
Abstract
Hyperuricemia, characterized by elevated serum urate levels, is a key factor in the pathogenesis of gout. URAT1 is essential for renal urate reabsorption and has emerged as a critical therapeutic target for managing hyperuricemia. However, the precise transport mechanism and the inhibitory effects of uricosuric drugs on URAT1 remain unclear. Here, we present structures of the double-mutant rat homolog of URAT1 in complex with its substrate urate, and the clinical drugs benzbromarone, lesinurad, verinurad, and sulfinpyrazone. The urate-bound structure elucidates key residues involved in recognizing urate, while the structures bound with drugs clearly demonstrate the distinct binding mode of each drug with URAT1. These drugs stabilize URAT1’s inward-facing state, blocking conformational transitions. Additionally, critical interactions essential for its conformational transition are identified. These findings provide a molecular framework for understanding the physiological function of URAT1 and for developing more efficacious therapies to treat hyperuricemia.
Similar content being viewed by others


Introduction
Uric acid, the end product of purine metabolism, circulates in the blood mainly as urate and is primarily eliminated through the kidneys and intestines1,2,3. Approximately two-thirds of urate is excreted via the kidneys, where urate initially passes through the glomerulus; however, only about 8–12% of the filtered urate is excreted in the urine4. The majority is reabsorbed into the bloodstream, primarily mediated by urate transporter 1 (URAT1)5,6. URAT1, located on the apical membrane of kidney proximal tubular cells, responsible for approximately 90% of urate reabsorption from the tubular fluid into the epithelial cells by exchanging urate with intracellular inorganic and organic anions4,5. Idiopathic renal hypouricemia is a hereditary disorder characterized by decreased serum uric acid levels and increased fractional excretion of uric acid5,7,8. The condition is primarily caused by loss-of-function mutations in URAT1 (encoded by SLC22A12), which impair uric acid reabsorption and lead to abnormally low uric acid levels in the blood4,8,9,10,11. This highlights the critical role of URAT1 in regulating uric acid levels and reabsorption in the kidneys4,9. However, due to the lack of structural information, the precise mechanisms by which these variants affect URAT1 function remain unclear.
Hyperuricemia, a progressive metabolic disorder characterized by elevated serum urate levels (SUA), affects ~47.2 million adults (21%) in the United States, with 27.9 million individuals (11.9%) having severe hyperuricemia (SUA > 7 mg/dL)12,13,14,15,16. This condition is associated with various health issues such as gout, chronic kidney disease, diabetes, hypertension, and vascular disorders6,17,18,19,20,21,22. The principal clinical manifestation of hyperuricemia is gout, which occurs when high levels of urate cause the deposition of monosodium urate crystals in one or more joints, leading to inflammation23,24. The prevalence of gout is about 4% in the United States, Europe, and Southeast Asia25. Reduced uric acid excretion, accounting for approximately 90% of cases, is a key factor in the disorder17,23,26,27. Consequently, increasing urate excretion by inhibiting the reabsorption process is a promising treatment strategy28,29. Since 90% of urate in the kidneys is reabsorbed by URAT1, the development of specific inhibitor targeting URAT1 has proven effective for treating gout30,31. URAT1 inhibitors, including benzbromarone, lesinurad, probenecid, and sulfinpyrazone, have been clinically approved for this purpose31,32,33,34,35. However, these medications can cause liver toxicity and other side effects36,37,38. The precise mechanisms through which these drugs bind to and inhibit URAT1 are crucial for future rational drug design and optimization and remain to be determined.
Here, we present high-resolution cryo-electron microscopy (cryo-EM) structures of URAT1 in complex with substrate uric acid, as well as the drugs benzbromarone, lesinurad, verinurad, and sulfinpyrazone. These structures provide detailed insights into the binding modes and inhibition mechanisms of these drugs, offering essential information for designing more effective URAT1 inhibitors with fewer side effects.
Results
Functional characterization and architecture of the URAT1
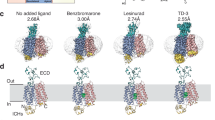
To investigate the binding modes of substrate urate and various uricosuric drugs with URAT1, we expressed URAT1 from Rattus norvegicus, which shares 74% sequence identity and 84% sequence similarity with human URAT1 (Supplementary Fig. 1). Previous literature has demonstrated that the double mutant N35S and Y365F in rat URAT1 enhances its substrate affinity and drug sensitivity to levels similar to those of human URAT139,40. Consequently, we introduced this double mutation into rat URAT1. Functional assays using [14C]-urate confirmed the activity of this construct, which also exhibited similar drug sensitivity to compounds such as benzbromarone, lesinurad, verinurad, and sulfinpyrazone, compared to human URAT1 (Supplementary Fig. 2A–E), consistent with earlier findings41. The size exclusion chromatography profile of the purified sample is symmetric, suggesting that the sample is homogeneous (Supplementary Fig. 2F, G). The modified construct, designated URAT1EM, was then used to prepare samples in complex with urate, benzbromarone, lesinurad, verinurad, and sulfinpyrazone. Single particle analysis provided cryo-EM maps of these complexes at resolutions of 3.5 Å, 3.5 Å, 3.6 Å, 3.5 Å, and 3.5 Å, respectively (Supplementary Figs. 3–7). These maps facilitated detailed model building and enabled a comprehensive elucidation of the binding modes of these ligands with URAT1 (Table S1).
The URAT1EM protein consists of 553 amino acids, with the structural model determined from a cryo-EM map covering 472 residues. Some flexible loop regions (residues 58–67, 100–110, and 325–346) are not clearly resolved, likely due to their flexibility. Both the N- and C-termini of URAT1EM are located on the cytoplasmic side of the plasma membrane (Fig. 1a, b). The transmembrane domain of URAT1EM features the characteristic fold of the major facilitator superfamily, which includes a core of 12 transmembrane helices (TM) organized into two pseudo-symmetrical domains: TM1-6 (N-domain) and TM7-12 (C-the membrane plane. A central cavity embedded within the membrane lies between these two domains, forming the substrate transport pathway (Fig. 1e). URAT1EM starts with a helix, NTH, parallel to the membrane plane, which extensively interacts with several short helices located between the TM6 and TM7, forming an intracellular domain (ICD) located underneath the N-domain. A prominent extracellular domain has also been determined, spanning residues A43 to S141 and connecting TM1 and TM2. This domain includes several short alpha helices and beta sheets. Two disulfide bonds, between C49 and C116, and C88 and C139, are identified (Fig. 1b). These disulfide bonds are conserved across the SLC22 family and are considered important for the structural integrity of the extracellular domain and transporter trafficking42.

a Cryo-EM maps of URAT1UA in LMNG detergent (gray). The N and C domains of URAT1EM are shown in green and orange, respectively. b Cartoon representation of URAT1EM in the same orientation and colors as in (A). Protein dimensions are indicated. Disulfide bonds in the extracellular domain are shown as sticks. c Cytoplasmic view of URAT1EM with TM helix numbers labeled. d Cryo-EM density of urate (UA) binding pocket. The cryo-EM densities are shown as mesh, with urate and surrounding residues shown as sticks. e Sagittal slice through an electrostatic surface potential of URAT1UA. The vertical distance from urate (UA) to cytoplasmic membrane plane is labeled. The urate molecular is shown as sphere. f Interaction of urate in binding pocket. The key residues are showed as sticks. g Comparative evaluation of the [14C] urate transportation activity of URAT1 mutants in relation to the URAT1EM. Data are mean ± s.e.m. from three independent assays, each with one measurement (n = 3). Comparison of URAT1EM with mutant constructs using an unpaired two-sided t-test showed significant differences (all P < 0.0001, denoted as ****).
Our structure of URAT1EM also provides a framework for understanding the effects of the disease-associated mutations. Beyond the nonsense mutations that lead to protein truncation, pathogenic missense mutations are distributed across the ECD, ICD, and TMD (Supplementary Fig. 8A). For instance, the R90 within the ECD forms electrostatic interactions with D118 and E114. The R90H would weakens these interactions, and consequently compromising the structural stability of the ECD (Supplementary Fig. 8B). Mutations such as V138M and G336R, which involve substituting smaller side chains with larger ones, are likely to disrupt structural packing, leading to the misfolding of URAT1 (Supplementary Fig. 8C, D). Additionally, the residue Q382 from the C-domain forms a hydrogen bond with N39 from the N-domain, which may help stabilize the inward-facing conformation. Substituting Q382 with leucine could disrupt this interaction, leading to an overstabilization of the outward-facing conformation and impaired URAT1 activity (Supplementary Fig. 8E).
Urate recognition by URAT1
To elucidate the structure of URAT1 bound to the substrate urate, we supplemented the purification process with 1 mM uric acid. Given the buffer pH of 7.5 and the pKa of uric acid (5.4), it is likely that urate associates with URAT1 (ref. 3). The urate-bound structure of URAT1 (URAT1UA) was resolved at a resolution of 3.5 Å, revealing the transporter in an inward-facing conformation (Fig. 1d, e and Supplementary Fig. 3). This central cavity, constituted by transmembrane segments TMs 1, 2, 4, 5, 7, 10, and 11, serves as the primary pathway for substrate translocation and is characterized by a highly positively charged electrostatic surface, which facilitate the binding and transport of negatively charged substrates such as urate.
Within the central cavity, we observed a prominent density, likely corresponds to urate molecule. The urate molecule is situated ~16 Å above the interior membrane surface (Fig. 1b–e). The binding site is primarily composed of hydrophobic residues that interact with urate, such as M214TM4, F360TM7, F365TM7, and F449TM10, while R477TM11 forms a crucial electrostatic interaction with the negatively charged part of urate molecule (Fig. 1f). Notably, F365 TM7 was changed from rat tyrosine to human phenylalanine in the construct, and this human phenylalanine residue confers higher affinity for urate40. Mutagenesis studies confirm the functional importance of these residues: the R477A mutation significantly reduces URAT1 activity, and M214A, F360A, F365A, and F449A mutations result in a substantial loss of function, retaining less than 30% of the URAT1EM construct activity (Fig. 1g and Supplementary Fig. 9). We also observed weaker density near the position discussed above, which partially overlaps with the urate-binding position observed in human URAT1 (ref. 43) (Supplementary Fig. 10A–C). Notably, the density of the nearby residue F241 is not well-defined, likely because its side chain adopts distinct conformations when stabilizing urate in various binding sites, supporting the notion of heterogeneous urate binding in the inward-facing conformation. This conformational flexibility of F241 suggests its involvement in urate binding, though detailed characterization through mutagenesis studies and quantitative affinity measurements will be needed to fully elucidate its functional role. Moreover, the complex structure of human URAT1 (R477S) bound with urate revealed that urate binds at a position distinct from the two locations discussed above44. These observations suggest that urate may adopt multiple binding modes and be highly dynamic and flexible within the intracellular cavity of URAT1. Similar observations have been reported45. This variability is likely due to the low affinity of the substrate for the intracellular cavity, a state that is ready to release the substrate.
URAT1 mediates urate transport through the exchange with counter-ions such as chloride or organic anions4,5. Although chloride ions were supplied in the buffer during protein purification. However, due to the moderate resolution of the cryo-EM map, the chloride binding site was not identified.
Binding site of benzbromarone
Benzbromarone, an effective uricosuric agent used primarily in the gout management, enhances urate excretion by inhibiting URAT146. Despite its restricted use in some regions due to hepatotoxicity concerns, benzbromarone remains valuable for patient’s intolerant to other gout therapies, such as allopurinol or febuxostat, or those with persistent hyperuricemia despite standard treatments46. To elucidate the inhibition mechanism of benzbromarone, we resolved the structure of URAT1EM in complex with benzbromarone (URAT1BEN) at 3.5 Å (Fig. 2a, b and Supplementary Fig. 4). The structure of URAT1BEN adopts an inward-facing conformation, characterized by a large solvent-accessible cavity from the cytoplasmic side, which accommodates a discernible non-protein density consistent with benzbromarone. The benzbromarone molecule is positioned ~18 Å from the cytoplasmic side, with its benzofuran group aligned nearly parallel to the membrane plane and the bromophenol group perpendicular (Fig. 2c). Notably, when we further analyzed the EM density of the benzbromarone, we found the benzofuran group is well-defined in the cryo-EM map, while the bromophenol group is less clearly resolved (Fig. 2a). Benzbromarone is primarily coordinated by a relatively conserved aromatic cluster consisting of F241, F360, F364, F365, and F449. F241 interacts with benzbromarone via π−π interactions, while Q245 forms a hydrogen bond with the oxygen atom of the benzofuran group (Fig. 2d and Supplementary Fig. 1). To validate the interaction between Q245 and benzbromarone, we designed the Q245H and Q245A mutations, but found that their transport activity was virtually abolished (Fig. 2e). However, in human URAT1, histidine occupies position 245, and the H245A mutant retained transport activity43. We speculate that this discrepancy in functional activity may arise from non-conserved neighboring residues altering local conformational dynamics, including perturbations in residue-residue interactions and cooperative helix motions within this structural domain (Supplementary Fig. 1). Additionally, the significantly higher transport rate of rat URAT1 may amplify the functional impact of these mutations40. However, these hypotheses remain speculative and warrant further investigation. The residue S35 is located close to the benzofuran group of benzbromarone. Notably, S35 was changed from rat asparagine to human serine in the construct, and this human serine residue leads to increased inhibitory potency of benzbromarone39. Substituting it with glutamine (S35Q), which has a larger side chain, may create steric hindrance with benzbromarone, resulting in a significant decrease in affinity (Fig. 2f). The side chain of F365 points towards the benzofuran group. Moreover, the F365Y mutation significantly reduces the inhibitory potency of benzbromarone, likely due to the introduction of the hydroxyl group of tyrosine directly clashing with benzofuran group of benzbromarone. These observations are consistent with previous findings39. In contrast to F365Y, the introduction of a hydroxyl group at other aromatic residues (F241Y, F360Y, F364Y, and F449Y) did not affect benzbromarone’s inhibitory potency, likely because these positions do not cause steric clashes with benzbromarone. M241 forms hydrophobic interactions with benzbromarone, and its mutation to glutamine does not significantly affect inhibitory potency, likely because this single substitution is not sufficient to effectively disrupt inhibitor binding. Although the residue Q473 is not directly involved in benzbromarone binding, mutating Q473 to histidine (Q473H) exhibits a slight reduction in its binding affinity, presumably due to introduced steric hindrance resulting from larger volume of histidine side chain. Furthermore, R477 forms electrostatic interactions with the bromophenol group of benzbromarone. Mutation of R477 to asparagine (R477N), which disrupts this interaction, significantly reduces benzbromarone binding affinity (Fig. 2f), aligning with structural observations and underscoring the critical role of R477 in stabilizing benzbromarone binding. The molecular dynamics simulations revealed that both the protein and ligand reached a stable state over the course of the simulation. The average Root-mean-square-deviations values for the protein and ligand remained below 4.0 Å and 2.0 Å, respectively, indicating minimal conformational fluctuations and high stability of the system (Supplementary Fig. 11B). These findings further support the binding mode of benzbromarone.

a The cryo-EM density of benzbromarone and surrounding residues is shown as mesh while the structure of which are presented as sticks. b Side view URAT1 in complex with benzbromarone. Electrostatic potential map of the cytoplasmic pocket is shown as a surface. URAT1EM is dipicted in sky-blue cartoon, while benzbromarone is shown as spheres with carbon, oxygen, and bromine atoms colored in deep teal, red, and green, respectively. c Coronal section view of electrostatic surface of URAT1BEN. Benzbromarone is represented as sphere. d Detailed view of the benzbromarone binding pocket. Surrounding residues are represented as sticks. Hydrogen bond is depicted as black dash line. e Comparative evaluation of the [14C] urate transportation activity of URAT1 mutants in relation to the URAT1EM. Data are mean ± s.e.m. from three independent assays, each with one measurement (n = 3). Comparison of URAT1EM with mutant constructs using an unpaired two-sided t-test showed significant differences (all P < 0.0001, denoted as ****). f Inhibition of 14C-urate transport by benzbromarone in URAT1EM and its mutants. Data are presented as normalized mean ± s.e.m. from three biologically independent assays (n = 3).
Previous literature has shown that human URAT1 exhibits a higher affinity for benzbromarone than OAT1-5. Sequence alignment and structural analysis of URAT1BEN reveal variations in residues within the binding pocket, which may explain its specificity (Supplementary Fig. 12). For example, the residue S35 in URAT1 is close to benzofuran group of benzbromarone, which is replaced by residues with larger side chains in other transporters, hindering benzbromarone association. Furthermore, the positions equivalent to F364 and F365 in URAT1 are occupied by tyrosine in other transporters. These variations may disrupt the hydrophobic environment critical for benzbromarone interaction. Additionally, F241 plays a crucial role in facilitating benzbromarone binding; however, it is replaced by W240 in OAT2 and L241 in OAT5, resulting in a reduced binding affinity of benzbromarone to these transporters (Supplementary Fig. 12).
Several analogs of benzbromarone have gained approval or are currently undergoing clinical evaluation47. These analogs maintain the core structural framework of benzbromarone and likely share a similar binding mode. For example, dotinurad, approved in Japan in 2020 for the treatment of gout and hyperuricemia, replaces both bromine atoms with more electronegative chlorine atoms48. We compared the structure of benzbromarone-bound URAT1 with the recent dotinurad-bound URAT1 structure43, revealing that both benzbromarone and dotinurad bind within the same pocket. The benzofuran group aligns well, while the bromophenol group exhibits conformational diversity, and the side chain of nearby Q473 is in a different rotamer (Supplementary Fig. 11A). Based on this structural comparison and cryo-EM map analysis of the two groups, we propose that the bromophenol group exhibits greater positional flexibility within the binding pocket compared to the benzofuran group, likely reflecting a higher degree of binding heterogeneity.
Inhibitory mechanisms of URAT1 by lesinurad and its derivative verinurad
Lesinurad, a representative URAT1 inhibitor, is approved for the treatment of hyperuricemia associated with gout by promoting the uric acid excretion31,34 (Fig. 3a). It exhibits a half-maximal inhibitory concentration (IC50) of 3.5 μM in inhibiting transport activity of human URAT139. To elucidate the inhibitory mechanisms of lesinurad, we determined the structure of URAT1EM in complex with lesinurad (URAT1LES) at a resolution of 3.6 Å (Supplementary Fig. 4). In the structure of URAT1LES, lesinurad is situated in a cavity that opens toward the cytoplasmic side, suggesting that the lesinurad stabilized the URAT1 in an inward-facing conformation (Fig. 3b). The binding pocket of lesinurad is formed by TMs 1, 5, 7, 10, and 11 located 16 Å deep from the intracellular leaf of the membrane plane (Fig. 3b, c). The orientation of lesinurad is approximately perpendicular to the membrane plane, with the naphthalene ring interacting with TMs 5, 7, and 10 and triazole moiety interacting with TMs 1 and 11. Specifically, the residue R477TM11 forms an electrostatic interaction with the oxygen atom on acetic acid group, while the residue S35TM1 forms a hydrogen bond with the nitrogen atom on triazole moiety. Additionally, the naphthalene moiety is stabilized by a π-π stacking with F241TM5 and a T-shape stacking with F360TM7. The residues F364TM7 and F449TM10 engage in hydrophobic interactions with this moiety as well (Fig. 3d).

a Chemical structure and cryo-EM density of lesinurad and verinurad. b Coronal section view of electrostatic surface of URAT1LES and URAT1VER. Benzbromarone is represented as spheres. c cytoplasmic view of URAT1 in complex with lesinurad and verinurad. d Detailed view of the lesinurad binding pocket. Surrounding residues are represented as sticks. Hydrogen bond is depicted as black dash line. Inhibition of 14C-urate transport by lesinurad e and verinurad h in URAT1EM and its mutants. Data are presented as normalized mean ± s.e.m. from three biologically independent assays (n = 3). IC50 values are calculated by fitting into a nonlinear regression model. f Detailed view of the verinurad binding pocket. Surrounding residues are represented as sticks. Hydrogen bond is depicted as black dash line. g Superposition between URAT1LES and URAT1VER binding pocket. The key residues are shown as sticks. Lesinurad and verinurad are colored purple and yellow, respectively.
Despite lesinurad is approved for the treatment of hyperuricemia associated with gout, its low affinity and short half-time (~5 h) requires high-dosage administration, which has led to a high rate of renal-related adverse effects49,50. Verinurad, developed by modifying the molecular structure of lesinurad—replacing the cyclopropyl group with a cyano group and the acetic acid group with a methylpropanoic acid group—exhibits approximately 140 times higher affinity for hURAT1 and a longer half-life than lesinurad39,51,52,53 (Fig. 3a). It is currently undergoing a phase II trials for the treatment of gout and asymptomatic hyperuricemia53. To investigate the molecular basis of higher affinity of verniurad for URAT1, we determined the structure of URAT1EM in complex with verinurad (URAT1VER) at resolution of 3.5 Å (Supplementary Fig. 6) and found that the binding pose of verinurad is similar to that of lesinurad. Molecular dynamics simulations were conducted to further validate the binding pose of the ligand. The results demonstrated a stable RMSD value for the ligand throughout the simulation trajectories (Supplementary Fig. 13), indicating consistent behavior of the ligand during the simulations. These findings suggest that verinurad maintains a stable binding state with URAT1EM. Comparison with the verinurad-bound hURAT1 structure (PDB: 9B1I) highlights minor differences in the orientation of the acene group and surrounding residue side chains43 (Supplementary Fig. 13B). The observed variation in the orientation of the acene group suggests greater positional flexibility and binding heterogeneity for this group, which is also evidenced by its weaker cryo-EM map density (Fig. 3a).
To validate the binding pocket of lesinurad and verinurad, we designed mutations to assess their effects on binding of these two drugs. We found that the S35Q and R477N mutations significantly reduced binding affinity, supporting the important role of these residues in lesinurad and verinurad binding. Mutations such as M214Q, F360Y, and F365Y disrupted hydrophobic interactions, resulting in varying degrees of reduced binding affinity. These observations highlight the importance of these residue in establishing a hydrophobic environment to accommodate inhibitors (Fig. 3e, h). The F241Y, F364Y, and F449Y mutations do not significantly impair binding affinity, likely because the structural perturbations induced by these individual mutations are not substantial enough to disrupt ligand recognition. Mutations at position D389 exhibit different effects on lesinurad and verinurad, suggesting that D389 residue may play nonequivalent roles in their binding. In specific, it stabilizes verinurad binding through electrostatic interaction with the positively charged cyano group. Substituting aspartate with the negatively charged glutamate (D389E) does not affect the binding affinity of verinurad. In contrast, neutralizing D389 (D389A) causes a substantial reduction in verinurad binding affinity. These observations suggest that the electrostatic interaction between verinurad and D389 is important for its binding. Unlike the positively charged cyano group in verinurad, the cyclopropyl group at the equivalent position in lesinurad is hydrophobic. The hydrophilic D389 residue may not be conducive to lesinurad binding due to an energetically unfavorable hydrophobic mismatch. The D389E mutation results in a significant decrease in lesinurad binding affinity, likely because the hydrophobic mismatch is strengthened as the cyclopropyl group and the side chain of D389 come closer. Moreover, substituting D389 with alanine (D389A) reduces this hydrophobic mismatch, thus improving lesinurad binding affinity.
Recognition of the sulfinpyrazone-binding site
Sulfinpyrazone, another FDA-approved drug used in the treatment of gout, has also been studied for its detailed binding mode with URAT154. It specifically inhibits both URAT1 with IC50 values of 32 μM39. To elucidate the binding mode of sulfinpyrazone, we determined the structure of URAT1EM in complex with sulfinpyrazone (URAT1SPZ) at resolution of 3.5 Å (Supplementary Fig. 7). Further inspection of the cryo-EM map revealed an isolated density at intracellular facing cavity, which aligns well with the structure of the sulfinpyrazone molecule (Fig. 4a and Supplementary Fig. 14A–C). The benzenesulfinyl group of sulfinpyrazone reaches the bottom of the intracellular cavity (18 Å deep), while the dione group extends towards the cytosol. Molecular dynamics simulations indicate that the overall binding of sulfinpyrazone to the protein remains relatively stable (Supplementary Fig. 14D). However, the clustering data mining technique generates seven main clusters and reveals that the benzenesulfinyl group shows significant variation across different clusters (Supplementary Fig. 14E, F), whereas the diphenylpyrazolidine group remains highly stable. These observations suggest that the benzenesulfinyl group exhibits more dynamic states compared to the deeply embedded diphenylpyrazolidine group. These findings are consistent with observations that the deeper sulfinpyrazone groups show stronger density than that of the benzenesulfinyl group near the intracellular side (Supplementary Fig. 14A–C). A noteworthy aspect of sulfinpyrazone’s binding mode is its unique spatial orientation compared to other ligands, such as benzbromarone, lesinurad, and probenecid. While these drugs are confined to the bottom of the pocket, sulfinpyrazone extends beyond this region. Its binding site stretches from the base of the cavity toward the intracellular domain, allowing it to engage in a broader range of interactions with the surrounding residues. Specifically, the oxygen atom on the diphenylpyrazolidine group forms a potential hydrogen bond with R477TM11 residue, while the oxygen atom on the benzenesulfinyl group forms a potential hydrogen bond with N237TM5 residue. Additionally, the dione group form extensive hydrophobic interactions with surrounding resides, including W357TM7, F360TM7, F364TM7, F365TM7, and F449TM10. The benzenesulfinyl group forms hydrophobic interactions with M215TM4, M222TM4, M234TM5, and M466TM11 based on structural analysis (Fig. 4d).

a Chemical structure of sulfinpyrazone. Cryo-EM density of sulfinpyrazone and surrounding residues is shown as mesh and the structure of sulfinpyrazone is shown as sticks. b Coronal section view of electrostatic surface of URAT1SPZ. Sulfinpyrazone is represented as spheres. c cytoplasmic view of URAT1 in complex with sulfinpyrazone. d Detailed view of the sulfinpyrazone binding pocket. Surrounding residues are represented as sticks. Hydrogen bond is depicted as black dash line. e Inhibition of 14C-urate transport by sulfinpyrazone in URAT1EM and its mutants. Data are presented as normalized mean ± s.e.m. from three biologically independent assays (n = 3). IC50 values are calculated by fitting into a nonlinear regression model.
To confirm the sulfinpyrazone binding pocket, we designed several mutations and conducted [14C]-urate transport assays to determine their effects on IC50 values. Mutations of F360 and F365 to tyrosine (F360Y, F365Y) significantly reduced the inhibitory potency, likely due to changes in hydrophobicity and steric hindrance introduced by the tyrosine hydroxyl group. Similarly, the F241Y mutation also decreased the binding affinity of sulfinpyrazone. In contrast, mutations F364Y and F449Y had minimal impact, likely because the introduced hydroxyl group in these mutants does not significantly alter the binding pocket’s hydrophobic character or the overall binding interaction with sulfinpyrazone. The R477N mutation disrupts a potential hydrogen bond with the oxygen atom of the diphenylpyrazolidine group in sulfinpyrazone (Fig. 4d), resulting in a marked reduction in binding affinity and supporting R477 is important in sulfinpyrazone binding. Residues M222, M234, and M466, located near the intracellular side close to the benzenesulfinyl group, exhibited minimal effects on sulfinpyrazone’s inhibitory effect when mutated to glutamine, suggesting that interactions between these residues and the benzenesulfinyl group may not be critical, which is consistent with observations that the benzenesulfinyl group is less tightly bound, as evidenced by the cryo-EM map and MD simulations (Supplementary Fig. 14). Residue N237, which forms a hydrogen bond with sulfinpyrazone, exhibited a slight reduction in binding affinity when mutated to alanine, highlighting the significance of this interaction. Additionally, residue W357, located farther from the sulfinpyrazone molecule, provides a hydrophobic environment that contributes to the stability and integrity of the binding pocket. When W357 was mutated to tyrosine or alanine, the binding affinity of sulfinpyrazone was slightly enhanced (Fig. 4e). This could be attributed to alterations in the side-chain interactions and possibly in the protein backbone, which may have modified the local environment of the binding pocket, leading to stronger interactions with sulfinpyrazone.
Conformational transition of URAT1
The URAT1 transporter belongs to the major facilitator superfamily (MFS), and operates through a rocker-switch mechanism, alternating the exposure of its substrate-binding site between the intracellular and extracellular environments to facilitate substrate transport55,56. To elucidate the mechanism basis underlying the conformational state transition of URAT1, we constructed an outward-facing homology model of URAT1 based on the structure of OCT3 in outward-facing conformation (PDB ID: 7ZH0)57. The inward-facing URAT1UA structure was superimposed on the outward-facing URAT1 model, using the N-domain as a reference, revealed significant rotational movements between these states (Supplementary Fig. S15A). In the inward-facing structure, Q149 forms a hydrogen bond with R487, a conserved interaction across different species of URAT1 (Supplementary Fig. 15B, C). Previous studies have shown that the R487A mutation decreases the maximum transport rate of human URAT1, supporting its potential role in the conformational transition58.
MFS transporters typically contain a highly conserved element, motif-A, which is crucial for stabilizing the outward-facing conformation59 (Supplementary Fig. 15F). In URAT1, the motif-A (G164HASD168R169FGR172R173) is located within the loop connecting TM2 and TM3. In the outward-facing URAT1 model, the small side chains of G146TM2 and G470TM11 enable tight helical packing of TM5 and TM11 (Supplementary Fig. 15D, E). Additionally, D168 engages in a charge-dipole interaction with the N-terminus of TM11. Additionally, a conserved ionic interaction between E223TM5 and R465TM11 further stabilizes the outward-facing structure (Supplementary Fig. 15E). These interactions are diminished in the inward-facing URAT1UA structure. To validate the significance of these interactions, we engineered mutations Q149A, D168A, E223A, R465, and R487A to disrupt these interdomain contacts. Almost all these mutations nearly abolished transport activity of URAT1EM (Supplementary Fig. 15G), demonstrating that these interactions are critical for the function of URAT1, probably due to impaired conformational transition and overstabilization of inward-facing conformation.
Discussion
In human, the loss of the uricase gene led to the inability to convert uric acid into the more excretable allantoin60. Consequently, serum uric acid levels are primarily regulated by renal excretion and reabsorption mechanisms3. The human URAT1 evolved to exhibit higher affinity and lower capacity for uric acid transport, a necessary adaptation to compensate for the loss of uricase activity40. This adaptation is primarily driven by the replacement of specific amino acid residues, the most significant of which is the presence of F365 in human URAT1, replaced by tyrosine in rURAT140. Structural analysis of URAT1UA revealed that the F365 residue forms hydrophobic interactions with the substrate. Mutating this residue to tyrosine may alter the properties of the binding pocket, thereby reducing substrate affinity40. Although the structure is stabilized in the inward-facing conformation, through sequence alignment and structural comparison, we identified critical interactions important for balance of transporter conformations. Mutations that disrupt these interactions lead to the abolishment of transport activity.
URAT1 inhibitors, including benzbromarone, sulfinpyrazone, and lesinurad, are key treatments for hyperuricemia and gout31,32,33,34,35, effectively inhibiting URAT1 despite their structural differences. We determined the structures of URAT1 bound with these drugs and elucidated the precise, distinct interactions that coordinate with these drugs. These structures adopt the same inward-open conformation, with the overall structures being superimposable. However, to accommodate the distinct drug molecules, slight displacements occur in the residue side chains around the binding site (Supplementary Fig. 16). Through systematic functional studies of these surrounding residues, we have elucidated the sophisticated recognition mechanism of URAT1’s binding pocket. URAT1 recognizes its endogenous substrate urate and various inhibitors through a highly conserved binding site, where two major types of interactions work synergistically: R477 provides polar binding forces through hydrogen bonding and electrostatic interactions, while the hydrophobic cluster (M214, F241, F360, F364, F365, and F449) establishes a stable hydrophobic binding environment. Notably, alanine mutations of these residues significantly affect urate transport activity, confirming their collective role in maintaining the structural integrity of the binding pocket. Tyrosine mutation studies further revealed that most positions (F241/F360/F364/F449) maintain inhibitor binding, demonstrating the dominant role of hydrophobic interactions, while F365Y mutation uniquely reduces affinity for both urate and inhibitors. Additionally, certain residues exhibit differential recognition of specific inhibitors. For instance, D389 shows dual functionality: it stabilizes verinurad binding through electrostatic interactions with its cyano group, while creating unfavorable hydrophobic clashes with lesinurad’s cyclopropyl moiety. These findings provide a detailed interaction map of URAT1’s binding pocket, offering clear molecular guidance for the design of targeted drugs against this transporter.
We aligned the outward-facing homology model of URAT1 (URAT1OUT) with the drug-bound inward-facing structures of URAT1 individually using their N-domains as a reference (Supplementary Fig. 17). This analysis revealed significant steric clashes between the drugs and the URAT1OUT structure, suggesting a potential inhibitory mechanism of drugs by obstructing its conformational transition from the inward-facing to the outward-facing state, thereby preventing the transporter from completing its functional cycle. Furthermore, URAT1 utilizes intracellular anions or organic anions to exchange extracellular urate into the cytosol4,5. The binding site for these drugs is located proximal to the potential counter-ion or organic anion pyrazinoic acid binding site43,44,61, which likely obstructs anion binding and therefore effectively impairs the transport activity of URAT1 (Supplementary Fig. 17B). However, these therapeutic agents face several significant challenges, including low binding affinity, limited selectivity for URAT1, and a relatively short half-life52,58,62. These limitations increase the risk of adverse effects and severely restrict their therapeutic potential and widespread clinical use. The structural insights gained here are of great importance in paving the way for rational drug optimization to enhance therapeutic efficacy. Taken together, we determined the structure of URAT1 in complex with the substrate urate and uricosuric drugs, shedding light on the understanding of its physiological role and the optimization of pharmacotherapeutic targeting of URAT1.
Methods
Expression and purification of URAT1
The full-length wild-type rat URAT1 (SLC22A12, Q3ZAV1) gene was cloned into the pEG BacMam vector, fused with an N-terminal superfolder GFP (sfGFP) and a Twin-Strep affinity tag. Two mutations, N35S and Y365F, were introduced into rat URAT1 for purification, based on previous findings indicating their importance in substrate and drug binding. Recombinant baculovirus was generated using the Bac-to-Bac system in IB905 Medium (YSK BIOSCIENCES, Zhejiang, China) cultured Sf9 cells and amplified to P3 generation. HEK293-F cells cultured in 293 F Hi-exp Medium (AC601501, Shanghai OPM Biosciences Co., Ltd.) were infected with 2% (v/v) P3 recombinant baculovirus at a density of ~2.5 × 106 cells/mL. After 12 h incubation at 37 °C with 5% CO2 and shaking, 10 mM sodium butyrate was added, and the temperature was reduced to 30 °C to enhance expression. Cells were harvested after 48 h by centrifugation at 3500 × g for 3 min, followed by flash-freezing in liquid nitrogen and storage at −80 °C.
For protein purification of urate-bound URAT1 complex, cell pellets were resuspended in purification buffer A [20 mM HEPES, 150 mM NaCl, pH 7.5, 1 mM urid acid, 2 μg/mL aprotinin (MedChemExpress), 1.4 μg/mL leupeptin (MedChemExpress), 0.5 μg/mL pepstatin A (MedChemExpress)]. The cell membrane was disrupted using a Dounce homogenizer, followed by centrifugation at 200,000 × g for 30 min to collect membrane fragments. Subsequently, the membrane fragments were solubilized by 1% (w/v) Lauryl Maltose Neopentyl Glycol (LMNG, Anatrace) and 0.2% (w/v) cholesteryl hemisuccinate (CHS, Anatrace) in buffer A at 4 °C for 2 h with gentle rotation. Following this, the insoluble cell debris was removed by centrifugation at 200,000 × g for 30 min, and the resulting supernatant was filtered through a 0.22 μm filter. The clarified supernatant was then applied to Streptactin Beads 4FF (Smart-Lifesciences)preequilibrated with buffer B [20 mM HEPES, 150 mM NaCl, pH 7.5, 1 mM urid acid, 0.05% (w/v) LMNG, 0.01% (w/v) CHS]. The beads were subsequently washed with five column volumes of wash buffer B supplemented with 2 mM ATP (Aladdin) and 5 mM MgCl2 to eliminate associated heat shock proteins. The URAT1 protein was eluted from Streptactin Beads using wash buffer B supplemented with 4 mM desthiobiotin (1169249, Leyan). Subsequently, it was incubated with His-tagged PPase at 4 °C for 90 min with gentle rotation to cleave off sfGFP. Following digestion, the protein sample was passed through a Ni-NTA column to remove PPase. The flow-through was collected and concentrated to 1 mL using a 50-kDa Millipore concentrator tube (Merck Millipore, Germany). Further purification was achieved through size exclusion chromatography (SEC) using a Superose 6 Increase 10/300 GL column (GE Healthcare, USA), equilibrated with buffer C [20 mM HEPES, 150 mM NaCl, pH 7.5, 1 mM urid acid, 0.002% (w/v) LMNG, 0.0004% (w/v) CHS]. The peak fractions were pooled and concentrated to 9 mg/mL. Before preparing cryo-EM sample, urid acid was added to the purified protein to a final concentration of 5 mM and incubated for 30 min.
For the purification of benzbromarone-bound, lesinurad-bound, verinurad-bound, and sulfinpyrazone-bound URAT1 complex, a similar purification process was employed. Instead of 1 mM urid acid, all buffers throughout the entire protein purification process contained 200 μM benzbromarone (MedChemExpress), 200 μM lesinurad (MedChemExpress), 40 μM verinurad (MedChemExpress), and 1 mM sulfinpyrazone (MedChemExpress), respectively. Additionally, 2 mM benzbromarone, 2 mM lesinurad, 800 μM verinurad, and 2 mM sulfinpyrazone was added before the preparation of the cryo-EM preparation. These protein samples were analyzed by SDS-PAGE, with molecular weight protein markers (New Cell & Molecular Biotech, P9008).
Cryo-EM sample preparation and data collection
For cryo-EM sample preparation, 3 μl of purified URAT1 sample was applied to the Quantifoil 1.2/1.3 μm holey carbon grids (300 mesh, copper) that have been glow-discharged in H2/O2 for 60 s. After having applied to the grids, the samples were blotted for 4.5 s with a Vitrobot (Vitrobot Mark IV; Thermo Fisher) after waiting for 5 s in the chamber at 4 °C with 100% humidity. The grids were then plunge-frozen in liquid ethane precooled by liquid nitrogen. Grids were imaged on a Titan Krios transmission electron microscope (Thermo Fisher Scientific) operated at 300 kV with a slit width of 10 eV on a GIF-Quantum energy filter. The movies were recorded using a K3 Summit direct electron detector camera (Gatan) at calibrated magnification of 105,000× in super-resolution counting mode, yielding a physical pixel size of 0.85 Å (0.425 Å super-resolution pixel size). Automated data collection were performed using EPU software. The total dose on the camera was set to 60 e−/Å2, and each image was fractioned into 32 subframes, recorded with a defocus value at a range of −1.0 to −2.0 μm.
Cryo-EM data processing
For the datasets of urate-bound URAT1, a total of 4078 movies were collected, followed by gain normalization and correction for beam-induced motion using MotionCor263. Subsequently, defocus parameters were estimated from the motion-corrected images utilizing patch CTF estimation in CryoSPARC464. Micrographs exhibiting severe contamination were removed through manual inspection. The remaining micrographs underwent particle picking using the blob picker, resulting in the selection of 2,728,392 particles. These particles underwent several rounds of 2D classification, where only the classes exhibiting distinct transmembrane helix features were retained in each iteration. This process culminated in the selection of 142,449 particles, which were then employed for ab-initio reconstruction to generate an initial map. The initial set of 216,482 particles was further subjected to multiple rounds of guided multi-reference 3D classification. During this step, the initial map served as a good reference map to guide the classification and facilitate the enrichment of homogeneous particles. Ultimately, 165,544 high-quality particles were selected for non-uniform refinement, yielding a reconstruction at 3.8 Å resolution. To enhance the map resolution, 3D classification without alignment was applied using a tight mask which exclude the micelles, resulting in 165,544 particles and a 3.5 Å map as determined by the gold-standard Fourier shell correlation criterion.
A similar strategy was applied in the data processing of URAT1BEN, URAT1LES, URAT1VER, and URAT1SPZ. Specifically, a total of 1,237,169/1,193,723/1,2654,709/1,088,732 particles were picked form 3218/5102/3967/2671 micrographs, respectively. After performing guided multi-reference 3D classification, 89,270/31,823/96,361/31,823 particles remained, yielding 3D reconstructions at 3.5 Å, 3.6 Å, 3.5 Å and 3.5 Å resolution respectively, according to the GSFSC criterion.
Model building
The atomic model of URATUA was built de novo using the 3.5 Å map in COOT65, enabled by the clear resolution of most side chains. Models for URAT1BEN, URAT1LES, URAT1VER, and URAT1SPZ were constructed by fitting the URAT1UA structure into the respective maps, followed by manual adjustments in COOT. Ligand geometry restraint files for urate, benzbromarone, lesinurad, verinurad, and sulfinpyrazone were generated from PubChem SDF files using the ELBOW module in PHENIX66. All inhibitors were manually fitted into corresponding map using COOT. The structures were refined using phenix.real_space_refine module and the model stereochemistry was validated by comprehensive validation (cryo-EM) tool in PHNIX.
All figures were prepared using open-source PyMOL67, UCSF Chimera68 or ChimeraX69.
Molecular dynamics simulations
All simulations were conducted using GROMACS version 2021.6 (ref. 70). We utilized the structure of the URAT-drug complexes as the initial model and prepared the system using CHARMM-GUI71. The URAT complex was inserted into a POPC lipid bilayer at positions calculated according to PPM 2.0 (ref. 72). The system was then solvated with TIP3P water73, and 150 mM NaCl was added to neutralize the system. The final system consisted of the URAT, drugs, POPC, water molecules, sodium ions, and chloride ions. The Amber14SB force field was applied to the protein and lipids74, while the corresponding GAFF2 force field was used for the drug molecules75. Each complex underwent production phase simulations of 3*150 ns under the NPT ensemble. The temperature and pressure coupling were achieved using the velocity-rescaling thermostat76 and the Parrinello-Rahman barostat77, respectively. For the calculation of long-range electrostatic interactions, the particle mesh Ewald method was employed. A cutoff of 9 Å was set for van der Waals interactions, incorporating a force-switching function at 9 Å. Additionally, the LINES algorithm78 was applied to keep hydrogen-containing bonds fixed during each 2 fs integration time step. The chosen force fields (Amber14SB/GAFF2) and explicit solvent/membrane model (POPC/TIP3P) have been extensively validated for simulating membrane protein-ligand interactions, ensuring sufficient accuracy for studying drug binding dynamics.
During the simulation process, the root means square deviation (R.M.S.D) of the non-hydrogen atoms for residues 2–42, 132–324, and 347–515 was calculated, excluding the highly flexible extracellular domains that are not related to drug binding. Three independent replicates with identical starting structures but different initial velocity distributions converged to consistent drug-binding conformations, confirming result robustness against initialization variability. The R.M.S.D for the drug was also calculated for its non-hydrogen atoms. The R.M.S.D calculations were performed using VMD (Visual Molecular Dynamics)79.
[14C]urate uptake assay
Urate uptake assays were performed as in previous studies with minor modifications. HEK293-F cells were infected with P2 baculovirus (1:100) prepared by Bac-to-Bac system (Invitrogen, USA). The cells were then cultured at 37 °C with 5% CO2 in suspension at 1 × g, followed by incubation for 16 h. 4 × 106 cells were harvested and then incubated with pre-warmed Cl−-free transport buffer (125 mM sodium gluconate, 4.8 mM potassium gluconate, 1.3 mM calcium gluconate, 1.2 mM KH2PO4, 1.2 mM MgSO4, 5.6 mM glucose, pH 7.4) for 5 min. For the inhibition assay, the buffer also contained specified concentrations of inhibitors. The uptake was initiated by adding 5 μM [14C]urate and then incubating at 28 °C for 2 min in a metal bath, falling within the initial rate kinetics range. To terminate the reaction, 500 μL of ice-cold transport buffer was added, followed by centrifugation at 18,000 × g for 15 s to collect the cells. The cells were lysed using 1% Triton X-100 and then transferred to scintillation fluid (Optiphase Supermix, PerkinElmer). Radioactivity counts were measured using a Hidex 300 SL instrument (HIDEX). The URAT1-specific transport activity counts were calculated by subtracting signals in control HEK293-F cells. For the inhibition assays, the signals were normalized to the sample without inhibitors. For the uptake assay, transport activities were normalized to the expression level of URAT1 assessed by HPLC. IC50 values were determined using nonlinear regression with the equation: Y = 100/(1 + 10^(X-LogIC50)) in GraphPad Prism 9.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The three-dimensional cryo-EM density maps have been deposited in the Electron Microscopy Data Bank under accession codes EMD-61191 (URAT1UA); EMD-61192 (URAT1BEN); EMD-61193 (URAT1LES; EMD-61194 (URAT1VER); and EMD-61195 (URAT1SPZ). The atomic coordinates have been deposited in the Protein Data Bank under accession codes 9J72 (URAT1UA); 9J73 (URAT1BEN); 9J74 (URAT1LES); 9J75 (URAT1VER); and 9J76 (URAT1SPZ). Previously published PDB codes referenced in this study include: 8WJQ (hURAT1); 8B1J (hURAT1(R477S)); 9B1G (dotinurad-bound human URAT1); 9B1I (verinurad-bound human URAT1); and 7ZH0 (OCT3). The molecular dynamics (MD) simulation dataset have been uploaded to Zenodo [https://doi.org/10.5281/zenodo.15490117]. The source data underlying Figs. 1q, 2f, 3e, h, 4e, and Supplementary Figs. 2A-E, 15G are provided in the Source Data file. Source data are provided with this paper.
References
Alvarez-Lario, B. & Macarron-Vicente, J. Uric acid and evolution. Rheumatology 49, 2010–2015 (2010).
Becker, B. F. Towards the physiological function of uric acid. Free Radic. Biol. Med. 14, 615–631 (1993).
Bobulescu, I. A. & Moe, O. W. Renal transport of uric acid: evolving concepts and uncertainties. Adv. Chronic Kidney Dis. 19, 358–371 (2012).
Enomoto, A. & Endou, H. Roles of organic anion transporters (OATs) and a urate transporter (URAT1) in the pathophysiology of human disease. Clin. Exp. Nephrol. 9, 195–205 (2005).
Enomoto, A. et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 417, 447–452 (2002).
So, A. & Thorens, B. Uric acid transport and disease. J. Clin. Investig. 120, 1791–1799 (2010).
Ichida, K. et al. Clinical and molecular analysis of patients with renal hypouricemia in Japan-influence of URAT1 gene on urinary urate excretion. J. Am. Soc. Nephrol. 15, 164–173 (2004).
Tasic, V. et al. Clinical and functional characterization of URAT1 variants. PLoS One 6, e28641 (2011).
Vazquez-Mellado, J. et al. Molecular analysis of the SLC22A12 (URAT1) gene in patients with primary gout. Rheumatology 46, 215–219 (2007).
Claverie-Martin, F. et al. URAT1 and GLUT9 mutations in Spanish patients with renal hypouricemia. Clin. Chim. Acta 481, 83–89 (2018).
Misawa, K. et al. Contribution of rare variants of the SLC22A12 gene to the missing heritability of serum urate levels. Genetics 214, 1079–1090 (2020).
Chen-Xu, M., Yokose, C., Rai, S. K., Pillinger, M. H. & Choi, H. K. Contemporary prevalence of gout and hyperuricemia in the United States and Decadal Trends: The National Health and Nutrition Examination Survey, 2007-2016. Arthritis Rheumatol. 71, 991–999 (2019).
Zhu, Y., Pandya, B. J. & Choi, H. K. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 63, 3136–3141 (2011).
Lozano, A. J. T. he & Daniel, K. Inouye college of pharmacy scripts. Hawai ‘i J. Med. Public Health 77, 261 (2018).
Li, L., Zhang, Y. & Zeng, C. Update on the epidemiology, genetics, and therapeutic options of hyperuricemia. Am. J. Transl. Res. 12, 3167 (2020).
Benn, C. L. et al. Physiology of hyperuricemia and urate-lowering treatments. Front. Med. 5, 160 (2018).
Richette, P. & Bardin, T. Gout. Lancet 375, 318–328 (2010).
Feig, D. I., Kang, D. H. & Johnson, R. J. Uric acid and cardiovascular risk. N. Engl. J. Med. 359, 1811–1821 (2008).
Kang, D. H. et al. A role for uric acid in the progression of renal disease. J. Am. Soc. Nephrol. 13, 2888–2897 (2002).
Annemans, L. et al. Gout in the UK and Germany: prevalence, comorbidities and management in general practice 2000-2005. Ann. Rheum. Dis. 67, 960–966 (2008).
Jin, M. et al. Uric acid, hyperuricemia and vascular diseases. Front. Biosci. (Landmark Ed.) 17, 656–669 (2012).
King, C. et al. Uric acid as a cause of the metabolic syndrome. Contrib. Nephrol. 192, 88–102 (2018).
Dalbeth, N. et al. Gout. Nat. Rev. Dis. Prim. 5, 69 (2019).
Gliozzi, M., Malara, N., Muscoli, S. & Mollace, V. The treatment of hyperuricemia. Int. J. Cardiol. 213, 23–27 (2016).
Dehlin, M., Jacobsson, L. & Roddy, E. Global epidemiology of gout: prevalence, incidence, treatment patterns and risk factors. Nat. Rev. Rheumatol. 16, 380–390 (2020).
Choi, H. K., Mount, D. B. & Reginato, A. M. American College of, P. & American Physiological, S. Pathogenesis of gout. Ann. Intern. Med. 143, 499–516 (2005).
Kuo, C. F., Grainge, M. J., Zhang, W. & Doherty, M. Global epidemiology of gout: prevalence, incidence and risk factors. Nat. Rev. Rheumatol. 11, 649–662 (2015).
Emmerson, B. T. The management of gout. N. Engl. J. Med. 334, 445–451 (1996).
Terkeltaub, R. Update on gout: new therapeutic strategies and options. Nat. Rev. Rheumatol. 6, 30–38 (2010).
Yang, Y. et al. Virtual screening and activity evaluation of human uric acid transporter 1 (hURAT1) inhibitors. RSC Adv. 13, 3474–3486 (2023).
Song, D., Zhao, X., Wang, F. & Wang, G. A brief review of urate transporter 1 (URAT1) inhibitors for the treatment of hyperuricemia and gout: current therapeutic options and potential applications. Eur. J. Pharm. 907, 174291 (2021).
Robbins, N., Koch, S. E., Tranter, M. & Rubinstein, J. The history and future of probenecid. Cardiovasc. Toxicol. 12, 1–9 (2012).
Lee, M. H., Graham, G. G., Williams, K. M. & Day, R. O. A benefit-risk assessment of benzbromarone in the treatment of gout. Was its withdrawal from the market in the best interest of patients?. Drug Saf. 31, 643–665 (2008).
Miner, J. N. et al. Lesinurad, a novel, oral compound for gout, acts to decrease serum uric acid through inhibition of urate transporters in the kidney. Arthritis Res. Ther. 18, 214 (2016).
Chen, Y., You, R., Wang, K. & Wang, Y. Recent Updates of Natural and Synthetic URAT1 Inhibitors and Novel Screening Methods. Evid. Based Complement. Altern. Med. 2021, 5738900 (2021).
Gillen, M. et al. Effects of renal function on pharmacokinetics and pharmacodynamics of lesinurad in adult volunteers. Drug Des. Dev. Ther. 10, 3555–3562 (2016).
Hautekeete, M. L. et al. Severe hepatotoxicity related to benzarone: a report of three cases with two fatalities. Liver 15, 25–29 (1995).
Strilchuk, L., Fogacci, F. & Cicero, A. F. Safety and tolerability of available urate-lowering drugs: a critical review. Expert Opin. Drug Saf. 18, 261–271 (2019).
Tan, P. K., Ostertag, T. M. & Miner, J. N. Mechanism of high affinity inhibition of the human urate transporter URAT1. Sci. Rep. 6, 34995 (2016).
Tan, P. K., Farrar, J. E., Gaucher, E. A. & Miner, J. N. Coevolution of URAT1 and uricase during primate evolution: implications for serum urate homeostasis and gout. Mol. Biol. Evol. 33, 2193–2200 (2016).
Tan, P. K., Ostertag, T. M. & Miner, J. N. Mechanism of high affinity inhibition of the human urate transporter URAT1. Sci. Rep. 6, https://doi.org/10.1038/srep34995 (2016).
Brast, S. et al. The cysteines of the extracellular loop are crucial for trafficking of human organic cation transporter 2 to the plasma membrane and are involved in oligomerization. FASEB J. 26, 976–986 (2011).
Dai, Y. & Lee, C. H. Transport mechanism and structural pharmacology of human urate transporter URAT1. Cell Res. 34, 776–787 (2024).
He, J. et al. Structural basis for the transport and substrate selection of human urate transporter 1. Cell Rep. 43, 114628 (2024).
Zhang, H., Cui, N., Ma, X. & Xu, H. E. https://doi.org/10.1101/2024.11.13.623519 (2024).
Azevedo, V. F., Kos, I. A., Vargas-Santos, A. B., da Rocha Castelar Pinheiro, G. & Dos Santos Paiva, E. Benzbromarone in the treatment of gout. Adv. Rheumatol. 59, 37 (2019).
Chen, X. et al. Novel natural scaffold as hURAT1 inhibitor identified by 3D-shape-based, docking-based virtual screening approach and biological evaluation. Bioorg. Chem. 117, 105444 (2021).
Kurihara, O., Yamada, T., Kato, K. & Miyauchi, Y. Efficacy of dotinurad in patients with severe renal dysfunction. Clin. Exp. Nephrol. 28, 208–216 (2023).
Perez-Ruiz, F. et al. Efficacy and safety of lesinurad for the treatment of hyperuricemia in gout. Drugs Context 8, 212581 (2019).
Fitz-Patrick, D. et al. Safety and efficacy of verinurad, a selective URAT1 inhibitor, for the treatment of patients with gout and/or asymptomatic hyperuricemia in the United States and Japan: findings from two phase II trials. Mod. Rheumatol. 29, 1042–1052 (2019).
Estiverne, C. & Mount, D. B. The management of gout in renal disease. Semin. Nephrol. 40, 600–613 (2020).
Zhao, Z. et al. Discovery of novel benzbromarone analogs with improved pharmacokinetics and benign toxicity profiles as antihyperuricemic agents. Eur. J. Med. Chem. 242, 114682 (2022).
Tan, P. K., Liu, S., Gunic, E. & Miner, J. N. Discovery and characterization of verinurad, a potent and specific inhibitor of URAT1 for the treatment of hyperuricemia and gout. Sci. Rep. 7, 665 (2017).
Jenkins, C., Hwang, J. H., Kopp, J. B., Winkler, C. A. & Cho, S. K. Review of urate-lowering therapeutics: from the past to the future. Front. Pharm. 13, 925219 (2022).
Jiang, D. et al. Structure of the YajR transporter suggests a transport mechanism based on the conserved motif A. Proc. Natl. Acad. Sci. 110, 14664–14669 (2013).
Drew, D., North, R. A., Nagarathinam, K. & Tanabe, M. Structures and general transport mechanisms by the major facilitator superfamily (MFS). Chem. Rev. 121, 5289–5335 (2021).
Khanppnavar, B. et al. Structural basis of organic cation transporter-3 inhibition. Nat. Commun. 13, 6714 (2022).
Zhao, Z. et al. Structural insights into the atomistic mechanisms of uric acid recognition and translocation of human urate anion transporter 1. ACS Omega 5, 33421–33432 (2020).
Jiang, D. et al. Structure of the YajR transporter suggests a transport mechanism based on the conserved motif A. Proc. Natl. Acad. Sci. USA 110, 14664–14669 (2013).
Roman, Y. M. The role of uric acid in human health: insights from the uricase gene. J. Pers. Med. 13, 1409 (2023).
Parker, J. L., Kato, T., Kuteyi, G., Sitsel, O. & Newstead, S. Molecular basis for selective uptake and elimination of organic anions in the kidney by OAT1. Nat. Struct. Mol. Biol. 30, 1786–1793 (2023).
Fitz-Patrick, D. et al. Safety and efficacy of verinurad, a selective URAT1 inhibitor, for the treatment of patients with gout and/or asymptomatic hyperuricemia in the United States and Japan: findings from two phase II trials. Mod. Rheumatol. 29, 1042–1052 (2018).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66, 213–221 (2010).
DeLano, W. L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 40, 82–49 (2002).
Pettersen, E. F. et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput Chem. 29, 1859–1865 (2008).
Lomize, M. A., Pogozheva, I. D., Joo, H., Mosberg, H. I. & Lomize, A. L. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376 (2012).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Maier, J. A. et al. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015).
Wang, J., Wang, W., Kollman, P. A. & Case, D. A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model 25, 247–260 (2006).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Parrinello, M. & Rahman, A. Crystal structure and pair potentials: a molecular-dynamics study. Phys. Rev. Lett. 45, 1196–1199 (1980).
Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 4, 116–122 (2008).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Acknowledgements
We thank B. Xu at Institute of Advanced Agricultural Sciences and X. Huang, B. Zhu, and other staff members at the Center for Biological Imaging (CBI), Core Facilities for Protein Science at the Institute of Biophysics, Chinese Academy of Science (IBP, CAS) for the support in cryo-EM data collection. We thank H. Zhang for his assistance in the [14C]urate uptake assays. We thank Yan Wu for his research assistant service. This work is funded by the Chinese Academy of Sciences Strategic Priority Research Program (Grant No. XDB37030304 to Y.Z.), Chinese National Programs for Brain Science and Brain-like Intelligence Technology (Grant No. 2022ZD0205800 to Y.Z.), the National Key Research and Development Program of China (Grant No. 2021YFA1301501 to Y.Z.), and the National Natural Science Foundation of China (Grant No. 92157102 to Y.Z.).
Author information
Authors and Affiliations
Contributions
Y.Z. conceived and supervised the project. Z.Y., N.L. carried out molecular cloning experiments, expressed and purified protein samples, and prepared samples for cryo-EM study. T.H. performed functional assays. R.L., J.Z., and Z.Y. carried out cryo-EM data collection. Z.Y. processed the cryo-EM data. Z.Y., T.H., Y. D., and P.Y. built the atomic model and analyzed the structures. Q.B. performed the molecular dynamics simulations. T.H., Z.Y., J.S., Q.M. and Q.C. prepared the figures. Y.Z., X.C.Z., Z.Y., T.H., and J.S. wrote and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yu, Z., Hu, T., Su, J. et al. Molecular mechanism of drug inhibition of URAT1. Nat Commun 16, 6551 (2025). https://doi.org/10.1038/s41467-025-61226-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61226-x
