
Abstract
Ni(Fe) (oxy)hydroxides have long been recognized as benchmark electrocatalysts for water oxidation in alkaline media. Maximizing their activity and longevity will be vital for facilitating scalable water electrolysis. However, progress is hindered by insufficient understandings of surface Fe dynamics and by the absence of effective regulatory approach. Here we show the surface Fe dynamics beyond the immediate vicinity of Ni(Fe) oxyhydroxides on working electrode but being remotely influenced by Fe diffusion and deposition on the counter electrode. While Fe could be highly active in reversible dynamics, disruptions in this process can result in significant activity decay due to the loss or deactivation of surface Fe species. On-site regulation of Fe dynamics is inspired by pre-catalysts. [Ni0.75Fe0.25]0.6Zn0.4Clx derives Ni(Fe) oxyhydroxide with enhanced performance due to Zn leaching and cation defects that improve Fe site accommodation. Theoretical studies suggest that Fe dissolution occurs when cation defects are lacking but shifts to Fe capture as defects increase, underscoring on-site defect manipulation to maintain the abundance of active Fe sites.
Similar content being viewed by others

Introduction
Water electrolysis is the main technology to produce clean and renewable H2 fuel, which, however, is limited for large-scale implementation to date due to its low energy efficiency and high capital cost1. Significant energy loss and kinetic limitation originate from the oxygen evolution reaction (OER) at the anode2,3. Under acidic conditions, the usage of precious metals in OER catalysts, like Iridium, is still indispensable for long-term performance. For obtaining sufficient OER catalysts, the metal scarcity is a main limitation for acidic OER targeting for hydrogen production at the terawatt scale4. Compared to acidic cases, OER in alkaline media can be enabled by more cost-effective electrocatalysts with only 3d transition metals, which notably mitigates the metal scarcity issue for OER catalysts5,6,7. To date, NiFe-based (oxy)hydroxides have been regarded as state-of-the-art for alkaline OER8,9,10,11.
It has been recognized for long that Fe incorporation in these catalysts stands in a pivotal role in the overpotential reduction of OER12,13,14. The role of Fe in Ni(Fe) (oxy)hydroxides has been extensively investigated15,16. Despite debates on whether Ni or Fe is the main active site for OER, introducing Fe to Ni oxyhydroxide, either through intentional material synthesis or incidental spiking, typically increases intrinsic activity, notably at very low Fe content17. The delivered OER current density of Ni(Fe)OOH shows a poor dependence on the nominal Fe abundance in Ni(Fe)OOH18. Recent studies have indicated that only Fe on the surface is active while becoming inactive in the bulk19,20. In addition, Fe dissolution from Ni(Fe)OOH during OER is significant, especially in Fe-free electrolyte21. During Fe dissolution, either the loss of the active Fe in the electrolyte or the Fe segregation in inactive form was proposed as the mechanism of the deactivation of Ni(Fe)OOH in alkaline OER22,23. The instability of Fe makes it nearly impossible for Ni(Fe) oxyhydroxides to work while maintaining original stoichiometric composition. At industrial-level current density, significant activity degradation was found for Ni(Fe) oxyhydroxides24,25. Therefore, fundamental insights about surface Fe dynamics are essential for developing robust Ni(Fe) (oxy)hydroxide electrocatalysts26. However, to date, it is still challenging to effectively regulate the surface Fe dynamics. Extensive effort is needed to gain more fundamental insights into the dynamics in terms of full life cycle of Fe.
In this study, we reveal the nature of the Fe dynamics to be non-localized, demonstrating that it extends beyond the anodic environment of Ni(Fe) oxyhydroxide and is remotely influenced by Fe diffusion and deposition on the counter electrode. When this remote connection is disrupted by anion exchange membrane (AEM) between the working and counter electrodes, significant activity degradation is observed for Ni(Fe) oxyhydroxide. While Fe site could be active in reversible dynamics, the capacity for Fe active sites in Ni(Fe) oxyhydroxide can be limited. This could be attributed to a questionable reversibility of Fe dynamics, especially in the case with AEM, and Ni(Fe) oxyhydroxides may experience significant Fe loss on the surface, followed by Fe segregation in an inactive form, thereby compromising the OER activity and stability. Under electrochemical condition, it will be highly desirable to achieve on-site regulation of Fe dynamics to facilitate the abundance and stability of active Fe sites.
We then propose pre-catalysts, advantageous in flexibility during electrochemical reconstruction, to facilitate the on-site regulation. After examining potential pre-catalysts, [Ni0.75Fe0.25]0.6M0.4Clx (M = Zn, V, Mo, and Cr), the Ni(Fe) oxyhydroxide derived from [Ni0.75Fe0.25]0.6Zn0.4Clx, demonstrates nearly complete Zn leaching and significant cation defects, facilitating a self-optimized surface chemistry rich with active Fe sites. Thus, its intrinsic OER performance is better than those oxyhydroxides prepared via direct synthesis and other pre-catalyst sources, as well as the benchmark Ni(Fe)-based electrocatalysts. An important cause for Fe dissolution is revealed the lattice oxygen participance in OER occurring when defects in Ni(Fe) oxyhydroxides are deficient, as evidenced by DFT simulation. Meanwhile, thermodynamic analysis suggests that when cation defects become excessive, they can serve as the host sites for capturing free Fe3+ with favorable energetics. A trade-off of Fe dynamics establishes between the Fe dissolution and capture, deciding a Ni(Fe) oxyhydroxide surface chemistry electrochemically. Therefore, we highlight the importance of taking the advantage of pre-catalysts for on-site defect manipulation, which is a promising approach to make more activity breakthroughs for Ni(Fe) (oxy)hydroxides.
Results and discussion
Activity decay of NiFe-LDH in varied cell configurations
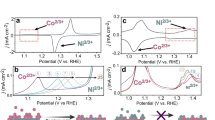
NiFe-layered double hydroxide (LDH) was synthesized with a nominal Fe/Ni ratio of 1:3 (i.e., Ni0.75Fe0.25-LDH), consistent with previously reported compositions showing favorable performance (Supplementary Fig. 1). We applied two different catalyst loadings on working electrode, which are 65 and 6.5 μgNiFe cm−2. Cyclic voltammetry (CV) was performed with Ni0.75Fe0.25-LDH in Fe-free 1 M KOH in a single cell without ion exchange membrane (Fig. 1a). After 100 cycles, the activity of high-loading Ni0.75Fe0.25-LDH was well maintained. In comparison, activity drop is notable during CVs when Ni0.75Fe0.25-LDH loading is low. This is consistent with past investigations of NiFe oxyhydroxides in Fe-free electrolyte, in which the activity degradation was explained by significant Fe dissolution22. The Fe dissolution reduces the surface active sites and decreases OER activity. Fe concentration is likely to reach the capacity of electrolyte when catalyst loading is high, which prevents further Fe dissolution. But at low catalyst loading, it is more likely to suffer from the low Fe surface abundance due to the Fe dissolution, which leads to significant activity degradation. Furthermore, we performed the same CV experiments in a H-cell with AEM between anode and cathode. Interestingly, as shown in Fig. 1b, Ni0.75Fe0.25-LDH showed notable activity degradation for both loadings. The activity degradation is more marked in H-cell with AEM compared to that in single cell (Fig. 1c). Activity retention at overpotential of 300 mV in H-cell with AEM was only about 30% for low-loading Ni0.75Fe0.25-LDH after 100 CVs.

a, b OER CVs (100 cycles) of high-loading and low-loading Ni0.75Fe0.25-LDH in (a) single cell without inter-chamber membrane and (b) H-Cell configuration with AEM in 1 M KOH at ambient temperature. High loading: 65 μgNiFe cm−2; low loading: 6.5 μgNiFe cm−2. Scan rate: 10 mV s−1. Non-iR corrected CVs are provided in the Supplementary Fig. 2. c Ni2+/Ni3+ oxidation peak shift along with 100 CVs of high-loading and low-loading Ni0.75Fe0.25-LDH under single cell configuration and H-Cell configuration with AEM. d Activity retention of Ni0.75Fe0.25-LDH at potential of 1.53 V vs. RHE in CV measurements. e CA measurement of low-loading Ni0.75Fe0.25-LDH in single cell, H-cell with CEM, and H-cell with AEM. f Activity retention of Ni0.75Fe0.25-LDH after 5-h CA measurement. The error bars represent standard deviation of at least three independent measurements.
Activity degradation during CV in the H-cell cannot be solely attributed to Fe dissolution. It is essential to consider that increased Fe incorporation into NiOOH can shift the Ni²⁺/Ni³⁺ oxidation peak anodically, providing insights into the Fe content in Ni(Fe)OOH22. The shifts in the Ni²⁺/Ni³⁺ oxidation peak relative to the 5th CV during the cycling of Ni0.75Fe0.25-LDH are shown in Fig. 1d. The original CVs for Fig. 1d can be found in Supplementary Fig. 3. In both single cell and H-cell with AEM, the peak shifts negatively during the first 20 cycles, indicating Fe dissolution. After approximately 25 CVs, the oxidation peak position remained almost constant in the single cell. However, in the H-cell with AEM, a notable positive shift in the oxidation peak was observed, especially for the low-loading Ni0.75Fe0.25-LDH, which can be attributed to Fe surface enrichment. Given the notable activity degradation in the H-cell, the accumulated Fe on the surface no longer acts as active sites but likely has detrimental effects on surface activity.
The stability of the OER was further investigated using chronoamperometry (CA) at a constant potential of 1.53 V vs. RHE for 5 h (shown in Fig. 1e). Low-loading Ni0.75Fe0.25-LDH was tested in both single cell and H-cell configurations. In the H-cell with an AEM, the activity degradation was more pronounced compared to the single cell, consistent with the CV results. The degradation under AEM configuration is confirmed with high-loading Ni0.75Fe0.25-LDH (Supplementary Fig. 4). Any change to the surface morphology of Ni0.75Fe0.25-LDH for activity degradation has been excluded (Supplementary Fig. 5). To exclude some effects such as those from electrolyte and bubbles on the performance degradation observed during CA, the following experimental procedures were conducted (Supplementary Fig. 6a). Prior to the CA test, the working electrode was cycled to reach a stable CV in a single cell. Afterward, the electrode was tested for CA under different cell configurations. After the CA test, the working electrode was placed back into a single cell with the electrolyte refreshed, and the CV was recorded again with all experimental conditions remaining the same as the initial CV test. CVs before and after CA are presented in Supplementary Fig. 6b–d. Notably, CVs of Ni0.75Fe0.25-LDH under the same experimental conditions showcase similar activity degradation as observed in CA, which confirms the activity degradation associated with the catalyst. To eliminate any physical effects from the membrane, CA was also measured in an H-cell with a cation exchange membrane (CEM). The activity degradation was notably mitigated with CEM compared to AEM. As summarized in the CA results (Fig. 1f), the activity retention in the H-cell with CEM was very similar to that in the single cell, in stark contrast to the results with AEM. The same CV experiments in a H-cell with CEM between anode and cathode were performed as well (Supplementary Fig. 7). The activity degradation for Ni0.75Fe0.25-LDH with CEM is similar to the ones in single cell without membrane. The key difference between using AEM and CEM is the type of ions exchanged between the anode and cathode. When Fe3⁺ dissolves into the electrolyte, it is more likely to pass through the CEM, while being blocked by the AEM. This suggests that the activity degradation could be closely related to the diffusion of Fe3⁺ between the anode and cathode.
Catalyst surface composition affected by Fe dynamics
Figure 2a summarizes the Fe/Ni ratio measured on Ni0.75Fe0.25-LDH by inductively coupled plasma mass spectrometry (ICP-MS) and scanning electron microscopy-energy dispersive X-ray analysis (SEM-EDX), in which ICP-MS indicates an overall composition while SEM-EDX offers insight into the near-surface composition. After performing 10 cycles for Ni0.75Fe0.25-LDH to reach a steady CV in single cell, ICP-MS results show a notable decrease in the Fe/Ni ratio compared to the pristine material, and even lower Fe/Ni ratio was detected by SEM-EDX, which suggests Fe loss from surface region. Afterwards, CA was conducted at 1.53 V vs. RHE for 5 h in single cell, H-cell with CEM, and H-cell with AEM. The surface Fe/Ni ratio showed small change after CA in both single cell and H cell with CEM, whereas a notable increase was observed after CA in the H-cell with AEM. Furthermore, X-ray photoelectron spectroscopy (XPS) was carried out to study the surface Ni and Fe species on Ni0.75Fe0.25-LDH after electrochemical reactions (Fig. 2b). After 10 CVs, XPS revealed very weak signal of Fe on the surface. Subsequent CA performance in single cell or H-cell with CEM did not increase the surface Fe species, while notable XPS signal of Fe 3p presented after CA performance in H-cell with AEM. The revealed surface Fe segregation is consistent with the anodic shift of Ni2+/Ni3+ oxidation peak observed during CVs in H-cell with AEM. Our findings suggest that the OER activity of Ni0.75Fe0.25-LDH may only relies on the minor existence of Fe active sites on the surface, while further increasing the surface Fe content during cycling does not increase the active sites but causes segregation, compromising the intrinsic activity.

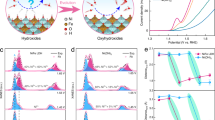
All measurements were conducted in 1 M KOH electrolyte at ambient temperature. a Fe/Ni elemental ratio of Ni0.75Fe0.25-LDH before and after different electrochemical experiments. b XPS spectra of Ni0.75Fe0.25-LDH before and after 5-h CA measurements in single cell, H-cell with CEM, and H-cell with AEM. All CA measurements were conducted on Ni0.75Fe0.25-LDH electrode after reaching steady CV by 10 cycles. c Deposition amount (ppb) of Ni and Fe on counter electrode after performing CA of Ni0.75Fe0.25-LDH at potential of 1.53 V vs. RHE for 5 h in single cell, H-cell with AEM, and H-cell with CEM. d Illustrations of surface Fe dynamics on NiFe oxyhydroxide with full or broken reversibility under alkaline condition. e Schematic illustration of Fe diffusion from NiFe oxyhydroxide and its deposition either on counter electrode or working electrode in single cell, H-cell with CEM, and H-cell with AEM.
Surface Fe segregation appears to be affected by the Fe deposition onto counter electrode. The deposited metals on the counter electrode (Pt) before and after 5-h CA measurement were collected by 1 M HNO3 and analysed by ICP-MS. As seen in Fig. 2c, Ni deposition on the counter electrode is negligible compared to Fe. The amount of Fe collected from the counter electrode was the highest in the single cell, followed by the H-cell with a CEM, and the lowest in the H-cell with an AEM. In addition to 1.53 V, metal deposition on the counter electrode was examined at lower (1.38 V) and higher (1.68 V) potentials relative to the Ni2+/Ni3+ transition (as shown in Supplementary Fig. 8). The amount of Fe increases as the potential rises from 1.38 V to 1.53 V, but remains nearly unchanged with further increase in potential (from 1.53 V to 1.68 V). Deposited Fe on counter electrode was confirmed by high-resolution transmission electron microscopy (HRTEM) (Supplementary Fig. 9). The Fe species on the counter electrode can segregate into the metallic phase under the reductive potential during hydrogen evolution. Additionally, it is evident that the diffusion of Ni to the counter electrode is negligible, consistent with the ICP results.
Fe deposition on counter electrode, surface Fe segregation on Ni0.75Fe0.25-LDH, and OER activity degradation in the three types of cells are strongly correlated, which gives important implications on surface Fe dynamics. This work identifies Fe dynamics extending beyond the microscopic region around the surface of NiFe oxyhydroxides, influencing the entire electrochemical system and are notably affected by macroscopic Fe diffusion behaviors. When Fe diffusion is impeded by the AEM, Fe tends to segregate as inactive species, thereby limiting the intrinsic surface activity.
The remote effect identified in this study is illustrated in Fig. 2d. Previous studies have considered surface Fe dynamics as a reversible exchange of Fe either in the host site or dissolved in the electrolyte, with a high exchange rate26. Fe with dynamic stability was demonstrated with high turnover frequency for OER26. With more experimental findings in this study, we realize that such an equilibrium of Fe dynamics could be fragile, and probably proceeding with broken reversibility. A plausible cause for this fragility is the significant release of Fe3+ from the bulk, which has been revealed in prior studies21. When the dissolved Fe3+ far exceeds the capacity of the host site on the surface, a significant portion of Fe may no longer participate in the surface Fe exchange. It is crucial to note that the electrolyte capacity for dissolved Fe3+ in alkaline is extremely low, Ksp (Fe(OH)3) ≈ 10−38. Hence, even in Fe-free electrolyte, the Fe3+ dissolution can quickly reach the electrolyte’s capacity. Beyond a threshold, significant Fe3+ can neither hosted by catalyst surface nor reside in the electrolyte, potentially leading to segregation of inactive Fe species.
Fe electrodeposition on counter electrode then becomes a main mechanism to accommodate the excessive Fe, maintaining the Fe dynamics on the surface of working electrode. As shown in Fig. 2e, such assistance from counter electrode deposition depends on successful diffusion of Fe3+ from working to counter electrode, which is feasible in both single cell and H-cell with CEM. Compared to the Fe segregation that is weak or negligible in both single cell and H-cell with CEM, it becomes more pronounced once the Fe diffusion is hampered by AEM, followed by diffused Fe3+ interacting directly with the OH- crossing the AEM from cathode side to quickly hydroxylate as inactive species.
But it should be cautious here about gradient pH near the working electrode. The local pH close to the OER electrode can be lower due to OER kinetics consuming significant OH- compared to regions farther from the OER electrode. Consequently, Fe3+ that dissolved from the surface of NiFe oxyhydroxide may be chemically limited by the elevated local pH when diffusing away from the working electrode and is likely to hydroxylate under alkalifying environment. As the hydroxylated Fe species (probably in FeOxHy form) become charge neutral, it loses the mobility in electric field, end up redepositing on working electrode. This could explain the slight increase of Fe/Ni ratio even when CA was performed in single cell and H-cell with CEM (Fig. 2a). The Fe segregation in long-term electrochemical performance cannot be fully excluded even assisted by counter electrode deposition.
OER activity can be associated with valence state of Fe. Existing experimental evidence suggests that high-valence-state Fe, particularly with a valence state higher than +3, such as Fe4+, is active under OER conditions. Relevant literature is summarized in Supplementary Table 1. In these reports, the existence of Fe4+ was typically found at the potential of working electrode much higher than the experimental potential employed in this work (1.53 V). In addition, according to the Pourbaix diagram (Supplementary Fig. 10), the theoretically stable Fe species at the potential of 1.53 V and pH of 14 predominantly exhibit a valence state of +3. Therefore, the diffused Fe species in this case is more likely to be Fe3+.
Elevating surface capacity for Fe active sites with pre-catalysts
The chemical and electrochemical stability of Fe surface sites lies in the essence of Fe dynamics. Loss or segregation of Fe notably compromises the activity. While past efforts tried to optimize NiFe (oxy)hydroxide by simply increasing the nominal Fe content in the catalysts, limited activity improvement was typically encountered beyond a Fe content threshold27, which probably is due to the finite capacity of catalytic surface to accommodate active Fe sites. To maximize the activity of NiFe (oxy)hydroxide, it is crucial to regulate Fe dynamics under operando condition rather than nominal Fe composition in the synthesis, to finalize an optimal surface environment for accommodating abundant Fe sites.
We performed a pre-catalyst strategy to create more cation defects in the derived NiFe oxyhydroxides (Fig. 3a), which revealed a close association with the stabilization of abundant surface Fe sites. Compared to directly synthesized NiFe-LDH, electrochemical reconstruction of NiFe precursors creates more defects. In this study, we employed chloride NiFe pre-catalysts for deriving NiFe oxyhydroxide. In addition, we included the pre-catalysts doped with some inexpensive metals that could leach out to create more cation defects in NiFe oxyhydroxide during electrochemical measurement. Representative XRD pattern for [Ni0.75Fe0.25]0.6Zn0.4ClX and Ni0.75Fe0.25Clx is shown in Supplementary Fig. 11. Among all doped metals (Zn, Cr, V, and Mo), Zn has been found almost completely leach out after only 10 CVs (Supplementary Fig. 12 and Table 2). The derived NiFe oxyhydroxides have been characterized by Raman spectroscopy, which validates the cation vacancies after Zn leaching (Supplementary Fig. 13).

a Schematic illustration of pre-catalyst strategy to introduce dynamic host on the surface for Fe. b OER steady CVs of [Ni0.75Fe0.25]0.6Zn0.4Clx measured in O2-saturated 1 M KOH in a single cell at ambient temperature, in comparison with Ni0.75Fe0.25Clx, and Ni0.75Fe0.25-LDH. Non-iR corrected CVs are showed in the Supplementary Fig. 14. c Activity retention during 200 CVs at potential of 1.53 V vs. RHE for [Ni0.75Fe0.25]0.6Zn0.4Clx, Ni0.75Fe0.25Clx, and Ni0.75Fe0.25-LDH. d TOF on the basis of both surface and bulk NiFe sites for [Ni0.75Fe0.25]0.6M0.4Clx (M = Zn, V, Mo, and Cr), Ni0.75Fe0.25Clx, and Ni0.75Fe0.25-LDH (high and low loading). Comparison with reported NiFe-based OER electrocatalysts including Ni-Fe-S48, NiFeZn-449, FeNi(MoO4)x50, Ni0.85Fe0.15/Au51, Ni0.67Fe0.33/C52, Ni0.67Fe0.33/rGO15, F-NiFe-A53, and Ni-MOF54 is shown in Supplementary Table 3. e Ni-L2,3 XAS for the derived NiFe oxyhydroxide from [Ni0.75Fe0.25]0.6Zn0.4Clx, Ni0.75Fe0.25Clx, and Ni0.75Fe0.25-LDH by 10 CVs in 1 M KOH. f Elemental ratio of Fe/Ni measured by ICP-MS and SEM-EDX for the derived NiFe oxyhydroxide from [Ni0.75Fe0.25]0.6Zn0.4Clx, Ni0.75Fe0.25Clx, and Ni0.75Fe0.25-LDH after 10 CVs.
The geometric OER activities of [Ni0.75Fe0.25]0.6Zn0.4Clx, Ni0.75Fe0.25Clx, and Ni0.75Fe0.25-LDH are compared in Fig. 3b. The NiFe oxyhydroxides derived by pre-catalysts were obtained by 10 CV cycles in 1 M KOH. Electrolyte was then refreshed to prevent the influence from the dissolved ions. Please note that the electrochemical surface areas of them are comparable after electrochemical conditioning, which can be indicated by the Ni redox surface area estimated from steady CV (Supplementary Fig. 15). Compared to the synthesized Ni0.75Fe0.25-LDH, the derived one from Ni0.75Fe0.25Clx shows improved activity. Further, with intentional Zn leaching through pre-catalyst electrochemical reconstruction, the OER activity of the derived NiFe oxyhydroxide is even boosted more. Moreover, we also performed CV cycling on [Ni0.75Fe0.25]0.6Zn0.4Clx, Ni0.75Fe0.25Clx, and Ni0.75Fe0.25-LDH. The activity retention along with CV cycles at potential of 1.53 V is summarized in Fig. 3c. Compared to Ni0.75Fe0.25-LDH that showed notable activity degradation, negligible activity loss during 200 CVs was found for the derived oxyhydroxide from [Ni0.75Fe0.25]0.6Zn0.4Clx and Ni0.75Fe0.25Clx, implying stable surface Fe chemistry with cation defects. The original CVs of [Ni0.75Fe0.25]0.6Zn0.4Clx and Ni0.75Fe0.25Clx are presented in Supplementary Fig. 16.
The intrinsic activities of the derived NiFe oxyhydroxides from varied precursors are presented by turnover frequency (TOF) at overpotential of 300 mV. Please see details of TOF evaluation in “Method” part. Both Ni and Fe sites are assumed as active metals in TOF evaluation. Figure 3d summarizes all estimated TOF values basing on surface metal sites and all metal sites of NiFe-based electrocatalysts. Please note that as the electrochemical surface area can be different for catalysts, it is important to carry out fair intrinsic activity comparison by such as TOF on surface-site basis (TOFsurface). Among all the pre-catalysts ([Ni0.75Fe0.25]0.6M0.4Clx), the ones doped with Zn, Cr, V, and Mo all outperform Ni0.75Fe0.25Clx. Of particular note is the activity found for the derived oxyhydroxide from [Ni0.75Fe0.25]0.6Zn0.4Clx, in which Zn has nearly completely leached out as evidenced by ICP-OES test (Supplementary Fig. 12 and Supplementary Table 2). In addition, the surface chemistry of [Ni0.75Fe0.25]0.6Zn0.4Clx before and after CA tests was characterized by XPS and SEM EDX. As shown in Supplementary Fig. 17, peaks around 1023 and 1046 eV are assigned to the Zn 2p3/2 and 2p1/2, respectively, which are notable for [Ni0.75Fe0.25]0.6Zn0.4Clx before electrochemical reconstruction. However, after electrochemical reconstruction, these Zn features completely disappear. Additionally, SEM-EDX analysis was conducted on [Ni0.75Fe0.25]0.6Zn0.4Clx before and after electrochemical reconstruction. As shown in the EDX mapping (Supplementary Fig. 18), a strong Zn signal is observed prior to reconstruction but becomes nearly undetectable afterward. These results suggest the nearly complete removal of Zn from the catalyst surface following electrochemical reconstruction.
In addition, in a comparison of intrinsic activity (TOFsurface), [Ni0.75Fe0.25]0.6Zn0.4Clx shows advantageous activity that comparably higher than referencing NiFe-based electrocatalysts. These findings indicate that the leaching of doped metals probably is the main origin of the elevated intrinsic activity of the derived NiFe oxyhydroxides. But we caution that not all activity difference here should be attributed to the leaching of doped metals only. The doped metals remaining in NiFe oxyhydroxides, even in trace amount, can probably influence the OER as well.
The derived oxyhydroxides from [Ni0.75Fe0.25]0.6Zn0.4Clx and Ni0.75Fe0.25Clx were studied by Ni L2,3-edge X-ray absorption spectroscopy (XAS), in comparison with directly synthesized Ni0.75Fe0.25-LDH. As shown in Fig. 3e, the spectra for [Ni0.75Fe0.25]0.6Zn0.4Clx, Ni0.75Fe0.25Clx, and Ni0.75Fe0.25-LDH are nearly identical, which indicates negligible electronic difference between the derived NiFe oxyhydroxides and the directly synthesized one. Thereby, the discovered big activity contrast could not be attributed to any changes in the electronic structures (e.g., oxidation state, spin state) of the NiFe oxyhydroxides. Fe/Ni elemental ratios of the derived oxyhydroxides from [Ni0.75Fe0.25]0.6Zn0.4Clx and Ni0.75Fe0.25Clx were measured by ICP-MS and SEM-EDX, which are shown for comparison with Ni0.75Fe0.25-LDH (Fig. 3f). Before elemental characterizations, 10 CV cycles had been performed on all samples in 1 M KOH to reach a steady surface chemistry state. The Fe/Ni ratio in [Ni0.75Fe0.25]0.6Zn0.4Clx, Ni0.75Fe0.25Clx, and Ni0.75Fe0.25-LDH shows consistent trend under both ICP-MS and SEM-EDX. The Fe/Ni ratio is relatively low in Ni0.75Fe0.25-LDH while comparatively high in the NiFe oxyhydroxides derived from [Ni0.75Fe0.25]0.6Zn0.4Clx. Please note that at only 10 CVs, Fe segregation has not yet happened or would not be significant. The revealed Fe/Ni ratio reflects the surface Fe abundance/deficiency right after electrochemical conditioning. Hence, the compositional analyses indicate a comparatively high abundance of Fe active sites at the surface region of the oxyhydroxide derived from [Ni0.75Fe0.25]0.6Zn0.4Clx. The trend in Fe/Ni elemental ratio is consistent with the order in intrinsic activity: [Ni0.75Fe0.25]0.6Zn0.4Clx > Ni0.75Fe0.25Clx > Ni0.75Fe0.25-LDH. The intrinsic activity improved by defects creation under pre-catalyst strategy can be associated with a regulation of surface Fe dynamics.
Theoretical insights into Fe dynamics affected by defects
In past study, T. Zhang and G.I.N. Waterhouse et al. directly synthesized the NiFe hydroxides doped with Zn2+ and Al3+, which similarly revealed the leaching phenomenon to create cation vacancy28. They attributed the stability improvement to relieved lattice distortion to increase the stability of FeO6 octahedra. With updated experimental findings about surface Fe dynamics, we provide additional explanation beyond existing understandings and reveal the role of defects in optimizing the surface Fe dynamics. We performed electrochemistry on 18O-isotope labelled NiFe oxyhydroxides derived from [Ni0.75Fe0.25]0.6Zn0.4Clx, Ni0.75Fe0.25Clx, and Ni0.75Fe0.25-LDH, and monitored the O2 chemistry using mass spectrometry (Fig. 4a). Please note that before isotope labelling, [Ni0.75Fe0.25]0.6Zn0.4Clx and Ni0.75Fe0.25Clx pre-catalysts have been cycled to reach a steady chemistry. The detected ratios of 34O2 (16O18O) to 32O2 (16O16O) as a function of applied potential are summarized in Fig. 4b. It is notable that the increased 34O2 signal was only observed for Ni0.75Fe0.25-LDH as the potential exceeded 1.6 V while 34O2 signal was nearly absence for the derived NiFe oxyhydroxides from [Ni0.75Fe0.25]0.6Zn0.4Clx and Ni0.75Fe0.25Clx. Thereby, the lattice oxygen exchange could be suppressed by the defects that are present in NiFe oxyhydroxides.

a Schematic illustration of the isotope experiment. b Ratios of 18O16O to 16O16O when performing CA at different potentials. c Pourbaix diagram of Ni0.75Fe0.25OOH at pH = 14. d The structure of 1OHtop-0.5Hbri configuration and possible active sites for OER. e Gibbs free energy differences of each elementary step of OER on bridge sites in original (left) and derived (right) Ni0.75Fe0.25OOH. Optimized structures of the original and derived Ni0.75Fe0.25OOH have been provided in Supplementary Data 1. f Gibbs free energy differences of each elementary step of OER on top sites in original (left) and derived (right) Ni0.75Fe0.25OOH. Optimized structures of the original and derived Ni0.75Fe0.25OOH have been provided in Supplementary Data 1. g Average formation energies of Ni vacancy at various coverage. Dia and near represent that the vacancies are distributed diagonally and adjacently, respectively. h Demonstration of Fe filling the defects in 1/2-dia structure and the corresponding ion replacement energies (Erep). Computational model has been provided in Supplementary Data 1. i Illustration of the relationship between vacancy content and Fe dynamics.
The lattice oxygen exchange on NiFe oxyhydroxides can be associated with the stability of Fe in oxyhydroxide. In past studies, Fe in NiFe oxyhydroxides has been found notably activating the dehydrogenation of ligand −OH to evolve ‘active oxygens’29. When lattice oxygen exchange is notable, it implies that these active oxygens may have participated in the OER, following lattice oxygen mediated mechanism (LOM). When lattice oxygen exchange happens on Fe sites locating in the interlayer basal plane, ligand −OH that have been reacted through LOM are difficult to be recovered immediately due to the limitation of OH- diffusion into the interlayers21. When excessive lattice oxygens are reacted while not refilled by OH-, it could create structural instability for the happening of bulk Fe dissolution along with acidifying local environment during OER30. It is thus crucial to limit the lattice oxygen exchange to mitigate Fe dissolution in our case.
To understand the effects of cation defects, we applied DFT calculations to examine the LOM pathways of OER on original oxyhydroxide (without vacancies) and the derived one (with vacancies). Surface configuration of Ni0.75Fe0.25OOH was determined by calculated Pourbaix diagram (Fig. 4c and Supplementary Table 4). Computational models for all surface configurations are shown in Supplementary Fig. 19. As shown in Fig. 4c, 1OHtop-0.5Hbri predominates at the operating potential (1.6 V vs RHE), which was thus selected for subsequent simulation. Two kinds of lattice oxygen have been examined to be involved in the LOM pathway: the bridge site and the top site, which are illustrated in Fig. 4d. The energetics and OER intermediate structures of the bridge site and the top site are demonstrated in Fig. 4e, f, respectively. All computational models are provided in Supplementary Data 1. For all pathways, the potential limiting steps for OER are revealed at the dehydrogenation process from the ligand oxygen, which can be assigned to the lattice oxygen activation31. The lattice oxygen activation shows the lowest energetic barrier (1.51 V) at the bridge site of Ni0.75Fe0.25OOH without vacancy. However, introducing vacancy to the system notably elevates the energetic barrier for the lattice oxygen activation to 1.92 eV. The lattice oxygen activation at top site is unfavorable for Ni0.75Fe0.25OOH both with and without vacancies, showing big energetic barrier of 1.72 and 1.79 V, respectively. Therefore, according to the energetics analysis, the LOM pathway should be predominated at bridge site on the Ni0.75Fe0.25OOH without cation vacancy, while becoming no longer favorable when cation vacancy is introduced through pre-catalyst approach. These results of theoretical analysis show good agreement with our 18O-labeling isotope experiment.
By suppressing lattice oxygen exchange, Fe in oxyhydroxide can maintain stable FeO6 framework, leading to less dissolution. In addition, NiFe oxyhydroxides may thermodynamically keep a trade-off between generating cation defects and filling the defects with free Fe3+ in electrolyte. We performed DFT-assisted thermodynamic analyses of the creation and filling of cation defects. Similar to the practice above, cation defects were created in NiFe oxyhydroxides. Reasonable surface cation defect densities are decided by analyzing vacancy formation energies (∆ENi-Vac). All Ni0.75Fe0.25OOH models with various Ni vacancies are shown in Supplementary Fig. 20, and their energy profiles are shown in Supplementary Table 5. The average formation energies at various coverage are summarized in Fig. 4g. We list all representative surface configurations, among which the defects have been progressively introduced until ∆ENi-Vac experiences a sudden drop. All structures have been fully relaxed after defects have been created. Considering the similar results of both 1/2 ML structures, 1/2-dia structure was studied as the representative to indicate the capability of accommodating Fe. On 1/2-dia structure, Fe ions fill into the cation defects progressively, and the corresponding energetics are illustrated in Fig. 4h. While DFT has certain limitations in dealing with the calculations of ions, thermodynamic analyses base on additional assumptions and iron replacement energy has been calculated accordingly. The details of calculations have been described in “Methods”. When filling one Fe ion in 1/2-dia structure, the iron replacement energy is calculated as −2.7 eV, including the energy of capturing a Fe3+ (*Fe) and generating one hole (*). Hole filling energy is estimated by subtracting the hole generation energy ∆ENi-Vac and the result turns out to be negative (−0.4 eV). Hence, one Fe filling should be thermodynamically favorable. However, filling with two Fe ions, the iron replacement energy is calculated positive as 0.1 eV, indicating limited thermodynamic ability to accommodate more Fe.
Overall, NiFe oxyhydroxides would keep a balanced concentration of surface Fe and defects (Fig. 4i). When defect is absent, Fe dissolution, possibly through the LOM pathway, creates surface cation defects. When the defects are overly generated, triggered by additional cation leaching (e.g., Zn2+ in this study), it may create more host sites on the surface for anchoring Fe3+ originally from the electrolyte or released from bulk. Therefore, by properly inducing defects through electrochemical reconstruction, the pre-catalyst strategy facilitates a self-optimization at the surface of NiFe oxyhydroxide, which can maintain abundant surface Fe active sites, elevating the intrinsic activity.
In summary, we have demonstrated a crucial role of Fe dynamics in Ni(Fe) (oxy)hydroxides. The effect of Fe dynamics has been found non-localized, which is not limited to the anodic environment of Ni(Fe) oxyhydroxides but remotely affected by Fe diffusion and deposition on counter electrode. Once the remote association is disrupted by AEM, significant Fe segregation on anode in inactive form has been found, leading to notable activity decay. Although Fe dynamics could be an origin of high reactivity of surface Fe, the fragile nature with questionable reversibility creates significant durability concern of these active Fe sites. The surface abundance of active Fe sites is more influenced by electrochemical conditioning rather than a nominal stoichiometric composition, emphasizing the need for on-site regulation for Fe dynamics.
Our pre-catalyst approach offers a solution by allowing the self-optimizing oxyhydroxide chemistry during electrochemical reconstruction. The [Ni0.75Fe0.25]0.6Zn0.4Clx pre-catalyst demonstrated nearly complete Zn leaching after electrochemical reconstruction, creating cation defects that enabled rich Fe active sites in the derived oxyhydroxide, and notably boost intrinsic OER performance. DFT studies provided additional insights about the roles of cation defects. Fe dissolution can be significant on Ni(Fe) oxyhydroxide without defects, triggered by lattice oxygen participance in OER, while Fe capturing become thermodynamically favorable with excessive cation defects. Therefore, with surface reconstruction of our pre-catalysts, the derived Ni(Fe) catalysts should have reached an ultimate surface with appropriate content of surface defects and Fe sites, at which there is no apparent loss or redeposition of Fe, i.e., achieving reversible Fe dynamics. An on-site regulation of Fe dynamics has been achieved by properly introducing cation defects under electrochemical conditions.
Overall, our study provided important fundamentals of Fe dynamics that is essential for the intrinsic activity of Ni(Fe) oxyhydroxides. A guidance of on-site regulation of Fe dynamics has been provided by showcasing the successful activity improvement through pre-catalyst approach, which opens more opportunities of activity breakthrough for Ni(Fe) oxyhydroxides to facilitate the scalable water electrolysis under alkaline media.
Methods
Materials
Toray 090 carbon paper and Nafion 212 PEM were purchased from Fuel Cell Store, US. The Alkymer W-25 AEM was purchased from EVE Fuel Cell Co., Ltd, China. Iron (III) nitrate nonahydrate (ACS reagent, 98%), Iron (III) chloride hexahydrate (ACS reagent, 97%), nickel (II) nitrate hexahydrate (crystals), nickel (II) chloride hexahydrate (99.9% trace metals basis), zinc chloride (reagent grade, ≥98%), vanadium (III) chloride (97%), chromium (III) chloride hexahydrate (purum p.a., ≥98.0% (RT)), potassium hydroxide (semiconductor grade, pellets, 99.99% trace metals basis), and sodium carbonate (ACS reagent, ≥99.5%) were purchased from Sigma-Aldrich. Molybdenum (V) chloride (99.5%) was purchased from Macklin. Ethanol (absolute, 99.9%), and isopropanol (Tech Grade) were purchased from Merck. Nafion perfluorinated resin (D520) was purchased from Adamas. Milli-Q deionized water (DI water, 18.2 MΩ·cm) was used in all experiments.
Ni0.75Fe0.25-LDH synthesis
The Ni0.75Fe0.25-LDH was prepared via a coprecipitation method using 0.1 M aqueous solutions of Ni(NO3)2 and Fe(NO3)3 as metal precursors. The synthesis was conducted at 80 °C. To begin, 10 mL of deionized water was added to a 0.1 M Na2CO3 solution, adjusted to pH 9.0. A mixed metal nitrate solution containing 0.3 M Ni(NO3)2 and 0.1 M Fe(NO3)3 was then slowly introduced into the carbonate solution. During the process, the pH was maintained around 9.0 by co-feeding 0.1 M Na2CO3 solution. The total volume (30 mL) of metal precursor solution and base was added over a span of 1.5 h. After precipitation, the suspension was centrifuged at 5000 rpm for 5 min to collect the solid product, which was subsequently dried at 120 °C for 48 h.
Materials characterization
The scanning electron microscopy (SEM) and SEM-energy-dispersive X-ray spectroscopy (SEM-EDS) images were obtained on FESEM JEOL JSM7800F PRIME equipped with EDS detector from Oxford Instruments. HRTEM was performed using a JEOL 2100 F transmission electron microscope with a 200 kV accelerating voltage. The Ni, Fe L-edge near-edge X-ray absorption fine-structure data were collected from the soft X-ray beamline of the Australian Synchrotron. The XPS was performed by Shimadzu Kratos Axis Supra with a monochromatic Al Kα X-ray source, and the binding energies were corrected using the C 1 s level at 284.6 eV as the reference. ICP-MS measurements were carried out on PerkinElmer Nexion 2000. The samples were dissolved in acid (HNO3) for two days and then were diluted with Milli-Q water to ensure the signal within appropriate emission intensity.
Electrodes preparation
The working electrodes were prepared using a drop-casting technique. To synthesize the NiFeCl precatalyst ink, NiCl2 and FeCl3 were dissolved in water with a molar ratio of 4:1 at a concentration of 5 mg mL−1. Separately, MClx (Zn, Cr, Mo, and V) were also dissolved in water at the same concentration. Then, 0.05 mL of the NiCl2/FeCl3 solution was diluted in an isopropanol/water mixture (v/v = 1:4), followed by the addition of 1 mg acetylene black (AB) and 0.05 mL of Na+-exchanged Nafion as a binder. For the [Ni0.75Fe0.25]0.6M0.4Clx precatalyst ink, additional MClx was added according to the target stoichiometry. For Ni0.75Fe0.25-LDH ink, either 2 mg or 0.2 mg of catalysts was mixed with 1 mg of AB, dispersed in an isopropanol/water mixture (v/v = 4:15), and supplemented with 0.05 mL Na+-exchanged Nafion. The final ink concentrations were 2 and 0.2 mg mL−1, referred to as high and low loading, respectively. All ink volumes were adjusted to 1 mL using isopropanol/water (v/v = 1:4) and homogenized via ultrasonication for 1 h. Glassy carbon electrodes (0.196 cm2) were polished using 50 nm α-Al2O3 and sonicated in isopropanol and water. Then, 10 μL of each ink was drop-cast onto the electrodes and dried overnight at room temperature. The final catalyst loadings were as follows: 5.4 μgNiFe cm−2 for Ni0.75Fe0.25Clx precatalyst, 6.5 μgNiFe cm−2 for 0.2 mg mL−1Ni0.25Fe0.75-LDH, and 65 μgNiFe cm−2 for 2 mg mL−1 Ni0.25Fe0.75-LDH. For [Ni0.75Fe0.25]0.6M0.4Clx pre catalyst, the final mass loading were 11.51 μgNiFeZn cm−2, 9.59 μgNiFeCr cm−2, 9.88 μgNiFeMo cm−2, and 9.53 μgNiFeV cm−2. To activate the electrodes, ten CVs were performed from 1.225 V to 1.575 V vs. RHE at a scan rate of 10 mV s−1.
Electrochemical characterization
Measurements were conducted in a three-electrode setup, with the prepared electrodes as working electrodes, a 1 × 2 cm2 platinum plate as the counter electrode, and a Hg/HgO (1 M KOH, MMO) reference electrode. The electrolyte was 1 M KOH (pH = 14.00 ± 0.01 measured using pH meter), stored at ambient temperature. The reference electrode was calibrated against a reversible hydrogen electrode (RHE) in the same electrolyte by measuring the open-circuit potential (OCP) of a platinum plate in H2-saturated 1 M KOH. Experiments were performed using a single chamber cell (SC) and a two-compartment H-cell (HC). Nafion 212 (PEM) and Alkymer W-25 (AEM) membranes separated the counter and working/reference electrodes. Each membrane was soaked in 1 M KOH for at least 24 h and rinsed before use. Electrolyte volumes were ~20 mL (SC) and ~30 mL (HC). All electrochemical measurements were carried out in O2- saturated 1 M KOH using a Metrohm AUTOLAB M204 potentiostat. All potentials were iR-corrected based on resistance values obtained from electrochemical impedance spectroscopy (EIS) conducted at OCP over a 100 kHz to 10 Hz range. The resistance (R) was extracted from the high-frequency intercept on the real axis of the Nyquist plot and used in the correction formula: Ecorrected = Emeasured − iR. Each experiment included a fresh EIS measurement to ensure accurate compensation. The conversion of potential from MMO to RHE was performed as follows: E (vs. RHE) = E (vs. MMO) + 0.098 + 0.059 × pH. Cyclic voltammetry (CV) was performed from 1.225 V to 1.575 V vs. RHE at a scan rate of 10 mV s−1. Chronoamperometry (CA) was conducted at 1.53 V vs. RHE. The turnover frequency (TOF) was calculated using TOF = (J × A)/(4Fn), where j is the current density (A cmdisk−2), A is the electrode area (0.196 cm2), F is the Faraday constant (96485 C mol−1), and n is the number of surface-active sites, determined by integrating the Ni2+/Ni3+ redox peaks from CVs and using the nominal Fe/Ni ratio to estimate Fe content (Supplementary Fig. 13).
Metal dissolution analysis
Ni and Fe deposition on the counter electrode was assessed by applying 50 μL of 2 mg mL−1 NiFe-LDH ink onto 1 × 1 cm2 carbon paper (100 μgNiFe-LDH cm−2). After every 5-h CA test, the counter electrode was soaked in 10 mL of 1 M HNO3 for 1 h. The resulting solutions were analyzed by ICP-MS to quantify dissolved Ni and Fe.
Isotope labeling experiment
To investigate oxygen origin, gas chromatography GC-MS experiments (Agilent 7890A-5977B) were performed. Ni0.75Fe0.25Clx, [Ni0.75Fe0.25]0.6Zn0.4Clx pre-catalysts and Ni0.75Fe0.25-LDH were sprayed onto 1 × 1 cm2 titanium felt (1.5 mg cm−2 loading), followed 20 CV cycles (1.22–1.57 vs. RHE, 10 mV s−1) to stabilize the surface. The samples were then subjected to CA at 1.6 V vs. RHE for 10 min in 0.1 M KOH prepared with H218O, followed by thorough rinsing with H216O. Subsequently, CA was performed at 1.4, 1.6, 1.8 V vs. RHE in 1.0 M K16OH for 3 min in an air-tight cell. The evolved gases were collected and analyzed via GC-MS at 110 °C with 3 mL min−1 airflow for 5 min.
DFT study
General parameters
Vienna Ab-initio Simulation Package (VASP)32,33,34 was implemented for all spin-polarized DFT calculations. A cut-off energy of 500 eV was applied for the plain-wave basis set to describe wavefunction. The projector augmented-wave35 pseudopotential was applied to approximate the interactions between the nuclei and core electrons. For geometry optimizations, the force and energy convergence tolerance were set to be 0.02 eV Å−1 and 1 × 10−5 eV, separately. The Perdew-Burke-Ernzerhof functional36 was selected to consider the exchange and correlation interactions of the electrons. The DFT + U method was employed to add penalty term on the strong interactions among the d electrons of Ni and Fe, where the corresponding effective Coulomb integrals (U-J) were set as 5.5 and 3.5 eV, separately37,38,39. 6 × 6 × 1 k-point mesh was used for both (100) and (001) unit cell calculations. As for the simulation of OER process, (001)-p(2 × 2) supercell was chosen for calculations.
OER process simulation & Pourbaix diagram
The OER performance is calculated as follows. As OER is a 4-electron process, it is divided into 4 steps with each step transferring one electron40.
Here, * represents the active site. The equilibrium potential is 1.23 V, assuming the Gibbs free energy changes equally within each step. The theoretical overpotential, which is the minimum potential exceeding the equilibrium potential that makes the reaction downhill, is then evaluated as
The corresponding step is termed as potential determining step. The computational hydrogen electrode (CHE)41 is applied to consider the chemical potential of the electrons and the protons. At standard conditions, i.e., pH = 0, p = 1 bar, 298.15 K and U = 0 V vs. standard hydrogen electrode, we have
A chemical potential equilibrium can be built as
Taking reaction (1) as an example, at standard condition, the Gibbs free energy change can be calculated as
Furthermore, at nonstandard conditions, pH and potential effects are corrected as follows
Consequently, the Gibbs free energy of reaction (1) at nonstandard conditions is
or
Where URHE stands for the potential versus reversible hydrogen electrode (RHE). Similarly, the energies of adsorbing different species with different coverage can be calculated to plot Pourbaix diagram.
As for the Gibbs free energy of adsorbed intermediates, we have
where E is the energy of the system calculated by VASP and TS and ZPE represents the entropy and zero-point energy contributions, respectively. Assuming that the ZPE change of the slab during OER process is negligible, only the ZPE of adsorbate is considered.
Fe capturing
To illustrate Fe capturing capability, we first construct Ni0.75Fe0.25OOH models with appropriate number of cation vacancies. According to past study, Zn2+ has been demonstrated to substitute the Ni2+ site in LDH42. The leaching of Zn2+ creates nominally Ni vacancies in Ni0.75Fe0.25OOH. We calculated the average formation energy of Ni vacancies at various coverage (∆ENi-Vac) as follows:
Here, n, En Ni Vacancies, Eslab, and µNi stands for the number of Ni vacancies, the energies of Ni0.75Fe0.25OOH with vacancies, pristine Ni0.75Fe0.25OOH, and the chemical potential of Ni, respectively. In detail, we use a quarter of energy Ni bulk (fcc) as the chemical potential of Ni (−5.872 eV). According to ∆ENi-Vac results, 1/2-dia was selected to evaluate the Fe capturing capability, as further increasing the vacancy to 2/3 causes significant ∆ENi-Vac increment. We realize the limitation of DFT calculations in dealing with the energy of ions, for which Eq. (13) is used for estimating the trend of energy variation along with Fe filling43,44,45,46.
Two Fe ions were filled into the vacancies, the exchange energy was calculated first.
Assuming the process:
Where * means a hole. The similar problem shows up for dealing with Fe3+. Hence, we introduce the exchange energy by considering the following process:
By adding electrons on both sides and taking the standard electrode potential into account, the energy of ions can be converted into the energy of metals. A similar method was used in the previous literature47.
It should be noted that for standard electrode potentials, the concentration of ions should be 1 M, which is not possible at pH = 14. Extra corrections must be applied based on Nernst equation.
The same approach can be applied on E(Ni/Ni3+), where E(Ni/Ni(OH)3) is calculated by E(Ni/Ni(OH)2) and E(Ni(OH)2/Ni(OH)3).
Then, the exchange energy of Eq. (16) can be calculated as
Similarly, when adding one Fe ion, the exchange energy can also be calculated through this method. By subtracting ∆ENi-Vac, the energy of filling one and two Fe ions can be compared.
Data availability
All data supporting the findings of this study are available within the article and its Supplementary Information/Source Data file, or can be obtained from the authors upon reasonable request. Source data are provided with this paper.
References
Li, A. et al. Atomically dispersed hexavalent iridium oxide from MnO2 reduction for oxygen evolution catalysis. Science 384, 666–670 (2024).
Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: insights into materials design. Science 355, eaad4998 (2017).
Suntivich, J., May, K. J., Gasteiger, H. A., Goodenough, J. B. & Shao-Horn, Y. A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles. Science 334, 1383–1385 (2011).
Kibsgaard, J. & Chorkendorff, I. Considerations for the scaling-up of water splitting catalysts. Nat. Energy 4, 430–433 (2019).
Magnier, L. et al. Fe–Ni-based alloys as highly active and low-cost oxygen evolution reaction catalyst in alkaline media. Nat. Mater. 23, 252–261 (2024).
Wu, T. et al. Spin pinning effect to reconstructed oxyhydroxide layer on ferromagnetic oxides for enhanced water oxidation. Nat. Commun. 12, 3634 (2021).
Wu, T. et al. Reconstruction of thiospinel to active sites and spin channels for water oxidation. Adv. Mater. 35, e2207041 (2023).
Roy, C. et al. Impact of nanoparticle size and lattice oxygen on water oxidation on NiFeOxHy. Nat. Catal. 1, 820–829 (2018).
He, Z. et al. Activating lattice oxygen in NiFe-based (oxy)hydroxide for water electrolysis. Nat. Commun. 13, 2191 (2022).
van der Heijden, O. et al. Li+ cations activate NiFeOOH for oxygen evolution in sodium and potassium hydroxide. Angew. Chem. Int. Ed. 63, e202318692 (2024).
Duan, J., Chen, S. & Zhao, C. Ultrathin metal-organic framework array for efficient electrocatalytic water splitting. Nat. Commun. 8, 15341 (2017).
Dionigi, F. et al. In-situ structure and catalytic mechanism of NiFe and CoFe layered double hydroxides during oxygen evolution. Nat. Commun. 11, 2522 (2020).
Trześniewski, B. J. et al. In situ observation of active oxygen species in Fe-containing Ni-based oxygen evolution catalysts: the effect of pH on electrochemical activity. J. Am. Chem. Soc. 137, 15112–15121 (2015).
Xu, Q. et al. Fluorination-enabled reconstruction of NiFe electrocatalysts for efficient water oxidation. Nano Lett. 21, 492–499 (2021).
Friebel, D. et al. Identification of highly active Fe sites in (Ni,Fe)OOH for electrocatalytic water splitting. J. Am. Chem. Soc. 137, 1305–1313 (2015).
Hu, C. et al. Surface-enhanced Raman spectroscopic evidence of key intermediate species and role of NiFe Dual-catalytic center in water oxidation. Angew. Chem. Int. Ed. 60, 19774–19778 (2021).
Trotochaud, L., Young, S. L., Ranney, J. K. & Boettcher, S. W. Nickel–iron oxyhydroxide oxygen-evolution electrocatalysts: the role of intentional and incidental iron incorporation. J. Am. Chem. Soc. 136, 6744–6753 (2014).
Stevens, M. B., Trang, C. D. M., Enman, L. J., Deng, J. & Boettcher, S. W. Reactive Fe-Sites in Ni/Fe (Oxy)hydroxide are responsible for exceptional oxygen electrocatalysis activity. J. Am. Chem. Soc. 139, 11361–11364 (2017).
Ou, Y. et al. Cooperative Fe sites on transition metal (oxy)hydroxides drive high oxygen evolution activity in base. Nat. Commun. 14, 7688 (2023).
Bao, F. et al. Host, suppressor, and promoter—The roles of Ni and Fe on oxygen evolution reaction activity and stability of NiFe alloy thin films in alkaline media. ACS Catal. 11, 10537–10552 (2021).
Chen, R. et al. Layered structure causes bulk NiFe layered double hydroxide unstable in alkaline oxygen evolution reaction. Adv. Mater. 31, 1903909 (2019).
Farhat, R., Dhainy, J. & Halaoui, L. I. OER catalysis at activated and codeposited NiFe-Oxo/hydroxide thin films is due to postdeposition surface-Fe and is not sustainable without Fe in solution. ACS Catal. 10, 20–35 (2020).
Kuai, C. et al. Phase segregation reversibility in mixed-metal hydroxide water oxidation catalysts. Nat. Catal. 3, 743–753 (2020).
Lu, X. & Zhao, C. Electrodeposition of hierarchically structured three-dimensional nickel–iron electrodes for efficient oxygen evolution at high current densities. Nat. Commun. 6, 6616 (2015).
Wu, L. et al. Boosting oxygen evolution reaction of (Fe,Ni)OOH via defect engineering for anion exchange membrane water electrolysis under industrial conditions. Adv. Mater. 35, 2306097 (2023).
Chung, D. Y. et al. Dynamic stability of active sites in hydr(oxy)oxides for the oxygen evolution reaction. Nat. Energy 5, 222–230 (2020).
Song, W., Xia, C., Zaman, S., Chen, S. & Xiao, C. Advances in stability of NiFe-based anodes toward oxygen evolution reaction for alkaline water electrolysis. Small 20, 2406075 (2024).
Peng, L. et al. Atomic cation-vacancy engineering of NiFe-layered double hydroxides for improved activity and stability towards the oxygen evolution reaction. Angew. Chem. Int. Ed. 60, 24612–24619 (2021).
Yang, C., Fontaine, O., Tarascon, J.-M. & Grimaud, A. Chemical recognition of active oxygen species on the surface of oxygen evolution reaction electrocatalysts. Angew. Chem. Int. Ed. 56, 8652–8656 (2017).
Monteiro, M. C. O. & Koper, M. T. M. Measuring local pH in electrochemistry. Curr. Opin. Electrochem. 25, 100649 (2021).
Grimaud, A. et al. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 9, 457–465 (2017).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Kresse, G. & Hafner, J. Abinitio molecular-dynamics for liquid-metals. Phys. Rev. B 47, 558–561 (1993).
Kresse, G. & Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Dai, W. et al. Electrochemical incorporation of heteroatom into surface reconstruction induced Ni vacancy of NixO nanosheet for enhanced water oxidation. J. Colloid Interface Sci. 608, 3030–3039 (2022).
Li, Y.-F. & Selloni, A. Mechanism and activity of water oxidation on selected surfaces of pure and Fe-doped NiOx. ACS Catal. 4, 1148–1153 (2014).
Lebreau, F., Islam, M. M., Diawara, B. & Marcus, P. Structural, magnetic, electronic, defect, and diffusion properties of Cr2O3: a DFT+ U study. J. Phys. Chem. C. 118, 18133–18145 (2014).
Norskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886–17892 (2004).
Rossmeisl, J., Qu, Z.-W., Zhu, H., Kroes, G.-J. & Nørskov, J. K. Electrolysis of water on oxide surfaces. J. Electroanalytical Chem. 607, 83–89 (2007.
Peng, L. et al. Atomic cation-vacancy engineering of NiFe-layered double hydroxides for improved activity and stability towards the oxygen. Evolution React. 60, 24612–24619 (2021).
Feng, Y. et al. Correlating DFT calculations with CO oxidation reactivity on Ga-doped Pt/CeO2 single-atom catalysts. J. Phys. Chem. C. 122, 22460–22468 (2018).
Hinuma, Y. et al. Density functional theory calculations of oxygen vacancy formation and subsequent molecular adsorption on oxide surfaces. J. Phys. Chem. C. 122, 29435–29444 (2018).
Wexler, R. B., Gautam, G. S., Stechel, E. B. & Carter, E. A. Factors governing oxygen vacancy formation in oxide perovskites. J. Am. Chem. Soc. 143, 13212–13227 (2021).
Zhang, X., Shen, J.-X., Turiansky, M. E. & Van de Walle, C. G. Minimizing hydrogen vacancies to enable highly efficient hybrid perovskites. Nat. Mater. 20, 971–976 (2021).
Li, A. et al. Enhancing the stability of cobalt spinel oxide towards sustainable oxygen evolution in acid. Nat. Catal. 5, 109–118 (2022).
Yin, Z. et al. In situ fabrication of a Ni–Fe–S hollow hierarchical sphere: an efficient (pre)catalyst for OER and HER. N. J. Chem. 45, 12996–13003 (2021).
Han, Y. et al. Self-adaption of Zn introduced Ni-Fe layered double hydroxide for efficient and durable oxygen evolution reaction electrocatalysis. Appl. Surf. Sci. 610, 155288 (2023).
Dastafkan, K. et al. Cosynergistic molybdate Oxo-anionic modification of FeNi-based electrocatalysts for efficient oxygen evolution reaction. Adv. Funct. Mater. 32, 2107342 (2022).
Batchellor, A. S. & Boettcher, S. W. Pulse-electrodeposited Ni–Fe (Oxy)hydroxide oxygen evolution electrocatalysts with high geometric and intrinsic activities at large mass loadings. ACS Catal. 5, 6680–6689 (2015).
Yin, S. et al. A highly efficient oxygen evolution catalyst consisting of interconnected nickel–iron-layered double hydroxide and carbon nanodomains. Adv. Mater. 30, 1705106 (2018).
Zhang, L. et al. Recycling spent ternary cathodes to oxygen evolution catalysts for pure water anion-exchange membrane electrolysis. ACS Nano 18, 22454–22464 (2024).
Meng, C. et al. A Ni-MOF nanosheet array for efficient oxygen evolution electrocatalysis in alkaline media. Inorg. Chem. Front. 8, 3007–3011 (2021).
Acknowledgements
T.W. thank the financial support from the NTU Presidential Postdoctoral Fellowship 2022 and Agency for Science, Technology and Research (A*STAR) through YIRG grant (M22K3c0100). Z.X. acknowledges the support from the Ministry of Education, Singapore through Tier 2 grant (MOE-T2EP10223-0006). Authors appreciate the Facility for Analysis, Characterisation, Testing and Simulation (FACTS) in Nanyang Technological University and Soft X-ray spectroscopy beamline at the Australian Synchrotron (ANSTO) for characterizations. Authors thanks Dr. Heijden, O. van der for discussing the electrochemical results.
Author information
Authors and Affiliations
Contributions
T.W. and Z.X. conceived the idea and designed the experiment. T.W. is responsible for pre-catalyst design. J.Z., Y.Z., and T.W. performed electrochemical measurements and data analysis. Y.Y. conducted the DFT studies. F.M. and Xin.L. conducted the isotope-labelling analysis. S.L. carried out SEM and EDX analyses. Xiao.L., S.Z., and T.W. performed the soft XAS experiment and analyses. A.Y. conducted FIB milling. X.R. conducted the Raman characterization and analysis. T.W., J.Z., and Y.Z. drafted the manuscript with contributions from all authors, and Q.X. and Z.X. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, J., Zhang, Y., Ye, Y. et al. Remote Iron dynamics of NiFe (oxy)hydroxides toward robust active sites in water oxidation. Nat Commun 16, 5601 (2025). https://doi.org/10.1038/s41467-025-60728-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60728-y
