
Abstract
mRNA modifications are vital in regulating cellular processes. Beyond N6-methyladenosine (m6A), most other internal mRNA modifications lack dedicated catalytic machinery and are typically introduced by tRNA-modifying enzymes. The distribution and stoichiometry of these modifications on mRNAs remain debated and require further validation. Furthermore, their precise function remains controversial due to the challenges of excluding the intricate combinational effects of tRNA modifications. Here, we biochemically validate that NSUN6, a tRNA structure-dependent methyltransferase, independently catalyzes 5-methylcytidine (m5C) formation with robust activity on mRNA by recognizing the CUCCA motif in a certain stem-loop structure. NSUN6 employs different strategies to recognize tRNA and mRNA substrates. By introducing mutations, we further separate its catalytic capabilities toward mRNA and tRNA revealing that NSUN6 promotes breast cancer cell migration depending on mRNA m5C modification. Mechanistically, a cohort of mRNAs involved in cell migration carries high levels of NSUN6-mediated site-specific m5C modification, thus being stabilized by the preferential binding of m5C readers YBX1 and YBX3. Moreover, introducing a single-site high-level m5C can significantly increase the stability of therapeutic mRNAs in cells. Our findings underscore the pivotal role of m5C-modified mRNAs in promoting breast cancer cell migration and their potential for therapeutic applications.
Similar content being viewed by others

Introduction
Up to now, a dozen internal mRNA modifications have been identified in eukaryotes, including N6-methyladenosine (m6A), 5-methylcytidine (m5C), N4-acetylcytidine (ac4C), pseudouridine (Ψ), and N1-methyladenosine (m1A). Among them, m6A and Ψ are the most abundant mRNA modifications. Notably, only m6A possesses mRNA-dedicated modifying machinery, whereas most other internal mRNA modifications are introduced by tRNA-modifying enzymes1,2,3,4. The catalytic mechanism of these enzymes toward mRNA substrates is largely elusive, and the authenticity of their role in mRNA modification remains debated5. Consequently, it is necessary to obtain biochemical confirmation of the authenticity and stoichiometry of site-specific modifications on mRNAs. Furthermore, non-m6A mRNA modifications have been reported to regulate various physiological and pathological processes6,7, but these functional studies primarily rely on the loss function of the corresponding modifying enzymes, which fails to separate the effects of mRNA substrates from tRNA substrates8. Still worse, understanding the regulatory role of mRNA modifications is obstructive due to the combinational effect of tRNA modifications in protein translation.
m5C is one of the longest-known RNA modifications widely distributed in various RNA species, including tRNA, mRNA, and rRNA9. It has been reported that the m5C modification plays roles in maintaining tRNA stability, enhancing translation efficiency and accuracy, and regulating the biogenesis of tRNA-derived small RNA10,11. With the advancements of m5C detection technologies based on high-throughput sequencing—such as RNA bisulfite sequencing (BS-seq), m5C-RNA immunoprecipitation (m5C-RIP), 5-azacytidine-mediated RNA immunoprecipitation (5-Aza-RIP), methylation-dependent individual-nucleotide resolution cross-linking and immunoprecipitation (miCLIP), and TET-assisted m5C-to-f5C oxidation with selective chemical labeling sequencing (m5C-TAC-seq)12,13,14,15,16—a comprehensive mapping of mRNA m5C modifications has been conducted in various species. However, due to the low-stoichiometry characteristic of m5C on mRNAs and the inherent limitations of these detection methods, discrepancies exist in the distribution and abundance of m5C identified across different studies17,18,19,20. Thus, it is imperative to validate the authentic sites and levels of mRNA m5C modification using biochemical methods.
In mammals, the m5C modification on RNA is catalyzed by the NOL1/NOP2/SUN (NSUN) family21 and DNA methyltransferase 2 (DNMT2)22. Recent studies have identified tRNA methyltransferases NSUN2 and NSUN6 as the primary ‘writer’ proteins responsible for mRNA m5C modifications23. A hypothetic explanation of mRNAs targeted by the tRNA-modifying machinery is that mRNAs mimic the sequence and/or structure of tRNA substrates. For instance, the TRMT6/TRMT61A-mediated catalysis of m1A requires the GUUCNANNC sequence along with a stem-loop, typically found within a conventional tRNA T-loop structure24. The induction of Ψ modification on mRNA by PUS7 hinges on the presence of a UGUAG consensus motif25, a characteristic shared by all canonical PUS7 tRNA substrates26. Similarly, the catalytic process of Ψ by PUS1 relies on specific structural features found within its tRNA substrates27,28. However, our previous biochemical and structural study illustrates that the full-length tRNA with an L-shaped tertiary structure is necessary for NSUN6 catalyzing tRNA substrates29,30. There is no clue how the tRNA tertiary structure-dependent tRNA-modifying enzymes catalyze mRNA substrates.
In human cells, thousands of m5C sites are identified on mRNA across various studies. The total abundance of m5C in mRNA is ~0.05%, with the median level of m5C methylation at each site around 20%16,18,31,32, which is significantly lower than the median level of 40% observed for m6A modifications33. Among these sites, NSUN6 is responsible for approximately two hundred mRNA m5C sites. Notably, more than 70% of NSUN6-mediated m5C sites on mRNA exhibit low methylation levels (<20%) as reported in multiple studies. In contrast, a small subset of NSUN6-mediated m5C sites displays high methylation levels ranging from 50% to 90%. Our previous work found that catalytically inactive mutation of NSUN6 is associated with neurodevelopmental disorders34. Another study showed that the knockdown of NSUN6 impairs the migration ability of breast cancer cells35. However, the precise molecular mechanism and function involved in m5C modification mediated by NSUN6 on mRNA remain to be elucidated.
Here, we determined that NSUN6 can independently catalyze high-level m5C modifications on mRNA substrates characterized by a stem-loop structure containing a C*UCCA motif, a loop size of 10–12 nucleotides, and non-G-C at the first two base pairs of the stem. Additionally, we identified that NSUN6 employed different strategies to recognize tRNA and mRNA substrates by utilizing distinct combinations of RNA-binding surfaces. We further found that NSUN6 promotes breast cancer cell migration dependent on its mRNA catalytic activity, catalyzing the methylation on a cohort of mRNAs characterized by high levels of m5C, which are extensively involved in cell migration. Moreover, we revealed that Y-box-binding protein 1 and 3 (YBX1 and YBX3) are ‘readers’ of m5C sites mediated by NSUN6 and stabilize the targeted mRNAs by preferentially recognizing m5C sites. Finally, we demonstrated that the stability of therapeutic mRNA could be effectively improved by introducing a single NSUN6-mediated high-level m5C site at the 3’ UTR. Collectively, our work provides a comprehensive mechanistic understanding of the biogenesis and function of human NSUN6-mediated m5C modifications on mRNA.
Results
NSUN6 catalyzes m5C modification on mRNA with high efficiency in vitro
Previous studies have identified the distribution and abundance of NSUN6-mediated m5C sites across various cell lines by high-throughput sequencing methods16,23,32,35,36. While the total number of mRNA substrates varied, a relatively limited subset of mRNAs exhibited high levels of m5C modification (Fig. 1a and Supplementary Fig. 1a). To explore NSUN6 catalytic activity toward these mRNA substrates, we set up in vitro methyl transfer activity assay and RNA Mass Spectrometry (RNA-MS) (Fig. 1b). First, we selected six candidate mRNAs—FURIN, REEP6, TIMM50, MARCKSL1, MAPK3, and PVRL2—which were shared in three or four datasets detected with high m5C levels (Fig. 1a). Then, we purified NSUN6 protein using affinity chromatography coupled with gel filtration (Supplementary Fig. 1b), and prepared the candidate mRNA substrates each approximately 100 nucleotides with m5C sites centrally located using in vitro transcription. The optimal tRNA substrate of NSUN6, tRNAThr(UGU), was used as a positive control37 (Fig. 1c). Our results showed that all examined mRNA substrates except MARCKSL1 could be methylated efficiently by NSUN6. Interestingly, NSUN6 exhibited comparable or even higher catalytic activity for certain mRNA substrates, such as FURIN, MAPK3 and PVRL2, than its activity on tRNAThr(UGU) (Fig. 1d and Supplementary Fig. 1c). Subsequently, we measured the steady-state kinetic parameters of NSUN6 for FURIN under the same condition. The results showed that the Km value for FURIN was 2.14 μM, which was lower than that for tRNAThr(UGU) (3.45 μM). Additionally, the kcat/Km value for FURIN was 0.36 min-1μM-1, which was approximately two folds for tRNAThr(UGU) (0.19 min-1μM-1) (Fig. 1e). These results further suggested that NSUN6 exhibited higher catalytic activity for specific mRNAs than tRNA. Moreover, RNA-MS results confirmed that NSUN6 generated modification on examined mRNA substrates is indeed m5C (Fig. 1f). Collectively, we found that NSUN6 independently catalyzes high levels of m5C on specific mRNAs in vitro, and emphasized that the catalytic activity of NSUN6 toward particular mRNA substrates is comparable to that for tRNA substrate.

a The Venn diagram shows the overlap of NSUN6-mediated mRNA m5C sites from different datasets23,32,35,36. The selected mRNAs with high-level m5C modifications, used in the following experiments, were labeled in red. b Flowchart of the in vitro methyl transfer activity assay and RNA mass spectrometry assay. The selected mRNAs, as shown in a, were produced through in vitro transcription using T7 RNA polymerase and subsequently incubated with purified NSUN6 in the reaction buffer. When 3H-SAM was used as the methyl donor, the reaction mixture was spotted onto filter paper, and the amount of radioactive [3H]-methyl-RNA was measured using a liquid scintillation counter. When SAM was used as the methyl donor, the reacted RNA was precipitated, dissolved, and digested into single nucleosides, and the target peak of nucleoside was detected using the RNA mass spectrometry system. c Schematic representation of transcribed tRNA and mRNAs. The candidate mRNAs were approximately 100nt in length, with m5C sites located in the middle. The position of m5C sites on the transcriptome were indicated by arrows, with the 5’ untranslated region (UTR) labeled in pink, the coding sequence (CDS) in black, and the 3’ UTR in blue. The m5C sites were highlighted in red. d The amount of methyl group incorporation into tRNAThr(UGU), FURIN, MAPK3, PVRL2, REEP6, TIMM50, and MARCKSL1 after incubation with NSUN6 for eight minutes. e The capacity of tRNAThr(UGU) and FURIN to be methylated by NSUN6 and the kinetic parameters of NSUN6 for tRNAThr(UGU) and FURIN in the methyl transfer assay. f Mass spectrometry of the nucleosides m5C (Q1/Q3 = 258.1/126.1) and A (Q1/Q3 = 268.1/136.2) in six mRNAs—FURIN, MAPK3, REEP6, PVRL2, TIMM50, and MARCKSL1—after incubation with or without NSUN6. Target m5C peaks were indicated by black triangles; n.d., not detected. The left vertical axis represents the intensity of A, and the right vertical axis represents the intensity of m5C. Data in d, and e are presented as the mean ± SD for three independent experiments. Source data are provided as a Source Data File.
The sequence and structural characteristics of mRNA substrates with high m5C levels recognized by NSUN6
Previous studies showed that NSUN6 mRNA substrates contain the C*UCCA motifs (* represents the modified C)23,35. However, with approximately two hundred thousand C*UCCA motifs present in human RNA transcripts, it remains unclear how NSUN6 selectively targets specific mRNA substrates and modifies them with high m5C levels. To investigate the structural features of the optimal substrates catalyzed by NSUN6, we performed selective 2’-hydroxyl acylation analyzed by primer extension (SHAPE) assays to resolve the RNA secondary structures38. Hydroxyl-selective electrophiles such as 1-methyl-7-nitroisatoic anhydride (1M7) prefer to form stable 2’-O-adducts with flexible nucleotides, and sites of 2’-O-adduct formation are identified as stops for primer extension by reverse transcriptase. Further, the SHAPE reactivity of each nucleotide site was analyzed by sequencing through high-resolution polyacrylamide gel electrophoresis, characterizing the structural features of substrate RNAs39 (Fig. 2a). The SHAPE-based secondary structure models revealed that all NSUN6 mRNA substrates exhibit a stem-loop structure, with the C*UCCA motif positioned in the loop region and the stem is formed by at least four base-pairs (Fig. 2b–e).

aThe flowchart of the SHAPE experiment. The SHAPE analysis of mRNA substrates of NSUN6—FURIN (b), MAPK3 (c), PVRL2 (d), and REEP6 (e), respectively. The left panel displayed the SHAPE reactivity of each detected nucleotide, and the right panel showed the SHAPE-based secondary structure model of the mRNA. The blue triangles indicated the target m5C sites catalyzed by NSUN6. Schematic diagrams showing the secondary structures of FURIN-100nt (f) and MAPK3-100nt (g), along with their 50nt-length and 25nt-length mutants. The m5C sites on the mRNAs were highlighted in red, and the UCCA motifs were labeled in blue. The amount of methyl moiety incorporated into FURIN-100nt (h), MAPK3-100nt (i), and their 50nt-length and 25nt-length mutants after incubation with NSUN6 for eight minutes. j Schematic diagram showing the secondary structure of FURIN-25nt, with mutations in the C*UCCA motif on the loop indicated in blue, and the m5C sites on the mRNAs were highlighted in red. k The amount of methyl moiety incorporated into FURIN-25nt and the FURIN-25nt mutants on the loop region after incubation with NSUN6 for eight minutes. Data in h, i, and k are presented as the mean ± SD for three independent experiments. p values are from one-way ANOVA followed by Tukey’s multiple comparisons test (h, i). ns no significance. Source data are provided as a Source Data File.
We further determined the minimal structural elements of mRNA substrates required by NSUN6. Using FURIN-100nt and MAPK3-100nt mRNAs as representative NSUN6 substrates, we truncated them to 50nt and 25nt shorter forms, preserving the stem-loop core containing the C*UCCA motif. The 25nt form retains a single stem-loop, while the 50nt form includes flanking sequences on both sides of the stem-loop (Fig. 2f, g). We determined the catalytic activity of NSUN6 on these truncated mRNAs and observed that the 25nt and 50nt truncated forms were catalyzed by NSUN6 with the same efficiency as their longer counterparts (Fig. 2h, i and Supplementary Fig. 1d, e). In addition, we relocated the C*UCCA motif to stem region while preserving the stem-loop structure of FURIN-25nt, and we found that this mutant (FURIN-CUCCA-stem) could not be methylated by NSUN6 (Supplementary Fig. 1f, g). The above results indicate that mRNA substrates with a single stem-loop structure, where the loop region contains the C*UCCA motif, are sufficient for catalysis by NSUN6.
SHAPE-based secondary structure of NSUN6 mRNA substrates showed that the loop size in the stem-loop varied from 10 to 13 nucleotides. mRNAs with higher m5C levels have loop sizes of 10 to 12 nucleotides, whereas REEP6, which has a lower level, has a loop size of 13 nucleotides (Fig. 2b–e). To investigate the optimal loop size of mRNA substrates catalyzed by NSUN6, we altered the loop sizes of FURIN-25nt by inserting or deleting nucleotides and subsequently measured the catalytic capability of NSUN6 toward these FURIN mutants (Supplementary Fig. 2a). The results showed that when the number of nucleotides in the loop was greater than 12 or fewer than 10, the catalytic activity of NSUN6 on these mutants was dramatically reduced (Supplementary Fig. 2b, c). Our findings indicate that mRNA substrates with loop sizes between 10 and 12 nucleotides are catalyzed more efficiently by NSUN6.
To further investigate the sequence features of NSUN6 mRNA substrates, we analyzed their sequence preference. We observed that the C*UCY(Y = C/U)A motif, present in all mRNA substrates, was located randomly on the loop, with no preference for other motifs. The first two base pairs in the stem preferred to be C-G or U-A, while the remaining base pairs could be any Watson-Crick base pair. First, we made mutations in the loop region of FURIN-25nt by individually substituting the “13UCCA16” sequence with three other kinds of nucleotides and assessed the catalytic capability of NSUN6 toward these mutants (Fig. 2j). Our data showed that U13, C14, and A16 are strictly required, as all the corresponding mutants could not be catalyzed by NSUN6. For C15, the catalytic activity of NSUN6 on the FURIN-25nt C15U mutant is about half that of the FURIN-25nt WT, while the C15A and C15G mutants lost the ability to be catalyzed by NSUN6 (Fig. 2k and Supplementary Fig. 2d–g). The above results suggest that the C*UCYA motif is necessary for NSUN6-targeted mRNAs, whereas the C*UCCA motif is the optimal one for NSUN6-targeted mRNA substrates. Further, we mutated the first two C-G base pairs in the stem of FURIN-25nt to other base pairs and determined the catalytic capability of NSUN6 toward these mutants (Supplementary Fig. 2h, k). Our results showed that NSUN6 could catalyze all these mutants. For the first base pair, NSUN6 exhibited nearly the same catalytic activity for the A-U and U-A mutants as for the FURIN-25nt WT, while its catalytic activity for the G-C mutants was significantly reduced (Supplementary Fig. 2i, j). Interestingly, the second base pair displayed a similar preference to the first (Supplementary Fig. 2l, m). These results suggest that the sequences of stem regions of NSUN6 mRNA substrates are relatively flexible.
Overall, we found that the minimal mRNA structure necessary for NSUN6 recognition is a single stem-loop containing the C*UCYA motif in the loop. Notably, mRNA substrates with high m5C levels are characterized by the C*UCCA motif within a loop of 10 to 12 nucleotides, along with non-G-C base pairs at the first two positions of the stem.
NSUN6 recognizes mRNA and tRNA substrates in different ways
Our previous study showed that NSUN6 catalyzes tRNA substrates requiring the unique L-shaped tertiary structure of tRNA, but is unable to recognize or catalyze tRNA minihelix containing the C*UCCA motif 30. In contrast to the stringent recognition conditions for tRNAs, NSUN6 recognized a stem-loop structure with the C*UCCA motif in the loop for mRNA substrates (Fig. 3a). We then investigated the mechanism by which the tRNA tertiary structure-dependent NSUN6 catalyzes mRNA substrates. The crystal structure of the NSUN6-tRNA complex revealed that a catalytic pocket formed by the methyltransferase (MTase) domain and the pseudouridine synthase and archaeosine transglycosylase (PUA) domain of NSUN6 is responsible for catalyzing m5C modification in the C*UCCA motif at 3’ end of the tRNA receptor stem (Fig. 3b, c)29. In addition, when NSUN6 recognizes the L-shaped tRNA, the positively charged patch 1 on the PUA domain binds to the D-stem of tRNA, while the positively charged patch 2 on the MTase domain binds to the acceptor stem of tRNA (Fig. 3d). The tRNA is anchored by the catalytic pocket and the two positively charged patches. NSUN6 mRNA substrates contain the C*UCYA motif that resembles the 3’ C*UCCA end of tRNA substrates. To investigate whether NSUN6 catalyzes the tRNA and mRNA substrates using the same catalytic pocket, we introduced a C75U mutation in tRNAThr(UGU) to generate 3’ C*UCUA end and measured the catalytic capability of NSUN6 toward this mutant (Fig. 3e). The results showed that the catalytic activity of NSUN6 for tRNAThr(UGU)-C75U was reduced to half that of tRNAThr(UGU) (Fig. 3f), which is similar to the effect of changing C*UCCA to C*UCUA in mRNA substrates. This suggests that NSUN6 recognizes the C*UCYA motif of tRNA and mRNA substrates using the same catalytic pocket. To further explore how NSUN6 binds to its mRNA substrates, we used AlphaFold3 to build the structural model of NSUN6 and FURIN-25nt complex40. The model of the NSUN6-mRNA complex shows that the C*UCCA motif binds to the catalytic pocket of NSUN6 in a manner similar to that of tRNA substrates (Fig. 3g).

a Schematic diagram showing the secondary structure of NSUN6 mRNA substrates (left), tRNAThr (middle), and tRNAThr-minihelix (right). m5C sites were labeled in red, and light-on represented substrates that could be catalyzed by NSUN6, while light-off represented those that cannot. b Crystal structure of the NSUN6-tRNA complex (PDB: 5WWS) in cartoon representation, with tRNA in gold and the C*UCCA motif marked in dark red. c The binding of tRNA (gold) to NSUN6, shown as an electrostatics surface representation. d The electrostatic surface of NSUN6 highlighting its two main positively charged patches 1 and 2, and the catalytic pocket. Key amino acids in these regions were labeled. e Schematic diagram showing the secondary structure of the tRNAThr(UGU) and the tRNAThr(UGU)-C75U mutant. The m5C site was highlighted in red, and the blue arrow indicated the mutant position. f The amount of methyl group incorporation into tRNAThr(UGU) and the tRNAThr(UGU)-C75U mutant after incubation with NSUN6 for eight minutes. g Structural model of NSUN6 and FURIN-25nt complex generated using AlphaFold3. NSUN6 was shown as an electrostatics surface representation, the loop region of FURIN-25nt (dark red) was combined with and the catalytic pocket of NSUN6 and the stem region of FURIN-25nt (gold) was close to the positively charged patch 2 on NSUN6. h FURIN-25nt contacted the positively charged patch 2 on the MTase domain (purple) of NSUN6, rather than patch 1 on the PUA domain (pink). The stem region of FURIN-25nt was in gold, and the loop region was in dark red. The amount of methyl moiety incorporated into FURIN-25nt (i) or tRNAThr(UGU) (j) after incubation with NSUN6 WT, NSUN6-K159A/R181A, NSUN6-R352A/K353A, and NSUN6-R352E/K353E mutants for eight minutes. k Schematic diagram showing how NSUN6 recognized mRNA and tRNA substrates differently. Data in f, i, and j are presented as the mean ± SD for three independent experiments. Source data are provided as a Source Data File.
Unlike the tRNA substrates, the positively charged patch 1 on NSUN6 does not contact with FURIN-25nt. Instead, the positively charged patch 2 on NSUN6 is in direct contact with the loop of the mRNA (Fig. 3h). Based on this model, we mutated key basic amino acid residues in positively charged patch 1 (K159 and R181) and patch 2 (R352 and K353) to generate NSUN6-K159A/R181A, NSUN6-R352A/K353A, and NSUN6-R352E/K353E mutants (Fig. 3d). Next, we determined the catalytic activity of these NSUN6 mutants for the FURIN-25nt and tRNAThr(UGU). Our data showed that, compared with NSUN6-WT, NSUN6-K159A/R181A double mutant completely lost catalytic activity toward tRNAThr(UGU) but retained similar catalytic efficiency for mRNA substrates (Fig. 3i, j and Supplementary Fig. 3a–d), indicating that the positively charged patch 1 is not involved in the NSUN6 recognition of mRNA substrates. However, the catalytic efficiency of the NSUN6-R352A/K353A mutant in patch 2 for both mRNA substrates and tRNAThr(UGU) decreased sharply compared to NSUN6-WT. Furthermore, the NSUN6-R352E/K353E mutant was unable to catalyze either mRNA substrates or tRNAThr(UGU) (Fig. 3i, j and Supplementary Fig. 3a–d). These results align closely with the observation from the predicted model of the NSUN6-mRNA complex. Together, our results suggest that NSUN6 recognizes tRNAs and mRNAs using different strategies by employing distinct combinations of RNA-binding surfaces (Fig. 3k). We provided a mechanism for how a multi-substrate methyltransferase recognizes tRNA and mRNA substrates.
NSUN6 promotes breast cancer cell migration dependent on its mRNA modification activity
The above findings prompt us to further investigate the distinct functions of NSUN6-mediated m5C modification in tRNA and mRNA substrates. NSUN6 is highly expressed in various cancers, making it a potential indicator for predicting survival and progonosis41. A previous study has shown that knocking down NSUN6 expression inhibits the migration ability of MDA-MB-231 cells35. Similarly, we knocked down the expression of NSUN6 in the BT-549 and MDA-MB-468 cell lines and found that the migration ability of these cells were significantly restricted, indicating that NSUN6 facilitates cell migration in several triple-negative breast cancer cells (Supplementary Fig. 4a, b). To elucidate the molecular mechanism of NSUN6 in breast cancer cell migration, we first constructed NSUN6 knockout (KO) cell lines in MDA-MB-231 cells (Supplementary Fig. 4c, d). The CCK-8 assays revealed that NSUN6 knockout did not affect cell proliferation (Supplementary Fig. 4e). However, transwell assays showed that cell migration was significantly attenuated in NSUN6 KO cells (Supplementary Fig. 4f, g). The migration ability of NSUN6 KO cells was restored by stable expression of wild-type NSUN6 (KO + NSUN6 WT). Notably, introducing the NSUN6 mutant (KO + NSUN6 K159A/R181A), which retained only the catalytic capability for mRNA, successfully restored migration ability. In contrast, rescuing with the NSUN6 mutant lacking catalytic capacity for both tRNA and mRNA (KO + NSUN6 C373A) failed to restore migration (Fig. 4a, b and Supplementary Fig. 4h). The above results suggest that NSUN6 promotes the migration of MDA-MB-231 cells through its mRNA modification activity.

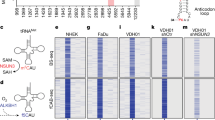
a Transwell migration assays in MDA-MB-231 WT cells, NSUN6 KO cells, and NSUN6 KO cells that stably expressed the empty vector (KO + EV), wild-type NSUN6 (KO + NSUN6-WT), an NSUN6 mutant with retained catalytic capability for mRNAs (KO + NSUN6-K159A/R181A), and an NSUN6 mutant lacking catalytic capability for both tRNAs and mRNAs (KO + NSUN6-C373A). For each group, four different fields were chosen and counted. b The statistical analysis of transwell migration assays in a. c Northern blot analysis of the stability of NSUN6 tRNA substrates in WT and NSUN6 KO MDA-MB-231 cells. The experiment was independently repeated three times, and the statistical analysis was shown in Supplementary Fig. 4i. d Northern blot analysis of in vivo aminoacylation level of NSUN6 tRNA substrates in WT and NSUN6 KO MDA-MB-231 cells. Samples were also deaminoacylated (DEA) by incubating with the alkaline buffer (pH 9.0) to obtain a fully uncharged tRNA control. The experiment was independently repeated three times, and the statistical analysis was shown in Supplementary Fig. 4j. e Volcano plots of differential expressed genes from RNA-seq data, comparing NSUN6 knockdown MDA-MB-231 cells (n = 2) to control cells (n = 2). The p values were calculated using a moderated t-test, and adjusted for multiple comparisons using the Benjamini-Hochberg false discovery rate (FDR) method. Significantly upregulated genes (FDR corrected p < 0.05) were colored in pink, and downregulated genes in blue. Labels of interesting genes that were mRNA substrates of NSUN6 were highlighted in orange, and those associated with cell migration in black. f The RT-qPCR analysis of the relative level of NSUN6 mRNA substrates in WT and NSUN6 KO MDA-MB-231 cells. g Gene Ontology analysis of downregulated genes in NSUN6 knockdown MDA-MB-231 cells from RNA-seq. The p-values were calculated using the hypergeometric test. h Detected m5C site distribution with different modification fractions in MDA-MB-231 mRNA. Of 1136 detected sites (binomial test, p < 10−6), 504, 204, and 47 showed modification fractions ≥10%, 20%, and 50%, respectively (n = 2). i Boxplot showing the change in m5C levels at all m5C sites following NSUN6 knockout in MDA-MB-231 cells. Boxes represent the 25–75th percentile (line at the median). j Detected NSUN6-mediated m5C site distribution with different modification fractions in MDA-MB-231 mRNA. Of 205 detected sites (binomial test, p < 10−6), 168, 76, and 18 showed modification fractions ≥10%, 20%, and 50%, respectively (n = 2). k Boxplot showing the change in m5C levels at NSUN6-mediated m5C sites following NSUN6 knockout in MDA-MB-231 cells. Boxes represent the 25th-75th percentile (line at the median). l Motif analysis showing enriched motifs at NSUN6-mediated mRNA:m5C sites in MDA-MB-231 cells. m Distribution of m5C sites along mRNA in MDA-MB-231 cells. n Pie chart displaying the proportion of m5C sites in MDA-MB-231 cells (n = 205). o Gene Ontology analysis of NSUN6-targeted genes from UBS-seq, showing the p-value for enrichment of biological process GO terms. Cell migration-related pathways were highlighted in red. The p-values were calculated using the hypergeometric test. p–s, The Venn diagram showing the overlap between NSUN6 mRNA substrates in MDA-MB-231 cells and total mRNAs detected in GEO: GSE49339 (p)47 and GEO: GSE102113 (r)48. The comparisons of mRNA half-lives between transcripts containing NSUN6-mediated m5C sites and all transcripts were reanalyzed from GEO: GSE49339 (q) and GEO: GSE102113 (s). Data in f is presented as the mean ± SD for three independent experiments, and data in i, and k are presented for two independent experiments. p-values are from two-tailed t test (b, f) or one-way ANOVA followed by Tukey’s multiple comparisons test (i, k). ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05. Source data are provided as a Source Data File.
To investigate the impact of NSUN6 on its tRNA substrates in MDA-MB-231 cells, we detected the steady-state and aminoacylation levels of NSUN6 tRNA substrates using Northern blot assays. The results showed that, compared to WT cells, there were no significant changes in either the stable levels or the aminoacylation levels of tRNAThr(UGU) and tRNACys(CGA) following the knockout of NSUN6 (Fig. 4c, d and Supplementary Fig. 4i, j). This suggests that, under normal culturing conditions, NSUN6-mediated m5C modification did not significantly affect the stability and aminoacylation levels of tRNA substrates in MDA-MB-231 cells.
Thus, we focused on the effects of NSUN6-mediated m5C modification on mRNA substrates for the subsequent studies. To explore the function of NSUN6-modified mRNA substrates, we performed RNA-seq transcriptomic analysis following NSUN6 knockdown in MDA-MB-231 cells (Supplementary Data 1). Compared to the control group, 265 genes were significantly downregulated, while 116 genes were significantly upregulated, with changes exceeding two fold (Fig. 4e). Subsequently, these results were validated by RT-qPCR. NSUN6 target mRNAs, including REEP6, FURIN, MAPK3, and PVRL2, exhibited reduced expression in NSUN6 KO MDA-MB-231 cells42,43,44,45 (Fig. 4f). Additionally, the mRNA levels of genes associated with cell migration, such as VTN, CEMIP, LOXL1, and PLOD2, were significantly downregulated (Supplementary Fig. 4k). Furthermore, we performed Gene Ontology (GO) analysis on RNA-seq data. The results showed that pathways related to cell migration, such as extracellular matrix (ECM) organization and extracellular structure, were the most highly enriched among the downregulated genes in NSUN6 knockdown MDA-MB-231 cells (Fig. 4g).
Next, we investigated how NSUN6-mediated m5C modification on mRNA affects cell migration in MDA-MB-231 cells. We first performed the ultrafast bisulfite sequencing (UBS-seq)32 to detect the mRNA m5C sites of this cell line, identifying the NSUN6-mediated sites by comparing detected mRNA m5C sites from WT and NSUN6 KO cells (Supplementary Data 2). In total, we identified 1136 m5C sites in poly(A) RNA from MDA-MB-231 cells. Among these, 504 sites exhibited m5C levels exceeding 10%, 204 sites had m5C levels surpassing 20%, and only 47 sites demonstrated m5C levels greater than 50% (Fig. 4h). The median overall m5C modification level in MDA-MB-231 WT cells was approximately 15%, whereas it dropped to about 5% in NSUN6 KO cells (Fig. 4i). Further analysis showed that NSUN6 catalyzes 205 out of the 1136 m5C sites in MDA-MB-231 cells (Fig. 4j). Remarkably, when NSUN6 was knocked out, the m5C level of poly(A) RNA significantly decreased (Fig. 4k), suggesting that NSUN6 is responsible for modifying the majority of high m5C-level mRNA sites in MDA-MB-231 cells. Notably, our sequencing results indicated a strong sequence preference for C*UCYA at NSUN6-dependent sites compared to all intracellular m5C sites (Fig. 4l), consistent with our biochemical validation. Furthermore, we analyzed the distribution of NSUN6-dependent sites along the transcripts and found that a large proportion were located in the coding sequence (CDS), accounting for 150 sites (73.07%). Additionally, approximately half of these sites was situated near the 3’ untranslated region (UTR). We also observed enrichment of NSUN6-mediated m5C sites in the 3’ UTR, comprising 46 sites (22.52%) (Fig. 4m, n). To investigate the biological function of NSUN6 substrates, we performed GO analysis on the NSUN6 target mRNAs. The results indicated that the NSUN6 mRNA substrates with m5C levels greater than 20% in the MDA-MB-231 cells exhibited significant enrichment in pathways associated with cell migration, including cell-substrate junction assembly, cell-substrate junction organization, and actin cytoskeleton reorganization (Fig. 4o). These data suggests that NSUN6 mRNA substrates are closely associated with cell migration.
Previous studies have revealed the important role of NSUN2-mediated m5C modification in maintaining mRNA stability31,46. To investigate the function of NSUN6-mediated m5C on mRNA substrates, we analyzed the overall half-lives of NSUN6 mRNA substrates using data from two independent public RNA stability datasets47,48. The analyses showed that mRNA substrates of NSUN6 exhibited longer half-lives compared to the average level observed across the datasets (Fig. 4p–s), implying that NSUN6-mediated site-specific m5C modification could also contribute to the increased mRNA stability.
A cohort of migration-related mRNAs with high m5C levels, mediated by NSUN6, regulates breast cancer cell migration
In the UBS-seq analysis of MDA-MB-231 cells, we observed that 18 mRNAs targeted by NSUN6 exhibited high m5C stoichiometries exceeding 50% (Fig. 4j). Notably, nine of these mRNAs have been reported to play roles in cell migration, including PVRL2 49, FURIN 50, MAPK3 51, MCAM 52, PXDN 53, TRAF7 54, IGFBP6 55, NCOR1 56, and LMAN1 57 (Fig. 5a and Supplementary Fig. 5). Using the in vivo RNA secondary structure databases58, we found that the sequence and structural characteristics around these mRNA m5C sites satisfied the NSUN6 preference for mRNA substrates with high m5C levels (Fig. 5a). Furthermore, we observed significant decrease in m5C levels of these migration-related mRNAs in NSUN6 KO cells (Fig. 5b). We further performed the in vitro methyl transfer activity assay to ascertain the catalytic activity of NSUN6 on these mRNAs. The results showed that NSUN6 exhibited higher catalytic activities on these mRNAs compared to tRNA positive control (Fig. 1d and Supplementary Fig. 6a). These findings suggest that NSUN6 contributes to the high m5C modification on a cohort of cell migration-related mRNAs in MDA-MB-231 cells.

a The left panel showed the m5C levels of NSUN6 mRNA substrates in MDA-MB-231 cells. mRNAs with m5C levels higher than 50% were highlighted in a blue dotted box. mRNAs involved in cell migration were labeled in pink, other mRNAs in grey. The right panel showed the secondary structures of cell migration-related NSUN6 mRNA substrates with high m5C levels, with m5C sites labeled in orange. Specific functions of these substrates were also listed in Supplementary Data 3. b Representative heatmaps of changed m5C levels of selected mRNAs between WT and NSUN6 KO MDA-MB-231 cells in UBS-seq. c The RT-qRCR analysis of the mRNA half-lives of FURIN, MAPK3, and MCAM in WT and NSUN6 KO MDA-MB-231 cells. d Transwell migration assays in MDA-MB-231-WT cells, NSUN6 KO cells, and NSUN6 KO cells stably expressing the empty vector (KO + EV), NSUN6 WT (KO + NSUN6-WT), FURIN (KO + FURIN), MAPK3 (KO + MAPK3), PVRL2 (KO + PVRL2), MCAM (KO + MCAM), and cells simultaneously expressing the above four genes (KO+ four mRNAs). For each group, five different fields were chosen and counted. e Statistical analysis of transwell migration assays in d. f Analysis of mRNA expression levels of NSUN6-targeted genes in patients, comparing those with low (first quartile, n = 36) and high (fourth quartile, n = 37) NSUN6 expression, was conducted using the TCGA online portal (https://www.cbioportal.org/). The top and bottom edges of the box represent the 75th and 25th percentiles, respectively. The central line in each box represents the median value, and the whiskers extend to the minimum and maximum values obtained in the dataset. g Kaplan–Meier survival analysis (http://gepia2.cancer-pku.cn/) of the relationship between the mRNA expression of NSUN6 (n = 67 (low)/67 (high)) and the overall survival time (OS) of triple-negative breast cancer patients. Data in c is presented as the mean ± SD for three independent experiments. p-values are from one-way ANOVA followed by Tukey’s multiple comparisons test (e) or two-tailed t test (f). ****p < 0.0001, ***p < 0.001, ns no significance. Source data are provided as a Source Data File.
Next, we investigated the impact of NSUN6-mediated high m5C modification on mRNA half-lives using RNA stability assays. The results showed that these NSUN6-modified mRNA substrates exhibited shortened half-lives following NSUN6 knockout in MDA-MB-231 cells (Fig. 5c). To illustrate the role of specific mRNAs with high m5C levels in NSUN6-mediated migration, we constructed stable cell lines expressing PVRL2, FURIN, MAPK3, and MCAM in NSUN6 KO MDA-MB-231 cells, restoring mRNA levels to resemble those in WT cells (Supplementary Fig. 6b). We then used transwell assays to assess their migration ability. The results showed that the migration ability of NSUN6 KO cells was significantly enhanced to varying extents by expressing these migration-related mRNAs with high m5C levels (Fig. 5d, e). Notably, when these four mRNAs with high m5C levels were simultaneously restored, the migration of NSUN6 KO cells was successfully recovered to that of the WT cells (Fig. 5d, e and Supplementary Fig. 6c). This suggests that NSUN6 promotes MDA-MB-231 cell migration by the coordination of several mRNA substrates, rather than a single target.
Next, we investigated the correlation between the expression levels of NSUN6 and these migration-related mRNAs in breast cancers. Using the TCGA online portal (https://www.cbioportal.org/), we performed an NSUN6-centered analysis. We selected the RNA-seq data from The Metastatic Breast Cancer Project and categorized samples by quartiles based on NSUN6 mRNA expression levels59. Our analysis unveiled a positive correlation between NSUN6 and eight of the nine migration-related mRNAs in metastatic breast tumors (Fig. 5f). Notably, Kaplan-Meier survival analysis (http://gepia2.cancer-pku.cn/)60 for expression of NSUN6 and the clinical behaviors of triple-negative breast cancer suggested high NSUN6 expression was significantly correlated with poor prognosis (Fig. 5g).
In summary, NSUN6 catalyzes high levels of m5C modifications on a cohort of migration-related mRNAs in breast cancer MDA-MB-231 cells. These site-specific m5C modifications enhance mRNA stability, thereby promoting breast cancer cell migration. Moreover, a significant correlation between NSUN6 expression and migration-related NSUN6 mRNA substrates was observed in clinical metastatic breast cancer samples, highlighting the potential role of NSUN6 in metastatic progression.
YBX1 and YBX3 are the readers of NSUN6-mediated mRNA:m5C and stabilize m5C-modified mRNA
To identify reader proteins that preferentially recognize NSUN6-mediated m5C modifications and regulate mRNA stability, we performed RNA pull-down experiments. We used transcribed FURIN-25nt, either with or without m5C modification, and incubated it with cytoplasmic protein lysate from MDA-MB-231 cells. The enriched proteins were then analyzed using mass spectrometry (Fig. 6a and Supplementary Data 4). Among the enriched proteins from our RNA pull-down experiments, we identified several potential readers, including YBX1 and YBX3. YBX1 is a known reader protein for m5C mediated by NSUN2 (Fig. 6b). To confirm the role of YBX1 and YBX3 as readers of NSUN6-mediated m5C on mRNA, we performed in vivo pull-down assays. The results demonstrated that YBX1 could be effectively pulled down by FURIN-25nt-m5C compared to its unmodified counterpart (Fig. 6c). Similarly, YBX3 showed a stronger binding affinity for FURIN-25nt-m5C than for the unmodified version (Fig. 6d). The Y-box protein family, which includes YBX1, YBX2, and YBX3, share similar sequence and structural features in the cold shock domain (CSD) that bind target RNA61 (Supplementary Fig. 7a). We examined the RNA-binding affinities of the YBX1 CSD (residues 48–159) for unmodified and m5C-modified FURIN-25nt using Biolayer interferometry (BLI). The YBX1 CSD exhibited higher binding affinity for m5C-modified mRNA (KD = 213.8 nM) compare to unmethylated mRNA (KD = 346.1 nM) (Fig. 6e). Similarly, the YBX3 CSD (residues 82-174) showed stronger binding affinity to FURIN-25nt-m5C (KD = 45.0 nM) than FURIN-25nt (KD = 87.5 nM) (Fig. 6f). The results indicated that both YBX1 and YBX3 have a stronger preference for NSUN6-methylated mRNAs than for unmodified ones. Previous structural studies revealed that the tryptophan residue (W65) in YBX1 CSD is crucial for recognizing NSUN2-catalyzed m5C modification31. To gain further insight into the binding details of YBX1 and YBX3 with NSUN6-methylated mRNA, we used AlphaFold3 to build the complex models of YBX1 CSD-PVRL2 and YBX3 CSD-PVRL2 (Fig. 6g and Supplementary Fig. 7b). The predicted models showed that W65 in YBX1 and W97 in YBX3 form π-π stacks with the m5C site (Fig. 6h and Supplementary Fig. 7c), suggesting that YBX1 and YBX3 preferentially bind to mRNA with NSUN6-mediated m5C compared to unmethylated mRNA. Subsequently, we analyzed the YBX1 and YBX3 target mRNAs from published CLIP datasets and found a significant overlap of 82% (160 out of 194) between the target mRNAs of YBX1 or YBX3 and the NSUN6-mediated m5C mRNAs identified through our UBS-seq analysis31,62 (Fig. 6i). These findings collectively suggest that YBX1 and YBX3 act as the reader proteins for NSUN6-mediated m5C modifications on mRNAs.

a Schematic diagram showing the flowchart of the RNA pull-down experiment. Biotin-labeled FURIN-25nt and FURIN-25nt-m5C were incubated with the cytoplasmic lysate of MDA-MB-231 cells. Proteins enriched by streptavidin were analyzed using mass spectrometry. b Scatter plot showing proteins binding to FURIN-25nt-m5C versus FURIN-25nt RNA in MDA-MB-231 cells. Potential reader proteins YBX1 and YBX3 were highlighted in red. Western blot analysis of the FLAG-YBX1 (c) or FLAG-YBX3 (d) pulled down by biotin-labeled FURIN-25nt or FURIN-25nt-m5C in HEK293T cells (left panels) and the corresponding quantification (right panels).Biolayer interferometry analysis of RNA binding affinity of the YBX1 CSD (e), and YBX3 CSD (f) with unmodified FURIN-25nt and FURIN-25nt-m5C. g The structural model of the YBX3 CSD and PVRL2-25nt complex generated using AlphaFold3. PVRL2-25nt was in dark blue, and the YBX3 CSD was in yellow. h Recognition of m5C by YBX3 in the AlphaFold3-generated YBX3 CSD and PVRL2-25nt complex. Hydrogen bonds were indicated with white dashes, and residues involved in the recognition of m5C were labeled. i The Venn diagram of the overlap between mRNA substrates of NSUN6 and mRNAs targeted by YBX1 and YBX3 from the published CLIP datasets31,62. The RT-qRCR analysis of mRNA half-lives following YBX1 (j) or YBX3 (k) knockdown in MDA-MB-231 cells. l The RT-qRCR analysis of mRNA half-lives in YBX1 knockdown MDA-MB-231 cells overexpressing either YBX1 WT or the YBX1 W65F mutant. m The RT-qRCR analysis of mRNA half-lives in YBX3 knockdown MDA-MB-231 cells overexpressing either YBX3 WT or the YBX3 W97F mutant. Data in c–f, and j–m are presented as the mean ± SD for three independent experiments. p-values are from two-tailed t test (c, d) or one-tailed t test (e, f). ***p < 0.001, **p < 0.01, *p < 0.05. n A simplified model shows the biogenesis and function of NSUN6-mediated high-stoichiometry m5C modifications on a cohort of cell migration-related mRNAs in breast cancer cells. Created in BioRender. Zhang, Y. (2025) https://BioRender.com/bk4f7pg. Source data are provided as a Source Data File.
Previous studies have identified that YBX family members generally function as RNA binding proteins (RBPs), stabilizing their bound mRNAs and modulating translation61,62,63,64,65. Additionally, YBX1 has been demonstrated as an m5C reader, regulating the stability of target transcripts by recognizing the m5C modifications mediated by NSUN2 in various cancers31,63,66,67. To assess their impact on NSUN6 mRNA substrates, we examined the stability of several mRNAs with high m5C levels in YBX1 knockdown or YBX3 knockdown MDA-MB-231 cells. The results revealed that the stabilities of these mRNAs were decreased upon knockdown of YBX1 or YBX3 (Fig. 6j, k and Supplementary Fig. 7d, e). To confirm the role of YBX1 and YBX3 in stabilizing mRNAs through the recognition of m5C modification, we overexpressed wild-type and recognition-deficient mutants of YBX1 (W65F) and YBX3 (W97F) in their respective knockdown MDA-MB-231 cells. Our data showed that the decreased mRNA half-lives caused by YBX1 knockdown were restored by YBX1 WT but not by the YBX1 W65F mutant (Fig. 6l and Supplementary Fig. 7f). Similarly, YBX3 WT, but not YBX3 W65F mutant, successfully reversed the decreased half-lives of FURIN and MAPK3 (Fig. 6m and Supplementary Fig. 7g). In summary, our findings suggest that NSUN6-mediated m5C modification on mRNAs is preferentially recognized by the reader proteins YBX1 and YBX3, leading to increased stability of these mRNAs.
Above, we demonstrated that NSUN6 catalyzes high-level m5C modifications on specific mRNAs by recognizing certain stem-loops with the C*UCCA motif. In breast cancer MDA-MB-231 cells, a cohort of cell migration-related mRNAs, which possess these sequence and structure characteristics, are decorated by NSUN6 with high-level, site-specific m5C modifications. These modifications subsequently enhance mRNA stability through recognition by m5C readers, such as YBX1 and YBX3 (Fig. 6n). The molecular mechanisms of NSUN6-mediated breast cancer cell migration that we have uncovered might provide a theoretical basis for the development of potential prognostic biomarkers and therapeutic targets. Inhibiting the mRNA modification function of NSUN6 is expected to be an effective strategy for hindering breast cancer cell migration.
NSUN6-mediated m5C modification could increase the stability of therapeutic mRNAs
Improving the stability and translational efficiency of therapeutic mRNAs is essential for enhancing their efficacy68. Introducing various modifications has been proven to significantly impact the stability of therapeutic mRNAs69. To investigate whether introducing a site-specific m5C modification mediated by NSUN6 could enhance the stability of therapeutic mRNA, we cloned the 1573nt-length 3’ UTR of FURIN and 716nt-length 3’ UTR of REEP6—both of which could be methylated by NSUN6—into dual-luciferase reporter vectors. Additionally, we created a control by mutating the methylation site from C to A (Fig. 7a). The results showed that the NSUN6-mediated m5C modification significantly increased the activity of the dual-luciferase reporter compared to the non-modified control (Fig. 7b). Furthermore, RT-qPCR assays revealed that the NSUN6-mediated m5C modification on the 3’ UTR of mRNA successfully increased mRNA stability (Fig. 7c). However, introducing the 3’ UTR containing FURIN or REEP6 did not enhance the stability of the mRNA in NSUN6 KO cells (Fig. 7d). These results suggest that introducing site-specific m5C modifications catalyzed by NSUN6 could improve mRNA stability and translation efficiency in vivo.

a Schematic representation of the 3’ UTRs of FURIN and REEP6, with WT or mutant (C-to-A) m5C sites of NSUN6, inserted downstream of the firefly luciferase reporter gene in a dual-luciferase reporter vector. b Relative luciferase activity in HEK293T cells transfected with the reporters shown in a. FURIN-WT vs FURIN-Mut, p = 0.0005; REEP6-WT vs REEP6-Mut, p = 0.0019. c RNA stability assays in HEK293T cells transfected with the reporters shown in a, measured by RT-qPCR. d RNA stability assays in WT and NSUN6 KO HEK293T cells transfected with the reporters shown in a, measured by RT-qPCR. e Schematic representation of the 25nt-length FURIN sequences inserted into the 3’ UTR of mRNA-1273 and cloned downstream of the firefly luciferase reporter gene in a dual-luciferase reporter vector. f Relative luciferase activity in HEK293T cells transfected with the reporters shown in e. mRNA-1273 vs mRNA-1273 + FURIN-25nt, p = 0.0002. g RNA stability assays in HEK293T cells transfected with the reporters shown in e, measured by RT-qPCR. h Schematic representation of transcribed mRNAs. i Relative luciferase activity in HEK293T cells transfected with the mRNAs shown in h. NC vs FURIN-25nt-WT, p = 0.0001. j RNA stability assays in HEK293T cells transfected with the mRNAs shown in h, measured by RT-qPCR. Data in b–d, f, g, i, and j are presented as the mean ± SD for three independent experiments. p-values are from two-tailed t test (b, f, i). ***p < 0.001, **p < 0.01, ns, no significance. Source data are provided as a Source Data File.
The mRNA-1273 vaccine, developed by Moderna, has demonstrated clinical effectiveness in preventing SARS-CoV-2 infection70. We inserted the FURIN-25nt sequence into the appropriate 3’ UTR region of mRNA-1273 to preserve the secondary structure of the FURIN-25nt, which could be catalyzed by NSUN6 (Fig. 7e). Our results showed that the introduction of FURIN-25nt sequence markedly improved both the translation efficiency and mRNA stability of mRNA-1273 (Fig. 7f, g). Additionally, we confirmed this effect using in vitro transcribed mRNAs (Fig. 7h). The results showed that adding the NSUN6-targeted FURIN-25nt sequence to the 3’ UTR of the mRNA-1273 enhanced its stability and translation efficiency in the cells, whereas mutating the catalytic site of FURIN-25nt did not (Fig. 7i, j). These results indicate that a single NSUN6-mediated m5C site with a high level can efficiently enhance the stability and translation efficiency of the mRNA vaccine, potentially offering strategies for improving the efficacy of therapeutic mRNAs.
Discussion
m6A is the most well-studied internal mRNA modification and possesses its dedicated modifying machinery. In contrast, most other internal mRNA modifications are low-abundant and primarily deposited by known tRNA-modifying enzymes. Most m5C modifications on mRNAs have been reported to be catalyzed by two tRNA-modifying enzymes, NSUN2 and NSUN616,18,20. In this study, we reveal that NSUN6 could independently and efficiently catalyze mRNA substrates, demonstrating comparable activity to the tRNA control. NSUN6 mRNA substrates with high m5C levels are characterized by a stem-loop structure containing the C*UCCA motif in the loop, with a loop size of 10-12 nucleotides, and non-G-C base pairs at the first two positions of the stem. A prevailing hypothesis suggests that multi-substrate modifying enzymes recognize different RNA substrates by mimicking partial sequence and structural features of non-coding RNAs, particularly tRNAs71. Our results indicate that while NSUN6 retains the same catalytic activity center, it employs distinct RNA-binding surfaces to accommodate mRNA or tRNA substrates. Notably, mRNA substrates share sequence features similar to those of tRNAs but possess a single stem-loop structure, which is entirely distinct from the tertiary structure of tRNAs. We propose that multi-substrate RNA-modifying enzymes, even those reliant on the tertiary structure of tRNAs, can adapt to various types of RNA substrates by utilizing different combinations of RNA-binding surfaces. This flexibility and adaptability of RNA-modifying enzymes could enhance the multifunctionality of these enzymes.
mRNA modifications regulate various aspects of mRNA metabolism. Dysregulation of RNA epigenetics has been implicated in the pathogenesis of various human diseases, including cancer6. However, understanding the function of non-m6A modifications on mRNA is particularly challenging due to the intricate combinatorial effects of tRNA modifications on translational regulation. Here, we found that NSUN6 promotes breast cancer cell migration through its mRNA catalytic capability, independent of its role in tRNA modification. The modification levels on mRNAs can significantly impact their functions, with higher levels of site-specific modifications potentially exerting more pronounced effects on mRNA metabolism. Cell migration is a complex and dynamic process co-regulated by the chemical and physical properties of multi-component structures, cellular components, and signaling pathways. Our findings revealed that many NSUN6-mediated mRNA substrates with high m5C levels are involved in processes related to cell migration (Supplementary Data 3). Restoring individual NSUN6 mRNA substrates with high m5C levels could partially rescue the migration ability of NSUN6 KO breast cancer cells to varying extents. Notably, the simultaneous restoration of four migration-related mRNA substrates fully restored the migration ability of NSUN6 KO cells to a level comparable to that of WT cells. This suggests that NSUN6 regulates cell migration not through a single driving gene, but through the coordinated effect of a cohort of mRNA substrates. The orchestration of a complex biological process by multiple related genes bearing the same mRNA modification could enhance systemic robustness and adaptability, ensuring proper and optimized function.
To date, several proteins have been identified as mRNA m5C readers. mRNA m5C modifications have been implicated in various biological processes and multiple diseases through the regulation of these reader proteins. A pivotal role of NSUN2-mediated m5C modification on mRNA is to stabilize RNA transcripts through binding to YBX1 and YBX231,46,62. Our findings suggest that NSUN6 increases the stability of mRNAs by recruiting two m5C readers-YBX1 and YBX3 by recognizing m5C sites. It is worth noting that, according to the sequence similarity inference from the UniProt Database, YBX3 could bind mRNAs containing the consensus motif 5’-UCCAUCA-3’, which shows a high degree of overlap with the mRNA substrates of NSUN6. We also observed that YBX3 has a higher binding affinity for both m5C-modified and unmodified RNA compared to YBX1 in vitro (Fig. 6e, f), which further indicates that YBX3 has a higher preference for NSUN6 mRNA substrates than YBX1. However, compared to YBX1, the low expression of YBX3 in cells restricts its ability to stabilize mRNAs. Although, YBX1 and YBX3 both contain a highly conserved CSD domain responsible for binding RNA, they have variable Ala/Pro-rich N terminal domain and C-terminal domain rich in arginine and aromatic amino acids (ARMs) and acidic amino acids (AcidMs) (Supplementary Fig. 7a), which may interact with different proteins to stabilize mRNAs. Interestingly, due to the transcriptional silencing of the zygotic genome during early developmental stages, extensive methylation, including m5C of maternal mRNAs, is critical for maintaining maternal messages46,72,73. Our work and previous studies indicate that m5C modifications on mRNAs, catalyzed by either NSUN2 or NSUN6, are preferentially recognized and bound by YBX family proteins, thereby conferring stability to target mRNAs.
The therapeutic applications of mRNA have generated considerable optimism in medicine. For therapeutic mRNAs to function effectively, they must have an extended half-life, which remains a key challenge. The stability and translation efficiency of mRNA is significantly impacted by its 5’ cap, 5’ UTR, CDS, 3’ UTR, and poly(A) tail68. Researchers strive to design these regulatory sequences to enhance the half-life and expression level of therapeutic mRNAs, including the addition of chemical modifications such as N1-methylpseudouridine (m1Ψ), 2-thiouridine (s2U), 5-methyluridine (m5U), and m5C69,74. Our study demonstrates that introducing a single NSUN6-mediated site-specific m5C modification to the 3’ UTR region effectively increases the expression of the mRNA-1273 vaccine for SARS-CoV-2 in human cells. This approach enhances the efficiency of therapeutic mRNAs by utilizing intracellular NSUN6 to catalyze specific sequences, without the need to synthesize modified mRNA sequences in vitro. However, additional validation is imperative to ascertain the effectiveness and safety of this modification. Our findings may offer potential strategies for designing therapeutic mRNAs.
Methods
Preparation of tRNA and mRNA substrates
The tRNA and mRNA substrates, including tRNAThr(UGU), FURIN, MAPK3, REEP6, PVRL2, TIMM50, MARCKSL1, MCAM, TRAF7, and IGFBP6, were generated by in vitro transcription. The corresponding DNA sequences were obtained from the GtRNAdb database75 and the UCSC Genome Browser database76. These genes were cloned and inserted into the pTrc99b vector (using BamHI and EcoRI as the restriction enzymes) and transcribed by T7 RNA polymerase. The RNAs were recycled by urea-denaturing polyacrylamide gel electrophoresis, extracted into 0.5 M sodium acetate (pH 5.2), precipitated with ethanol at −20 °C overnight, dissolved in 5 mM MgCl2, annealed at 85 °C for 5 min, and cooled naturally to room temperature to ensure the correct RNA structure formation. Other mutants based on FURIN and MAPK3 were generated using the Vazyme Mut Express II Fast Mutagenesis Kit V2, and the FURIN-25nt and MAPK3-25nt was linked with a hammerhead ribozyme sequence to improve the transcription efficiency77. The concentrations of these RNAs were measured using UV absorbance at 260 nm and calculated according to the molar absorption coefficient.
Protein expression and purification
The gene encoding human NSUN6 was inserted into pET22b, which has a C-terminal His6-tag, using NdeI and XhoI as the restriction enzymes. The pET22b-NSUN6 plasmid was transformed into Escherichia coli Rosetta, and 0.2 mM IPTG was used with culturing at 18 °C overnight to induce the expression of the recombinant NSUN6. Cells were harvested and resuspended in lysis buffer consisting of 20 mM Tris-HCl pH 7.0, 500 mM NaCl, 5 mM MgCl2, 5 mM DTT, 10 mM imidazole, and 10% glycerol, then sonicated with 1 mM PMSF in the ice-water bath. The lysate was centrifugated at 16,000 g at 4 °C for 30 min, and the supernatant was incubated with Ni2+-NTA-agarose beads (QIAGEN). The beads were sequentially washed with lysis buffer, 1 M NaCl, 15 mM imidazole, and 25 mM imidazole, and 250 mM imidazole was used to elute NSUN6 with His6-tag. The purified protein was dialyzed against a solution containing 20 mM Tris-HCl pH 7.0, 300 mM NaCl, 5 mM MgCl2, and 2 mM DTT, concentrated, and stored at −20 °C in 50% glycerol.
The mutant proteins NSUN6-K159A/R181A, NSUN6-R352A/K353A, and NSUN6-R352E/K353E were constructed based on the pET22b-NSUN6 plasmid using the Vazyme Mut Express II Fast Mutagenesis Kit V2, and the purification method was the same as that for NSUN6 WT.
The DNA sequences encoding the human YBX1 CSD (residues 48–159) or YBX3 CSD (residues 82–174) were inserted into the pET28a-SUMO vector, which includes an N-terminal SUMO tag. The recombinant plasmids were transformed into Escherichia coli Rosetta cells, and protein expression was induced with 0.2 mM IPTG overnight at 18 °C. Cells expressing recombinant YBX1 CSD were harvested and resuspended in lysis buffer containing 10 mM Tris-HCl pH 7.5, 100 mM NaCl, 2 mM DTT, 5 mM imidazole, and 10% glycerol. Similarly, cells expressing recombinant YBX3 CSD were resuspended in lysis buffer containing 20 mM Tris-HCl pH 8.0, 500 mM NaCl, 2 mM DTT, 5 mM imidazole, and 10% glycerol. The proteins were purified with Ni2+-NTA-agarose beads (Smart-Lifesciences), followed by cleavage of the SUMO tag using Ulp1 protease. Further purification to remove nucleic acids was performed using the HiTrap SP HP column (Cytiva) with buffer A (20 mM citrate buffer pH 5.5, 100 mM NaCl, and 2 mM DTT) and buffer B (20 mM citrate buffer pH 5.5, 1 M NaCl, and 2 mM DTT). The purified proteins were concentrated and stored in a buffer containing 10 mM Tris-HCl pH 7.0, 100 mM NaCl, and 1 mM DTT.
RNA methyltransferase activity assay in vitro
To measure the methyl transfer activity of NSUN6 on tRNA and mRNA substrates, 5 μM RNAs were used. The reactions were performed at 37 °C under the same conditions in a 25 μL reaction mixture containing 200 μM [3H]-SAM, 50 mM Tris-HCl pH 7.0, 100 mM NaCl, 10 mM MgCl2, 100 μg/mL BSA, and 5 mM DTT. Reactions were initiated by the addition of 0.5 μM NSUN6 protein. At time intervals ranging from 2 to 8 min, 5 μL aliquots were removed and applied to glass fiber filter discs, which were then soaked in 5% trichloroacetic acid to precipitate the labeled methylated RNA. After washing, the amount of radioactive [methyl-3H] RNA on each disc was measured using the LS6500 Liquid Scintillation Counter (Beckman).
RNA mass spectrometry analysis
400 ng of RNA was hydrolyzed using 4 mM NH4OAc, benzonase, phosphodiesterase I, and bacterial alkaline phosphatase in a total volume of 20 μL for over 12 h at 37 °C. As previously reported30, the hydrolysis solution was diluted with acetonitrile and applied to RNA-Mass Spectrometry (RNA-MS). The specific nucleoside-to-base ion mass transitions under this condition were m5C (Q1/Q3 = 258.1/126.1) and A (Q1/Q3 = 268.1/136.2).
Selective 2’-hydroxyl acylation analyzed by primer extension (SHAPE)
For SHAPE analysis, the SHAPE handle (5’-CCGAUCCGCUUCGGCGGAUCCAAAUCGGGCUUCGGUCCGGUUC-3’) was added to the 3’ end of 100nt RNA substrates. Transcribed and purified RNA samples were annealed in a buffer containing 5 mM MgCl2 by heating at 85 °C for 5 min and then slowly cooling to room temperature. These RNAs were further purified using the Mono Q anion exchange column (Cytiva). SHAPE modification, reverse transcription, capillary electrophoresis, data processing, and normalization were performed largely as previously described, using 1-methyl-7-mitroisatoic anhydride (1M7) as the 2’-hydroxyl-selective electrophile39. Raw traces from fragment analysis were analyzed using QuShape78.
Cell culture
The MDA-MB-231, HEK293T, BT-549, and MDA-MB-468 cell lines were obtained from the cell resource center of the Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China. The cells were cultured in DMEM Media (Corning), supplemented with 10% FBS (Lonsera), and kept under 5% CO2 at 37 °C.
RNA interference and RNA transfection
The double-stranded RNA oligonucleotides (short interfering RNA, siRNA) used for RNA interference were obtained from GenePharma. The transfection of siRNA was performed using Lipofectamine™ RNAiMAX (Thermo Scientific). The transfection of mRNA was performed using Hieff Trans® mRNA Transfection Reagent (Yeasen) following the manufacturer’s protocol.
Generation of NSUN6 knockout cell lines and western blotting
NSUN6 knockout MDA-MB-231 cells were generated using sgRNAs targeting the exon of NSUN6. The sgRNAs were designed using http://crispr.mit.edu and cloned into the pX330-mcherry vector.
sgRNA-1: 5’-GGCGGTTAAACTTGATATGG-3’
sgRNA-2: 5’-TAGCACGTGCACTATAACAC-3’
5 μg of each plasmid expressing sgRNA were transfected into MDA-MB-231 cells using Lipofectamine 2000 (Thermo Fisher) and cultured for 24 h. Single cells significantly expressing red fluorescence protein were sorted into 96-well plates, and the expression of NSUN6 was identified using western blotting. The cells were collected, rinsed with PBS solution, lysed using RIPA buffer (Merck), supplemented with Protease Inhibitor Cocktail (MedChemExpress) on ice for 30 min, centrifugated at 12,000 g at 4 °C for 15 min, and the supernatant was mixed with SDS sample buffer. The samples were separated using 10% SDS-PAGE, transferred to a 0.22 µm-PVDF membrane. Antibodies of NSUN6 (Abclonal) and β-Actin (Abcam) were used to probe the respective proteins. The membrane was blocked with 5% (w/v) non-fat milk in PBST (0.05% Tween-20) for 30 min at room temperature, incubated with the primary antibody diluted in 5% (w/v) non-fat milk overnight at 4 °C, washed three times with PBST for 10 min each, incubated with the proper secondary antibody at room temperature for 1 h, and washed three times with PBST for 10 min each. The membrane was incubated with chemiluminescent substrates and imaged using the Amersham Imager 680 (GE).
Cell Counting Kit-8 assay
Cell proliferation was determined using the Cell Counting Kit-8 (Beyotime). Briefly, 4 × 103 WT and NSUN6 KO MDA-MB-231cells were seeded in a 96-well flat-bottomed plate under normal culture. At days 1, 2, 3, 4, 5, and 6, 10 μL of CCK-8 was added into each well and the cells were incubated for 2 h. The optical density at 450 nm (OD450) was measured using a microplate reader (Tecan Spark). The experiments were repeated for three times.
Transwell cell migration assay
Cells with the indicated treatment were starved in serum-free DMEM for 16 h. 24-well 8 μm pore transwell polycarbonate membrane inserts from Corning were used. For each insert, 5 × 104 cells were seeded in serum-free media in the upper chamber, and the chamber was then transferred to a well containing complete DMEM with 10% FBS. Cells were incubated at 37 °C for 24 h. The membranes were stained with 1% crystal violet (Sigma), and the remaining cells were counted. Five fields of view were counted for each membrane.
Total RNA isolation
Total RNA was extracted using TRIzol reagent (TIANGEN), purified with chloroform, and centrifugated at 12,000 g at 4 °C for 15 min. The supernatant was mixed with an equal volume of isopropanol and precipitated at −80 °C for 30 min. After centrifugation, the precipitates were washed twice with 75% ethanol, dissolved in DEPC-treated water, and stored at −80 °C.
Northern blotting
Total RNA was electrophoresed through a 12% polyacrylamide gel containing 8 M urea and then transferred to a nylon membrane (MILLIPORE). The membrane was crosslinked at 254 nm, with an energy of 150 mJ/cm2 using UV Stratalinker 1800. The membrane was then pretreated with ULTRAhyb Ultrasensitive Hybridization Buffer (Thermo Fisher) at 55 °C for 2 h. Digoxigenin (DIG)-labeled probes were denatured and added to the hybridization buffer to hybridize at 55 °C for 12 h. The membrane was washed at 55 °C and then incubated in blocking buffer (Roche) for 1 h at room temperature. The Anti-Digoxigenin-AP antibody (Roche) was added and incubated for 30 min. The membrane was washed twice with wash buffer (100 mM maleic acid, 150 mM NaCl, and 0.3% (v/v) Tween-20, adjust the pH to 7.5), with each wash lasting for 15 min, and then incubated in detection buffer (100 mM NaCl and 100 mM Tris-HCl pH 9.0). Lastly, the membrane was incubated with CDP-Star (Roche), and signal intensities were detected using the Amersham Imager 680 (GE). The probes are listed in Supplementary Data 5.
Analysis of aminoacyl-tRNA
Total RNA from each sample was extracted using TRNzol reagent (TIANGEN), purified with chloroform, and centrifugated at 12,000 g at 4 °C for 15 min. The supernatant was mixed with a one-tenth volume of 3 M sodium acetate (pH 5.0) and three volumes of ethanol, then precipitated at −80 °C overnight. The deaminoacylated RNA sample was dissolved with DEPC-treated water, while other RNA samples were dissolved in 0.1 M sodium acetate (pH 5.0). The deaminoacylated RNA sample was treated with Tris-HCl pH 9.0 at 37 °C for 30 min as a control. A total of 2 µg RNA was mixed with 2×RNA loading buffer (0.1 M sodium acetate pH 5.0, 90% glycerol, 0.03% xylene cyanide, and 0.03% bromophenol blue) and 10% deionized formamide. The samples were fractionated by electrophoresis and run at a constant 18 W on a gel containing 8 M urea, 10% polyacrylamide, and 0.1 M sodium acetate (pH 4.6) at 4 °C. The gel was electroblotted onto a nylon membrane at 250 mA for 90 min. After crosslinking, the membrane was pre-hybridized at 55 °C for 2 h and hybridized with a Digoxin-probe at 55 °C overnight.
RT-qPCR
First-strand complementary DNA (cDNA) was synthesized by reverse transcription of 1 μg RNA using HiScript III RT SuperMix for qPCR ( + gDNA wiper) (Vazyme). qPCR was carried out using ChamQ Universal SYBR qPCR Master Mix (Vazyme) and mRNA expression was normalized to the reference genes, ACTB (encoding β-actin). The primers used in all qPCR assays are listed in Supplementary Data 5.
Poly(A) tail RNA isolation
Two rounds of poly(A) enrichment were conducted using VAHTS mRNA Capture Beads (Vazyme). In brief, 50 μL of mRNA Capture Beads were mixed with 12.5 μg of total RNA in 50 μL of RNase-free ddH2O. The samples were incubated at 65 °C for 5 min and then at 25 °C for 5 min. The beads were resuspended with 200 μL Beads Wash Buffer from the kit. The beads were resuspended in 50 μL of Tris Buffer from the kit and incubated at 80 °C for 2 min, followed by mixing with 50 μL of Beads Binding Buffer from the kit. After incubating at room temperature for 5 min, the beads were washed with 200 μL of Beads Wash Buffer again. The beads were eluted with 10.5 μL of RNase-free H2O.
RNA-Seq and data analysis
mRNA from WT and two NSUN6 knockdown MDA-MB-231 cell lines were used to construct the library using the NEBNext Ultra RNA Library Prep Kit. Each library was sequenced on Illumina NovaSeq sequencers, following the manufacturer’s directions (Illumina). Each cell sample had two biological replicates. Paired-end 100-cycle sequencing reads were first trimmed for adapters using trim_galore. Then alignment to the reference genome hg38 was performed using STAR79. For differential gene expression analysis, HTSeq were used to estimate the counts based on GENCODE80,81. After normalization by TMM, Voom was used to identify differential expressed genes82.
Poly(A) RNA UBS-seq
Bisulfite treatment was performed using UBS-2 buffer as previously described method in ref. 32, with some modification. In brief, 45 µL of UBS-2 reagent was preheated to 98 °C for 5 min in a PCR instrument, and then approximately 100 ng of poly(A) RNA in 5 µL of water was added and mixed well. The mixture was incubated at 98 °C for 9 min with the lid temperature set to 105 °C. After cooling to room temperature, desulphonation was accomplished on the Zymo-Spin IC columns following the manufacturer’s protocol. The quantity of the converted RNA was determined using the Qubit. Libraries were then constructed using the Stranded mRNA-seq Lib Prep Module for Illumina (ABclonal). Libraries were sequenced at Majorbio. In brief, libraries first underwent quality control assessment using Agilent TapeStation. The libraries that passed quality control were sequenced on the HiSeq X (Illumina) to produce paired-end 150-bp reads. Each cell sample had two biological replicates.
UBS-seq data analysis
The UBS-seq data was analyzed using the previously described pipeline18, with some modifications. In brief, we performed trimming of adapter sequences and low-quality reads with catadapt83 and trimmomatic84. Paired reads and unpaired reads were then mapped separately to C-T, A-G transformed human genome with HISAT285. Unmapped and multiple mapped reads were further mapped to a C-to-T converted transcriptome by Bowtie286. The transcript coordinates were adapted to match the genomic locations based on the GTF annotation. We analyzed the paired reads and unpaired reads, and merged them into the total BAM files. We set the criteria for potential m5C modification site calling to have a coverage of C + T above 20 and a base quality score above 30. We used a binomial model to assess the p-value for each potential m5C site. For m5C sites identified by UBS-seq, the flanking sequence of each site was extracted from the reference genome. Motifs were discovered and plotted with MEME87.
Isolation of cytoplasmic fractions
Separation of cytoplasmic extracts was performed using low-permeability buffer (20 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 1 U/μL RNase inhibitor (Thermo Fisher), and 1× protease inhibitor cocktail (EDTA-free)), followed by the addition of NP-40 to a final concentration 5% and vortex for 10 s. Centrifuge lysates at 3000 g for 10 min at 4 °C. The supernatant contains cytosol fraction.
Biotin-labelled RNA oligonucleotides preparation
First, 20 µg of RNA oligonucleotides with (Oligo-m5C) or without m5C (Oligo-C) were treated with 40 µL of 25 mM KIO4 in the dark at room temperature for 1 h. The sequence of the oligonucleotide was 5’-GGAGGCCACCUXUCCAAGGGCUUCU-3’ (X = C or m5C). Then, the reaction was terminated with 25 µL of 50% ethylene glycol, and DEPC-treated water was added to bring the final volume to 300 µL. The RNA oligonucleotides were precipitated by adding 1/10 volume of 3 M NaAc and three volumes of ethanol at −80 °C for 1 h. The pellets were separated by centrifugation at 13,000 g for 30 min at 4 °C, washed once with 75% ethanol, and air-dried at room temperature. Next, the RNA oligonucleotides were dissolved in 100 µL of 10 mM biotinamidocaproyl hydrazide solution (Sigma) and incubated at 37 °C in the dark for 2 h. Afterward, 100 µL of 0.2 M NaBH4 and 200 µL of 1 M Tris-HCl pH 8.0 were added and incubated in an ice bath for 30 min. Then, add 1/10 volume of 3 M NaAc and three volumes of ethanol to precipitate the RNA oligonucleotides at −80 °C for 1 h. The pellets were centrifuged at 13,000 g for 30 min at 4 °C, washed once with 75% ethanol, and dissolve in 20 µL of DEPC-treated water before being stored at −80 °C.
RNA pull-down and mass spectrometry analysis
In vivo RNA pull-down assays were carried out using MDA-MB-231 cell cytoplasmic fractions as previously described31. Biotin-labeled RNA oligonucleotides were incubated with cytoplasmic fractions for 4 h at 4 °C under gentle rotation with streptavidin magnetic beads (MedChemExpress). Beads were washed three times with wash buffer (50 mM Tris-HCl pH 7.5, 250 mM NaCl, 0.4 mM EDTA, 0.1% NP-40, 1 mM DTT, 1 U/μL RNase inhibitor (Thermo Fisher), and 1× protease inhibitor cocktail (EDTA-free)). The beads-bound proteins were eluted with 0.1 M glycine (pH 2.0) and subjected to mass spectrometry analysis using an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher).
Biolayer interferometry assay
The RNA-binding affinities of the YBX1 CSD and YBX3 CSD were measured using the Biolayer Interferometry system (Octet RED96) with a running buffer containing 10 mM Tris HCl 7.0, and 100 mM NaCl. Streptavidin-coated biosensors (Octet SA biosensor) were loaded with 0.5 μM biotinylated FURIN-25nt or NSUN6-methylated FURIN-25nt for 1 min. The sensors were then incubated with various concentrations (0, 0.3125, 0.625, 1.25, 2.5, and 5 μM) of the purified YBX1 CSD or YBX3 CSD for 2 min, followed by 3 min of dissociation. The data were analyzed, and the binding parameters were determined using the software supplied by the manufacturer.
RNA stability assay
WT and NSUN6 KO MDA-MB-231 cells were seeded into 6-well plates to reach 50% confluency after 24 h. Cells were treated with 5 µg/mL actinomycin D (MedChemExpress) and collected at indicated time points. The total RNA was extracted by using TRIzol (TIANGEN) and analyzed by RT-qPCR. The turnover rate and half-lives of mRNA were estimated according to a previous report88. As actinomycin D treatment results in transcription stalling, the change in mRNA concentration at a given time (dC/dt) is proportional to the constant of mRNA decay (Kdecay) and the mRNA concentration (C), leading to the following equation:
Thus, the mRNA degradation rate Kdecay was estimated by:
To calculate the mRNA half-life (t1/2), when 50% of the mRNA is decayed (that is, C/C0 = 1/2), the equation was:
From where:
Dual-luciferase reporter assay
The pmirGLO Dual-Luciferase Expression Vector (Promega) containing sequences for firefly luciferase (F-luc) and Renilla luciferase (R-luc), was used to construct the reporter plasmids. The 3’ UTR of FURIN and REEP6 were inserted into the 3’ UTR of the firefly luciferase gene, respectively. The m5C modification site in the 3’ UTR of FURIN and REEP6 was mutated to A and inserted into the 3’ UTR of the firefly luciferase reporter gene as negative controls. 40 ng of each reporter plasmid was transfected into HEK293T cells in a 24-well plate using Lipofectamine 2000. After 24 h, the cells in the 24-well plate were assayed using the Dual Luciferase Reporter Gene Assay Kit (YEASEN). R-luc activity was used to normalize F-luc activity to evaluate the translation efficiency of the reporters.
Statistical analysis
All Statistics were performed using GraphPad Prism Software (v10.2.0, La Jolla, CA, USA). Error bars represent mean ± SD for at least three independent experiments. Comparisons between two groups were analyzed using unpaired Student’s t-test, for more groups, statistical analysis was performed using one-way ANOVA with Tukey’s multiple comparisons test.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available from the corresponding authors upon request. The RNA-seq and UBS-seq data used in this study are available in the Gene Expression Omnibus database under accession code GSE279973 and GSE280051. Source data for the figures and Supplementary Figs. are provided as a Source Data file. Source data are provided with this paper.
References
Gilbert, W. V., Bell, T. A. & Schaening, C. Messenger RNA modifications: form, distribution, and function. Science 352, 1408–1412 (2016).
Frye, M., Jaffrey, S. R., Pan, T., Rechavi, G. & Suzuki, T. RNA modifications: what have we learned and where are we headed?. Nat. Rev. Genet. 17, 365–372 (2016).
He, C. Grand challenge commentary: RNA epigenetics?. Nat. Chem. Biol. 6, 863–865 (2010).
Roundtree, I. A., Evans, M. E., Pan, T. & He, C. Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200 (2017).
Wiener, D. & Schwartz, S. The epitranscriptome beyond m(6)A. Nat. Rev. Genet. 22, 119–131 (2021).
Frye, M., Harada, B. T., Behm, M. & He, C. RNA modifications modulate gene expression during development. Science 361, 1346–1349 (2018).
Delaunay, S., Helm, M. & Frye, M. RNA modifications in physiology and disease: towards clinical applications. Nat. Rev. Genet. 25, 104–122 (2024).
Barbieri, I. & Kouzarides, T. Role of RNA modifications in cancer. Nat. Rev. Cancer 20, 303–322 (2020).
Blanco, S. & Frye, M. Role of RNA methyltransferases in tissue renewal and pathology. Curr. Opin. Cell Biol. 31, 1–7 (2014).
Schaefer, M. et al. RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev. 24, 1590–1595 (2010).
Muthukumar, S., Li, C. T., Liu, R. J. & Bellodi, C. Roles and regulation of tRNA-derived small RNAs in animals. Nat. Rev. Mol. Cell Biol. 25, 359–378 (2024).
Edelheit, S., Schwartz, S., Mumbach, M. R., Wurtzel, O. & Sorek, R. Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria, archaea, and yeast Reveals m5C within Archaeal mRNAs. PLOS Genet. 9, e1003602 (2013).
Hussain, S. et al. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 4, 255–261 (2013).
Khoddami, V. & Cairns, B. R. Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat. Biotechnol. 31, 458–464 (2013).
Cui, X. et al. 5-Methylcytosine RNA methylation in arabidopsis thaliana. Mol. Plant 10, 1387–1399 (2017).
Lu, L. et al. Base-resolution m(5)C profiling across the mammalian transcriptome by bisulfite-free enzyme-assisted chemical labeling approach. Mol. Cell 84, 2984–3000 e2988 (2024).
Legrand, C. et al. Statistically robust methylation calling for whole-transcriptome bisulfite sequencing reveals distinct methylation patterns for mouse RNAs. Genome Res. 27, 1589–1596 (2017).
Huang, T., Chen, W., Liu, J., Gu, N. & Zhang, R. Genome-wide identification of mRNA 5-methylcytosine in mammals. Nat. Struct. Mol. Biol. 26, 380–388 (2019).
Zhang, Z. et al. Systematic calibration of epitranscriptomic maps using a synthetic modification-free RNA library. Nat. Methods 18, 1213–1222 (2021).
Liu, J. et al. Developmental mRNA m(5)C landscape and regulatory innovations of massive m(5)C modification of maternal mRNAs in animals. Nat. Commun. 13, 2484 (2022).
Reid, R., Greene, P. J. & Santi, D. V. Exposition of a family of RNA m(5)C methyltransferases from searching genomic and proteomic sequences. Nucleic Acids Res. 27, 3138–3145 (1999).
Goll, M. G. et al. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311, 395–398 (2006).
Liu, J. et al. Sequence-and structure-selective mRNA m(5)C methylation by NSUN6 in animals. Natl. Sci. Rev. 8, nwaa273 (2021).
Safra, M. et al. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature 551, 251–255 (2017).
Safra, M., Nir, R., Farouq, D., Vainberg Slutskin, I. & Schwartz, S. TRUB1 is the predominant pseudouridine synthase acting on mammalian mRNA via a predictable and conserved code. Genome Res. 27, 393–406 (2017).
Behm-Ansmant, I. et al. The Saccharomyces cerevisiae U2 snRNA:pseudouridine-synthase Pus7p is a novel multisite-multisubstrate RNA:Psi-synthase also acting on tRNAs. RNA 9, 1371–1382 (2003).
Carlile, T. M. et al. mRNA structure determines modification by pseudouridine synthase 1. Nat. Chem. Biol. 15, 966–974 (2019).
Sibert, B. S. & Patton, J. R. Pseudouridine synthase 1: a site-specific synthase without strict sequence recognition requirements. Nucleic Acids Res. 40, 2107–2118 (2012).
Liu, R. J., Long, T., Li, J., Li, H. & Wang, E. D. Structural basis for substrate binding and catalytic mechanism of a human RNA:m5C methyltransferase NSun6. Nucleic Acids Res. 45, 6684–6697 (2017).
Long, T. et al. Sequence-specific and shape-selective RNA recognition by the human RNA 5-methylcytosine methyltransferase NSun6. J. Biol. Chem. 291, 24293–24303 (2016).
Chen, X. et al. 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat. Cell Biol. 21, 978–990 (2019).
Dai, Q. et al. Ultrafast bisulfite sequencing detection of 5-methylcytosine in DNA and RNA. Nat. Biotechnol. 42, 1559–1570 (2024).
Liu, C. et al. Absolute quantification of single-base m(6)A methylation in the mammalian transcriptome using GLORI. Nat. Biotechnol. 41, 355–366 (2023).
Mattioli, F. et al. Biallelic variants in NSUN6 cause an autosomal recessive neurodevelopmental disorder. Genet Med 25, 100900 (2023).
Selmi, T. et al. Sequence- and structure-specific cytosine-5 mRNA methylation by NSUN6. Nucleic Acids Res 49, 1006–1022 (2021).
Fang, L. et al. CIGAR-seq, a CRISPR/Cas-based method for unbiased screening of novel mRNA modification regulators. Mol. Syst. Biol. 16, e10025 (2020).
Haag, S. et al. NSUN6 is a human RNA methyltransferase that catalyzes formation of m5C72 in specific tRNAs. RNA 21, 1532–1543 (2015).
Wilkinson, K. A., Merino, E. J. & Weeks, K. M. Selective 2’-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 1, 1610–1616 (2006).
Mortimer, S. A. & Weeks, K. M. A fast-acting reagent for accurate analysis of RNA secondary and tertiary structure by SHAPE chemistry. J. Am. Chem. Soc. 129, 4144–4145 (2007).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Lou, N., Gu, X., Fu, L., Li, J. & Xue, C. Significant roles of RNA 5-methylcytosine methylation in cancer. Cell Signal 126, 111529 (2025).
Hu, L. et al. LOXL1 modulates the malignant progression of colorectal cancer by inhibiting the transcriptional activity of YAP. Cell Commun. Signal 18, 148 (2020).
Saito, K. et al. PLOD2-driven IL-6/STAT3 signaling promotes the invasion and metastasis of oral squamous cell carcinoma via activation of integrin beta1. Int. J. Oncol. 58, 9 (2021).
Domanegg, K., Sleeman, J. P. & Schmaus, A. CEMIP, a Promising Biomarker That Promotes the Progression and Metastasis of Colorectal and Other Types of Cancer. Cancers (Basel) 14, 5093 (2022).
Zhang, Y., Zhang, W., Zhang, R. & Xia, Y. Knockdown of FBLN2 suppresses TGF-β1-induced MRC-5 cell migration and fibrosis by downregulating VTN. Tissue Cell 81, 102005 (2023).
Yang, Y. et al. RNA 5-Methylcytosine Facilitates the Maternal-to-Zygotic Transition by Preventing Maternal mRNA Decay. Mol. Cell 75, 1188–1202.e1111 (2019).
Wang, X. et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014).
Arango, D. et al. Acetylation of Cytidine in mRNA Promotes Translation Efficiency. Cell 175, 1872–1886.e1824 (2018).
Rossignoli, A. et al. Poliovirus Receptor-Related 2: A Cholesterol-Responsive Gene Affecting Atherosclerosis Development by Modulating Leukocyte Migration. Arterioscler Thromb. Vasc. Biol. 37, 534–542 (2017).
Chen, C. et al. ERBB3-induced furin promotes the progression and metastasis of ovarian cancer via the IGF1R/STAT3 signaling axis. Oncogene 39, 2921–2933 (2020).
Xia, Y. T. et al. Suppression of migration and invasion by taraxerol in the triple-negative breast cancer cell line MDA-MB-231 via the ERK/Slug axis. PLoS One 18, e0291693 (2023).
Du, X., Zhang, Q., Wang, S., Chen, X. & Wang, Y. MCAM is associated with metastasis and poor prognosis in osteosarcoma by modulating tumor cell migration. J. Clin. Lab Anal. 36, e24214 (2022).
Jayachandran, A. et al. Identifying and targeting determinants of melanoma cellular invasion. Oncotarget 7, 41186–41202 (2016).
Zhang, Q., Zhang, X. & Dong, W. TRAF7 contributes to tumor progression by promoting ubiquitin-proteasome mediated degradation of P53 in hepatocellular carcinoma. Cell Death Discov. 7, 352 (2021).
Fu, P., Yang, Z. & Bach, L. A. Prohibitin-2 binding modulates insulin-like growth factor-binding protein-6 (IGFBP-6)-induced rhabdomyosarcoma cell migration. J. Biol. Chem. 288, 29890–29900 (2013).
Manjur, A. et al. BCOR modulates transcriptional activity of a subset of glucocorticoid receptor target genes involved in cell growth and mobility. J. Steroid Biochem. Mol. Biol. 210, 105873 (2021).
Du, Q. et al. Bioinformatics analysis of LMAN1 expression, clinical characteristics, and its effects on cell proliferation and invasion in glioma. Brain Res 1789, 147952 (2022).
Spitale, R. C. et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature 519, 486–490 (2015).
de Bruijn, I. et al. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res 83, 3861–3867 (2023).
Tang, Z., Kang, B., Li, C., Chen, T. & Zhang, Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res 47, W556–w560 (2019).
Kleene, K. C. Y-box proteins combine versatile cold shock domains and arginine-rich motifs (ARMs) for pleiotropic functions in RNA biology. Biochem J. 475, 2769–2784 (2018).
Cooke, A. et al. The RNA-Binding Protein YBX3 Controls Amino Acid Levels by Regulating SLC mRNA Abundance. Cell Rep. 27, 3097–3106.e3095 (2019).
Kwon, E. et al. The RNA-binding protein YBX1 regulates epidermal progenitors at a posttranscriptional level. Nat. Commun. 9, 1734 (2018).
Cai, Y., Li, N. & Li, H. YBX2 modulates mRNA stability via interaction with YTHDF2 in endometrial cancer cells. Exp. Cell Res. 427, 113586 (2023).
Li, Y. et al. TET2-mediated mRNA demethylation regulates leukemia stem cell homing and self-renewal. Cell Stem Cell 30, 1072–1090.e1010 (2023).
Yu, T. et al. THOC3 interacts with YBX1 to promote lung squamous cell carcinoma progression through PFKFB4 mRNA modification. Cell Death Dis. 14, 475 (2023).
Zhang, G. et al. NSUN2 stimulates tumor progression via enhancing TIAM2 mRNA stability in pancreatic cancer. Cell Death Discov. 9, 219 (2023).
Metkar, M., Pepin, C. S. & Moore, M. J. Tailor made: the art of therapeutic mRNA design. Nat. Rev. Drug Discov. 23, 67–83 (2024).
Qin, S. et al. mRNA-based therapeutics: powerful and versatile tools to combat diseases. Signal Transduct. Target Ther. 7, 166 (2022).
Baden, L. R. et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 384, 403–416 (2021).
Grozhik, A. V. & Jaffrey, S. R. Shrinking maps of RNA modifications. Nature 551, 174–176 (2017).
Walser, C. B. & Lipshitz, H. D. Transcript clearance during the maternal-to-zygotic transition. Curr. Opin. Genet. Dev. 21, 431–443 (2011).
Barckmann, B. & Simonelig, M. Control of maternal mRNA stability in germ cells and early embryos. Biochim Biophys. Acta 1829, 714–724 (2013).
Taibi, T., Cheon, S., Perna, F. & Vu, L. P. mRNA-based therapeutic strategies for cancer treatment. Mol. Ther. 32, 2819–2834 (2024).
Chan, P. P. & Lowe, T. M. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 44, D184–D189 (2016).
Lee, B. T. et al. The UCSC Genome Browser database: 2022 update. Nucleic Acids Res 50, D1115–d1122 (2022).
Fechter, P., Rudinger, J., Giegé, R. & Théobald-Dietrich, A. Ribozyme processed tRNA transcripts with unfriendly internal promoter for T7 RNA polymerase: production and activity. FEBS Lett. 436, 99–103 (1998).
Karabiber, F., McGinnis, J. L., Favorov, O. V. & Weeks, K. M. QuShape: rapid, accurate, and best-practices quantification of nucleic acid probing information, resolved by capillary electrophoresis. Rna 19, 63–73 (2013).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012).
Anders, S., Pyl, P. T. & Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Bailey, T. L., Johnson, J., Grant, C. E. & Noble, W. S. The MEME Suite. Nucleic Acids Res. 43, W39–W49 (2015).
Chen, C. Y., Ezzeddine, N. & Shyu, A. B. Messenger RNA half-life measurements in mammalian cells. Methods Enzymol. 448, 335–357 (2008).
Acknowledgements
This work is supported by National Key Research and Development Program of China (2021YFA1100800 to R.L., 2020YFA0803400 to R.L.); National Natural Science Foundation of China (82450109 to R.L., 32022040 to R.L.); This work is also supported by the Shanghai Frontiers Science Center for Biomacromolecules and Precision Medicine in ShanghaiTech University. We thank the Molecular and Cell Biology Core Facility (MCBCF), the Multi-Omics Core Facility (MOCF), and the Molecular Imaging Core Facility (MICF) at the School of Life Science and Technology, ShanghaiTech University, as well as the Discovery Technology Platform (ShanghaiTech University, SIAIS), for providing technical support. We also thank Jing Chen (ShanghaiTech University) for her technical assistance in the experiments related to breast cancer cell migration.
Author information
Authors and Affiliations
Contributions
R.L., Y.Z. and C.L. designed the experiments and wrote this manuscript. Y.Z., and C.L performed and analyzed most of these experiments. Y.J.Z. analyzed the UBS-seq results. H.L., J.L. and Q.X. contributed to the protein purification and in vitro methyltransferase activity assay. W.Z. and Y.X. contributed to research exploration. W.H. and Q.Z. contributed to the analysis of RNA secondary structure. B.X. contributed to the analysis of RNA-seq data. E.W. oversaw the study. R.L. provided conceptual insight.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yun-Gui Yang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, YY., Li, CT., Zhou, YJ. et al. A cohort of mRNAs undergo high-stoichiometry NSUN6-mediated site-specific m5C modification. Nat Commun 16, 6119 (2025). https://doi.org/10.1038/s41467-025-60873-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60873-4
