
Abstract
P-glycoprotein (P-gp) is a crucial drug efflux transporter in the gastrointestinal tract, reducing drug uptake and expelling harmful xenobiotics to prevent pathological changes. Current P-gp enhancers primarily increase P-gp expression, requiring 1–3 days, thus missing the critical rescue window for acute poisoning. This study identifies glucosamine (GlcN) as a potent P-gp activator that swiftly enhances drug efflux, significantly reducing drug absorption without altering P-gp expression levels. GlcN directly binds to P-gp, boosting its transport efficiency. Only GlcN with a polymerization degree below 5 can activate P-gp, whereas higher polymerized chitooligosaccharides enhance drug absorption. Additionally, GlcN activation of P-gp has significant implications for cellular metabolism by expelling xenobiotics and metabolic by-products, maintaining cellular homeostasis. Our findings suggest GlcN’s potential as an effective antidote for paraquat poisoning and offer a detoxification strategy. This research provides a foundational understanding for developing improved detoxification agents and metabolic modulators.
Similar content being viewed by others


Introduction
P-glycoprotein (P-gp), a crucial membrane protein with efflux properties, is expressed in human and animal intestinal, renal, hepatic, and brain endothelial cells1,2,3,4,5. Alongside factors such as potential of hydrogen (pH) levels, enzymatic degradation, tight junction integrity, and the mucus layer, P-gp plays a vital role in safeguarding the body by expelling toxins from cells into the extracellular environment6,7,8. Currently, research on P-gp mainly focused on developing inhibitors that suppress its efflux transport function. These inhibitors aim to enhance the effective uptake of drugs in malignant cells or pathogenic bacteria, thereby improving therapeutic outcomes. This plays a crucial role in overcoming tumor and bacterial resistance, enhancing clinical efficacy, and protecting human health9,10,11,12,13,14,15. However, P-gp, as an intrinsic efflux system in the body, also plays a critical role in clearing harmful substances and protecting the body. Therefore, exploring the activation mechanisms of P-gp is not only of scientific importance but also holds potential application value in detoxification research16,17,18,19,20.
It is well known that exposure of cells to various environmental stressors, including drugs, cytokines, and reactive oxygen species, can upregulate the expression of the multidrug resistance protein 1 (MDR1) gene, which in turn induce a significant increase in P-gp protein levels15,21,22. Despite the existence of a diverse range of P-gp inducers, clinical detoxification treatment is still compromised by the limited time-effectiveness of these inducers. In situations requiring emergency intervention, it is crucial for inducers to act swiftly, within minutes. However, commonly used P-gp inducers such as rifampicin, dexamethasone, and curcumin generally take 1–3 days to induce increase P-gp expression16,23,24,25,26. The emergence of a new category of compounds known as P-gp activators, which rapidly enhance the efflux transport efficiency of P-gp by binding to it and altering its conformation without affecting its expression level, offers promising prospects for rapid detoxification through enhanced P-gp efflux16,27,28,29. However, research on P-gp activators is limited, highlighting the urgent need for safe and effective P-gp activators in the clinical treatment of patients suffering from poisoning.
In this work, we make a surprising discovery: Glucosamine (GlcN) acts as a potent inhibitor of drug absorption in the intestine, contrary to our initial hypothesis aimed at exploring the correlation between the intestinal absorption-enhancing effects of chitooligosaccharides (COS, polymers of GlcN) and the degree of polymerization of COS30. More importantly, it was demonstrated that GlcN’s activation of P-gp efflux transport activity is the direct mechanism underlying its inhibition of drug intestinal absorption. The cellular thermal shift assay and “inside-out” inverted vesicle model confirmed that GlcN directly binds to P-gp, enhancing its efflux transport efficiency without altering P-gp protein levels. Gene knockout experiments further validated GlcN’s role in reducing drug absorption mediated by P-gp. Additionally, the correlation between the polymerization degree of COS and P-gp activation ability was revealed with surface plasmon resonance assays, and it shows that only COS with a low polymerization degree (degree of polymerization <5) can bind to P-gp. Furthermore, pharmacokinetic and survival rate experiments demonstrated GlcN’s efficacy in alleviating toxic reactions in paraquat-intoxicated rats, leading to improved survival rates and underscoring its potential as a safe and effective detoxifying agent. In summary, this study identifies the activating effect of GlcN on P-gp, explaining its impact on drug intestinal absorption. It provides a theoretical foundation for GlcN’s physiological effects research and its rational applications, as well as offering possibilities for detoxification research based on the P-gp efflux effect.
Results
Discovery and confirmation of GlcN’s inhibitory effect on oral drug absorption
Drawing from our prior investigations, COS were observed to enhance the oral absorption of BCS 3 drugs (biopharmaceutical classification system)30. However, upon delving deeper into the relationship between the polymerization degree of COS and its absorption-enhancing properties, we found that the monosaccharide GlcN significantly inhibits drug intestinal absorption. To further elucidate the nonspecific effects of GlcN on drug absorption in the intestine, we conducted pharmacokinetic studies on four structurally distinct drugs (Capecitabine, Acyclovir, Cimetidine, Oxybutynin). The study commenced by establishing the methodology and standard curves for determining the plasma concentration of each drug (Supplementary Fig. 1, 2 and Supplementary Table 1, 2). Subsequently, pharmacokinetic analysis revealed a significant inhibition of oral absorption for all four drugs when co-administered with GlcN. Specifically, the area under the curve (AUC), representing the relative bioavailability, decreased by >40% compared to the corresponding control groups (Fig. 1a–e and Supplementary Table 3). Compatibility studies showed that GlcN did not form micelles or precipitates with various drugs, and had no significant effect on the stability of the drug within 6 h (Supplementary Fig. 3). Furthermore, the impact of dosage and exposure duration of GlcN’s inhibition effect on drug intestinal absorption was investigated using an everted intestinal sac model31,32,33. It was observed that the inhibitory effect of GlcN on drug transport in the intestine increases with concentration, reaching a plateau peak at a dose of 250 mg/kg. GlcN’s inhibitory effect also shows time dependency. Pre-incubation of the intestine with GlcN for up to 20 min enhances the inhibitory effect as the incubation time lengthens. However, prolonged incubation (40 min) reverses the inhibitory effect. Therefore, incubating the intestine with 250 mg/kg of GlcN for 20 min resulted in the lowest transport rate (Supplementary Fig. 4 and Supplementary Table 4, 5). These results indicate that GlcN’s impact on drug intestinal transport is potent, rapid, reversible, and nonspecific. We speculate that this broad drug inhibition may be attributed to alterations in intestinal barrier permeability, such as changes in tight junction function, mucus layers, and P-gp activity (Fig. 1f)7,34,35,36,37,38.

a–d The pharmacokinetics and AUC of capecitabine (a), acyclovir (b), cimetidine (c), and omeprazole (d) were examined in Sprague-Dawley (SD) rats treated with or without GlcN (500 mg/kg). e The percentage decrease in the bioavailability of the four drugs, normalized to their respective control groups. f Schematic of inhibition of drug intestinal absorption by oral GlcN. Data are means ± Standard Error of the Mean (SEM) from six (n = 6) biological independent replicates in (a−d) p-values were calculated using two-tailed Student’s t-test. ***p < 0.001, ****p < 0.0001. Created in BioRender. Qinghua, W. (2025) https://BioRender.com/c20pkwq.
Enhanced P-gp efflux: GlcN’s role in inhibiting drug intestinal absorption
To elucidate the mechanism underlying GlcN’s inhibition of drug intestinal absorption, we investigated its effects on three key intestinal barrier functions: the intestinal mucus layer, tight junctions between intestinal epithelial cells, as well as metabolic enzymes and drug efflux pumps within intestinal epithelial cells18,38. It was observed through a rheometer that when co-incubated in vitro, GlcN had no effect on the viscosity of rat intestinal mucus, indicating that GlcN cannot alter the thickness or structure of the mucus layer (Fig. 2a)39. Transmission electron microscopy (TEM) images revealed that the tight junctions between intestinal epithelial cells remained unchanged before and after oral administration of GlcN (Fig. 2b and Supplementary Fig. 5a). Since GlcN concentrations below 50 mM did not induce significant cytotoxicity in Caco-2 cells (Supplementary Fig. 5b), a transwell model was constructed to examine the effect of GlcN on P-gp function (Supplementary Fig. 5c)40,41,42. Notably, the addition of GlcN significantly inhibited the transport of capecitabine from the apical side (AP) to the basolateral side (BL), as indicated by a marked reduction in its apparent permeability coefficient (Papp, AP-BL) (Fig. 2c). Simultaneously, GlcN significantly enhanced the transport of capecitabine from the BL to the AP side, with a significant increase in Papp (BL-AP) (Supplementary Fig. 5d). The effect of the P-gp inhibitor verapamil on capecitabine transport was the opposite of that observed with GlcN. When both verapamil and GlcN were present, the antagonistic effect was predominantly driven by verapamil. Importantly, the efflux ratio (ER) of capecitabine (Papp (BL-AP) / Papp (AP-BL)) was >2, suggesting that capecitabine is likely affected by P-gp-mediated efflux. After the addition of GlcN, the efflux ratio increased to >8, indicating that GlcN induces a strong efflux of capecitabine, which may be a crucial factor influencing its absorption and bioavailability (Supplementary Table 6). This implies that GlcN’s inhibitory effect on drug absorption may be linked to its impact on P-gp efflux function43,44,45.

a Viscosity of rat intestinal mucus treated with or without GlcN (50 mg/mL for 30 min) was examined by rheometer. b The tight junctions between intestinal epithelial cells treated with GlcN (250 mg/kg for 20 min) were detected by TEM. Scare bar, 1 μm. The image only represents the results of one biological sample. c Transcellular transport rate of capecitabine in Caco-2 cells treated with the indicated drugs was examined by transwell model. n = 3 biologically independent samples. d Schematic of validating the interaction between GlcN and P-gp through cell uptake and gene knockout. Created in BioRender. Qinghua, W. (2025) https://BioRender.com/c20pkwqe, f Accumulation (e) and efflux (f) of Rh123 in Caco-2 cells treated with or without GlcN at the indicated concentrations were determined by a multimode reader (Ex = 485 nm, Em = 528 nm). n = 6 biologically independent samples. g Expression of P-gp in the indicated Caco-2 cells was determined by immunoblot. h Cellular uptake of Rh123 in control and P-gp silenced Caco-2 cells treated with or without GlcN at the indicated concentrations was determined (Ex = 485 nm, Em = 528 nm). n = 6 biologically independent samples (After-silencing, p-value for Verapamil = 0.3034, 1 mM = 0.9996, 10 mM = 0.7150, 15 mM = 0.9993). i, k Pharmacokinetic behavior of digoxin in wild-type (wt) FVB mice (i) and abcb1(-/-) mice (k) in the presence or absence of GlcN (500 mg/kg). n = 6 biologically independent mice. j Schematic of the abcb1 gene knockout process in mice. Created in BioRender. Qinghua, W. (2025) https://BioRender.com/c20pkwq. Data are means ± SEM and p-values were calculated using one-way ANOVA in c−h. **p < 0.01, ***p < 0.001, ****p < 0.0001, NS represent not significant.
To validate this hypothesis, cellular uptake and gene knockout experiments were conducted (Fig. 2d). Rhodamine 123 (Rh123), a fluorescent dye that can penetrate cell membranes, was used to evaluate P-gp efflux activity5,46,47,48. When co-incubated with GlcN, there was a notable and dose-dependent reduction in Rh123 concentration inside Caco-2 cells. The most pronounced inhibitory effect was observed at 10 mM GlcN, leading to ~40% more Rh123 was excreted from the cells (Fig. 2e). However, when verapamil was added concurrently, this inhibitory effect was reversed, and a noticeable increase in intracellular Rh123 concentration was observed (Supplementary Fig. 5e). Furthermore, Rh123-preloaded Caco-2 cells were exposed to GlcN or glucose (Glu). Compared to the control group, GlcN notably reduced the fluorescence intensity of intracellular Rh123, while the addition of glucose had no discernible effect on Rh123 transport (Fig. 2f). Similarly, when verapamil (VRP) was added simultaneously, the enhancing effect of GlcN on the efflux of Rh123 was reversed (Supplementary Fig. 5f). This suggests that GlcN does not serve as an energy source to promote the efflux of Rh123 from the cells, but rather may be related to its interaction with P-gp49,50.
Furthermore, to verify the ligand-receptor relationship between GlcN and P-gp, P-gp expression in Caco-2 cells was silenced using short interfering RNA (siRNA), and confirmed by western blot analysis showing reduced P-gp protein expression (Fig. 2g and Supplementary Fig. 5g)51,52,53,54. Subsequently, P-gp knockdown Caco-2 cells were co-incubated with Rh123 and GlcN, revealing that GlcN, at various concentrations, hardly had any effect on the intracellular accumulation level of Rh123 (Fig. 2h). Crucially, this interaction between the ligand and receptor was further confirmed through in vivo experiments. Digoxin is a typical P-gp substrate drug, and its absorption in the organism is influenced by P-gp55,56. A method for detecting digoxin levels in vivo has been established (Supplementary Fig. 5h). GlcN significantly inhibited the oral absorption of digoxin in wild-type FVB mice (Fig. 2i). However, in abcb1(-/-) mice with P-gp gene knockout, the addition of GlcN does not affect the in vivo pharmacokinetic behavior of digoxin (Fig. 2j, k). Similarly, the previously observed inhibitory effect of GlcN on the intestinal absorption of capecitabine, acyclovir, cimetidine, and oxcarbazepine were also abolished in abcb1(-/-) mice (Supplementary Fig. 6). This indicates that GlcN affects drug absorption in the body via its interaction with P-gp (Fig. 2i–k and Supplementary Table 7)57. In summary, GlcN was observed to enhance P-gp efflux activity, effectively reducing drug absorption across intestinal cells and consequently leading to a significant decrease in the bioavailability of these drugs in the body.
GlcN functions as an activator of P-gp
Current research suggests that P-gp enhancement can occur through two distinct pathways: P-gp inducers and P-gp activators, which increase protein expression level or activate transport efficiency, respectively16. Given GlcN’s rapid and reversible activation properties on P-gp, we hypothesized its potential as a P-gp activator. Thus, the investigation focused on GlcN’s binding ability to P-gp and its impact on P-gp transport efficiency at a fixed protein level. The cellular thermal shift assay (CETSA) assessed the binding capability between drug molecules and target proteins by the enhanced thermal stability of the molecule-protein binding complex58,59,60. Inside the cells, when a drug binds to the target protein, the protein’s structure becomes more stable, making it less prone to denaturation at elevated temperatures. As the temperature increases, unbound proteins gradually denature and precipitate, whereas drug-bound proteins remain more stable at the same temperature. P-gp extracted from Caco-2 cells exhibited significant degradation above 54.4 °C, reaching nearly complete degradation beyond 68.0 °C. However, GlcN incubation noticeably enhanced P-gp’s thermal stability, retaining over 70% integrity at 68 °C (Fig. 3a, b and Supplementary Fig. 7, a–f). Additionally, a recombinant “inside-out” inverted vesicle model was utilized to study GlcN’s impact on P-gp transport efficiency. This classical model reflects transport efficiency per unit protein quantity with fixed P-gp expression on the vesicle surface (Fig. 3c)61,62,63. The uptake ratio of the NC group (blank buffer) transporter was 4.28 ( > 2.0), while the inhibition rate of the PC group (Cyclosporine A, CsA, P-gp inhibitor) was 87.1% (>50%). These collectively validate the functionality of the transporter assay system, ensuring that the results reliably represent the transport activity of P-gp. The transport activity of the GlcN group was ~1.48 times higher than that of the control group. However, this enhancement was completely abolished upon co-treatment with CsA (Fig. 3d). These suggesting that GlcN can rapidly enhance P-gp transport activity without altering P-gp expression levels. Therefore, these findings demonstrate that GlcN can function as an activator of P-gp. Furthermore, the impact of GlcN on P-gp-mediated ATPase activity and the cytochrome enzyme CYP3A4 was explored, as P-gp transport relies on ATP energy supply64, and potential co-regulation between P-gp and CYP3A4 has been reported65,66. The transport of digoxin, a P-gp substrate, is accompanied by a significant increase in P-gp associated ATPase activity, which supplies the energy for efflux. Conversely, the P-gp inhibitor verapamil markedly inhibits this activity. GlcN boosted ATPase activity within a concentration range of 1 μM to 20 mM, reaching its peak effect at 10 μM GlcN, where it doubled the activity compared to baseline (p < 0.001) (Fig. 3e). Moreover, during a 30 min incubation period, GlcN concentrations ranging from 1 μM to 20 mM rapidly increased intracellular CYP3A4 enzyme activity. The most significant enhancement occurred at 100 μM, resulting in a 301% boost compared to the control group (p < 0.001) (Supplementary Fig. 8a). While rifampicin, a CYP3A4 inducer67, and ketoconazole, a CYP3A4 inhibitor68,69, showed slight stimulatory effects on CYP3A4 activity during short-term stimulation, their long-term effects varied significantly. After 48 h of induction, rifampicin increased CYP3A4 enzyme activity to 516% of the control group, while ketoconazole inhibited activity by 47%. Notably, GlcN exhibited a promotive effect on CYP3A4 enzyme activity, with the most pronounced enhancement observed at 1 mM, resulting in a 388% increase compared to the control group (p < 0.001) (Fig. 3f). In summary, GlcN has been demonstrated to act as a P-gp activator, swiftly enhancing P-gp’s efflux transport activity through direct binding without altering its expression.

a Thermal stability of P-gp treated with or without GlcN (10 mM for 30 min) at indicated temperatures was determined by CETSA, each experiment was repeated three times independently with similar results. b The protein content of CETSA results was quantified, normalized to 45 °C. n = 3 biologically independent experiments. c Schematic of recombinant “inside-out” inverted vesicle model to determine the P-gp transport efficiency at a fixed protein level. Created in BioRender. Qinghua, W. (2025) https://BioRender.com/c20pkwq. d Uptake activity of P-gp treated with or without GlcN (10 mM for 5 min) was determined by recombinant “inside-out” inverted vesicle model. n = 3 independent experiments. e P-gp mediated ATPase activity in Caco-2 cells treated with the indicated conditions was determined in the presence or absence of sodium orthovanadate by ATPase test kit. n = 3 biologically independent samples. f CYP3A4 activity in Caco-2 cells treated with or without GlcN at indicated concentrations was determined by P450-Glo™ CYP3A4 Assay and Screening System. n = 6 biologically independent samples. Data are means ± SEM and p-values were calculated using one-way ANOVA in b−f. *p < 0.05, ***p < 0.001, NS represent not significant.
The polymerization degree of GlcN is crucial for its binding to P-glycoprotein
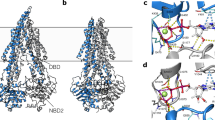
Given the discovery of the potential of GlcN as a P-gp activator, we are keen to investigate how its degree of polymerization relates to its influence on drug absorption. A series of structure-activity relationship studies were conducted to further explore why the physiological effects of GlcN on drug absorption shift from inhibition to enhancement as its degree of polymerization increases. A P-gp protein model (Protein data bank, PDB ID: 4Q9H), representing an inward-facing conformation, was selected for molecular docking simulations based on its prior use in studies of P-gp activators70. The results revealed that GlcN and COS with a degree of polymerization of 6 or lower can effectively bind to P-gp (Supplementary Table 8). We also compared the calculated binding energies of the inhibitor verapamil and the substrates digoxin and quinidine, finding that the calculated binding energy of GlcN with P-gp is slightly lower than that of verapamil and P-gp substrates. This also explains why, in the presence of both verapamil and GlcN, the antagonistic effect is dominated by verapamil (Supplementary Table 9). The three-dimensional interaction diagram between GlcN and P-gp illustrates GlcN binding at the helix crossover, a region within one of the two transmembrane domains (TMD) of P-gp responsible for drug transport channels. The interaction detail diagram reveals that GlcN forms hydrogen bonds with Y303, Q-721, N-835, Q-834, and Q986 on P-gp, with no hydrophobic interactions or π-Stacking observed. This may be due to the relatively low number of non-polar atoms in GlcN. However, it is worth noting that hydrogen bonds are the primary interaction force between small molecules and proteins, and they are also one of the strongest non-covalent interactions (Fig. 4a, b and Supplementary Fig. 8b). More interestingly, by comparing the primary structure of P-gp from UniProt, we found that the sequences at the binding sites for GlcN—Y303, Q721, N835 and Q834 are conserved across species. Additionally, although asparagine (N) is present at position 986 in rat P-gp, both the glutamine (Q) and asparagine (N) residues exposed at this position have amide side chains. At this site, GlcN binds to P-gp by forming hydrogen bonds with the amide group. This suggests that the homologous P-gp model used in this study is suitable for investigating GlcN binding to P-gp (Supplementary Fig. 8c). This interaction was further validated through surface plasmon resonance (SPR) experiments71,72, where COS with a degree of polymerization <5 exhibited binding affinity with P-gp similar to that of GlcN. However, COS with a degree of polymerization exceeding 5 ceased to exhibit significant binding interactions with P-gp. Notably, chitotriose (COS3) displayed the strongest affinity for P-gp (KD = 0.2756 mM), even surpassing the positive control drug verapamil (Fig. 4c and Supplementary Fig. 8, d–i and Supplementary Table 10).

a, b Three-dimensional overall view (a) and partial view (b) of the interaction between GlcN and P-gp protein (PDB code: 4Q9H). c Affinity between P-gp and COS with different degrees of polymerization (10 mM) was determined by SPR. d, e Pharmacokinetics (d) and AUC (e) of capecitabine in rats treated with COS (250 mg/kg) with indicated degrees of polymerization. n = 6 biologically independent mice. f Schematic of the varied effects of COS with different degrees of polymerization on drug intestinal absorption. Created in BioRender. Qinghua, W. (2025) https://BioRender.com/c20pkwq. Data are means ± SEM and p-values were calculated using one-way ANOVA in d and e. *p < 0.05, ***p < 0.001, ****p < 0.0001.
The influence of polymerization degree on physiological effects was further investigated through pharmacokinetic studies. Compared to the previously established impact of GlcN on capecitabine pharmacokinetics in results section30, it was found that COS with P-gp affinity, such as chitobiose (COS2), chitotriose (COS3), and chitotetraose (COS4), inhibited the intestinal absorption of capecitabine. Notably, chitotriose, exhibiting the strongest P-gp affinity, displayed the most pronounced inhibitory effect on capecitabine intestinal absorption (AUC only 12.65% of the control group). Additionally, it was observed that chitopentaose (COS5) facilitated the intestinal absorption of capecitabine. This is consistent with our prior research demonstrating that COS with higher polymerization degrees enhance drug absorption by compacting the intestinal mucus layer (Fig. 4, d, e and Supplementary Table 11)30. In summary, it has been demonstrated that the degree of polymerization of GlcN is a key factor influencing its binding capacity with P-gp, leading to different physiological effects of COS with varying degrees of polymerization (Fig. 4f).
GlcN’s potential to serve as an antidote for paraquat
To leverage the discovery of GlcN’s potential as a P-gp activator, we attempted its application in the oral detoxification of paraquat (PQ). PQ is the most lethal herbicide globally, upon entering the human body, it rapidly induces multi-organ damage and failure, with no effective antidote or treatment currently available73. Research indicates that PQ is a substrate of P-gp, suggesting that activating P-gp could be a potential therapeutic approach for PQ poisoning (Fig. 5a)70,74. Patients who orally ingest paraquat often develop severe pulmonary fibrosis and respiratory failure, which led us to examine GlcN’s effect on paraquat resistance in intestinal cells (Caco-2) and lung cells (A549), GlcN significantly enhanced the resistance of both cell lines to paraquat, with a notable increase in cell viability. This protective effect was also reversed by verapamil. Importantly, the resistance mediated by GlcN was superior to that induced by dexamethasone, a previously studied P-gp activator, although statistical significance was not observed (Supplementary Fig. 9a,b). The transport of paraquat in the P-gp recombinant inside-out vesicle model demonstrates that the enhanced cellular resistance to paraquat arises from GlcN-induced activation of P-gp efflux activity. P-gp inhibitors significantly suppressed paraquat transport, whereas the P-gp activator GlcN enhanced its transport (Supplementary Fig. 9c). In vivo studies yield similar results, and a method for determining paraquat plasma concentrations was established (Supplementary Fig. 9d). Pharmacokinetic studies revealed that co-administration of GlcN and PQ resulted in a more pronounced inhibition of PQ’s intestinal absorption in rats compared to the positive drug dexamethasone (DEX)75. This led to a significant reduction in both the maximum plasma concentration (41.01% of control) and bioavailability (34.41% of control) of PQ (Fig. 5b and Supplementary Table 12). Similarly, GlcN significantly reduced PQ absorption in FVB mice, but had no substantial effect on PQ pharmacokinetics in abcb1(-/-) mice (Fig. 5c, d). Moreover, the impact of GlcN on the survival rate of PQ-intoxicated rats and PQ-intoxicated mice was further investigated. All poisoned rats without administration died within 6 days. Conversely, rats administered with GlcN or DEX orally, 2 h post-intoxication, exhibited survival rates of 60% and 30%, respectively, over a 15 days duration (Fig. 5e). Moreover, GlcN improves the survival rate of PQ-intoxicated FVB mice (Fig. 5f), whereas it fails to significantly rescue the survival of PQ-intoxicated abcb1(-/-) mice. Although DEX shows a statistically significant extension in survival time, it does not improve the survival rate within 15 days. Additionally, there is no notable difference between GlcN and DEX in terms of improving the survival rate of PQ-intoxicated mice (Fig. 5g). This indicates that GlcN possesses the capability to mitigate PQ toxicity, offering potential benefits for the further treatment of PQ poisoning patients. Given GlcN’s inherent safety profile, it shows promise for further development as a paraquat detoxification agent.

a Schematic of GlcN increasing the survival rate of paraquat-poisoned animals. Created in BioRender. Qinghua, W. (2025) https://BioRender.com/c20pkwq. b−d Pharmacokinetics of paraquat in SD rats (b), wt FVB mice (c) and abcb1(-/-) mice (d) treated with or without GlcN (250 mg/kg in SD rat and 500 mg/kg in mice). n = 6 biologically independent experiments. e−g Survival rate of paraquat-poisoned rats (e), paraquat-poisoned FVB mice (f) and paraquat-poisoned abcb1(-/-) mice (g) treated with GlcN (250 mg/kg in SD rat and 500 mg/kg in mice) or dexamethasone (100 mg/kg) 2 h post-poisoning. n = 10 biologically independent samples. Data are means ± SEM in b−d. p-values were calculated using Simple survival analysis (Kaplan-Meier) in e, f and g. *p < 0.05, **P < 0.01, NS represent not significant.
Discussion
P-gp, also known as abcb1 protein, is a transmembrane glycoprotein encoded by the multidrug resistance 1 (MDR1) gene, with a molecular weight of ~170 kDa. As the first identified member of the ATP-binding cassette (ABC) transporter superfamily with representative characteristics in humans, P-gp is widely present on the surface of epithelial cells in the human intestine and blood-brain barrier. Functioning as a robust drug efflux pump, P-gp possesses multiple cooperatively functioning drug-binding sites, relies on ATP energy, and can rapidly pump a large number of drug molecules out of cells in a non-specific manner, thereby protecting cells from the intrusion of harmful external molecules2,16. Although numerous studies attribute multidrug resistance (MDR) in tumor cells to the overexpression and activation of P-gp9, it is crucial to acknowledge that P-gp plays a vital role in normal organisms as a mechanism for self-protection by actively expelling harmful xenobiotics. In particular, in the intestines, this protection holds significant importance as it prevents harmful xenobiotics from being absorbed into the bloodstream or organs16. Research on P-gp inducers is a common detoxification strategy that relies on P-gp efflux activity, as many drugs or environmental factors can upregulate P-gp expression. However, protein expression involves transcription, translation, modification, and expression processes, which can be time-consuming, whereas detoxification demands swift effectiveness. The identification of P-gp activators introduces a fresh alternative74. P-gp contains a pair of ATP-binding domains located in the cytoplasm and two sets of transmembrane domains (TMDs), each comprising six transmembrane α-helices76. Biochemical analyses indicate that all transmembrane helices are involved in substrate binding, with the volume covered by the TMDs being larger than that of any known abcb1 substrate. The TMD dimer in abcb1 contains highly mixed substrate binding sites, and increasing experimental and computational data suggest that P-gp contains at least two distinct drug binding and transport sites, which interact with each other in a cooperative manner16,27,70. Recent studies suggest that P-gp contains a central binding cavity within the transmembrane domain and peripheral sites composed of shallow hydrophobic pockets. These sites exhibit dynamic conformational cooperativity, whereby substrate binding—such as rhodamine at the central site—may induce local conformational changes (e.g., alterations in the hydrophobic microenvironment) that facilitate substrate interaction or release at peripheral sites. This cooperativity underlies the broad substrate specificity and efficient transport function of P-gp. Regrettably, progress in the research on P-gp activators has been limited, and drugs known to activate P-gp, such as dexamethasone and amphotericin, may pose safety risks to humans when used long-term or in large quantities, thus restricting their application77,78. Regrettably, progress in the research on P-gp activators has been limited, and drugs known to activate P-gp, such as dexamethasone and amphotericin, may pose safety risks to humans when used long-term or in large quantities, thus restricting their application. Therefore, the development of safe and effective P-gp activators is significantly important in detoxification research. In our study, GlcN was surprisingly found to have a significant inhibitory effect on drug absorption. More importantly, we demonstrated that this inhibitory effect is related to GlcN’s ability to activate P-gp. GlcN can rapidly enhance P-gp’s efflux transport activity by directly binding to P-gp without altering its expression levels, indicating that GlcN can serve as a P-gp activator. Although further experiments are needed to gain broad recognition of GlcN as a P-gp activator, such as utilizing structural biology to verify the specific binding sites of GlcN on P-gp and to understand the conformational changes induced by GlcN on P-gp.
Notably, our research may also clarify the debated physiological effects of COS. While COS are widely acknowledged as bioadhesive absorption enhancers, controversy persists regarding their physiological effects79,80. Some studies argue that only chitobiose (COS-2) and chitotriose (COS-3) can be absorbed in the gastrointestinal tract81, while others suggest that higher degrees of polymerization (DP) in COS (DP > 6) exhibit superior anti-tumor effects82,83. The crux of these disputes lies in the challenges associated with obtaining large quantities of single-component COS due to the inherent difficulty in isolating and purifying them. Typically, studies on the physiological activity of COS employ mixtures of various polymerization degrees, leading to controversies regarding the absorption forms, transmembrane transport types, and characteristics of COS. This significantly hampers in-depth investigations into the physiological mechanisms of COS. Furthermore, during the production process, it is challenging to completely eliminate GlcN, an unavoidable byproduct in COS production. Commercially available COS often contains residual GlcN due to difficulties in purification. Additionally, COS is prone to hydrolysis, generating GlcN during prolonged storage80. Therefore, the contrasting impact of GlcN and COS on drug intestinal absorption, as observed in this study, may offer a potential explanationfor the divergent physiological activities of COS reported in various studies. While we speculate that GlcN and low-degree COS (DP < 5) may compress the thickness of the intestinal mucus layer to facilitate the penetration of drug molecules through the layer, those molecules absorbed by epithelial cells are extruded extracellularly due to the increased efflux transport activity of P-gp activated by GlcN. This results in a reduction in the transcellular transport of drugs. Conversely, high-degree COS (DP > 5), with their larger molecular structure and stronger intermolecular hydrogen bonding, are hindered from accessing the binding pocket of P-gp. These differences, stemming from varying degrees of polymerization, lead to discrepancies in the physiological effects between GlcN and high-degree COS. Additionally, in vitro experiments revealed that higher concentrations of GlcN can induce significant changes in solution osmolarity, which may affect cell hydration and drug absorption efficiency. According to the literature84,85,86,87, cells maintain optimal function when osmolarity is within the range of 280–320 mOsmol/kg, which typically results in ideal drug uptake. Osmolarity differences beyond this range can disrupt the cell’s osmotic balance, with a 20–40 mOsmol/kg change in osmotic pressure potentially impacting cell hydration and drug absorption. Therefore, the in vitro data supporting our findings on GlcN’s activation of P-gp activity were limited to concentrations below 20 mM (ΔOsmolality < 20 mOsmol/kg) (Supplementary Fig. 9d).
PQ is a herbicide primarily composed of N,N’-dimethyl-4,4’-bipyridinium. It commonly exists in commercial products in two forms: dichloride and dimethyl sulfate salt. PQ is highly toxic to humans, and there is no specific antidote available. Upon entering the human body, PQ rapidly induces multi-organ damage and failure, particularly causing rupture, bleeding, and edema in alveolar and bronchial epithelial cells, leading to progressive pulmonary fibrosis and respiratory failure. Clinical cases have shown that the mortality rate of paraquat poisoning patients is closely related to the concentration of paraquat in their blood88. Therefore, reducing the absorption into the bloodstream and tissue accumulation of PQ is of paramount significance. The use of GlcN has been shown to promote the efflux of PQ by P-gp, leading to a reduction of ~60% in its peak plasma concentration and bioavailability. Compared to activators like dexamethasone, GlcN exhibits higher biocompatibility, allowing for long-term and large-scale use. Hence, GlcN holds substantial potential for development as a detoxifying agent based on its P-gp efflux activity. This finding establishes a crucial theoretical foundation for understanding GlcN’s role in drug metabolism and absorption, opening avenues for further exploration of its therapeutic significance. In conclusion, this discovery establishes a crucial theoretical foundation for understanding GlcN’s role in drug metabolism and absorption, opening avenues for further exploration of its therapeutic significance.
Methods
Cell lines
Caco-2 were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Dingguo Changsheng biotechnology, China) containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37 °C supplied with 5% CO2. MCF-7 and MCF-7/ADR were gifts from our collaborators as indicated in the Acknowledgments.
Animals
Six to eight weeks old female SD rat and FVB mice were purchased from Chongqing ensiweier Co., Ltd., China. Abcb1a/Abcb1b-KO(FVB) mice (Cat. NO. NM-KO-191211) were purchased from Shanghai Model Organisms Center, Inc. All animals were bred in the Experimental Animal Center of School of Pharmacy, Southwest University. All procedures meet relevant ethical regulations for animal experiments and research, and have been approved by the Institutional Animal Care and Use Committee of Southwest University (IACUC-20241108-01, IACUC-20240727-01, IACUC-20241108-02).
In addition, our collaborators conducted pharmacokinetic studies of digoxin in abcb1-/- knockout mice (supervised by Animal Core Facility of Nanjing Medical University, SYXK-2023-0029), as noted in the Acknowledgments.
Pharmacokinetic studies
Animals were fasted for 12 h before drug administration, with free access to water. Post-administration, blood samples were collected from the retro-orbital venous plexus. After processing the plasma samples, drug concentrations in the blood were analyzed using HPLC or LC-MS. The detailed experimental protocol can be found in the Supplementary Information.
Intestinal transport rate assays
After being fasted for 12 h, virgin 6–8 week-old SD rats were sacrificed, and ~10 cm of the duodenum was collected. The segment was placed in Tyrode’s solution at 37°C, flushed with 95% oxygen, and gently rinsed to remove contents. The intestinal segment was everted, filled with Tyrode’s solution, tied at both ends, and immersed in Tyrode’s solution containing the drug. Samples were taken at 5, 10, 15, 30, 45, 60, and 90 min, and add an equal amount of blank Tyrode’s solution. After the experiment is completed, the intestinal segment is longitudinally cut open, the length and inner diameter are measured, the surface area of the intestinal segment is calculated, and the drug concentration of the sampled solution is detected by HPLC.
The calculation formula for the drug absorption rate is as follows: The cumulative drug absorption amount Q (μg) = (C1 + C2 + C3 + …+Cn − 1) × Vsampling + Cn × Vblank; The absorption rate K (μg·min⁻¹·cm⁻²) = dT × AdQ
Cn, represents the actual concentration at each sampling time point, in μg/mL; Vsampling, is the liquid volume during actual sampling, in mL; Vblank, is the volume of blank Tyrode’s solution added to the intestinal segment, in mL; T is the sampling time, in min; and A is the cross-sectional area of the small intestine, in cm².
Viscosity measurement and TEM observation
After fasting for 12 h, virgin 6–8 week-old SD rats were sacrificed, and a segment of the small intestine was gently flushed with 37 °C PBS to remove contents. The intestinal mucus was collected and purified by adding 0.1 M NaCl (5 mL per 1 g of mucus), then stirred at 4 °C for 1 h. Afterward, the mixture was centrifuged at 10 °C for 2 h, and the supernatant was discarded to obtain purified mucus. A 10 mL portion of the purified mucus was mixed with 10 mL of GlcN (10 mg/mL) and incubated at 37 °C for 30 min. The viscosity was measured using a rheometer (Discovery HR-1, TA Instruments). The rheometer was set to flow scanning mode, with a temperature of 25 °C, a 30 s equilibration time, and a shear rate range from 0.1 to 1000 1/s. The mucus sample was placed on the sample plate, and its viscosity was measured using a 40 mm parallel plate.
SD rats fasted for 12 h were orally administered GlcN (250 mg/kg) and sacrificed 20 min later. The intestines were collected for TEM observation (JEM-1400, JEOL) of the tight junctions between epithelial cells.
Cell viability and transcellular transport assays
1 × 104 cells/well were seeded into 96-well plates and treated with different concentrations of GlcN for 24 h or 48 h. The cell viability was subsequently determined by Cell Counting Kit-8 (GlpBio). Caco-2 cells (6 × 105 cells/well) were seeded in a transwell plate (Corning) for 21 days until the TEER was above 600 Ω·cm2.
For transcellular transport assays. AP → BL side: Remove the original culture medium, wash twice with HBSS, and then add fresh HBSS to equilibrate in the incubator for 30 min. Discard the original HBSS, add 0.5 mL of drug-containing HBSS (pre-warmed to 37 °C) to the apical compartment as the donor side, and add 1.5 mL of HBSS (pre-warmed to 37 °C) to the basolateral compartment as the receiver side. Incubate in a shaking incubator at 37 °C to complete the transport test. Sample 0.5 mL from the receiver side (basolateral compartment) at 15, 30, 45, 60, 90, 120, 180, and 240 min post-dosing for analysis, and replenish the basolateral compartment with 0.5 mL of fresh HBSS after each sampling. Add 2 volumes (i.e., 1 mL) of methanol to each sample to precipitate proteins, vortex for 30 s, and then centrifuge at 10,000 g for 5 min. Analyze the supernatant using HPLC to determine the drug concentration.
BL → AP side: Remove the original culture medium, wash twice with HBSS, and then add fresh HBSS to equilibrate in the incubator for 30 min. Discard the original HBSS, add 0.5 mL of blank HBSS (pre-warmed to 37 °C) to the basolateral compartment as the donor side, and add 1.5 mL of drug-containing HBSS (pre-warmed to 37 °C) to the apical compartment as the receiver side. Incubate in a shaking incubator at 37 °C to complete the transport test. Sample 0.25 mL from the receiver side (apical compartment) at 15, 30, 45, 60, 90, 120, 180, and 240 min post-dosing for analysis, and replenish the apical compartment with 0.25 mL of fresh HBSS after each sampling. Add 2 volumes (i.e., 0.5 mL) of methanol to each sample to precipitate proteins, vortex for 30 s, and then centrifuge at 10,000 g for 5 min. Analyze the supernatant using HPLC to determine the drug concentration.
Accumulation and efflux of Rh123
Caco-2 cells were seeded into 96-well plates (6 × 105 cells/well) and cultured overnight. For accumulation assays, cells were washed with HBSS for 2 times, add the drug-containing medium and incubate for 30 min. After incubation, discard the drug-containing medium, wash three times with ice-cold HBSS (without phenol red), and add 100 μL of ice-cold HBSS (without phenol red) to each well. Keep the 96-well plate on ice until measurement.
For efflux assays, cells were washed with HBSS for 2 times and then incubated with 4 μM Rh123 for 30 min to complete the accumulation of Rh123. Discard the Rh123-containing medium and wash three times with ice-cold HBSS to stop the transport of Rh123. Add fresh drug-containing medium and incubate for 45 min. The concentration of GlcN used was the optimal inhibitory concentration (10 mM) determined in the accumulation experiment, and the concentration of verapamil was 100 μM. Wash three times with ice-cold HBSS (without phenol red), and add 100 μL of ice-cold HBSS (without phenol red) to each well. Keep the 96-well plate on ice until measurement.
The intracellular fluorescence intensity of Rh123 in each well is detected using a multimode reader to represent the intracellular concentration of Rh123. For detection with the multimode reader, set the excitation wavelength (Ex) to 485 nm and the emission wavelength (Em) to 528 nm.
SiRNA
Caco-2 cells were seeded into 6-well plates (2 × 105 cells/well) and cultured with antibiotic free culture medium for 24. Dilute the siRNA products (siNC as the negative control and three siPgp sequences from Tsingke, China) with 250 μL of Opti-MEM medium. Similarly, dilute 5 μL of Lipofectamine™ 3000 transfection reagent (Thermo Fisher) with 250 μL of Opti-MEM medium. Mix gently and let sit at room temperature for 5 min. Then, combine the siRNA and transfection reagent, mix gently, and let sit at room temperature for 20 min. Add 500 μL of the transfection complex to each well of a 6-well plate and add 1.5 mL of culture medium, mixing thoroughly. Incubate for 6 h, then replace with fresh culture medium and continue incubation for 72 h before assessing protein expression levels.
Primers for siPgp: (forward: GGAUGAAAGUAUACCUCCATT, reverse: UGGAGGUAUACUUUCAUCCTT), (forward: CUAAUAGAAGUGAUAUCAATT, reverse: UUGAUAUCACUUCUAUUAGTT), (forward: CGCUACUGAAGCAAUAGAATT, reverse: UUCUAUUGCUUCAGUAGCGTT).
Immunoblot
Proteins obtained from cell lysates (containing protease inhibitor cocktail) were quantified using a BCA protein assay kit (Dingguo Changsheng biotechnology, China) and normalized for protein content. Protein samples were mixed with SDS-PAGE sample buffer and denatured at 37 °C for 45 min. After denaturation, the samples were cooled to room temperature, centrifuged at 10,000 g for 10 min at 4 °C, and the supernatant was collected. The protein samples were then separated by SDS-PAGE and transferred to a PVDF membrane. Immunoblotting was performed with the indicated antibodies, and visualization was carried out using the Tanon 5200 chemiluminescent imaging system. Grayscale values were quantified using ImageJ.
The following antibodies were used: P glycoprotein Polyclonal antibody (1:1000, Proteintech, 22336-1-AP); Anti-rabbit IgG, HRP-linked Antibody (1:2000, Cell Signaling Technology, #7074); Recombinant Anti-beta Actin antibody (Mouse mAb) (1:3000, Servicebio, GB15001-100); Anti-mouse IgG, HRP-linked Antibody (1:2000, Cell Signaling Technology, #7076). ExcelBand™ Enhanced 3-color Regular Range Protein Marker (9–180 kDa) (SMBIO, PM2510) was used for protein size marker.
CETSA assays
CETSA is based on the principle of protein thermal stability. Inside the cells, when a drug binds to the target protein, the protein’s structure becomes more stable, making it less prone to denaturation at elevated temperatures. As the temperature increases, unbound proteins gradually denature and precipitate, whereas drug-bound proteins remain more stable at the same temperature. By measuring the differences in protein degradation at different temperatures, we can infer whether the drug can bind to the protein. The Caco-2 cell suspension was lysed by repeated freeze-thaw cycles with liquid nitrogen after adding a protease inhibitor cocktail. The lysate was then centrifuged at 12,000 g for 20 min at 4 °C, and the supernatant was collected. The supernatant was divided into two equal portions: one portion was mixed with PBS containing GlcN (10 mM), and the other portion was mixed with an equal volume of PBS. Both mixtures were incubated at room temperature for 30 min. After incubation, each mixture was divided into eight aliquots, which were then individually heated for 3 min at a series of gradient temperatures (45.0, 46.8, 49.8, 54.4, 59.9, 64.8, 70.0 °C). Following heating, the samples were centrifuged at 15,000 g for 20 min at 4 °C, and the supernatant was collected. Protein content was then analyzed using western blotting. The quantification method used in the CESTA experiment is as follows: The Western blot image was opened using ImageJ software and converted to 8-bit (Image > Type > 8-bit). The background grayscale of the entire image was normalized to eliminate background interference (Process > Subtract Background), and the image colors were inverted (Edit > Invert). The measurement parameters were set (Analyze > Set Measurements), including Area, Mean Gray Value, and Integrated Density. The measurement units were set to “pixels” (Analyze > Set Scale, changing the Unit of length from inches to pixels). The bands to be measured were circled in the image, and the integrated optical density was measured. We have added the quantification approach in Methods section.
P-gp transport efficiency assays
A recombinant “inside-out” inverted vesicle model (GenoMembrane, Japan) was utilized to investigate the P-gp transport efficiency at a fixed P-gp expression level on the vesicle surface. These vesicles are derived from sf9 cells transfected with the human P-gp gene for expression. The vesicles were pre-incubated with 10 mM GlcN or 20 μM cyclosporin A (PC group) in reaction buffer equipped with the vesicle system for 5 min. Subsequently, the vesicles were incubated with the probe substrate N-methylquinidine for 30 min under conditions of either ATP or AMP addition. The assay was terminated using pre-chilled buffer B2, and the experimental samples were transferred to a 96-well filter plate, vacuum-filtered, and washed repeatedly five times with 0.2 mL pre-chilled buffer B2. The vesicles on the filter plate were lysed with 50 μL of 80% methanol, centrifuged at 600 g for 2 min, and the filtrate was collected. This lysing and centrifugation process was repeated. The collected filtrates were combined, mixed thoroughly, and diluted with 200 μL of methanol for quantification of N-methylquinidine content using LC-MS/MS. The Uptake Activity (pmol/min/mg) and Uptake Ratio were calculated using the following formulas:
Enzyme activity determination
For P-gp-mediated ATPase activity assays, Caco-2 cells were seeded into 96-well plates (6 × 105 cells/well) and treated with different concentrations of GlcN for 30 min. Cells were then collected using a cell scraper and prepared as a suspension of 107 cells/mL. The cell suspension was sonicated to break the cells, and the resulting sample solution was used to measure ATPase activity using an ultramicro ATPase assay kit (Nanjing jiancheng, China). The activity of P-gp-mediated ATPase is reflected by the difference in ATPase activity in the presence or absence of sodium orthovanadate.
For CYP3A4 enzyme activity assays, Caco-2 cells were seeded into 96-well plates (6 × 105 cells/well) and treated with different concentrations of GlcN for 30 min or 48 h. Additionally, Caco-2 cells treated with 1 μM ketoconazole or 25 μM rifampicin were used as controls. CYP3A4 enzyme activity in cells was measured using the P450-Glo™ CYP3A4 Assay with Luciferin-IPA (Promega, V9001), following the protocol provided in the instruction manual, fter treatment, cells were washed with PBS three times, then 50 μL of medium containing 3 μM Luciferin-IPA (diluted 1:1000) was added, and incubated for an additional 60 min in the incubator. Following the incubation, 25 μL of the medium was transferred to a white opaque well plate, and 25 μL of the Luciferin-IPA detection reagent was added. The plate was then incubated at room temperature for 20 min, and luminescence was measured using a multimode plate reader.
Molecular docking simulation
Docking simulations were performed using a homology-modeled mouse abcb1 structure (PDB code: 4Q9H, representing an inward-facing conformation) with GlcN or COS (3D structures downloaded from the PUBCHEM database). Water molecules, salts, and other small molecules were removed from the receptor protein using PyMol 2.5. The small molecules were energy-minimized using the MMFF94 force field. Molecular docking simulations and affinity scoring were carried out with AutoDock Vina 1.1.2 in global search mode, and the lowest-scoring docking conformation was considered the binding conformation. Binding modes were subsequently visualized and analyzed using PyMol 2.5.
SPR assays
SPR assays were conducted with assistance from our collaborators, as acknowledged in the Acknowledgments section. The specific procedures were as follows:
MCF-7 human breast cancer cells and MCF-7/ADR human breast cancer cells resistant to doxorubicin were cultured in DMEM medium containing 10% fetal bovine serum and 1% penicillin-streptomycin. The MCF-7/ADR cell culture medium contained 2 μM doxorubicin. Cells were harvested using a cell scraper, and cell membranes were extracted using an ultrasonic method. The supernatant was collected after centrifugation at 1000 g for 5 min, followed by centrifugation at 12000 g for 30 min at 4 °C to obtain the cell membrane precipitate. The wet weight of the cell membrane was determined, and an appropriate amount of PBS was added to resuspend the membrane, resulting in a concentration of 80 mg/mL. An equal volume of 5% styrene maleic acid (SMA) copolymer solution was added to obtain a final membrane concentration of 40 mg/mL and SMA concentration of 2.5% (w/v). After shaking at room temperature for 4 h, the mixture was centrifuged (12000 g, 30 min), and the supernatant was collected to obtain SMALP-MCF-7 and SMALP-MCF-7/ADR liposomes. The extraction of P-gp by SMA membrane liposomes was analyzed by western blot. Qualified SMALP liposomes were coupled to L1 chips, and the chips were washed with HBS-N buffer at a flow rate of 10 μL/min. Isopropanol: 50 mM NaOH (2:3) was used as the regeneration solution, with an injection time of 15 s and two washes. SMALP-MCF-7 (fc1) and SMALP-MCF-7/ADR (fc4) solutions were injected at a flow rate of 2 μL/min for 15 min per injection, and the final coupling amount was recorded. PBS buffer was prepared, and the test substance stock solution was serially diluted with buffer to obtain a concentration gradient. The Kinetics/Affinity program was used for injection, with a sample flow rate of 10 μL/min, injection time of 60 s, and dissociation time of 120 s. Data analysis was performed using Biacore T200 Evaluation Software, appropriate concentration ranges and binding curves were selected, reference channels and zero concentration were subtracted, and fitting analysis was performed using kinetic or steady-state models to obtain affinity values and parameters.
Survival period assays
SD rats (350 mg/kg) or wt FVB mice and abcb(-/-) mice (400 mg/kg) were orally administered paraquat to induce poisoning. After 2 h of poisoning, the rats were orally administered distilled water, dexamethasone, or GlcN, and the survival of the rats was recorded daily over a 15 day observation period.
Statistics and reproducibility
No statistical methods were used to pre-determine the sample sizes, the sample sizes were determined based on general principles of bio-sample analysis. No data were excluded from the analyses. The data are presented as mean ± SD, as indicated in each figure legend. Statistical analyses were conducted using GraphPad Prism software (version 8.0). For multiple group comparisons, one-way ANOVA followed by a multiple-comparison test was used, while a two-tailed t-test was employed for comparisons between two groups. P-values < 0.05 were considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data from these studies are contained within this paper. Source data are provided with this paper.
References
Juliano, R. L. & Ling, V. A surface glycoprotein modulating drug permeability in chinese hamster ovary cell mutants. Biochim. Biophys. Acta (BBA) Biomembranes 455, 152–162 (1976).
Ambudkar, S. V., Kimchi-Sarfaty, C., Sauna, Z. E. & Gottesman, M. M. P-glycoprotein: from genomics to mechanism. Oncogene 22, 7468–7485 (2003).
Katz, D. A., Murray, B., Bhathena, A. & Sahelijo, L. Defining drug disposition determinants: a pharmacogenetic-pharmacokinetic strategy. Nat. Rev. Drug Discov. 7, 293–305 (2008).
Aburto, M. R. & Cryan, J. F. Gastrointestinal and brain barriers: unlocking gates of communication across the microbiota-gut-brain axis. Nat. Rev. Gastroenterol. Hepatol. 21, 222–247 (2024).
Chaudhary, P. M. & Roninson, I. B. Expression and activity of P-glycoprotein, a multidrug efflux pump, in human hematopoietic stem cells. Cell 66, 85–94 (1991).
Stamatovic, S. M., Blasig, I. E., Keep, R. F. & Andjelkovic, A. V. Endocytosis of tight junction proteins: a pathway for barrier remodeling. In Tight Junctions (ed. Gonzalez-Mariscal, L.) 299-321 (Springer International Publishing, Cham, 2022).
Chelakkot, C., Ghim, J. & Ryu, S. H. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp. Mol. Med. 50, 1–9 (2018).
Doo, M.-H. et al. Effect of nonylphenol ethoxylates (NPEs) on barrier functions of epithelial cell membranes: Opening of tight junctions and competitive inhibition of P-gp-mediated efflux. Int. J. Pharm.302, 145–153 (2005).
Mirzaei, S. et al. Advances in understanding the role of P-gp in doxorubicin resistance: Molecular pathways, therapeutic strategies, and prospects. Drug Discov. Today 27, 436–455 (2022).
Das, T. et al. Therapeutic strategies to overcome taxane resistance in cancer. Drug Resist. Updates 55, 100754 (2021).
Engle, K. & Kumar, G. Cancer multidrug-resistance reversal by ABCB1 inhibition: a recent update. Eur. J. Med. Chem. 239, 114542 (2022).
Dong, J. et al. Medicinal chemistry strategies to discover P-glycoprotein inhibitors: an update. Drug Resist. Updates 49, 100681 (2020).
Yao, X. et al. Acid-degradable hydrogen-generating metal-organic framework for overcoming cancer resistance/metastasis and off-target side effects. Adv. Sci. 9, 2101965 (2022).
Patra, H. K. & Turner, A. P. F. The potential legacy of cancer nanotechnology: cellular selection. Trends Biotechnol. 32, 21–31 (2014).
Pearson, C. K. & Cunningham, C. Multidrug resistance during cancer chemotherapy. Trends Biotechnol. 11, 511–516 (1993).
Silva, R. et al. Modulation of P-glycoprotein efflux pump: Induction and activation as a therapeutic strategy. Pharmacol. Therap.149, 1–123 (2015).
Sharom, F. J. Multidrug resistance protein: P-glycoprotein. In Drug Transporters 223-262 (2007).
Fromm, M. F. Importance of P-glycoprotein at blood–tissue barriers. Trends Pharmacol. Sci. 25, 423–429 (2004).
Schinkel, A. H. The physiological function of drug-transporting P-glycoproteins. Semin. Cancer Biol. 8, 161–170 (1997).
Aday, S., Cecchelli, R., Hallier-Vanuxeem, D., Dehouck, M. P. & Ferreira, L. Stem cell-based human blood-brain barrier models for drug discovery and delivery. Trends Biotechnol. 34, 382–393 (2016).
Zhou, S. F. Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica 38, 802–832 (2008).
Hu, Z., Jin, S. & Scotto, K. W. Transcriptional activation of the MDR1 gene by UV irradiation: role of NF-Y and Sp1. J. Biol. Chem. 275, 2979–2985 (2000).
Elmeliegy, M., Vourvahis, M., Guo, C. & Wang, D. D. Effect of P-glycoprotein (P-gp) inducers on exposure of P-gp substrates: Review of clinical drug-drug interaction studies. Clin. Pharmacokinet. 59, 699–714 (2020).
Gant, T. W., Oconnor, C. K., Corbitt, R., Thorgeirsson, U. & Thorgeirsson, S. S. In vivo induction of liver P-glycoprotein expression by xenobiotics in monkeys. Toxicol. Appl. Pharmacol. 133, 269–276 (1995).
Zhao, J. Y., Ikeguchi, M., Eckersberg, T. & Kuo, M. T. Modulation of multidrug resistance gene expression by dexamethasone in cultured hepatoma cells. Endocrinology 133, 521–528 (1993).
Juan, H. et al. P-gp induction by curcumin: an effective antidotal pathway. J. Bioequivalence Bioavailability https://doi.org/10.4172/jbb.1000165 (2013).
Sterz, K., Möllmann, L., Jacobs, A., Baumert, D. & Wiese, M. Activators of P-glycoprotein: structure–activity relationships and investigation of their mode of action. ChemMedChem 4, 1897–1911 (2009).
Kondratov, R. V., Komarov, P. G., Becker, Y., Ewenson, A. & Gudkov, A. V. Small molecules that dramatically alter multidrug resistance phenotype by modulating the substrate specificity of P-glycoprotein. Proc. Natl Acad. Sci. USA 98, 14078–14083 (2001).
García-Rico, R. O. et al. Heterotrimeric G protein alpha subunit controls growth, stress response, extracellular protease activity, and cyclopiazonic acid production in Penicillium camemberti. Fungal Biol. 121, 754–762 (2017).
Qi, Y. et al. Chitooligosaccharide reconstitutes intestinal mucus layer to improve oral absorption of water-soluble drugs. J. Control. Release 360, 831–841 (2023).
Alam, M. A., Al-Jenoobi, F. I. & Al-mohizea, A. M. Everted gut sac model as a tool in pharmaceutical research: limitations and applications. J. Pharm. Pharmacol. 64, 326–336 (2012).
Wu, Z. et al. Oral mitochondrial transplantation using nanomotors to treat ischaemic heart disease. Nat. Nanotechnol. 19, 1375−1385 (2024).
Barthe, L., Bessouet, M., Woodley, J. F. & Houin, G. The improved everted gut sac: a simple method to study intestinal P-glycoprotein. Int. J. Pharm. 173, 255–258 (1998).
Chu, J. N. & Traverso, G. Foundations of gastrointestinal-based drug delivery and future developments. Nat. Rev. Gastroenterol. Hepatol. 19, 219–238 (2022).
Martel, J. et al. Gut barrier disruption and chronic disease. Trends Endocrinol. Metab. 33, 247–265 (2022).
Tajik, N. et al. Targeting zonulin and intestinal epithelial barrier function to prevent onset of arthritis. Nat. Commun. 11, 1995 (2020).
Tong, L. et al. Milk-derived extracellular vesicles protect intestinal barrier integrity in the gut-liver axis. Sci. Adv. 9, eade5041 (2023).
Schoultz, I. & Keita, Å.V. The intestinal barrier and current techniques for the assessment of gut permeability. Cells 9, 1909 (2020).
Cai, P. C. et al. Air–liquid intestinal cell culture allows in situ rheological characterization of intestinal mucus. APL Bioeng. 8, 026112 (2024).
Li, Y. et al. Establishment of an astrocyte-like cell line from the brain of tilapia (Oreochromis niloticus) for virus pathogenesis and a vitro model of the blood–brain barrier. J. Fish. Dis. 45, 1451–1462 (2022).
Song, P. et al. Estrogen receptor β inhibits breast cancer cells migration and invasion through CLDN6-mediated autophagy. J. Exp. Clin. Cancer Res. 38, 354 (2019).
Gleeson, J. P. et al. Development of physiologically responsive human iPSC-derived intestinal epithelium to study barrier dysfunction in IBD. Int. J. Mol. Sci. 21, 1438 (2020).
Barrand, M. A., Rhodes, T., Center, M. S. & Twentyman, P. R. Chemosensitisation and drug accumulation effects of cyclosporin A, PSC-833 and verapamil in human MDR large cell lung cancer cells expressing a 190k membrane protein distinct from P-glycoprotein. Eur. J. Cancer 29, 408–415 (1993).
Verschraagen, M., Koks, C. H. W., Schellens, J. H. M. & Beijnen, J. H. P-glycoprotein system as a determinant of drug interactions: the case of digoxin–verapamil. Pharmacol. Res. 40, 301–306 (1999).
Bansal, T., Mishra, G., Jaggi, M., Khar, R. K. & Talegaonkar, S. Effect of P-glycoprotein inhibitor, verapamil, on oral bioavailability and pharmacokinetics of irinotecan in rats. Eur. J. Pharm. Sci. 36, 580–590 (2009).
Doyle, L. A. et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 95, 15665–15670 (1998).
Zaman, G. J. et al. The human multidrug resistance-associated protein MRP is a plasma membrane drug-efflux pump. Proc. Natl. Acad. Sci. USA 91, 8822–8826 (1994).
Leith, C. P. et al. Frequency and clinical significance of the expression of the multidrug resistance proteins MDR1/P-glycoprotein, MRP1, and LRP in acute myeloid leukemia. A southwest oncology group study. Blood 94, 1086–1099 (1999).
Collett, A., Tanianis-Hughes, J. & Warhurst, G. Rapid induction of P-glycoprotein expression by high permeability compounds in colonic cells in vitro: a possible source of transporter mediated drug interactions?. Biochem. Pharmacol. 68, 783–790 (2004).
Silva, R. et al. Colchicine effect on P-glycoprotein expression and activity: in silico and in vitro studies. Chem. Biol. Interact. 218, 50–62 (2014).
Zhang, Y. et al. The inhibitory and combinative mechanism of HZ08 with P-glycoprotein expressed on the membrane of Caco-2 cell line. Toxicol. Appl. Pharmacol. 274, 232–239 (2014).
Klein, S. et al. Functionalized silicon quantum dots tailored for targeted siRNA delivery. Biochemm. Biophys. Res. Commun. 387, 164–168 (2009).
Watanabe, T., Onuki, R., Yamashita, S., Taira, K. & Sugiyama, Y. Construction of a functional transporter analysis system using MDR1 knockdown Caco-2 cells. Pharm. Res. 22, 1287–1293 (2005).
Tariq, I. et al. Downregulation of MDR 1 gene contributes to tyrosine kinase inhibitor induce apoptosis and reduction in tumor metastasis: a gravity to space investigation. Int. J. Pharm. 591, 119993 (2020).
Schinkel, A. H. et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc. Natl. Acad. Sci. USA 94, 4028–4033 (1997).
Schinkel, A. H., Wagenaar, E., Mol, C. A. & van Deemter, L. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J. Clin. Invest. 97, 2517–2524 (1996).
Schinkel, A. H., Wagenaar, E., van Deemter, L., Mol, C. A. & Borst, P. Absence of the mdr1a P-glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J. Clin. Investig. 96, 1698–1705 (1995).
Molina, D. M. et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87 (2013).
Wang, S. et al. Structure-based design, synthesis, and biological evaluation of new triazolo[1,5-a]pyrimidine derivatives as highly potent and orally active ABCB1 modulators. J. Med. Chem. 63, 15979–15996 (2020).
Wang, S. et al. Discovery of the triazolo[1,5-a]pyrimidine-based derivative WS-898 as a highly efficacious and orally bioavailable ABCB1 inhibitor capable of overcoming multidrug resistance. J. Med. Chem. 64, 16187–16204 (2021).
Jia, Y.-M. et al. Multidrug resistance–associated protein 3 is responsible for the efflux transport of curcumin glucuronide from hepatocytes to the blood. Drug Metab. Dispos. 48, 966 (2020).
Ji, J.-Z. et al. Mrp3 transports clopidogrel acyl glucuronide from the hepatocytes into blood. Drug Metab. Dispos. 46, 151 (2018).
Syed, S. K., Christopherson, R. I. & Roufogalis, B. D. Reversal of vinblastine transport by chlorpromazine in membrane vesicles from multidrug-resistant human CCRF-CEM leukaemia cells. Br. J. Cancer 78, 321–327 (1998).
Sugano, K. et al. Coexistence of passive and carrier-mediated processes in drug transport. Nat. Rev. Drug Discov. 9, 597–614 (2010).
Watkins, P. B. The barrier function of CYP3A4 and P-glycoprotein in the small bowel. Adv. Drug Deliv. Rev. 27, 161–170 (1997).
Silva, R. et al. Induction and activation of P-glycoprotein by dihydroxylated xanthones protect against the cytotoxicity of the P-glycoprotein substrate paraquat. Arch. Toxicol. 88, 937–951 (2014).
Nilles, J., Theile, D., Weiss, J., Haefeli, W. E. & Ruez, S. Lack of CYP3A4 protein induction despite mRNA induction in primary hepatocytes exposed to rifabutin as a possible explanation for its low interaction risk in vivo. Archives Toxicol. 98, 2541−255 (2024).
Wen, J., Xiao, Y., Zhao, M., Yang, C. & Hu, W. Effects of clarithromycin and ketoconazole on FK506 metabolism in different CYP3A4 genotype recombinant metabolic enzyme systems. Curr. Drug Metab. 25, 174–177 (2024).
Vu, L. et al. Effects of paroxetine, ketoconazole, and rifampin on the metabolism of eliglustat, an oral substrate reduction therapy for Gaucher disease type 1. Mol. Genet. Metab. Rep. 22, 100552 (2020).
Martins, E. A.-O. X. et al. Newly synthesized oxygenated xanthones as potential p-glycoprotein activators: in vitro, ex vivo, and in silico studies. Molecules 24, 707 (2019).
Cao, Y. et al. Surface plasmon resonance biosensor combined with lentiviral particle stabilization strategy for rapid and specific screening of P-Glycoprotein ligands. Anal. Bioanal. Chem. 413, 2021–2031 (2021).
Cao, Y. et al. Screening potential P-glycoprotein inhibitors by combination of a detergent-free membrane protein extraction with surface plasmon resonance biosensor. Acta Pharm. Sin. B 12, 3113–3123 (2022).
Majidi, M. Therapeutic strategies in managing acute paraquat poisoning: a review study: therapeutic strategies in paraquat poisoning. Int. J. Med. Toxicol. Forensic Med. 11, 33633 (2021).
Semeniuk, M. et al. Protective effect of genistein pre-treatment on paraquat hepatotoxicity in rats. Toxicol. Appl. Pharmacol. 426, 115636 (2021).
Dinis-Oliveira, R. J. et al. P-glycoprotein induction: an antidotal pathway for paraquat-induced lung toxicity. Free Radic. Biol. Med. 41, 1213–1224 (2006).
Jin, M. S., Oldham, M. L., Zhang, Q. & Chen, J. Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 490, 566–569 (2012).
Shapiro, A. B. & Ling, V. Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur. J. Biochem. 250, 130–137 (1997).
Culbertson, A. T. & Liao, M. Cryo-EM of human P-glycoprotein reveals an intermediate occluded conformation during active drug transport. Nat. Commun. 16, 3619 (2025).
Hu, L., Sun, Y. & Wu, Y. Advances in chitosan-based drug delivery vehicles. Nanoscale 5, 3103–3111 (2013).
Sangnim, T. et al. Chitosan in oral drug delivery formulations: A review. Pharmaceutics 15, 2361 (2023).
Chen, A.-S. et al. Pharmacokinetics of chitobiose and chitotriose administered intravenously or orally to rats. Biol. Pharm. Bull. 28, 545–548 (2005).
Zhai, X. et al. The impact of chitooligosaccharides and their derivatives on the in vitro and in vivo antitumor activity: A comprehensive review. Carbohydr. Polym. 266, 118132 (2021).
Huang, X., Jiao, Y. & Zhou, C. Impacts of chitosan oligosaccharide (COS) on angiogenic activities. Microvasc. Res. 134, 104114 (2021).
Stephen, R. L., Novak, J. M., Jensen, E. M., Kablitz, C. & Buys, S. S. Effect of osmotic pressure on uptake of chemotherapeutic agents by carcinoma cells. Cancer Res. 50, 4704–4708 (1990).
Halabian, M., Beigzadeh, B. & Siavashi, M. A numerical study on the effect of osmotic pressure on stress and strain in intercellular structures of tumor tissue in the poro-elastic model. Meccanica 56, 2471–2486 (2021).
Wennerström, H. & Oliveberg, M. On the osmotic pressure of cells. QRB Discov. 3, e12 (2022).
Zheng, S., Li, Y., Shao, Y., Li, L. & Song, F. Osmotic Pressure and Its Biological Implications. Int. J. Mol. Sci. 25, 3310 (2024).
Lin, X.-H. et al. Association between liberal oxygen therapy and mortality in patients with paraquat poisoning: a multi-center retrospective cohort study. PLoS ONE 16, e0245363 (2021).
Acknowledgements
We thank Professor Zhuo Wang (Institute of Process Engineering, Chinese Academy of Sciences) for providing COS samples, which were used in SPR experiments and pharmacokinetic studies evaluating the effects of COS with varying degrees of polymerization on capecitabine. We are also grateful to Professor Hongguang Xie (Nanjing Medical University) for conducting the pharmacokinetic study of digoxin in abcb1(-/-) knockout mice, and to Professor Yan Cao (Second Military Medical University) for assistance with SPR-related experiments. We further acknowledge Professor Lin Cheng (West China Hospital, Sichuan University) for support with molecular docking analysis, and express our special thanks to Dr. Honghong Zhan (College of Pharmaceutical Sciences, Southwest University) for his insightful advice and technical guidance on molecular docking studies. This research was financially supported by National Key Research and Development Program of China (2021YFD1800900), Chongqing Science and Technology Commission (CSTB2023NSCQ-JQX0002), Special Fund for Youth Team of Southwest University (SWU-XJLJ202306), Chongqing Natural Science Foundation (CSTB2024NSCQ-MSX0547), Science and Technology Innovation Key R&D Program of Chongqing (CSTB2024TIAD-STX0038).
Author information
Authors and Affiliations
Contributions
L.L., Q.W. and Y.Q. conceived this research. Q.W., X.L., Q.W., P.J., M.J. and J.M. designed and performed the experiments and was involved in all aspects of this study. L.L. and Q.W. drafted the manuscript. Q.W., M.J., S.H., H.S. and Y.Y. critically revised the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Laust Moesgaard, Gergely Szakács, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, Q., Wang, Q., Luo, X. et al. Glucosamine activates intestinal P-glycoprotein inhibiting drug absorption. Nat Commun 16, 6100 (2025). https://doi.org/10.1038/s41467-025-61437-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61437-2
