase MnOOH phase (JCPDS no. 12-0733) at ~43.24°, which is convincing evidence of the intercalation and deintercalation of H+ ions and closely dependent on the contents of H+ ions in the MnO2 lattice49. The disappearance of the MnOOH phase is accompanied by the emergence of a small peak at 10.21°, which may result from the migration of Mn3+ ions from the MnO2 plane into the interlayer space, creating metal vacancies52,53. The presence of this peak is likely influenced by the relatively small ionic radius of high-spin Mn3+ (0.64 Å)54. The overall peak alterations are reversible in the extraction procedure. However, no apparent peak variations appear in the SR-XRPD patterns of the bare MnO2 electrode (Supplementary Fig. 20) during the H+/Zn2+ insertion/extraction. There is an inconspicuous reversible peak shift from 43.16° to 43.19° in the bare MnO2 patterns, suggesting the slight shrinkage of (220) facets and the layered [MnO6] plane in ZnxMn2O4 phase (for example, ZnMn2O4 JCPDS no. 12-0733). The strain distribution of Zn2+-inserted MnO2/graphene superlattice was analyzed in Supplementary Fig. 21 and Supplementary Note 7. After full discharge, although MnO2 converts into ZnxMn2O4, the superlattice retains its 2D structure and biaxial strains, with tensile strain in-plane and compressive strain out-of-plane, without localized strain accumulation. The overall strain is reduced to the range of −0.2% to 0.2%, indicating that the pre-introduced biaxial strain effectively mitigates Zn2+ insertion-induced stress.
It is evident that the long-range biaxial lattice strains induced by the f-CJTE play a crucial role in mitigating the dissolution of Mn and maintaining the structural integrity of the electrode material during the H+/Zn2+ insertion/extraction. The deterioration of the pristine MnO2 structure is illustrated in Fig. 5d. In this system, protons infiltrate the MnO2 layers through their edges, concurrently subjecting the [MnO6] cells on the surface to direct attack (Step I), as reported in previous studies55. Consequently, the resultant MnOOH species are rapidly corroded when exposed to the corrosive proton-rich environment. Simultaneously, the migration of Mn3+ from the MnO2 plane to the interlayer space, and eventually to the surface and edges, further destabilizes the local structure. This corrosion process ultimately leads to the irreversible transformation of MnO2 layers into soluble Mn2+ ions and MnO2 fragments (Step II). In stark contrast, the MnO2 layers within the MnO2/graphene superlattice are effectively shielded, thanks to the pre-existing lattice strain, as illustrated in Fig. 5e. When confronted with attempts at H+ insertion, the out-of-plane compressive strain primarily regulates the intercalation of H+ ions at the edge sites. Consequently, the MnOOH phases gradually develop toward the central regions, inducing a lattice mismatch at the interface between MnO2 and MnOOH. The presence of graphene and the associated compressive strain help confine Mn3+ migration locally, preventing its movement toward the surface and edges, where further corrosion occurs. Simultaneously, the formation of layered ZnxMn2O4 may also introduce lattice distortion due to the increased concentration of Jahn-Teller Mn3+ ions within the Zn2+ insertion56. In both scenarios, stress propagates from the edges inward, but the pre-existing in-plane tensile strain effectively counteracts these stresses, thereby stabilizing the superlattice structure throughout reversible cycles.
We further investigated the kinetics of bare MnO2 and MnO2/graphene superlattice electrodes, as shown in Supplementary Fig. 22 and Supplementary Table 4, and discussed in Supplementary Note 8. The MnO2/graphene superlattice electrodes deliver a better rate performance than the bare MnO2 electrodes at various specific currents. The MnO2/graphene superlattice electrodes deliver specific capacities of about 180 and 130 mAh g−1 at high rates of 5 and 10 C, respectively. In contrast, the bare MnO2 electrodes only achieve 100 and 50 mAh g−1 at the same specific currents. The superior rate performance of MnO2/graphene superlattice electrodes is attributed to the high electronic conductivity of the graphene matrix and, more importantly, their enhanced ion transport kinetics, supported by a series of electrochemical characterization techniques, including galvanostatic intermittent titration technique, cyclic voltammetry (CV), and electrochemical impedance spectra. The superior kinetics stem from the much lower Zn2+ ion diffusion barrier in the MnO2/graphene superlattice, as shown by DFT calculations (Supplementary Fig. 23 and Supplementary Note 9).
Discussion
In this study, the introduction of long-range biaxial strain, consisting of in-plane tensile and out-of-plane compressive stains, in MnO2 layers was achieved by constructing a superlattice structure via electrostatic self-assembly of monolayer-stacked δ-MnO2 and p-Gr nanosheets. Within the superlattice structure, the strong charge transfer between MnO2 and graphene induces a significant valence decrease of Mn ions and, consequently, a fully cooperative Jahn-Teller effect on MnO2 layers. Both nano- and macro-scale characterizations, including TEM and Raman spectra techniques, confirmed the existence of long-range biaxial strains along the entire superlattice. When applying MnO2/graphene superlattice electrodes in aqueous zinc-ion batteries, this introduced long-range biaxial strain efficiently protects the Mn sites in [MnO6] octahedra from H+ corrosion, offsets the Zn2+ insertion-induced stress and maintains the integrity of δ-MnO2. Hence, the MnO2/graphene superlattice exhibited good cycling stability over 5000 cycles at 5 C. Ionic diffusion kinetics are also enhanced. This work provides an innovative strategy to construct long-range strains via the cooperative Jahn-Teller effect and opens a promising prospect for regulating ion insertion-induced stresses in electrode materials for battery applications.
Methods
Preparation of δ-MnO2 nanosheets
Two precursor solutions were prepared prior to synthesis. Solution A comprised 0.6 M tetramethylammonium hydroxide (TMAOH, 25 wt.% in H2O, Sigma-Aldrich) and 3 wt.% hydrogen peroxide (H2O2, 30 wt.%, Sigma-Aldrich), while solution B consisted of freshly prepared 0.3 M MnCl2 (99%, Sigma-Aldrich) in deionized water. Under constant stirring at ambient temperature, 20 mL of solution A was gradually introduced into 10 mL of solution B. The resulting dark brown suspension was stirred in air for 12 h. The precipitate was isolated by centrifugation at 10,000 rpm (10,621 × g) for 30 min and washed three times with deionized water under the same conditions to eliminate excess TMA+ ions. The final product was redispersed in water and centrifuged at 5000 rpm (2655 × g) for 10 min to collect a reddish-brown supernatant, representing a colloidal suspension δ-MnO2 nanosheets.
Preparation of positively charged graphene nanosheets
Graphene oxide was converted to positively charged graphene via surface functionalization with PDDA. A 500 mL suspension of graphene oxide (0.2 mg mL−1), synthesized using a modified Hummer’s method26, was refluxed with 3.75 mL of PDDA solution (20 wt.% in H2O, Sigma-Aldrich) and 100 μL hydrazine hydrate (N2H4·H2O, 50–60 wt.%, Sigma-Aldrich) at 90 °C for 3 h. The product was collected by centrifugation at 10,000 rpm for 30 min to remove unbound PDDA. The residue was redispersed in water and subjected to a second centrifugation at 5000 rpm for 10 min to yield a stable suspension of positively charged graphene nanosheets.
Preparation of MnO2/graphene superlattices
The δ-MnO2 nanosheets and positively charged graphene suspensions were mixed at a mass ratio of ~2.8:1 (δ-MnO2:graphene), based on a previously reported area-matching model26, and stirred at room temperature. Electrostatic self-assembly occurred rapidly, forming a flocculent precipitate within seconds. The composite was recovered by centrifugation at 5000 rpm for 5 min, followed by washing with deionized water to remove residual ions and unbound components.
Materials characterizations
XRD measurements were carried out using a Bruker D8 Discover diffractometer with Cu Kα radiation (λ = 1.5406 Å). Data were collected over a 2θ range of 2–80° with a scan rate of 0.5° min−1 and a step size of 0.04°. AFM via a Park XE7 was applied to characterize thicknesses and lateral sizes. SEM via a Zeiss Supra 55VP and TEM via a FEI Tecnai G2 F30 S-TWIN were carried out to elucidate morphology at a lower resolution, while FEI Titan Cubed Themis G2 300 was used to observe atomic-level STEM images. SEM-EDX data were collected with the beam voltage of 30 keV. XPS measurements were performed using a Themo Scientific ESCALAB 250Xi spectrometer equipped with a monochromatic Al Kα X-ray source (hυ = 1486.6 eV). Powder samples were mounted on conductive carbon tape and transferred to the XPS chamber through ambient air. The binding energies were calibrated using the C 1 s peak at 284.8 eV to correct for surface charging. A Renishaw inVia Raman microscope obtained Raman spectra. The Raman mapping was conducted in areas with the size of 52 × 52 μm and the collecting increment of 4 μm. An Agilent Cary 60 UV-Vis spectroscopy tested ultraviolet-visual spectra. An SDT-2960 system performed TGA in air from room temperature to 800 °C with a temperature rising rate of 5 °C min−1.
Magnetic measurements
Magnetic characterization was conducted using a Quantum Design MPMS-3 system to evaluate both temperature- and field-dependent magnetization behaviours. Powder samples were loaded into gelatin capsules, which were secured in the magnetometer using a non-magnetic straw holder. For ZFC measurements, samples were first cooled to 1.8 K in the absence of an external magnetic field. Upon reaching this temperature, a magnetic field (H) was applied, and the magnetization (M) was recorded during subsequent warming to 300 K. FC measurements were obtained by cooling the sample under the same applied field from room temperature to 1.8 K, followed by magnetization recording over the same temperature range.
Cell assembly and electrochemical measurements
To prepare electrode slurries, active materials, carbon black (Super P, Timcal) and polyvinylidene fluoride (PVDF, Mw ~1,000,000, Arkema) were manually ground at a mass ratio of 7:2:1 in N-methyl-2-pyrrolidone (NMP, 99.5%, Sigma-Alrich) solvent with an approximate solvent-to-solid ratio of 2:1 by weight. The mixture was then single-side coated on hydrophobic carbon cloth current collectors (thickness of 454 µm, ELAT LT1400, FuelCellsEtc) with a blade and dried under vacuum at 100 °C overnight. The active material-coated carbon clothes were punched into 12 mm by using a precision disc cutter (MTI Corporation), and the average active material mass loading is ~2.5 mg cm−2. The Zn||MnO2 coin cells were assembled by Zn foil (thickness of 0.1 mm, 99.99%, SCI Materials Hub) counter electrodes, glass fibre separators (GF/D, Whatman), and working electrodes with ~75 μL 2 M ZnSO4 (99.0%, Sigma-Aldrich) + 0.1 M MnSO4 (99%, Sigma-Alrich) electrolyte, in which Zn foil was washed with ethanol, dried under vacuum, and then punched into disc with the diameter of 12 mm. The assembled batteries were tested at 25 ± 5 °C in the ambient condition without climatic/environmental chamber. For the electrochemical measurements, CV at various scanning rates (0.1–1.0 mV s−1) in a potential window of 0.8–1.8 V vs. Zn/Zn2+ was performed on a Bio-Logic VMP3 workstation. All galvanostatic tests were accomplished on LAND battery testers (1 C = 308 mA g−1), including galvanostatic discharge/charge profiles at 1 C, cycling life tests at 1 C and 5 C (including a ten-cycle activation at 1 C), and rate performance at various specific currents (0.2–5 C) between 0.8 V and 1.8 V. 3–6 of coin cells were tested for each single electrochemical experiment, and the presented data in the manuscript represent a statistical analysis. Electrochemical impedance spectroscopy (EIS) measurements were conducted at open circuit potential using a Bio-Logic VMP3 electrochemical workstation for cells cycled ten and 100 cycles at a specific current of 1 C. A potentiostatic signal with an AC amplitude of 5 mV was applied over a frequency range of 10 mHz to 100 kHz, with six data points collected per decade.
In situ UV-vis Tests
The schematic illustration of the in situ UV-vis setup is shown in Supplementary Fig. 13. The bare MnO2 and MnO2/graphene superlattice electrodes were coated onto hydrophobic carbon clothes using the same methods as mentioned above. The prepared electrodes were cut into pieces of 4 × 30 mm with an active material mass loading of ~2.5 mg cm−2 and positioned at the side of UV-vis cuvettes, secured with Kapton tape. For static in situ UV-vis characterisation, the electrodes were immersed in the electrolyte (2 M ZnSO4 + 0.1 M MnSO4) with a relatively higher ratio of m(active materials):V(electrolytes) = 1 mg:0.5 mL compared to the standard conditions used in aqueous ZIBs (1 mg:0.03 mL in this work). The cuvette was then placed inside the UV-vis spectrometer chamber for measurement at 25 ± 5 °C in the ambient condition. For in situ UV-vis tests under electrochemical control, zinc metal anodes were used and fixed in place. A Neware battery tester was employed to supply a constant current and record electrochemical data. The electrolyte ratio remained the same as in the static tests. Once the UV-vis test commenced, a constant current of 1 C (1 C = 308 mA g−1) was applied between 0.8 and 1.8 V to the home-made two-electrode cell for 20 cycles. The UV-vis spectra were automatically recorded within the 200–800 nm range, with a scanning speed of 300 nm min−1 and a measurement interval of 10 min.
In situ SR-XRPD Characterizations
The mass loading of the electrodes for in situ SR-XRPD was ~3.0 mg cm−2 with carbon clothes as current collectors. The working electrodes were assembled with Zn foil anodes and glass fibre separators in designed coin cells, which had windows on both cathode and anode parts and sealed by Kapton tapes. The in situ SR-XRPD characterizations were conducted at the PD Beamline in the Australian Melbourne Synchrotron. The wavelength was 0.6883 Å using the NIST LaB6 600b standard reference materials. The coin cells were placed on a homemade stage for testing with galvanostatic cycling at a rate of 0.25 C using a Neware battery tester at 25 ± 5 °C in the ambient condition. The PD data collection was scanned within 1 min twice with an interval of 8 min.
Computational methods
All the spin-polarized DFT57 computations involved in this work were performed using plane-wave Vienna Ab initio Simulation Package58,59. The projector augmented wave pseudopotential approach60 was used to reflect the core and valence electrons of each element. The climbing image nudged elastic band (CI-NEB) method61 was used to calculate the ion diffusion property. The exchange-correlation interactions were described by generalized gradient approximation62 of the Perdew-Burke Enzerhor functional63, and the plane-wave cutoff energy was set to 450 eV. Considering the long-range dispersion effect, empirical DFT-D3 scheme64 was adopted to treat the van der Waals correction, and only the Gamma point was considered in the first Brillouin zone because of the large enough crystal lattice of heterojunction. The energy and residue force were converged until 10−4 eV and 0.02 eV Å−1, respectively. Notably, in terms of electronic calculations, DFT plus Hubbard method (DFT + U)65 was employed with 4.0 eV U value66 added to the 3d orbital of Mn. The charge distribution was analysed by the Bader population using code developed by Henkelman et al. 67.
Data availability
All data that support the finding of this study are presented in the manuscript and Supplementary Information, or are available from the corresponding author upon request. Source data are also provided with this paper. Source data are provided with this paper.
References
Tarascon, J.-M. & Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 414, 359–367 (2001).
Sun, Y., Liu, N. & Cui, Y. Promises and challenges of nanomaterials for lithium-based rechargeable batteries. Nat. Energy 1, 16071 (2016).
Choi, J. W. & Aurbach, D. Promise and reality of post-lithium-ion batteries with high energy densities. Nat. Rev. Mater. 1, 16013 (2016).
Tang, B., Shan, L., Liang, S. & Zhou, J. Issues and opportunities facing aqueous zinc-ion batteries. Energy Environ. Sci. 12, 3288–3304 (2019).
Balke, N. et al. Nanoscale mapping of ion diffusion in a lithium-ion battery cathode. Nat. Nanotechnol. 5, 749–754 (2010).
Sun, J. et al. A phosphorene-graphene hybrid material as a high-capacity anode for sodium-ion batteries. Nat. Nanotechnol. 10, 980–985 (2015).
Wang, S., Zhao, S., Guo, X. & Wang, G. 2D material-based heterostructures for rechargeable batteries. Adv. Energy Mater. 12, 2100864 (2021).
Peng, X. et al. Strain engineering of two-dimensional materials for energy storage and conversion applications. Chem. Synth. 3, 46 (2023).
Zhu, X. et al. LiMnO2 cathode stabilized by interfacial orbital ordering for sustainable lithium-ion batteries. Nat. Sustain. 4, 392–401 (2020).
Yang, Y. et al. Strain engineering by atomic lattice locking in P2-type layered oxide cathode for high-voltage sodium-ion batteries. Nano Energy 76, 105061 (2020).
Xiong, P. et al. Strain engineering of two-dimensional multilayered heterostructures for beyond-lithium-based rechargeable batteries. Nat. Commun. 11, 3297 (2020).
Wang, L., Liu, T., Wu, T. & Lu, J. Strain-retardant coherent perovskite phase stabilized Ni-rich cathode. Nature 611, 61–67 (2022).
Muralidharan, N. et al. Tunable mechanochemistry of lithium battery electrodes. ACS Nano 11, 6243–6251 (2017).
Muralidharan, N., Carter, R., Oakes, L., Cohn, A. P. & Pint, C. L. Strain engineering to modify the electrochemistry of energy storage electrodes. Sci. Rep. 6, 27542 (2016).
Qi, Y. et al. Recent progress in strain engineering on van der Waals 2D materials: Tunable electrical, electrochemical, magnetic, and optical properties. Adv. Mater. 35, 2205714 (2023).
Gehring, G. A. & Gehring, K. A. Co-operative Jahn-Teller effects. Rep. Prog. Phys. 38, 1–89 (1975).
Goodenough, J. B. Jahn-Teller phenomena in solids. Annu. Rev. Mater. Sci. 28, 1–27 (1998).
Li, X. et al. Direct visualization of the Jahn–Teller effect coupled to Na ordering in Na5/8MnO2. Nat. Mater. 13, 586–592 (2014).
Kim, W. J. et al. Geometric frustration of Jahn-Teller order in the infinite-layer lattice. Nature 615, 237–243 (2023).
Xiong, T., Zhang, Y., Lee, W. S. V. & Xue, J. Defect engineering in manganese-based oxides for aqueous rechargeable zinc-ion batteries: a review. Adv. Energy Mater. 10, 2001769 (2020.
Lanson, B., Drits Victor, A., Feng, Q. & Manceau, A. Structure of synthetic Na-birnessite: evidence for a triclinic one-layer unit cell. Am. Mineral. 87, 1662–1671 (2002).
Liu, S. et al. Reviving the lithium-manganese-based layered oxide cathodes for lithium-ion batteries. Matter 4, 1511–1527 (2021).
Huang, Z.-F., Du, F., Wang, C.-Z., Wang, D.-P. & Chen, G. Low-spin Mn3+ ion in rhombohedral LiMnO2 predicted by first-principles calculations. Phys. Rev. B 75, 054411 (2007).
Heo, J. et al. Suppressing charge disproportionation of MnO2 cathodes in rechargeable zinc ion batteries via cooperative Jahn-Teller distortion. Batter. Supercaps 4, 1881–1888 (2021).
Deng, W. et al. Zn2+ induced phase transformation of K2MnFe(CN)6 boosts highly stable zinc-ion storage. Adv. Energy Mater. 11, 2003639 (2021).
Xiong, P., Ma, R., Sakai, N. & Sasaki, T. Genuine unilamellar metal oxide nanosheets confined in a superlattice-like structure for superior energy storage. ACS Nano 12, 1768–1777 (2018).
Xiong, P. et al. Two-dimensional unilamellar cation-deficient metal oxide nanosheet superlattices for high-rate sodium ion energy storage. ACS Nano 12, 12337–12346 (2018).
Xiong, P. et al. Interface modulation of two-dimensional superlattices for efficient overall water splitting. Nano Lett. 19, 4518–4526 (2019).
Indra, A. et al. CrO4 distortion-driven ferroelectric order in (R,Y)CrO4(R=Sm,Gd,and Ho): a new family of multiferroics. Phys. Rev. B 98, 014408 (2018).
He, J. et al. Induction and control of room-temperature ferromagnetism in dilute Fe-doped SrTiO3 ceramics. Appl. Phys. Lett. 107, 012409 (2015).
Kumar, S., Singh, A. K. & Pandey, D. Investigation of new magnetoelastic and magnetic transitions accompanied with magnetoelectric coupling in 0.1BiFeO3-0.9 Sr(Fe0.5Nb0.5)O3 multiferroic. J. Phys. Condens. Matter 32, 105401 (2020).
Shen, X.-F. et al. A magnetic route to measure the average oxidation state of mixed-valent manganese in manganese oxide octahedral molecular sieves (OMS). J. Am. Chem. Soc. 127, 6166–6167 (2005).
Zhu, H. et al. Birnessite-type MnO2 nanowalls and their magnetic properties. J. Phys. Chem. C112, 17089–17094 (2008).
Li, W. et al. Performance modulation of a-MnO2 nanowires by crystal facet engineering. Sci. Rep. 5, 8987 (2015).
Yang, X., Makita, Y., Liu, Z.-H., Sakane, K. & Ooi, K. Structural characterization of self-assembled MnO2 nanosheets from Birnessite manganese oxide single crystals. Chem. Mater. 16, 5581–5588 (2004).
Hÿtch, M., Snoeck, E. & Kilaas, R. Quantitative measurement of displacement and strain fields from HREM micrographs. Ultramicroscopy 74, 131–146 (1998).
Tanzi, M. C., Farè, S. & Candiani, G. in Foundations of Biomaterials Engineering (eds Maria Cristina Tanzi, Silvia Farè, & Gabriele Candiani) Ch. 2, 105-136 (Academic Press, 2019).
Ma, S. et al. First principles calculation of mechanical, dynamical and thermodynamic properties of MnO2 with four crystal phases. J. Alloy. Compd. 852, 157007 (2021).
Khomskii, D. & Kugel, K. Elastic interactions and superstructures in manganites and other Jahn-Teller systems. Phys. Rev. B 67, 134401 (2003).
Khomskii, D. I. & Streltsov, S. V. Orbital effects in solids: basics, recent progress, and opportunities. Chem. Rev. 121, 2992–3030 (2021).
Post, J. E., McKeown, D. A. & Heaney, P. J. Raman spectroscopy study of manganese oxides: layer structures. Am. Mineral. 106, 351–366 (2021).
Julien, C. et al. Raman spectra of birnessite manganese dioxides. Solid State Ion. 159, 345–356 (2003).
Soldatova, A. V. et al. Biogenic and synthetic MnO2 nanoparticles: size and growth probed with absorption and Raman spectroscopies and dynamic light scattering. Environ. Sci. Technol. 53, 4185–4197 (2019).
Xie, Q. et al. Alkali ions pre-intercalation of δ-MnO2 nanosheets for high-capacity and stable Zn-ion battery. Mater. Today. Energy 24, 100934 (2021).
Badger, R. M. A relation between internuclear distances and bond force constants. J. Chem. Phys. 2, 128 (1934).
Herschbach, D. R. & Laurie, V. W. Anharmonic potential constants and their dependence upon bond length. J. Chem. Phys. 35, 458–464 (1961).
Hardcastle, F. D. & Wachs, I. E. Determination of molybdenum-oxygen bond distances and bond orders by Raman spectroscopy. J. Raman Spectrosc. 21, 683–691 (1990).
Pan, H. et al. Reversible aqueous zinc/manganese oxide energy storage from conversion reactions. Nat. Energy 1, 16039 (2016).
Sun, W. et al. Zn/MnO2 battery chemistry with H+ and Zn2+ coinsertion. J. Am. Chem. Soc. 139, 9775–9778 (2017).
Huang, J. et al. Polyaniline-intercalated manganese dioxide nanolayers as a high-performance cathode material for an aqueous zinc-ion battery. Nat. Commun. 9, 2906 (2018).
Tan, Y. et al. Reaction kinetics in rechargeable zinc-ion batteries. J. Power Sources 492, 229655 (2021).
Gao, P. et al. The critical role of point defects in improving the specific capacitance of δ-MnO2 nanosheets. Nat. Commun. 8, 14559 (2017).
Gaillot, A.-C., Drits, V. A., Manceau, A. & Lanson, B. Structure of the synthetic K-rich phyllomanganate birnessite obtained by high-temperature decomposition of KMnO4. Microporous Mesoporous Mater. 98, 267–282 (2007).
Renaudin, G., Gomes, S. & Nedelec, J. M. First-row transition metal doping in calcium phosphate bioceramics: a detailed crystallographic study. Materials 10, 92 (2017).
Asl, H. Y. & Manthiram, A. Proton-induced disproportionation of Jahn-Teller-active transition-metal ions in oxides due to electronically driven lattice instability. J. Am. Chem. Soc. 142, 21122–21130 (2020).
Alfaruqi, M. H. et al. A layered δ-MnO2 nanoflake cathode with high zinc-storage capacities for eco-friendly battery applications. Electrochem. Commun. 60, 121–125 (2015).
Hohenberg, P. & Kohn, W. Density functional theory (DFT). Phys. Rev. 136, B864 (1964).
Blöchl, P. E., Jepsen, O. & Andersen, O. K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 49, 16223 (1994).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133 (1965).
Payne, M. C., Teter, M. P., Allan, D. C. & Arias, T. & Joannopoulos, a. J. Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients. Rev. Mod. Phys. 64, 1045 (1992).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Anisimov, V. I., Zaanen, J. & Andersen, O. K. Band theory and mott insulators: Hubbard U instead of stoner I. Phys. Rev. B 44, 943 (1991).
Sun, C., Wang, Y., Zou, J. & Smith, S. C. A formation mechanism of oxygen vacancies in a MnO2 monolayer: a DFT + U study. Phys. Chem. Chem. Phys. 13, 11325–11328 (2011).
Henkelman, G., Arnaldsson, A. & Jónsson, H. A fast and robust algorithm for bader decomposition of charge density. Comput. Mater. Sci. 36, 354–360 (2006).
Acknowledgements
Thanks for the support from Associate Professor Peng Li from Nanjing University of Aeronautics and Astronautics, Professor Qiaobao Zhang from Xiamen University, and Dr. Zefu Huang from University of Technology Sydney. This work was financially supported by the Australian Research Council (ARC) through the Discovery Projects (DP210101389 and DP230101579) and ARC Research Hub for Integrated Energy Storage Solutions (IH180100020). Part of the experiment was carried out at the PD Beamline (M18798 and M20371) of the Australian Melbourne Synchrotron. G. W. would like to acknowledge the support provided by The Royal Society, UK through the Royal Society Wolfson Visiting Fellowship (RSWVF\VF\R3\233017).
Author information
Authors and Affiliations
Contributions
S.W. and X.G. conceived the concept. S.W. performed the synthesis and electrochemical tests and wrote the manuscript. X.G., J.S., D.L. and L.G. conducted the morphology characterizations. A.A. proceeded with magnetic tests. Y.L. and Q.G. conducted the in situ synchrotron PD measurements. C.S. did the theoretical calculations. K.H., S.L., R.R.N., A.G. and G.W. and all other authors helped analyze the data, provide critical feedback, and revise the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, S., Guo, X., Huang, K. et al. Cooperative Jahn-Teller effect and engineered long-range strain in manganese oxide/graphene superlattice for aqueous zinc-ion batteries. Nat Commun 16, 5191 (2025). https://doi.org/10.1038/s41467-025-60558-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60558-y
