
Abstract
Plant residue decomposition is critical for carbon cycling in terrestrial ecosystems. Nitrogen (N) availability alters this process through orchestrating the microbial community, yet the mechanisms remain elusive. By investigating the wheat residue decomposition process and the microbial succession under different N input levels in agricultural fields, we find that higher N availability accelerates residue breakdown mainly at the early stage by promoting the rapid colonization of a soil-derived microbial consortium with key interactions. Metabolic potential evaluations show that the Bacillus decomposers harbor diverse carbohydrate-active enzymes that degrade cellulose and hemicellulose, whereas the non-decomposer Staphylococcus sciuri efficiently transports and consumes downstream sugar products. Synthetic communities combined with omics techniques confirm that the N-enriched non-decomposer S. sciuri restricts the growth of weak decomposers through sugar depletion, thereby restructuring the community dominated by strong decomposers. This shift increases the residue decomposition rate by 16.77% under N fertilization. Our results highlight the important role of usually overlooked fast-growing non-decomposers in agricultural soil carbon cycling.
Similar content being viewed by others

Introduction
Global agricultural land generates approximately 4 billion tons of various crop residues per year1. Crop residue return is an important agricultural management practice to enhance soil carbon (C) sequestration, which has a profound impact on climate change and food security2,3. A global-scale analysis of long-term (≥4 years) and short-term (<4 years) residue return experiments indicated that residue incorporation into cropland resulted in 14.9% and 5.1% increases in soil organic C (SOC) content and crop yield, respectively4. Because of the high ratio of C:nitrogen (N) (C:N > 70:1) inherent in lignocellulosic materials, C availability increases in residue-amended soils, which stimulates microbes to mineralize soil organic matter (C:N = ~17:1) to obtain N to sustain their ecological stoichiometry (C:N = ~7:1) (i.e., a priming effect)5,6. This microbial N mining may lead to soil N restriction, resulting in competition for N between crops and microbes7. Thus, the combined application of N fertilizers and crop residues represents a promising strategy to balance the C:N ratio to allow for both microbial decomposition and crop growth5. Studies have indicated that N fertilization not only accelerates the decomposition of crop residues into newly formed SOC by stimulating microorganisms, but also reduces the priming effect, thereby increasing net SOC accumulation8,9. Therefore, it is imperative to understand how external N supply affects in situ crop residue decomposition dynamics and the underlying microbial mechanisms.
Previous studies have demonstrated the sensitivity of the residue decomposition process to N application from local to global scales, and proposed several mechanisms mainly from the perspectives of soil properties, extracellular enzyme activity, community composition, and functional genes10,11,12,13,14. A higher N deposition rate continuously increased the litter decomposition rate over 2 years in a semi-arid grassland by increasing the soil bacteria:fungi ratio and manganese availability10. Global meta-analyses and experimental studies have consistently indicated that N fertilization imparts shifting effects on the dynamics of residue decomposition, with the positive and negative effects usually occurring at the early and later stages, respectively11,12,13. This opposite influence has been largely attributed to the contrasting effects of N fertilization on the activity of stage-specific extracellular enzymes. For example, N fertilization was found to increase the activity of cellulase by 15.2% while decreasing the activity of lignin-modifying enzymes by 12.8%14. However, there is a paucity of information on the interactions among microbes that participate in this process, a crucial aspect considering that plant residue decomposition primarily depends on microbial interactions rather than the capabilities of individual species.
In natural environments, soil-dwelling microorganisms gradually establish on the residue’s surface and assemble into consortia, where each member is specialized in a subtask to synergistically perform the complete depolymerization and degradation of lignocellulose15,16. Some cooperation strategies have been identified through top-down enrichment and bottom-up synthetic community studies, such as mutualism and division of labor17,18. For example, Pluralibacter gergoviae in a lignin-degrading microbial community is responsible for lignin degradation and provides succinate and malate to non-degraders, whereas the non-degraders supply glycerol, alanine, and aspartate to support P. gergoviae’s growth and further stimulate lignin degradation19. Moreover, cross-feeding between primary decomposers and downstream intermediate users can alleviate the reduction in the enzymatic hydrolysis rate of lignocellulose caused by excessive accumulation of metabolites such as monosaccharides and disaccharides (i.e., end-product inhibition)20. Previous studies on long-term N deposition and the composition of bacterial communities in different coastal sediments and seawater have reported that higher nutrient availability favors fast-growing bacteria and changes the intensity and complexity of microbial interactions21,22. More specifically, there are more ecological interactions among species in nutrient-rich environments, where competition plays a more dominant role21. Because the combined application of N fertilizer and plant residues creates a more nutrient-rich microenvironment compared with the surrounding soil, we hypothesize that N-stimulated fast-growing species may rapidly colonize the residue surface and compete for nutrients with other residue-inhabiting community members, thereby having a cascade effect on residue decomposition.
In the present study, we attempt to determine the comprehensive responses of residue-inhabiting microbial communities in terms of membership, enzyme strategies, and interactions to N fertilization on a temporal scale, and how these dynamic responses affect wheat residue decomposition in an agricultural ecosystem (Supplementary Fig. 1). For these purposes, we first conduct a 187 days litterbag experiment to examine the decomposition process of wheat residues under field conditions with three N input levels. We find that the N-induced accelerated breakdown of residues mainly occurred at the early stage of decomposition. Amplicon and metagenomic analyses confirm that the N-enriched genera Bacillus and Staphylococcus occupy a broad niche in the early-stage residue-inhabiting microbial communities. Following large-scale strain screening, we identify the non-decomposer Staphylococcus sciuri and a decomposer community consisting of four Bacillus strains from 19 isolated strains that were enriched at the early stage. By performing different omics analyses (i.e., transcriptomics, metabolomics, and amplicon analyses), laboratory decomposition experiments, and synthetic communities (SynComs), we demonstrate that the N-enriched, fast-growing non-decomposer S. sciuri restricts the growth of weak decomposers through sugar depletion, resulting in the restructuring of a community dominated by strong decomposers, which facilitates residue decomposition under N fertilization. The results of our study reveal details of the interactions between decomposers and the usually overlooked fast-growing non-decomposers, and how they contribute to residue decomposition in agricultural soil systems.
Results
Impacts of N fertilization on wheat residue decomposition
To detect the effects of N application on wheat residue decomposition, we conducted a wheat residue bag experiment under field conditions with three urea input levels (N0, no N input; N1, low N input (180 kg hm−2); N2, high N input (240 kg hm−2)) (Supplementary Fig. 2). After 187 days of field incorporation, the cumulative decomposition rate of wheat residue was significantly increased by N fertilization (two-way ANOVA, P < 0.01), where its values in the N1 and N2 treatments were 1.10 times and 1.06 times higher than those in the N0 treatment, respectively (Fig. 1a). The residue cumulative decomposition rate was well fitted to a two-phase exponential decay model characterized by early fast (i.e., SpanFast) and later slow (i.e., SpanSlow) degradation periods (R2 ≥ 0.99; Fig. 1a and Supplementary Table 1). The SpanFast period was prolonged by N application, with its duration under N0, N1, and N2 conditions being 28 days, 41 days, and 39 days, respectively (gray boxes in Fig. 1a). We further obtained the fitted real-time decomposition rates based on the two-phase exponential decay model (Fig. 1b). Input of N significantly increased the residue decomposition rate by 10–12% before 57–70 days, but exerted a negative effect at the later stage, consistent with our actual measured decomposition rates (bar plots in Fig. 1b). Indeed, scanning electron microscopy (SEM) analyses showed that the physical structure of wheat residues was more disrupted in the N1 and N2 treatments than in the N0 treatment, especially at the early stage (Fig. 1c and Supplementary Fig. 3). These results indicate that the N-induced accelerated breakdown of residues mainly occurred at the early stage of decomposition.

a Cumulative decomposition rate over time under N0, N1, and N2 fertilizer levels. N0, no N input; N1, low N input (180 kg hm−2); N2, high N input (240 kg hm−2). Data were fitted to a two-phase exponential decay model. Gray boxes represent the rapid decomposition period of wheat residue derived from the two-phase exponential decay model (i.e., SpanFast). Two-way ANOVA in conjunction with Fisher’s LSD test was applied to test the effects of N fertilization, decomposition time, and their interaction on the wheat residue decomposition rate. b Simulated decomposition rate derived from the two-phase exponential decay model. Inset at the top of (b) indicates the measured decomposition rate. Statistical significance is based on one-way ANOVA followed by Fisher’s LSD test. Data in (a, b) are mean values ± standard deviation (n = 3). c SEM images of the structure of wheat residues after 0 days, 14 days, 62 days, and 187 days of degradation in N0, N1, and N2 treatments. SEM images of samples collected at all seven time points during decomposition are shown in the Supplementary Information. Asterisks indicate significance levels: *P < 0.05, **P < 0.01, and ****P < 0.0001. ns not significant. Source data are provided as a Source Data file.
Succession of wheat residue-inhabiting microbial diversity, taxonomic, and functional compositions over decomposition time
Using qPCR, amplicon sequencing, and metagenomics, we investigated the diversity, absolute abundance, and taxonomic and functional composition of residue-inhabiting microbial communities during decomposition. Microbial taxonomic and phylogenetic diversity showed a pronounced increasing trend over time (two-way ANOVA, P < 0.01; except for fluctuating fungal Shannon diversity) (Supplementary Fig. 4). N fertilization did not exert a strong effect on microbial alpha diversity. However, under three N levels, we observed clear gradual successions of the structures of residue-inhabiting bacterial (R2 = 41.72%) and fungal (R2 = 21.34%) communities (Adonis, P < 0.001; Supplementary Fig. 5). The decomposition rate of wheat residues was associated more with the bacterial community (R2 = 17.10–32.74%) than with the fungal community (R2 = 13.00–13.30%), especially at the early stage (Supplementary Table 2).
We performed FEAST analyses to quantitatively evaluate the impact of N fertilization on the colonization process of microbiotas from different sources. Without applying N fertilizers, 79.48% of the residue-inhabiting bacterial microbiota and 92.89% of the residue-inhabiting fungal microbiota at 7 days were still derived from the original wheat residues (Fig. 2a). Conversely, following the application of N fertilizers, the soil-derived and unknown-source-derived microorganisms rapidly colonized the residue, accounting for 58.28–97.15% of bacterial and fungal communities at 7 days depending on the N input levels. Meanwhile, the contents of ammonia N (NH4+) and nitrate N (NO3−) in the N-amended residue bags only remained at high levels for the first 7 days and were rapidly consumed thereafter (Supplementary Fig. 6). These results demonstrate that exogenous N input accelerated the transmission and colonization of soil-derived microbiotas on the wheat residues at the early stage.

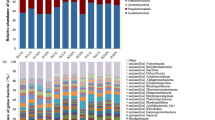
N0, no N input; N1, low N input (180 kg hm−2); N2, high N input (240 kg hm−2). a Area plots showing the potential source of residue-inhibiting microbial communities as determined by fast expectation-maximization microbial source tracking (FEAST) analysis. b Temporal dynamics of bacterial 16S rRNA gene and fungal ITS region absolute abundance under N0 and N2 treatments. The bars indicate mean values, and the error bars indicate standard deviation (n = 9). Statistical significance is based on Kruskal-Walli’s test and Mann–Whitney test. Different letters and asterisks indicate significance levels: **P < 0.01, ***P < 0.001, and ****P < 0.0001. ns, not significant. c, d Relative abundance of the top 16 bacterial genera (c) and top 11 fungal genera (d) as determined by amplicon sequencing analysis (A) and metagenomic sequencing analysis (M). Proteobacteria are shown at the class level. Firm Firmicutes, Beta Betaproteobacteria, Gamm Gammaproteobacteria, Alph Alphaproteobacteria, Acti Actinobacteria, Doth Dothideomycetes, Euro Eurotiomycetes, Sord Sordariomycetes, Pucc Pucciniomycetes. Black arrows indicate genera significantly enriched under the N2 treatment at the early stage that were detected by A or M. The statistical analyses were performed using a one-sided t-test (n = 3). e Enrichment and depletion of carbohydrate active enzymes (CAZymes) modules in N2 compared with N0 treatment as determined by differential abundance analysis (n = 3). Differentially abundant CAZyme modules of different categories were individually color-coded. Vertical lines represent the absolute value of fold change = 2. Bar plot shows genus-level taxonomic distributions associated with genes encoding enriched CAZyme families. GH glycoside hydrolase, GT glycosyl transferase, CE carbohydrate esterase, AA auxiliary activities, CBM carbohydrate-binding module, PL polysaccharide lyase. Source data are provided as a Source Data file.
The absolute and relative abundance of the residue-inhabiting microbial communities in the N0 and N2 treatments were further analyzed to determine which species’ colonization was promoted by N application. The bacterial loads were remarkably enriched by N fertilization and reached their highest values on 7 days (2.78 × 1010 copies g–1 residue; Fig. 2b). In contrast, the absolute abundance of fungi was not stimulated by N fertilization and tended to increase during the residue decomposition period (Fig. 2b). The microbiota in original wheat residues were dominated by the members of bacterial phylum Gammaproteobacteria including the main genera Pseudomonas, Pantoea, and Enterobacter (Fig. 2c), and by the members of the fungal class Dothideomycetes including the main genus Alternaria (Fig. 2d). These original microorganisms persistently dominated the community on the wheat residues in the first 7 days when no N fertilizer was applied. However, N fertilization resulted in larger changes in microbial succession in the first 7 days and to a lesser extent at the later stage. Notably, on 7 days, two predominant bacterial genera belonging to Firmicutes, Bacillus and Staphylococcus, accounted for 60.15–70.66% of the total bacterial relative abundance in the N-amended wheat residues, as confirmed by the data of amplicon and metagenomics (black arrow in Fig. 2c).
To determine the ability of the residue-inhabiting microbiotas to degrade wheat residues, we predicted their carbohydrate-active enzymes (CAZymes) on the basis of the metagenomics data. During the decomposition process, the number of CAZyme families showing significant differences in abundance between the N0 and N2 treatments gradually decreased, from 198 on 7 days, to 31 on 14 days, 46 on 62 days, and 2 on 187 days (Fig. 2e). Focusing on the significantly enriched sets at the early stage (7 days and 14 days), most of the CAZyme families targeted the main components of wheat residues, such as lignin, hemicellulose, and cellulose (Supplementary Data 1). We further observed a clear difference in microbial taxa associated with the enriched CAZyme genes between the early and later stages (bar plots in Fig. 2e). At the early stage, the enriched CAZyme families were mainly derived from bacterial genera, such as Staphylococcus (57.30%) and Bacillus (12.38%) on 7 days, and Brevundimonas (39.55%) and Brachybacterium (19.80%) on 14 days. At the later stage, the enriched CAZyme families were mainly produced by fungal genera, such as Scedosporium (16.32%) and Chaetomium (9.47%) on 62 days, and Fusarium (29.85%) and Aspergillus (23.87%) on 187 days. Overall, these results show that the N-shaped bacterial consortium at the early stage mainly drove the metabolism of carbohydrates in wheat residues.
CAZymes and potential metabolic pathways of sugars produced from lignocellulose degradation at the early stage
Given that the N-induced bacterial consortium contributed more to wheat residue decomposition than did fungi at the early stage, we further recovered bacterial genomes from the metagenomics data, enabling the analysis of metabolic potential and interactions at the species level. Based on metagenomic binning, we obtained 68 high-quality metagenome-assembled genomes (MAGs) (>80% completeness and <10% contamination), about half of which showed <95% inter-species ANI values, implying potential novel species (Fig. 3a and Supplementary Data 2). The MAGs belonging to Gammaproteobacteria accounted for 48.69% of total reads in N0 (vs 2.76% in N2) on 7 days, while the MAGs belonging to Firmicutes occupied 67.54% of total reads in N2 (vs 4.47% in N0). Of the 13 Firmicutes MAGs, six MAGs were identified as Bacillus and Staphylococcus genera and exhibited a high relative abundance in N2 on 7 days, especially the overrepresented MAG2 belonging to Staphylococcus sciuri (newly named Mammaliicoccus sciuri) that accounted for 47.54% of total reads (Fig. 3a), consistent with our results based on the taxonomic composition of the residue-inhabiting community (Fig. 2c).

a Phylogenetic tree of 68 recovered MAGs from wheat residues under N0 and N2 treatments. N0, no N input; N2, high N input (240 kg hm−2). Clades are colored according to taxonomic phylum. Proteobacteria are shown at the class level, and MAGs belonging to the genera Staphylococcus and Bacillus are highlighted with blue bold font. Green solid blocks represent novel MAGs (i.e., MAGs with <95% inter-species ANI values). Heatmap shows the relative abundance of MAGs over 187 days under N0 and N2 fertilizer levels. Outside blue and yellow bars represent the completeness and contamination of MAGs, respectively. b Bar plot represents the contribution of Bacillus and Staphylococcus to functional profiles in terms of CAZymes and sugar metabolism. Baci. Bacillus, Stap. Staphylococcus. c Heatmap indicates the number of genes encoding CAZymes related to lignocellulose degradation. GH glycoside hydrolase, CE carbohydrate esterase. Source data are provided as a Source Data file.
Focusing on the six N-stimulated Bacillus and Staphylococcus MAGs, we analyzed their annotations to determine the content and diversity of CAZymes falling into the functional categories of lignocellulases. In total, the number of CAZyme genes encoding hemicellulases and cellulases was higher in the Bacillus MAGs than in the Staphylococcus MAGs (Fig. 3b and Supplementary Data 3). The Bacillus MAGs possessed genes encoding diverse CAZymes involved in all key steps of cellulose and hemicellulose degradation, whereas Staphylococcus could not hydrolyze these substrates (Fig. 3c and Supplementary Fig. 7). For example, genes encoding endo-glucanase, exo-glucanase, endo-xylanase, and endo-mannanase (GH48, GH9, GH51, GH16_21, GH10, GHGH11, GH30_8, and GH5), which are responsible for the initial enzymatic step of cellulose and hemicellulose, were only present in the Bacillus MAGs. The Staphylococcus MAGs included more genes encoding GH1, which hydrolyze downstream sugar products such as cellobiose, xylobiose, and mannobiose, suggesting their stronger capacity for sugar product utilization. We thus further determined the contributions of the Bacillus and Staphylococcus species corresponding to the six MAGs to the metabolism of sugar products by taxonomically classifying assembled contigs-based KEGG genes in the whole metagenomics gene set (Supplementary Data 4, 5). An overwhelming contribution of Staphylococcus to sugar metabolic potential was observed (Fig. 3b). Compared with the Bacillus MAGs, the Staphylococcus MAGs had a higher abundance of genes involved in the phosphotransferase system (PTS), a major carbohydrate transport system that catalyzes the uptake of numerous monosaccharides, disaccharides, and sugar derivatives and their conversion into their respective phosphoesters, and downstream pathways such as glycolysis, the TCA cycle, and pyruvate metabolism (Supplementary Fig. 8). These results suggest that there might be cooperation between the workhorses of decomposition (e.g., Bacillus) and non-decomposers with a potentially high capacity to consume the product sugars (e.g., Staphylococcus).
Interactions between the N-shaped Bacillus decomposers and Staphylococcus non-decomposers confirmed by SynComs
To explore the potential interactions between Bacillus decomposers and Staphylococcus non-decomposers, we first conducted a large-scale strain screening of residue-inhabiting microbiota at the early stage (7 days and 14 days) using six media. A total of 167 unique isolates belonging to four phyla and 13 families were collected (Supplementary Data 6). We further cross-referenced the 16S rRNA gene of these 167 strains against the time-based OTU biomarkers identified by LEfSe analysis of the amplicon data (Fig. 4a). We obtained 19 bacterial strains that were enriched at the early stage, which accounted for 68.20% and 43.31% of the relative abundance of the residue-inhabiting community on 7 days and 14 days, respectively (Fig. 4b and Supplementary Data 7). The degradation capacity of the 19 strains was determined through decomposition experiments and culture on CMC-Congo red medium (Supplementary Fig. 9). As expected, four of the five N-enriched Bacillus were equipped with the metabolic pathway to depolymerize wheat residues (i.e., strains AZ50, AZ52, AZ103, and AZ290), whereas N-enriched Staphylococcus sciuri AZ288 lacked this metabolic pathway (Fig. 4c). This was further confirmed by the identification of CAZymes through whole genome sequencing (Supplementary Data 8). Among the four Bacillus decomposers, B. safensis AZ103 and B. altitudinis AZ290 exhibited stronger decomposition ability (i.e., strong decomposers) than B. tequilensis AZ50 and AZ52 (i.e., weak decomposers) (Fig. 4c).

a Results of matching strains to biomarkers obtained from LEfSe analysis. The inner column corresponds to individual isolates. Heatmap represents differences in relative abundance over incubation times under no N input (N0), low N input (180 kg hm−2; N1), and high N input (240 kg hm−2; N2). Strains belonging to the genera Staphylococcus and Bacillus decomposers are shown in red. Nineteen strains were grouped into two categories: (1) enriched by N input (N+; n = 8); (2) depleted or unresponsive to N input (N−; n = 11). b Pie plots showing the relative abundance of matching microbes at different sampling times during decomposition. c Decomposition capacity of 19 strains. The bars and error bars indicate mean values and standard deviation, respectively (n = 3). Statistical significance is based on one-way ANOVA followed by Tukey’s test. d Residue decomposition rate after 10 days by four Bacillus decomposer co-cultured with autoclaved or active non-decomposer S. sciuri AZ288, respectively. Statistical analyses were conducted by one-sided t-test (n = 6). e Decomposition capacity of synthetic communities (SC) that consist of different decomposers in residue-N-supplemented media. Statistical significance is based on one-way ANOVA followed by Tukey’s test (n = 4). Boxplots in d, e display the median and interquartile range. Upper and lower whiskers extend to data no more than 1.5× the interquartile range from the upper and lower edges of the box, respectively. f, g Community structure and composition of SC1 with and without S. sciuri AZ288 in N-supplemented and N-free media after 10 days of residue decomposition. Data in (f, g) were analyzed by permutational multivariate analysis of variance and Mann–Whitney test, respectively (n = 3). Different letters and asterisks in (c–g) indicate significance levels: *P < 0.05 and ****P < 0.0001. ns not significant. Source data is provided as a Source Data file.
Next, we mixed each of the four Bacillus decomposer strains with the non-decomposer S. sciuri AZ288 and cultured them in residues-NH4+-supplemented medium. After 10 d of incubation, the decomposition abilities of the weak decomposers (B. tequilensis AZ50 and AZ52) were significantly suppressed by S. sciuri AZ288, with their degradation rates decreasing by 44.34% and 57.05%, respectively (t-test, P < 0.05; Fig. 4d). However, both strong decomposers maintained robust degradation abilities, even in the presence of S. sciuri AZ288 (Fig. 4d), implying that S. sciuri AZ288 exerts completely different effects on strong and weak decomposers. By testing SynComs consisting of different decomposers, we found that the inclusion of weak decomposers significantly decreased the decomposition efficiency of the SynComs by ~21.38% (Fig. 4e). Interestingly, the addition of the non-decomposer S. sciuri AZ288 restored the residue degradation efficiency of SynComs to a level similar to that of SynComs consisting only of strong decomposers (12.91–16.77%; Fig. 4e). These results suggest that S. sciuri AZ288 may restrict the growth of the weak decomposers under N-fertilized conditions, thereby allowing the stronger decomposers to primarily accomplish decomposition. To test this hypothesis, we analyzed the community dynamics of SynComs containing four Bacillus strains, with and without S. sciuri AZ288. The addition of S. sciuri AZ288 significantly altered the community structure and composition of Bacillus decomposers only in the presence of N source (Fig. 4f, g). Specifically, the non-decomposer S. sciuri AZ288 was able to reshape the decomposer community dominated by a weak decomposer (93.17% of AZ50) into one dominated by strong decomposers (53.65% of AZ103 and 19.93% of AZ290), as evidenced by both colony morphological characteristics (Supplementary Fig. 10) and sequencing data (Fig. 4g).
Mechanisms by which non-decomposer S. sciuri AZ288 restricts the growth of weak decomposers during residue decomposition
Finally, we attempted to decipher the mechanisms by which the non-decomposer S. sciuri AZ288 restricts the growth of the weak decomposers B. tequilensis AZ50 and AZ52 during residue decomposition. First, we checked whether this inhibition is caused by S. sciuri AZ288 secreting specific inhibitory molecules. Neither the S. sciuri AZ288 solution (Supplementary Fig. 11a) nor the filtrate of the culture treated with both Bacillus decomposers and S. sciuri AZ288 (Supplementary Fig. 11b, c) showed any inhibitory effect on the four Bacillus decomposers. Thus, we further investigated whether the inhibitory phenomenon was caused by nutrient competition. By performing transcriptomics analyses, we found that the downregulated KEGG orthologs (KOs) in weak decomposers after S. sciuri AZ288 addition were distributed in several sugar uptake and utilization pathways such as ‘phosphotransferase system (PTS)’, ‘fructose and mannose metabolism’, and ‘pentose and glucuronate interconversions’ (Fig. 5a). Among the PTS pathways, multiple genes encoding proteins responsible for the uptake of specific sugars were significantly depleted in the weak decomposers (B. tequilensis AZ50 and AZ52) rather than strong decomposers (Fig. 5b and Supplementary Data 9). Metabolomics analyses confirmed that several downstream metabolites of sugar, such as N-acetyl-D-glucosamine, fructosyl-lysine, N-acetylmuramate, and fructose-6P, were depleted in the co-culture of the weak decomposers with S. sciuri AZ288 (Fig. 5c and Supplementary Data 10). These results suggest that the non-decomposer S. sciuri AZ288 inhibits the growth of weak decomposers through sugar competition.

a Enrichment analysis of downregulated KOs in four Bacillus decomposers in co-culture vs monoculture. Three replicates (n = 3) were used for transcriptomics analyses. Sugar-related pathways downregulated in weak decomposers are shown in red font. b Metabolic pathways involved in sugar utilization were reconstructed based on differentially expressed genes between co-culture and monoculture. Gene symbols and metabolites are labeled with KEGG designations. Blank boxes indicate absent genes or those with no significant difference in transcript levels between the two groups. The color bar represents gene expression level. c Sugar-related metabolites showing significant differences in abundance between the two groups. Six replicates (n = 6) were used for metabolomics analyses. Blank boxes indicate absent metabolites or those with no significant difference in abundance between the two groups. d Growth of four Bacillus decomposers and S. sciuri AZ288 in liquid medium containing glucose, fructose, mannose, or cellobiose. Asterisks indicate significance levels (one-way ANOVA followed by Tukey’s test (n = 12)): ****P < 0.0001. e Decomposition rate of wheat residue after 10 days by decomposer alone, the co-culture of decomposer and S. sciuri AZ288 (+AZ288), and the co-culture of decomposer and S. sciuri AZ288 with the addition of exogenous sugar (+sugar). Image at the bottom showing decomposition of residue with the addition of mannose. Images of residue decomposed with the addition of fructose and cellobiose are shown in Supplementary Fig. 12. The SynComs contain four Bacillus decomposers. Different letters indicate significance levels (one-way ANOVA followed by Tukey’s test (n = 3)). Boxplots in (d, e) display the median and interquartile range. Upper and lower whiskers extend to data no more than 1.5× the interquartile range from the upper and lower edge of the box, respectively. Source data is provided as a Source Data file.
To confirm this speculation, we first tested the growth of the five strains in the presence of downstream product sugars, such as mannose from hemicellulose, cellobiose from cellulose, and fructose isomerized from glucose. Notably, S. sciuri AZ288 showed the strongest growth in N-supplemented media with different sugars, exceeding the average growth of the other four decomposers by 97.04% (Fig. 5d). This is consistent with our findings based on the reconstructed MAGs that the Staphylococcus non-decomposers showed stronger sugar uptake capacity than that of the Bacillus decomposers (Fig. 3b). Interestingly, the addition of excess fructose, mannose, or cellobiose was almost sufficient to restore the residue degradation efficiency of the weak decomposers B. tequilensis AZ50 and AZ52 in the presence of S. sciuri AZ288 (Fig. 5e and Supplementary Fig. 12). However, the opposite phenomenon was observed in the co-cultures of S. sciuri AZ288 with B. safensis AZ103, B. altitudinis AZ290, or SynComs consisting of four decomposers. This result indicated that excess sugars impair the strong decomposers. Altogether, these results demonstrate that the fast-growing non-decomposer S. sciuri AZ288 indeed limits the growth of weak decomposers by sugar depletion, providing a sufficient ecological niche for the strong decomposers and allowing them to maintain their robust decomposition capacity.
Discussion
In the present study, N fertilization increased wheat residue decomposition in situ (Fig. 1a). Such an increase is a result of high N availability leading to a higher decomposition rate during the early stage (Fig. 1b), consistent with the findings of previous studies conducted in multiple ecosystems11,12,13. For example, the addition of ammonium nitrate increased the cellulose decomposition of leaf litter at the early stage in an evergreen broadleaf forest11. These common outcomes suggest that the accelerating effects of N inputs on plant residue decomposition at the early stage may be independent of the ecosystem, N form, and residue type.
In a unique habitat like decomposing plant residues, microbial subgroups can be deterministically selected from the surrounding soil to degrade residues23. Consistent with this theory, our FEAST results showed a progressive colonization of soil-derived microbiota on the surface of wheat residues (Fig. 2a). More importantly, this colonization process was considerably promoted by N fertilization at the early stage. For example, the proportion of residue-derived microbiota remained high at 79.48–92.89% on 7 days without N application, while the soil-derived microbiota only accounted for 3.42–5.23%. However, this situation was completely changed by N fertilization, where the soil-derived and unknown-source-derived microbiotas accounted for an overwhelming majority of the microbial community (87.68–97.15%) on 7 days. This N-stimulated striking acceleration in soil microbe colonization could be an important reason for the prominent higher decomposition rate of wheat residues at the early stage. According to the qPCR, amplicon, and metagenomics results, bacteria, rather than fungi, contributed more to the increased residue decomposition rate at the early stage under sufficient N supply (Fig. 2 and Supplementary Table 2). This may be related to the differences in nutritional requirements and substrate preferences between bacteria and fungi24,25. More specifically, bacteria typically have higher nutrient requirements than fungi24, so they grow more quickly and occupy a more extensive niche on decomposing residues in the presence of easily available C and N sources25,26. Thus, bacteria are likely responsible for the decomposition of water-soluble compounds, cellulose, and hemicellulose in residues at the early stage27. In contrast, fungi typically have lower nutrient requirements and a higher capacity to degrade recalcitrant lignified structures at the later stage28.
In this study, we found that the soil-derived genera Bacillus and Staphylococcus in the phylum Firmicutes were significantly increased by N fertilization and accounted for 60.15–70.66% of the residue-inhabiting microbiota on 7 day (Fig. 2c). Because the widely distributed Bacillus is well known to secrete various hydrolytic enzymes, members of this genus are recognized as versatile decomposers of complex carbohydrates and safe organisms for industrial enzyme production29,30. Based on metagenomics binning and CAZymes annotation analyses, we detected Bacillus genes encoding diverse functional CAZymes involved in the entire process of cellulose and hemicellulose degradation (Fig. 3c). Therefore, Bacillus species can be expected to function as “workhorses” to reduce the recalcitrance of lignocellulose during the early stage of decomposition. This is further confirmed by the fact that four isolated N-enriched Bacillus decomposers possessed the strongest decomposition ability (Fig. 4c). Unexpectedly, we found that Staphylococcus sciuri accounted for a large proportion of the residue-inhabiting microbial community on 7 days (Fig. 3a). Usually, lignocellulose degradation is not a common property of Staphylococcus species in the environment, although some members of this genus identified from insects’ gut are able to hydrolyze lignocellulose31,32. This is consistent with the results of metagenomics binning and isolation analyses, which showed that neither Staphylococcus MAGs nor isolated Staphylococcus sciuri AZ288 were able to metabolize lignocellulose (Figs. 3c, 4c). Although relatively rare, a few studies have uncovered the positive roles of non-decomposers from the perspective of mutualism and division of labor between decomposers and non-decomposers19,33. For example, in a bacterial consortium enriched through the course of 40 transfers, the bidirectional cross-feeding between the lignin-degrader P. gergoviae and non-degraders fostered P. gergoviae’s growth and further stimulated lignin degradation19. In the consolidated bioprocessing of lignocellulosic feedstocks, the non-decomposer Lactobacillus pentosus, which consumes cellobiose, was able to alleviate the inhibitory effect of the over-accumulation of cellobiose on the lignocellulolytic activity of Trichoderma reesei, which is deficient in β-glucosidase production33. However, the interactions between N-stimulated Bacillus decomposers and the non-decomposer S. sciuri, as well as their impacts on residue decomposition in agricultural ecosystems, have not been explored in previous studies.
It is a widely accepted fact that fertilization provides sufficient N sources for decomposers, thereby promoting the decomposition of crop residues with a high ratio of C:N (>70:1) in agricultural soils5. Surprisingly, our study indicates that N-enriched copiotrophic non-decomposers also play a key role in this process by exerting different effects on two types of Bacillus with varying decomposition capabilities. Specifically, we found that the non-decomposer S. sciuri AZ288 restricted the growth of weak decomposers in the presence of N sources, thereby reshaping the decomposer community into one dominated by strong decomposers during residue decomposition (Fig. 4g). This regulatory process increased the decomposition rate by 16.77% (Fig. 4e). It has been reported that inhibitory effects between microorganisms are usually a result of inhibitory molecules produced by specific bacteria34,35. Our multi-omics technologies identified a mechanism by which the non-decomposer S. sciuri AZ288 inhibits the growth of weak decomposers by sugar depletion, rather than through the production of inhibitory factors. In line with this mechanism, the transcriptomics data revealed significant down-regulation of series of genes in weak decomposers related to subunits of PTS system and sugar metabolism when S. sciuri AZ288 was present (Fig. 5b). This implied that the various sugars produced from residue degradation were rapidly assimilated by S. sciuri AZ288, as it has a strong uptake capacity for sugars under N-supplemented conditions (Figs. 3b, 5d). Accordingly, the metabolomics analysis also showed that sugar products (e.g., N-acetyl-D-glucosamine and fructose-6P) were markedly depleted in the co-cultures of weak decomposers and S. sciuri AZ288 (Fig. 5c). The inhibition of the degradation efficiency of weak decomposers by S. sciuri AZ288 was reversed upon the addition of sugar sources (Fig. 5e), further confirming the mechanism of sugar consumption. Such restriction of microbial growth as a result of sugar depletion by specific strains has been detected in some studies focused on gut pathogens36,37. For example, the bacterial consortium restricted the growth of vancomycin-resistant Enterococcus through nutrient depletion, specifically by reducing the levels of fructose36.
The strong decomposers were not inhibited by the N-enriched non-decomposers, most likely because they were able to quickly break down residue to access available nutrients and produce large amounts of monosaccharides and disaccharides. However, an excess of sugar slowed the decomposition capacity of strong decomposers (Fig. 5e). In general, over-accumulation of lignocellulose degradation intermediates (such as cellobiose) causes end-product inhibition, which significantly inhibits the rates of enzyme-catalyzed lignocellulose hydrolysis20. Therefore, we speculated that the high sugar uptake capacity of S. sciuri AZ288 may maintain the decomposition ability of strong decomposers. This was further supported by the observation that in the S. sciuri AZ288-reconstructed community dominated by strong decomposers, the addition of exogenous sugars negatively affected residue decomposition (Fig. 5e). Overall, our results show that the fast-growing non-decomposer S. sciuri AZ288 restricts the growth of weak decomposers through sugar depletion, leading to the restructuring of a community dominated by strong decomposers that maintain robust degradation abilities. However, in the traditional method of top-down enrichment, these non-decomposers are first filtered out by the selection media with specific substrates like carboxymethyl cellulose (CMC), and therefore, their key roles have been overlooked in the design of SynComs. Consequently, their role in enhancing residue decomposition under N fertilization has never been paid attention to. In the present study, the application of six media enabled us to obtain residue-inhabiting microbiota as comprehensively as possible and permitted exploration of interactions between N-enriched decomposers and non-decomposers.
In conclusion, our study demonstrates that N fertilization accelerates wheat residue breakdown mainly at the early stage by promoting rapid colonization of key soil-derived microbial consortium on the residue surface. In this consortium, the N-enriched, fast-growing non-decomposer S. sciuri restricts the growth of weak decomposers through sugar depletion, resulting in the restructuring of a community dominated by strong decomposers that retain robust degradation abilities (Fig. 6). Thus, in addition to providing an abundant N source for decomposers, the N-shaped fast-growing copiotrophic bacteria are also a significant factor in accelerating residue decomposition. This relationship represents a pathway that is usually overlooked by the biotechnology community when designing processes for the complete decomposition of crop residues. Future research is needed to explore the potential of this N-induced microbial consortium to regulate the incorporation of residue into soil C pools, ultimately developing new microbial strategies to strengthen the C sequestration capacity of crop residue-returned agricultural ecosystems.

Nitrogen-rich environment favors the Bacillus decomposers and fast-growing non-decomposer S. sciuri. Under nitrogen fertilization, the fast-growing non-decomposer S. sciuri restricts the growth and decomposition ability of weak decomposers through sugar depletion, resulting in the restructuring of a community dominated by strong decomposers, thereby facilitating residue decomposition.
Methods
Study site description
The field study site is located in Yuanyang, Henan Province (34.78° N, 113.67° E) on the North China Plain, which is the primary cultivation area for wheat and maize in China and produces 47% of the national total of crop residues. The common cropping system in this area is wheat–maize rotation. The wheat and maize growing seasons are from early October to early June of the following year, and from the end of June to the end of September, respectively. The wheat and maize residues after harvesting were incorporated into the soil at a rate of 7000 kg hm−2 in June 2016. The site had a warm climate with an annual average temperature of 16.42 °C and annual average precipitation of 590.18 mm. The soil type was classified as Calcaric Fluvisol according to the Harmonized World Soil Database (v1.2). At the beginning of the experiment, the soil had a pH of 8.5, 10.81 g kg−1 SOC, 1.41 g kg−1 total N, 27.20 mg kg−1 available phosphorus, and 117.64 mg kg−1 available potassium (Supplementary Table 3).
Field experiment on wheat residue decomposition
Treatments with three N fertilizer (urea) rates (N0, no N input; N1, low N input (180 kg hm−2); N2, high N input (240 kg hm−2)) were established in the study field in June 2016 (Supplementary Fig. 2). A total of nine plots were arranged in a completely randomized block design with each N-input treatment replicated three times (Supplementary Fig. 13). All individual plots were separated by a buffer zone.
To characterize the influence of N fertilization on wheat residue degradation, we established a litterbag experiment in the agricultural plots under different N addition rates. Briefly, wheat residues were collected after harvesting wheat. The wheat residues were air-dried, cut into 5–8 cm lengths, and loaded into nylon bags with a mesh size of 0.05 mm (15 cm in length and 10 cm in width). This small mesh size allowed free access of microorganisms from the soil and excluded all meso- and macrofauna, which necessarily means that our study focused on microbially mediated decomposition12. Litterbags containing 10 ± 0.03 g wheat residues were then separately buried in the plots corresponding to N0, N1, and N2 at a depth of 10 cm during the maize growing season. According to the ratio of N fertilizer rates to the amount of incorporated wheat residues in the field (i.e., 7000 kg hm−2), we added 25.71 mg g−1 and 34.29 mg g−1 N fertilizer into the residue bags buried in the corresponding N1 and N2 plots, respectively. Litterbags were retrieved at seven sampling times (Supplementary Fig. 2): in 2017 at the onset of the experiment (0 day), on 27 June (7 days), 04 July (14 days), 29 July (39 days), 21 August (62 days), 28 September (100 days), and 24 December (187 days). At each sampling time, two litter bags were taken from each plot and transported on ice to the laboratory. One litterbag was used to determine the decomposition rate, and the other was used to analyze mineral N content and microbial communities. In total, 126 litterbags were collected from the N0, N1, and N2 plots (3 N input rates × 3 replicate plots × 7 sampling times × 2 uses). Soil samples were collected at the onset of the experiment (0 day) and on 24 December (187 days).
Decomposition rate of wheat residue and mineral N content in wheat residue bags
After removing the roots and soil from the surface of the litterbags, all litterbags were carefully washed three times with distilled water. The retrieved wheat residues were then oven-dried to a constant weight at 65 °C to determine the remaining dry mass. The decomposition rate of wheat residues was calculated based on the dry weight loss as follows: (dry weight of initial residue − dry weight of final residue)/dry weight of initial residue. The residue decomposition data were fitted to a two-phase exponential decay model using GraphPad Prism 8.0.0 (GraphPad Software, San Diego, CA, USA, https://www.graphpad.com/).
To determine the effect of N fertilization on the physical structure of wheat residues, the ultrastructural changes and surface characteristics of wheat residues on 0 day, 7 days, 14 days, 39 days, 62 days, 100 days, and 187 days under N0, N1, and N2 conditions were assessed by SEM (Hitachi S4800, Tokyo, Japan). To determine the contents of NH4+ and NO3–, 1 g fresh wheat residue was extracted with 5 mL 1 M KCl, and then their concentrations were determined using a flow injection autoanalyzer (FLA star 5000 Analyzer, Foss, Denmark).
DNA extraction, qPCR, amplicon sequencing, and processing of sequencing data
The DNA was extracted from wheat residues and soil samples with the Fast DNA Spin Kit (MP Biomedicals, Solon, OH, USA). To prevent exogenous DNA from contaminating the environmental samples, the consumables used for DNA extraction were sterilized, and the personnel performing the test wore gloves. To estimate the absolute abundances of 16S rRNA gene and ITS region in the residue samples, we performed qPCR by using 338F/806R and ITS1F/ITS2R primer sets, respectively (Supplementary Table 4). The qPCR reaction and amplification, and data analysis are described in the Supplementary Information.
The V3–V4 region of the bacterial 16S rRNA gene and the fungal ITS region were amplified using the 338F/806R and ITS1F/ITS2R primer pairs, respectively. Each DNA extract was diluted 10-fold, and then amplified in duplicate along with a negative PCR control (no template added) to verify that the PCR products were not contaminated. The total PCR reaction volume was 50 µL, which contained 25 µL of 2× EasyTaq PCR SuperMix (TransGen Biotech, China), 2.5 µL of each primer (10 µM), 2.5 µL of 10-fold diluted DNA template, and 17.5 µL of sterilized water. The PCR amplification program is described in detail in the Supplementary Information. If there was no visible amplification from the negative control, the duplicate PCR products were mixed. The mixed PCR products were subsequently measured using a Quant-iT™ PicoGreen™ dsDNA Assay (Thermo Fisher Scientific, Waltham, MA, USA), and 120 ng of each PCR product was pooled. Sequencing libraries were generated using an Illumina TruSeq DNA PCR-Free Library Preparation Kit (Illumina, San Diego, CA, USA) and then sequenced on the miseqPE300 platform.
QIIME v.1.9.138, QIIME2-2020.839, VSEARCH v.2.12.040, USEARCH v.11.041, and mothur v.1.40.442 were used to process the sequencing data. Briefly, paired-end sequencing data were confirmed by FastQC v.0.10.143 and merged using VSEARCH v.2.12.0. Merged reads were assigned and demultiplexed to each sample according to unique barcodes using QIIME v.1.9.1 and QIIME2-2020.8. Unique reads were obtained using scripts to remove primers and low-quality sequences (fastx_filter) and to identify non-redundant reads (derep_fulllength). The unique reads were clustered into operational taxonomic units (OTUs) at the 97% threshold by VSEARCH v.2.12.0. The representative sequences were picked by USEARCH v.11.0, and the OTU table was generated using VSEARCH (usearch_global). Bacterial and fungal OTUs were taxonomically assigned to the SILVA 13844 and UNITE45 databases, respectively, using the classify.seqs command in mothur v.1.40.4. The OTUs classified as chloroplast, mitochondria, eukaryote, cyanophyta, cyanobacteria, cercozoan, and protista were removed from the dataset. Finally, the bacterial and fungal OTU tables were rarefied to ensure an even sampling depth.
Metagenomics sequencing and read analysis
To explore the impacts of N fertilization on the functional composition of the wheat residue-inhabiting microbial community, we analyzed the samples collected on 0 day, 7 days, 14 days, 62 days, and 187 days under N0 and N2 conditions. Before preparing metagenomics sequencing libraries, the contamination, purity, and concentration of DNA samples were tested. The contamination of DNA was checked by 1% agarose gel electrophoresis. The purity of DNA was analyzed using a nanophotometer (Implen, Westlake Village, CA, USA). The concentration of DNA was determined using a Qubit DNA Assay Kit with a Qubit 2.0 fluorometer (Invitrogen, Carlsbad, CA, USA). Metagenomics sequencing libraries were constructed using NEXTFLEX Rapid DNA-Seq Kits (Bioo Scientific, Austin, TX, USA) and sequenced on the Illumina HiSeq 2500 platform.
After quality filtering using Fastp v.0.20.0 (https://github.com/OpenGene/fastp), the reads were assembled using MEGAHIT v.1.1.2 with a minimum contig size of 300 bp46. Open reading frames (ORFs) were predicted from these assembled contigs using MetaGene47, and the genes with nucleic acid lengths longer than 100 bp were selected and translated into amino acid sequences. The gene sequences were then clustered by CH-HIT v.4.6.1 (http://www.bioinformatics.org/cd-hit) with 90% sequence identity and 90% coverage to create gene sets. The high-quality reads were mapped to the non-redundant gene catalog to estimate gene abundance using SOAPaligner v.2.21 (http://soap.genomics.org.cn). To obtain species taxonomic annotations, representative sequences of the non-redundant gene catalog were aligned against the NR database in NCBI using Diamond (parameters: blastp, e-value ≤ 1e − 5)48. To obtain information about CAZyme modules, the gene sets were compared with the CAZy database v.849 using hmmscan v.8 (https://www.ebi.ac.uk/Tools/hmmer/search/hmmscan) with an e-value cutoff of ≤1e − 5. The CAZyme-associated genes were classified into six modules: GH, GT, CE, AA, PL, and CBM. The KEGG pathway annotations were conducted using Diamond with searches against the KEGG database v. 0.8.35 at the optimized e-value threshold of 1e − 548.
Genome binning and analysis
To generate MAGs, all bacterial assembled contigs were binned using concoct, maxbin2, and metabat250,51,52 within MetaWRAP v.1.3.253. The DAS Tool was then applied to screen for the highest quality MAGs across the three binned sets54. The completeness and contamination of MAGs were evaluated using CheckM v.1.1.355. All MAGs were pooled and then de-replicated with dRep (parameters: -comp 80 -con 10 -sa 0.95)56, resulting in 68 high-quality MAGs that were unique at the species level for further analyses.
To calculate the coverage of each of the de-replicated MAGs in each metagenome sample, all raw reads from that sample were mapped using Bowtie 2 to a concatenated fasta file including the 68 MAGs57. Taxonomic assignment of the MAGs was achieved using GTDB-TK v.1.3.0 with release 95 of the GTDB database58. A phylogenetic tree including all 68 MAGs was constructed using FastTree v.2.1.1159 and visualized using interactive Tree of Life (iTOL)60.
Gene annotation of the 68 MAGs was performed using Prokka v.1.14.661. Genes encoding CAZymes in MAGs were identified by their protein family domains using Diamond with a cutoff e-value of ≤ 1e − 548. Genes annotated as AA, GH, and CE were divided into ligninase, cellulase, and hemicellulase categories based on the CAZy database49. The metabolic pathways related to sugar utilization of MAGs were constructed based on information at KEGG using KOFAM v.1.3.0 with an e-value below the 1e − 5 threshold62. To identify the contributions of MAGs to sugar metabolism, we screened the species corresponding to the MAGs from the whole set of genes recovered from the assembled contig and then counted the abundance of KEGG genes related to sugar metabolism contained in them.
Statistical analysis
All statistical analyses and plots were generated with R and GraphPad Prism 8.0.0, unless otherwise noted. Data normality was checked by the Shapiro–Wilk test or Kolmogorov-Smirnov test in GraphPad Prism 8.0.0. One-way and two-way ANOVA in conjunction with post hoc Tukey’s or Fisher’s LSD test, or one-sided t-test were used for normally distributed data. In contrast, the Kruskal–Walli’s test, Mann–Whitney test, or Scheirer–Ray–Hare test, followed by a Dunn’s post-test correction, were employed for non-normally distributed data. A P value of < 0.05 was considered statistically significant. Three alpha diversity indexes, including species richness, Shannon index, and Faith’s phylogenetic diversity (Faith’s PD) were calculated using the vegan and picante packages63,64. Beta diversity indexes were calculated using QIIME2-2020.839. We performed a non-metric multidimensional scaling (NMDS) ordination of Bray–Curtis distance matrices to reveal the microbial community structure. The effects of N fertilization, decomposition time, and decomposition rate on microbial beta-diversity were assessed by a permutational multivariate analysis of variance (Adonis) in the vegan package. We performed the fast expectation-maximization microbial source tracking (FEAST) method to determine the potential origin of the wheat residue-inhabiting microbiota65. The difference in microbial absolute abundance was assessed by a nonparametric test. We used the DESeq2 package to identify CAZymes showing a significant difference in abundance between the N0 and N2 regimes66. The linear discriminant analysis (LDA) effect size (LEfSe) algorithm was performed to screen for biomarkers (i.e., OTUs) along longitudinal trends. Biomarkers identified from the LEfSe analysis were used to match subsequent isolates.
Isolation of bacteria from the litter bags
Bacterial strains were isolated from the litterbags at the early stage of decomposition (7 days and 14 days). Briefly, 1 g of wheat residue was cut into 1 cm pieces with sterilized scissors, and the pieces were transferred to a triangular flask containing 200 mL sterile saline. The flask was incubated with shaking at 180 rpm at 28 °C for 1 h. To isolate strains, six gradient dilutions (10−1, 10−2, 10−3, 10−4, 10−5, and 10−6) of wheat residue suspensions were incubated onto CMC, LB, CBA, R2A, TSB, and TSA-YE media and cultured for 3–4 days at 28 °C (Supplementary Data 11). After incubation, colonies were selected based on their characteristics and colony morphology and were purified by triple serial colony isolation. The isolates were analyzed by PCR using 27F/1492R primer pairs, and then the PCR products were subjected to Sanger sequencing. The 16S rRNA gene sequences of the isolates were compared with available 16S rRNA gene sequences in the NCBI using the BLAST algorithm. In total, we isolated 167 unique bacterial strains. These isolates were further identified by comparing their 16S rRNA V3–V4 regions against the biomarkers (from LEfSe analysis) using UCLUST with 98.65% similarity as the threshold67. This threshold has been reported to accurately distinguish between two species. A total of 19 unique strains enriched at the early stage were selected for further analysis. Cladograms were visualized using iTOL v6.660. The isolated cultures were stored in 30% (v/v) glycerol at ‒80 °C.
We used two methods to evaluate the lignocellulose degradation capacity of the 19 strains. Each strain was inoculated into liquid medium containing wheat residues. Liquid medium without the addition of strains served as a control. The strains with a significantly higher residue decomposition rate than that of the control were identified as decomposers. CMC-Congo red medium was used to confirm the lignocellulose-degrading capacity of each strain by measuring the diameter of the clear hydrolysis zone surrounding the bacterial colonies. The 19 isolated strains could be grouped into the following two subcategories according to their response to N fertilizers: (1) N-enriched decomposers (n = 8); (2) N-depleted or unresponsive decomposers (n = 11) (Supplementary Data 7).
Laboratory experiment of wheat residue decomposition by SynComs
Decomposition experiments of wheat residues using SynComs were conducted to explore the potential interactions between N-enriched Bacillus decomposers and non-decomposers. For this purpose, we used the following two different liquid media: (1) inorganic salt medium without N source (yeast extract 0.1 g L–1, peptone 0.1 g L–1, K2HPO4 2.0 g L–1, MgSO4·7H2O 0.3 g L–1, pH 7); (2) inorganic salt medium with (NH4)2SO4 added as an NH4+ source. The N sources were added at a rate of 0.135% (w/v).
The wheat residues were alkalized by soaking in 1% NaOH (w/v) solution for 24 h, rinsed with distilled water until the pH returned to neutral, and oven-dried at 70 °C. Then, 1.2 g oven-dried wheat residues were cut into 0.5–1 cm pieces, transferred to 100 mL triangular flasks, and sterilized by autoclaving for subsequent wheat residue decomposition experiments. Each 1.2 g sterilized wheat residue sample was incubated in liquid medium at a rate of 2% (w/v), and each SynCom was inoculated into liquid medium at a rate of 5% (v/v). For each SynCom, bacterial suspensions were mixed in equal amounts to a final OD600 of ~0.05. To simulate the early decomposition of wheat residue, the liquid media were incubated with shaking at 180 rpm at 30 °C for 10 days.
After 10 days of incubation, the remaining wheat residues were collected using an iron 30-mesh filter, washed three times with sterile water, oven-dried at 80 °C for 2 days, and then weighed. The liquid medium was transferred to a 50-mL sterile centrifuge tube and centrifuged at 1000×g for 10 min. The supernatant was transferred to a new centrifuge tube and centrifuged at 2000×g for 10 min. The supernatants were stored at ‒80 °C for subsequent metabolomics analysis. Meanwhile, the biomass pellet was collected for subsequent amplicon sequencing and transcriptomics analysis.
Changes in SynCom's structure
To investigate the impact of the non-decomposer S. sciuri on the community structure of Bacillus decomposers, we performed amplicon sequencing on the biomass pellet collected from the residue decomposition experiment. The analysis of amplicon data followed similar methods as those described above. The changes in the relative abundance of each strain were analyzed using Mann–Whitney test.
Microbial antagonism experiment
Two methods were used to determine whether the non-decomposer S. sciuri AZ288 secretes specific inhibitory molecules to restrict the growth of weak decomposers. Firstly, the Oxford cup method was used for the analysis of the non-decomposer solution. Briefly, 20 µL of decomposer suspension (OD600 = 1) was spread evenly on plates, and then three sterilized Oxford cups were placed on each plate. The three cups were inoculated separately with 200 µL of non-decomposer culture, ampicillin (positive control), and sterilized water (negative control). Secondly, the decomposition liquid treated with decomposer alone, or with both the decomposer and non-decomposer, was filtered to remove living cells (0.22 µm filter). The effect of decomposition liquid on the growth of decomposers was tested by the Oxford cup method and by in vitro assays. The detailed method is provided in the Supplementary Information.
Bacterial whole genome analysis
Genomic DNA was extracted from Bacillus decomposers and one non-decomposer S. sciuri AZ288, using the TIANamp Bacteria DNA Kit (TIANGEN, Beijing, China) according to the manufacturer’s protocol. The genomic DNA was sequenced using a combination of PacBio Sequel IIe and Illumina Novaseq 6000 sequencing platforms by the Majorbio Biopharm Technology Co., Ltd. (Shanghai, China). The raw sequencing reads were subjected to quality filtering using fastp v.0.23.0 (https://github.com/OpenGene/fastp). Then, the clean short reads and HiFi reads were assembled to construct complete genomes using Unicycle v.0.4.868. The coding sequences (CDs) of chromosomes and plasmids were predicted using Prodigal v.2.6.369 or GeneMarkS v.4.370, respectively. The predicted CDs were annotated at the CAZy database v.8 using sequence alignment tools such as Diamond v.0.8.35.
Transcriptomics profiling and analysis
Total RNA was extracted from the biomass pellet using the CTAB method, and genomic DNA was removed. Only high-quality RNA samples were used to construct sequencing libraries. Detailed methods for library construction are included in the Supplementary Information. The paired-end RNA-seq libraries were sequenced using the Illumina Novaseq 6000 instrument by Majorbio Biopharm Technology Co., Ltd. (Shanghai, China). Using Bowtie 2, high-quality reads in each sample were mapped to the reference genomes of specific strains57. Gene expression levels were estimated by the transcript per million mapped reads (TPM) method by using RSEM. DESeq2 was used to detect differentially expressed genes between different treatment groups66. Genes with fold change > 2 or < 0.5, and P < 0.05 (BH method for P value adjustment) were considered to be differentially expressed genes. The KEGG enrichment analysis revealed which biological metabolic pathways were significantly changed with the differentially expressed genes.
Metabolomics profiling and analysis
Supernatants from decomposition experiments inoculated with different SynComs were subjected to untargeted metabolic analysis by liquid chromatography mass spectrometry (LC-MS). Detailed methods for metabolomics are included in the Supplementary Information. The raw data were converted into the common format by Progenesis QI software (Waters, Milford, USA). The metabolites were identified by searching the database, and the main databases were the CAZy, HMDB, Metlin (https://metlin.scripps.edu/), and the self-compiled Majorbio Database (MJDB) of Majorbio Biotechnology Co., Ltd. (Shanghai, China). The processed data were analyzed using the ropls package71, where data were subjected to orthogonal partial least-squares discriminant analysis (OPLS-DA). Seven-fold cross-validation and response permutation testing were used to evaluate the robustness of the model. The metabolites with VIP > 1, P value < 0.05 (generated by Student’s t-test) were determined as significantly different metabolites based on the variable importance in projection (VIP) obtained by the OPLS-DA model.
Validation experiment of sugar depletion mechanism
To verify that the non-decomposers indeed inhibit weak decomposers through sugar consumption, we conducted an exogenous sugar addition experiment. The residue decomposition experiment included three treatments: (1) addition of the decomposers alone; (2) addition of the co-culture of decomposers and non-decomposers; and (3) addition of the co-culture of decomposers and non-decomposers, with 1 mL 0.15 g/mL sugar added after 2 days of decomposition. This was intended to compensate for the existing nutrient depletion. After 10 days of cultivation, the residue decomposition rate was calculated, and one-way ANOVA was conducted to evaluate the differences in decomposition rates among the different treatments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw reads of amplicon for the decomposition experiment and SynComs experiment have been deposited in the Sequence Read Archive under BioProject PRJNA1263673 and PRJNA1226766, respectively. Raw reads of metagenomics and transcriptomics have been deposited in the Sequence Read Archive under BioProject PRJNA1071817 and PRJNA1240452, respectively. The Whole Genome Shotgun data have been deposited at DDBJ/ENA/GenBank under the accession PRJNA1240023. The metabolomics data have been deposited in the National Genomics Data Center under BioProject PRJCA037298. Source data are provided with this paper.
Code availability
All source code and scripts are available on GitHub (https://github.com/PlantNutrition/Meiling_2025_NC).
References
Lal, R. World crop residues production and implications of its use as a biofuel. Environ. Int. 31, 575–584 (2005).
Lal, R. Soil carbon sequestration impacts on global climate change and food security. Science 304, 1623–1627 (2004).
Chabbi, A. et al. Aligning agriculture and climate policy. Nat. Clim. Change 7, 307–309 (2017).
Xia, L. et al. Trade-offs between soil carbon sequestration and reactive nitrogen losses under straw return in global agroecosystems. Glob. Change Biol. 24, 5919–5932 (2018).
Liu, J. et al. Crop residue return sustains global soil ecological stoichiometry balance. Glob. Change Biol. 29, 2203–2226 (2023).
Wang, X. et al. Nitrogen transfer and cross-feeding between Azotobacter chroococcum and Paracoccus aminovorans promotes pyrene degradation. ISME J. 17, 2169–2181 (2023).
Chen, R. et al. Soil C and N availability determine the priming effect: microbial N mining and stoichiometric decomposition theories. Glob. Change Biol. 20, 2356–2367 (2014).
Fang, Y., Nazaries, L., Singh, B. K. & Singh, B. P. Microbial mechanisms of carbon priming effects revealed during the interaction of crop residue and nutrient inputs in contrasting soils. Glob. Change Biol. 24, 2775–2790 (2018).
Liang, Z. et al. Effect of the combined addition of mineral nitrogen and crop residue on soil respiration, organic carbon sequestration, and exogenous nitrogen in stable organic matter. Appl. Soil Ecol. 171, 104324 (2022).
Hou, S. et al. Increasing rates of long-term nitrogen deposition consistently increased litter decomposition in a semi-arid grassland. N. Phytol. 229, 296–307 (2021).
Peng, Y. et al. Nitrogen addition slows litter decomposition accompanied by accelerated manganese release: A five-year experiment in a subtropical evergreen broadleaf forest. Soil Biol. Biochem. 165, 108511 (2022).
Sun, T., Dong, L., Wang, Z., Lü, X. & Mao, Z. Effects of long-term nitrogen deposition on fine root decomposition and its extracellular enzyme activities in temperate forests. Soil Biol. Biochem. 93, 50–59 (2016).
Gill, A. L., Schilling, J., Hobbie, S. E. & Liu, L. Experimental nitrogen fertilisation globally accelerates, then slows decomposition of leaf litter. Ecol. Lett. 24, 802–811 (2021).
Chen, J. et al. A keystone microbial enzyme for nitrogen control of soil carbon storage. Sci. Adv. 4, eaaq1689 (2018).
Lindemann, S. R. et al. Engineering microbial consortia for controllable outputs. ISME J. 10, 2077–2084 (2016).
Wahdan, S. F. M. et al. Future climate conditions accelerate wheat straw decomposition alongside altered microbial community composition, assembly patterns, and interaction networks. ISME J. 17, 238–251 (2023).
Lin, L. Bottom-up synthetic ecology study of microbial consortia to enhance lignocellulose bioconversion. Biotechnol. Biof. Biop. 15, 14 (2022).
Shahab, R. L. et al. A heterogeneous microbial consortium producing short-chain fatty acids from lignocellulose. Science 369, eabb1214 (2020).
Peng, Q. et al. Modeling bacterial interactions uncovers the importance of outliers in the coastal lignin-degrading consortium. Nat. Commun. 16, 639 (2025).
Andric, P., Meyer, A. S., Jensen, P. A. & Dam-Johansen, K. Reactor design for minimizing product inhibition during enzymatic lignocellulose hydrolysis: II. Quantification of inhibition and suitability of membrane reactors. Biotechnol. Adv. 28, 407–425 (2010).
Dai, T. et al. Nutrient supply controls the linkage between species abundance and ecological interactions in marine bacterial communities. Nat. Commun. 13, 175 (2022).
Ma, X. et al. Long-term nitrogen deposition enhances microbial capacities in soil carbon stabilization but reduces network complexity. Microbiome 10, 112 (2022).
Bao, Y. et al. Straw chemistry links the assembly of bacterial communities to decomposition in paddy soils. Soil Biol. Biochem. 148, 107866 (2020).
Zhou, Z., Wang, C., Zheng, M., Jiang, L. & Luo, Y. Patterns and mechanisms of responses by soil microbial communities to nitrogen addition. Soil Biol. Biochem. 115, 433–441 (2017).
Meidute, S., Demoling, F. & Bååth, E. Antagonistic and synergistic effects of fungal and bacterial growth in soil after adding different carbon and nitrogen sources. Soil Biol. Biochem. 40, 2334–2343 (2008).
Debray, R. et al. Priority effects in microbiome assembly. Nat. Rev. Microbiol. 20, 109–121 (2022).
Zhong, Y. et al. Microbial community assembly and metabolic function during wheat straw decomposition under different nitrogen fertilization treatments. Biol. Fert. Soils 56, 697–710 (2020).
Bastian, F., Bouziri, L., Nicolardot, B. & Ranjard, L. Impact of wheat straw decomposition on successional patterns of soil microbial community structure. Soil Biol. Biochem. 41, 262–275 (2009).
Ma, C. et al. Molecular mechanisms underlying lignocellulose degradation and antibiotic resistance genes removal revealed via metagenomics analysis during different agricultural wastes composting. Bioresour. Technol. 314, 123731 (2020).
Danilova, I. & Sharipova, M. The practical potential of Bacilli and their enzymes for industrial production. Front. Microbiol. 11, 1782 (2020).
Dar, M. A. et al. Exploring the region-wise diversity and functions of symbiotic bacteria in the gut system of wood-feeding termite, Coptotermes formosanus, toward the degradation of cellulose, hemicellulose, and organic dyes. Insect Sci. 29, 1414–1432 (2022).
Mamtimin, T. et al. Gut microbiome of mealworms (Tenebrio molitor Larvae) show similar responses to polystyrene and corn straw diets. Microbiome 11, 1–20 (2023).
Shahab, R. L., Luterbacher, J. S., Brethauer, S. & Studer, M. H. Consolidated bioprocessing of lignocellulosic biomass to lactic acid by a synthetic fungal-bacterial consortium. Biotechnol. Bioeng. 115, 1207–1215 (2018).
Pang, Z. et al. Microbiota-mediated nitrogen fixation and microhabitat homeostasis in aerial root-mucilage. Microbiome 11, 85 (2023).
Kim, S. G. et al. Microbiota-derived lantibiotic restores resistance against vancomycin-resistant Enterococcus. Nature 572, 665–669 (2019).
Isaac, S. et al. Microbiome-mediated fructose depletion restricts murine gut colonization by vancomycin-resistant Enterococcus. Nat. Commun. 13, 7718 (2022).
Oliveira, R. A. et al. Klebsiella michiganensis transmission enhances resistance to Enterobacteriaceae gut invasion by nutrition competition. Nat. Microbiol. 5, 630–641 (2020).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335 (2010).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584 (2016).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microb. 75, 7537–7541 (2009).
Andrews, S. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2012).
Abarenkov, K. et al. The UNITE database for molecular identification of fungi–recent updates and future perspectives. N. Phytol. 186, 281–285 (2010).
Li, D., Liu, C., Luo, R., Sadakane, K. & Lam, T. W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Noguchi, H., Park, J. & Takagi, T. MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 34, 5623–5630 (2006).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Drula, E. et al. The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res. 50, D571–D577 (2022).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014).
Wu, Y., Simmons, B. A. & Singer, S. W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607 (2016).
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 1–13 (2018).
Sieber, C. M. et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 3, 836–843 (2018).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868 (2017).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. G. T. D. B. Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36, 1925–1927 (2020).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490 (2010).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Aramaki, T. et al. KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252 (2020).
Dixon, P. Vegan, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930 (2003).
Kembel, S. W. et al. Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26, 1463–1464 (2010).
Shenhav, L. et al. FEAST: fast expectation-maximization for microbial source tracking. Nat. Methods 16, 627–632 (2019).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014).
Kim, M., Oh, H. S., Park, S. C. & Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351 (2014).
Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13, e1005595 (2017).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinforma. 11, 1–11 (2010).
Besemer, J. & Borodovsky, M. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 33, W451–W454 (2005).
Thevenot, E. A. ropls: PCA, PLS (-DA) and OPLS (-DA) for multivariate analysis and feature selection of omics datad. R Package Version (2016).
Acknowledgements
This research was financially supported by the National Key R&D Program of China (2022YFD1900900 (C.A.)), the National Natural Science Foundation of China (32322076 (C.A.) and 32272817 (C.A.)), the China Agriculture Research System (CARS-23-B04 (C.A.)), the Smart Fertilization Project (05) (C.A.), the Center for Science in Arable Land (CAAS-CSAL-202401 (C.A.)), and the Agricultural Science and Technology Innovation Program (CAAS-ZDRW202308 (C.A.)). We are grateful to Laurent Philippot, from Université Bourgogne Franche-Comté, for constructive suggestions on this manuscript. We thank Peng Xu for his assistance in metagenomic data analysis and genome binning. We thank Jennifer Smith, PhD, from Liwen Bianji (Edanz) (www.liwenbianji.cn/) for editing the English text of a draft of this manuscript.
Author information
Authors and Affiliations
Contributions
C.A. conceived the study and supervised the project. M.Z., L.Z., J.L., S.H., S.W. and Y.Z. performed the experiments. W.Z. reviewed the manuscript. M.Z. and C.A. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, M., Zhang, L., Li, J. et al. Nitrogen-shaped microbiotas with nutrient competition accelerate early-stage residue decomposition in agricultural soils. Nat Commun 16, 5793 (2025). https://doi.org/10.1038/s41467-025-60948-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60948-2
