
Abstract
Glycans modify protein, lipid, and even RNA molecules to form the regulatory outer coat on cells called the glycocalyx. The changes in glycosylation have been linked to the initiation and progression of many diseases. Herein, we report a DIA-based glycomic workflow, termed GlycanDIA, to identify and quantify glycans with high sensitivity and precision. The GlycanDIA workflow combines higher energy collisional dissociation (HCD)-MS/MS and staggered windows for glycomic analysis, which facilitates the sensitivity in identification and precision in quantification compared to conventional glycomic methods. To facilitate its use, we also develop a generic search engine, GlycanDIA Finder, incorporating an iterative decoy searching for confident glycan identification from DIA data. Our results demonstrate that GlycanDIA can distinguish glycan composition and isomers from N-glycans, O-glycans, and human milk oligosaccharides (HMOs), while it also reveals information on low-abundant modified glycans. With the improved sensitivity and precision, we perform experiments to profile N-glycans from RNA samples, which have been underrepresented due to their low abundance. Using this integrative workflow to unravel the N-glycan profile in cellular and tissue glycoRNA samples, we find that RNA-glycans have different abundant forms as compared to protein-glycans and there are also tissue-specific differences, suggesting their distinct functions in biological processes.
Similar content being viewed by others


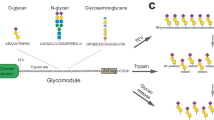
Introduction
Glycosylation is a major modification of proteins and lipids, and the glycosylated RNAs have also recently been found on the cell membrane1,2. Glycans are fundamental for cellular functional processes, including cell adhesion, cell signaling, immunological response, and cancer metastasis3. The extension of oligosaccharide chains involves a complicated competition between various glycosyltransferases, leading to the observed heterogeneity of glycan structures. Glycans are historically analyzed using nuclear magnetic resonance (NMR) or lectin-array4,5. However, the high sample demand or the lack of sequence information for each glycan is not ideal for comprehensive analysis. Therefore, developing new techniques to characterize the glycan chains is essential to understanding the roles of glycosylation in cell physiology.
Mass spectrometry (MS)-based methods emerged as a powerful tool and have significantly advanced glycomic analysis, providing information about glycan composition, structure, and abundance6. The biosynthetic pathway of glycans often yields glycan isomers, and tandem MS is required for their differentiation and determination. Data-dependent acquisition (DDA) has been the predominant approach for resolving glycan information, wherein only the most abundant precursor ions (top N) are selected for tandem MS/MS (MS2) analysis (Fig. 1A)7,8. This feature from DDA-based methods often leads to underrepresented and inconsistent detection of low-abundance molecules. This is particularly important in material-limited contexts with clinical samples or places where glycan levels are low, like RNA. For example, to investigate the glycan alterations during embryonic development, millions of sorted embryonic cells are collected, while limited glycan information is acquired using the DDA-based method9. To improve the detection, reducing end labeling and derivatization have been employed to enhance ionization efficiency in the analysis, while this requires extra processing steps, causing potential sample loss and generating issues in isomer separation10. To tackle these challenges, targeted analysis, such as multiple reaction monitoring (MRM), has been employed for glycomics, which relies on the product ions from different glycans and provides reliable quantitative results due to higher specificity and less variation of the fragment ions11. However, only specific glycoforms of interest can be monitored, which is not optimal for glycan discovery in novel samples such as glycosylated RNA. Data-independent acquisition (DIA)-based methods have been introduced, which do not rely on precursor ion selection12; instead, all precursors within a predefined mass window are fragmented simultaneously. Because DIA can generate an unbiased and comprehensive dataset, it has been more recently widely applied to the characterization of peptides, lipids, metabolites, and nucleosides13,14,15. The development of DIA has also been reported for intact glycoproteomic analysis16,17,18,19. However, to our knowledge, the application of DIA in analyzing released glycan has not been established, primarily due to two technical challenges: (1) the lack of optimized glycomics DIA instrumentation methods, and (2) the absence of bioinformatics tools for data interpretation.

A In the conventional data-dependent acquisition (DDA)-based method (left), a few most abundant precursor ions are respectively, selected within a small mass window (typically 0.7-2 m/z) and are further fragmented. DDA ideally only generates the tandem MS/MS (MS2) information for specific ions. During the data-independent acquisition (DIA) (right), pre-defined wide isolation windows are applied without regard to the abundance of precursor ions, and all precursor ions within the isolation window are fragmented in MS2. DIA often yields mixed MS2 information from different ions. B To cover all the N-glycans using GlycanDIA, the released N-glycans from different cells were mapped, and all their masses were within the range of 500–2000 m/z. Importantly, the precursor masses from 600 to 1800 m/z cover all glycan species. C The illustration of staggered windowing schemes in GlycanDIA. The first cycle consisted of one MS1 scan and different DIA scans across the mass range of selection. The second cycle consisted of another MS1 scan and different DIA scans that staggered the first-round window. Dashed lines indicate when an isolation window range is repeated. The legend for monosaccharide units and annotations can be found in Supplementary Table 4. Source data are provided as a Source Data file.
In this work, we establish a DIA-based glycomic workflow, termed GlycanDIA, utilizing higher energy collisional dissociation (HCD)-MS/MS and staggered DIA windows at the positive mode for glycomic analysis. The generated data can be analyzed manually using MS1-centric and MS2-centric strategies, while we further develop a search engine, GlycanDIA Finder, to perform automated data analysis with iterative decoy searching. This integrated workflow outperforms the conventional glycomic methods in both identification numbers and the precision of quantification. We have examples that show GlycanDIA enables generalizable glycomic analysis covering different types of oligosaccharides, including N-glycans, O-glycans, and human milk oligosaccharides (HMOs), while retaining the ability to discriminate different glycan isomers (including composition and linkage isomers) and empowering the information of low-abundant modified species. Furthermore, to illustrate the advantage of the GlycanDIA workflow, we apply this workflow to measuring N-glycans on RNA extracted from cultured human cell lines and mouse tissues. This application yields comprehensive and quantitative information about the N-glycan landscape of RNA extracts from different samples. The GlycanDIA workflow can be broadly applied for monitoring glycosylation status in different biological conditions, providing researchers with an additional avenue in glycobiology research.
Results
Establishment of GlycanDIA workflow
Porous graphitic carbon (PGC) can resolve native glycans with different degrees of polymerization and subtypes (such as fucosylation and sialylation) based on molecular size, hydrophobicity, and polar interactions20. Therefore, PGC has been demonstrated as one of the best chromatography methods for glycomic analysis. Importantly, glycans isomers and anomers have different structures, and they can be efficiently separated using PGC, which can largely reduce co-eluted species from the chromatography aspect. Electrospray ionization in positive mode reveals the comprehensive profile of glycans, allowing for the identification of different glycan subtypes, including sialylated glycans and sulfated glycans, although with reduced efficiency6. We therefore employed our analysis using the PGC column with positive MS mode.
To find the optimal fragmentation energy for the analysis, we first optimized the normalized collision energy (NCE) for HCD fragmentation. Examples shown in Supplementary Figs. 1–3, the signals corresponding to the sequence-defining fragments of the represented high mannose, fucosylated, and sialylated glycans first increased with rising NCE due to more efficient fragmentation of the precursor ions, while the larger fragment ions decreased when the collision energy was set to be greater than 25% due to over-fragmentation. As a result, 20% NCE was selected as the optimal energy for generating the best sequence information with high efficiency, while retaining more information about specific large fragments. We did not employ stepped HCD as it leads to ion signal dilution through more fragmentation pathways and longer dynamic cycle time. In addition, although cross-ring fragments can be generated from glycans, the majority of fragment ions (> 99.5%) were generated from glycosidic bond cleavages (Supplementary Fig. 4). This result is in agreement with previous observations21.
DIA-based methods fragment all precursor ions from an isolation window, generating highly multiplexed fragment ion spectra. It was therefore critical to optimize the mass window scheme for the glycan analysis. We mapped the previously profiled N-glycans from eight cell lines that employed DDA-MS to acquire the general N-glycan distributions22,23. As shown in Fig. 1B, we found that the N-glycan precursors allocate to a wide mass-to-charge range (500–2000 m/z). Interestingly, the precursor range of 600–1800 m/z covers all major glycan species, while an extended mass range has been shown to improve identification in EThcD-based glycoproteomic analyses24. After selecting the precursor window, we evaluated different strategies to set up the mass window scheme, including fixed DIA, staggered DIA, multiplexed (MSX) DIA, and variable DIA. We balanced the size of the isolation window and the cycle of scans (the total counts of isolation windows) in different DIA strategies. As shown in Supplementary Figs. 5–8, staggered and variable DIA showed better results in generating fewer interfering ions. It was also noted that the loop counts (window numbers) for GlycanDIA are larger than that of the common DIA for proteomics. This is due to the full width at half maximum (FWHM) of the peaks for most glycans eluting from the PGC column being more than 0.3 minutes (Supplementary Fig. 9A). As a result, most glycan compounds had sufficient data points (~10) for constructing Gaussian peaks (Supplementary Fig. 9B) using the 24 m/z staggered method, which yielded higher precision for quantification. The representative scan scheme of staggered windowing and detailed parameters are shown in Fig. 1C and Supplementary Table 1, respectively. Meanwhile, although the variable windowing showed promising results in quantification, the method was cumbersome to set up and adjust using the manufacturer’s software; therefore, we used 24 m/z staggered with 50 windows for further analysis. Furthermore, we examined the frequency of N-glycan precursors (from a general library) across a 12 m/z interval spanning 600–1800 m/z, independent of LC separation, to evaluate the selected MS window. As shown in Supplementary Fig. 10, the 936–948 m/z region exhibited the highest precursor density (21 glycans). Analysis of theoretical fragments revealed that, aside from small universal fragments (e.g., 163 and 204 m/z), fragments larger than 500 m/z were shared by limited glycans (less than 5), and each glycan had more than 2 featured fragment ions. These results confirm that a 24 m/z staggered DIA window is optimal for GlycanDIA.
In DDA-MS, only one precursor is theoretically picked and fragmented per MS/MS scan, whereas DIA contains complicated MS2 information due to the collection of fragmentation from all coeluting compounds within a specified mass window. To decipher the glycan information from the DIA data, we used the MS1-centric method in the GlycanDIA workflow (Fig. 2A). To illustrate this strategy, we took the N-glycan Hex(4)HexNAc(4)Fuc(0)Neu5Ac(1) (4401 for short) as an example, and the possible precursor ion masses were calculated and specifically extracted from the MS1 level. After locating the peak, the product ion yielded from this glycan (e.g., 1258.45 m/z) was extracted from two MS2 spectra obtained from the window containing the precursor ion (876–900 m/z and 864–888 m/z) to confirm the fragmentation and one spectrum from a nearby window (888–912 m/z) as a negative control. As a result, only the 876–900 m/z and 864–888 m/z windows showed extracted product ions coeluting with the precursor ion, and multiple product ions were extracted simultaneously to validate the sequence of the glycan. In addition, we also employed a complementary MS2-centric strategy to find the glycans via glycan product ions starting from the MS2 spectrum. For example, 292.10 m/z corresponding to Neu5Ac was first extracted from the 864–888 m/z window. After examining the same ion from the two adjacent MS2 spectra, 876–900 m/z and 852–876 m/z, we found that only the MS2 from 876–900 m/z contained the 292.10 m/z fragment ion and coeluted with the 864–888 m/z window. This result means the precursor of this product ion fell into the overlap of the two windows, 876–888 m/z. After extracting the MS1 spectrum from 876 to 888 m/z, the doubly charged monoisotopic ion at 885.8257 m/z was noted, and the glycan composition, 4401, can be determined and validated by revisiting the MS2 information. Collectively, the two peaks corresponding to Hex(4)HexNAc(4)Fuc(0)Neu5Ac(1) anomers were identified (Fig. 2B), and the two strategies enable the determination of the glycan information from different directions, which provides complementary and synchronized information.

A To analyze GlycanDIA data, the MS1-centric method needs firstly to calculate the precursor ion mass and monitor it from the MS1 level (885.82 m/z, z = 2, in this example). Subsequently, the product ion yielded from this glycan (e.g., 1258.45 m/z) was also monitored from the MS2 spectra that isolated the precursor ion. A series of product ions were extracted simultaneously to validate the glycan sequence. The MS2-centric strategy in the GlycanDIA data analysis strategy relied on the glycan signature ions from an MS2 spectrum. B The peak alignment between glycan precursor isotopic masses at the MS1 level (left) and a series of glycan product ions at the MS2 level (right). The blue, purple, and red colors correspond to 885.82, 886.33, and 886.83 m/z, respectively. The product ions are color-coded to facilitate visualization. C More than 270 different N-glycans were identified using GlycanDIA, while the conventional DDA-based method only identified 200 glycans. Blue color indicates DDA, and yellow indicates GlycanDIA. The error bar showed the standard deviation of identified glycans from three technical replicate (n = 3) of the N-glycans mixture released from RNase B and human sera using two methods. Data are presented as mean ± SD. D The glycan quantification in MS1 and MS2 levels was evaluated. E GlycanDIA covered all the glycans identified from DDA runs. F Three heavy isotope-labeled N-glycan standards were mixed with the N-glycan pool released from RNase B and human sera. The GlycanDIA provides more consistent glycan identification compared to the DDA-based method. G The GlycanDIA also generates less variations in quantitative results. Boxes show 25th–75th percentiles, center lines represent medians, whiskers indicate min–max range from ten technical replicates (n = 10) of standard heavy isotope-labeled N-glycans using two methods. H Extracted compound chromatogram (ECC) shows the elution profile of 15 HMO standards. The full name of HMO standards can be found in Supplementary Table 2. I The example GlycanDIA results of two HMO isomers, 3FL and 2’FL. The legend for monosaccharide units and annotations can be found in Supplementary Table 4. Source data are provided as a Source Data file.
Development of GlycanDIA Finder as a computational software
To meet the need for computational tools that streamline data interpretation and extend the application of the workflow, we developed a generic search engine, GlycanDIA Finder, upon Python and the matchms library25. This in-house developed software is based on the MS1-centric method, which allows for automated and high-throughput compositional analysis on DIA-MS-based glycomics spectra. As illustrated in Supplementary Fig. 11A, pre-filtering of spectra information is first performed using set parameters, including the mass tolerance, intensity threshold, and maximum charge range. Next, the software uses an input glycan library containing glycan composition and their identified fragments to identify the glycans from both MS1 and MS2 levels. We incorporated Glycan Product Generator, which calculates possible glycan fragmentations based on the input structure and outputs the glycans in a format that can be directly imported into GlycanDIA Finder search and further Skyline evaluation. Finally, the apex of glycan peaks can be assigned by realigning the information from MS1 and MS2, and the aligned tandem-MS spectra are saved in mgf format for further annotation. Moreover, the add-on mass feature allows analysis of modified glycans in samples, such as labeled, derivatized, and reduced glycans.
The models for removing false-positive results from glycomics data are hampered by the complicated non-linear glycan structures. Hong and co-workers introduced a powerful de novo glycan structure identification tool, GlycoDeNovo2, that identifies glycan compositions and branching structures using a p-value scoring system26. More recently, Liu et al. also developed GlycoNote, which assigns putative glycan compositions based on DDA data and employs the random mass shift algorithm to filter false positives27. Therefore, we applied random mass shift as it has been further demonstrated in DIA glycoproteomics using GproDIA and has helped to successfully improve glycan identification confidence16. We optimized parameters adopted from GlycoNote and embedded them in a decoy mode of the software. Two parameters, coverage of intensity (Cov. Int.) and coverage of sequence (Cov. Seq.), were calculated to represent the relative abundance of matched fragments and fragmentation efficiency. For each aligned tandem MS, 100 decoy sets were generated, and we estimated the average distribution of Cov. Int. and Cov. Seq. is 0.1% and 7%, respectively, by iteratively searching against the decoy database under a 2% false discovery rate (FDR) as an optimized solution in both reproducibility and search time (Supplementary Fig. 11B and Supplementary Fig. 12). We further integrated glycan molecule feature detecting glycan isotope peaks and monitoring possible misidentification. As shown in Supplementary Data 6, we demonstrated high confidence in the software’s identification performance after manual cross-verification of glycans identified by the software.
GlycanDIA empowers sensitivity and precision for glycomic analysis
To illustrate the performance of the GlycanDIA workflow in characterizing glycans, we have employed the analysis to characterize N-glycans and HMOs in their protonated native forms and O-glycans in their reduced forms. First, we characterized the N-glycans released from a mixture of well-characterized glycoprotein sources (RNase B and human sera) as the standard20,28. As a result, the GlycanDIA approach outperformed DDA in terms of both identification power and quantification precision. Specifically, DIA consistently mapped over 270 different glycans (including isomers) from three replicates, while DDA only identified around 200 glycans (Fig. 2C) and all the glycans annotated from DDA analyses were able to be identified using GlycanDIA (Fig. 2E). As the examples shown in Supplementary Fig. 13, tandem MS/MS information was generated across the two Hex(3)HexNAc(5) and four Hex(5)HexNAc(4)Neu5Ac(1) peaks, while only one and three MS2 events were triggered in DDA, respectively, causing lower identification numbers using DDA.
Next, we examined the quantitative performance of different glycans from the released standards, and we found significant improvements in the quantification precision in terms of linearity (R2 value) and coefficients of variation (CVs). Specially, decent linearities were achieved after a 1000-fold dilution for most glycans using GlycanDIA (Supplementary Fig. 14 and Supplementary Data 7), and the CVs of the technical replicates were less than 5% on average when using the more selective fragment ion measurements compared to the precursor signals typically employed in DDA quantification (Fig. 2D). Notably, we evaluated the quantitative performance of different fragment ions from different glycans and observed that larger ions exhibited better linearity, which is consistent with the theoretical predictions, as larger fragments generate lower matrix interferences in GlycanDIA analysis (Supplementary Data 8). We also compared the GlycanDIA to the previously established nanoLC-Time-of-Flight (TOF) system; as shown in Supplementary Fig. 15, the glycan peak was barely quantifiable after a 100-fold dilution of the standard using the TOF system, while the detection and quantification were both significantly improved with GlycanDIA (Supplementary Fig. 16).
To further demonstrate the outperformance of GlycanDIA for glycomic analysis, we spiked three standard heavy isotope-labeled N-glycans into the N-glycan mixture (released from RNase B and human sera). We then analyzed the sample with ten continuous runs using the DDA or GlycanDIA method. As a result, at least one isotope-labeled standard glycan was not identified from 60% of DDA runs (6 out of 10), that is, only 40% of DDA runs could detect all three glycan standards from the complex mixture; however, all ten GlycanDIA runs consistently revealed the fragmentation information for the identification (Fig. 2F). Notably, the GlycanDIA method also provided more consistent stable quantitative results, while the conventional DDA method displayed limitations in such low concentrations of glycans in terms of a high level of quantification variability (Fig. 2G). These results demonstrate the congenital limitation of DDA in abundance-dependent identification and MS1-based quantification, while the features of DIA ignore the intensity of analytes and enable MS2-based quantification. Overall, the GlycanDIA workflow can increase the number of glycan identifications by 25% over the conventional DDA method while generating more sensitive and consistent quantitative results.
GlycanDIA is generalizable for characterizing different glycans
Besides N-glycans, analysis of other types of glycans, particularly HMOs and O-glycans, is also critical for investigating different biological processes6. Therefore, we examined the generalizability of GlycanDIA for characterizing HMOs and O-glycans. To adapt the method, we merely changed the mass window range to 300–1600 m/z because HMOs and O-glycans are relatively smaller compared to N-glycans. As shown in Fig. 2H, 15 different HMO standards can all be monitored at the femtomole level. We then monitored the HMOs from the human milk sample; the signal of 163.06 m/z and 204.08 m/z at the MS2 level shows the general level of hexose and N-acetylhexosamine-containing HMOs (Supplementary Fig. 17A). As a result, more than 70 HMOs can be confidently quantified (Supplementary Fig. 17B). We also investigated the GlycanDIA on O-glycans released from bovine mucin. As shown in Supplementary Fig. 18A and B, more than 35 O-glycans were identified, and the tandem MS signal of reduced N-acetylhexosamine (224.11 m/z) aligned the O-glycan profiles, which confirmed O-glycans were generated from O-GalNAc-linked mucins. Surprisingly, the sulfated O-glycans normally have a lower response in the positive mode compared to the negative mode29; however, several sulfated O-glycans can still be identified from GlycanDIA, which suggests the sensitivity of the method. Overall, the results showed the exceptional sensitivity and precision of GlycanDIA for glycomic analysis.
Compositional and structural glycan isomers can be determined using GlycanDIA
Glycan isomers with different composition and structure are important for their biological functions, and tandem MS information is necessary as a layer of evidence for obtaining specific isomer information and differentiating glycan isomers. First, the combination of different monosaccharides often generates the same masses for some glycans, known as the glycan composition isomer. For example, the mass of one fucose adding one N-glycolylneuraminic acid (Neu5Gc) equals one hexose adding one Neu5Ac (453.14 Da). To evaluate the performance of GlycanDIA for these composition isomers, we selected the fetal bovine serum sample containing both Neu5Ac and Neu5Gc and released the N-glycans. As shown in Supplementary Fig. 19A, six peaks corresponding to three major glycan groups were found after extracting the 1120.90 m/z ion at the MS1 level. Three different N-glycan compositions, Hex(6)HexNAc(4)Fuc(1)Neu5Ac(1) (6411), Hex(5)HexNAc(4)Fuc(0)Neu5Ac(1)Neu5Gc(1) (54011), and Hex(5)HexNAc(4)Fuc(2)Neu5Ac(0)Neu5Gc(1) (54201) can have a same mass at 1120.90 m/z. The monoisotopic masses of 6411 and 54201 are exactly the same, but 54011 differs by around 0.5 at charge state 2. After looking into the MS1 spectra at different times, the monoisotopic peaks 1120.91, 1120.91, and 1290.39 m/z were identified at 29, 35, and 39 minutes, respectively. The MS2 spectrum at 39 minutes showed the diagnostic peaks of both Neu5Ac and Neu5Gc, suggesting the glycan with 54011 compositions (Supplementary Fig. 19B). On the other hand, the peaks around 29 and 35 minutes contained Neu5Ac fragments, while they did not contain any Neu5Gc ions, indicating the peaks belong to the glycan 6411. Furthermore, the two 6411 isomer structures can be proposed based on their fragments and the ratio between the fragmented antenna with and without (e.g., 512.20 and 657.24 m/z, respectively) informed the peak at 29 minutes is the 6411 glycan with antenna fucose and the other peak (at 35 min) is more likely to be the glycan with core fucose structure instead.
In addition, although HCD with positive MS mode is perceived to only reveal limited linkage information, studies have demonstrated the distinguishment of structural isomers under this condition30. Therefore, to evaluate the performance of GlycanDIA for distinguishing glycan structural isomers, we comprehensively investigated whether different glycans, including HMO, O-glycans, N-glycans, and sialic acid linkage, can be differentiated using the DIA MS2 information. We first checked the two common isomeric HMOs, 2’-fucosyllactose (2’FL) and 3-fucosyllactose (3FL), from the HMO standard pool. As shown in Fig. 2I, they can be distinguished based on the relative abundance of lactose fragments (325.11 and 343.12 m/z). Indeed, 325.11 m/z was used to monitor 3FL during the targeted analysis31. For O-glycans, Xu et al. previously annotated O-glycan Hex(2)HexNAc(2)Fuc(1)Neu5Ac(1) isomers based on their signature ions generated from the different positions of fucose and sialic acid32. We examined whether this information could be resolved in GlycanDIA and found a series of aligned fragments that determined its structure, especially the signature ion 657.23 m/z (Supplementary Fig. 18C, D), which confirmed the performance of GlycanDIA for determining O-glycan structural isomers. Furthermore, we investigated a more complicated case, in which different Man8 glycans from the RNase B protein was characterized and analyzed20. As shown in Supplementary Fig. 20, GlycanDIA provides characteristic fragment information that aids in differentiating the six Man8 isomers and anomers. Importantly, different Man8 glycans can be quantified using their product ions with high linearity. This indicates that our method can provide reliable identification and quantification for glycan isomers. Recently, Pett et al. showed the assignment of α2,3-/α2,6-linked sialic acid from glycoproteomic analysis33. To investigate if GlycanDIA data can disclose the α2,3- and α2,6-sialylated glycan isomers, we examined the N-glycan isomers, Hex(5)HexNAc(4)Neu5Ac(1) (5401), that have been well-characterized using PGC (Supplementary Fig. 21A)34. Remarkably, by normalizing the intensity of fragments to the general 366.14 m/z fragment, the relative abundance of several fragment ions displayed different preferences in our dataset. Similar to the observation from glycoproteomic results, α2,3-linked glycans generated more abundant product ions at 292.10 and 274.09 m/z compared to α2,6-sialylated glycans (Supplementary Fig. 21B). Besides, when sialic acid is located at the 3’ arm, it produces generic higher antenna ions (such as 1055.37, 893.32, 749.28, and 690.24 m/z) compared to 6’ arm-sialic acid. A more general inspection of different glycan isomers is required for further validation, while our results suggest that GlycanDIA can generate much useful information for determining different glycan compositional and structural isomers.
The preceding examples relies on isomer resolution in chromatography, which may not always be possible. To evaluate scenarios where isomers are not fully separated, we analyzed two Hex(5)HexNAc(5)Fuc(1)Neu5Ac(1) isomers, as shown in Supplementary Fig. 22, DIA acquired MS2 spectra across both apex regions, while PGC chromatography partially resolved these isomers. These spectra contained fragment ions for the differentiation (e.g., 1607.59 m/z). In contrast, traditional DDA failed to resolve the isomers, as only one MS2 scan was triggered between the peaks. This highlights an advantage of GlycanDIA as its systematic MS2 acquisition provides continuous spectral coverage. Meanwhile, incorporating ion mobility as an additional dimension of separation is expected to reduce co-elution and co-isolation, thereby enhancing the accuracy and reliability of glycan identification and quantification in future work.
Identification of modified glycans using GlycanDIA
Similar to proteins, glycans can be modified, for example, by acetylation, sulfation, and phosphorylation; however, due to the low abundance of these modifications, a specific enrichment method is normally required for their mapping35. Data generated from the GlycanDIA workflow theoretically contains the PTM information because the DIA-based method can fragment all the ions within a selection window. To investigate if the modified glycans can be directly identified from the unenriched samples using GlycanDIA, we examined the MS2 ions at 699.24 m/z, which corresponds to the signature ions produced from the acetylated sialic acid. As shown in Supplementary Fig. 23, various fragment peaks of acetylated sialylated glycans were identified from the data, and one of the spectra corresponding to Hex(5)HexNAc(4)Neu5Ac(1) glycan was shown as an example. Notably, the level of sialic acid acetylation is three orders of magnitude less than the unmodified sialic acid based on the ratio between their signature ions, and such a low-abundant modification can barely be identified using the traditional DDA-based method. These results emphasize the significant advantage of GlycanDIA and the potential to acquire missing information when the dataset is re-interpreted later.
Revealing the landscape of N-glycans from RNA extracts
We recently found the 3-(3-amino-3-carboxypropyl)uridine (acp3U) as a site of attachment of N-glycans in glycoRNA and the function of glycoRNA in the cell-penetrating peptide entry36. However, the profile of RNA N-glycan information is still limited to the low level of glycosylation on RNAs (approximately 20 pmol per μg of total RNA)2. The GlycanDIA workflow has the substantial advantage of accurately identifying and quantifying low-abundance glycans. Therefore, we applied GlycanDIA to characterize the N-glycans released from RNA extracts (Supplementary Fig. 24). To ensure the purity of the RNA, we treated the crude RNAs from TRIzol extraction with DNase, mucinase, and protease to ensure the quality of glycoRNA samples. This is important to rule out cross-contamination from potential glycoproteins, and no proteins were notable from the SDS-PAGE gel with 5 μg loaded RNA using Coomassie staining (Supplementary Fig. 25A). We further performed proteomic analysis, and only detected common contaminant peptides corresponding proteins, such as keratins, and no glycoproteins or RNA-binding proteins (RBPs) were identified, which suggests the high specificity of our glycoRNA preparation (Supplementary Data 9).
We then monitored the glycans released from either RNAs or glycoproteins in HeLa and HEK 293 T cells. After fractionating the total RNA into large and small RNAs based on size (Supplementary Fig. 25B), the signature glycan signals were observed from the small RNA fraction, while no significant glycans were identified from the large RNA fraction (Supplementary Fig. 26). We annotated the glycan profile of glycoRNA using glycan references obtained in previous studies, aligning retention times on PGC chromatography and matching fragmentation patterns, while the total abundance of N-glycans can be represented using the EIC of 204.08 m/z from MS/MS. As a result, the N-glycans released from the HEK 293 T and HeLa RNA extracts showed an overall different profile compared to those from the glycoproteins (Fig. 3A and Supplementary Figs. 27 and 28A). Although both RNA and protein samples were dominated by fucosylated glycans, their relative abundance is different. Meanwhile, the RNA sample contained more sialofucosylated (containing both sialic acid and fucose) glycans, and the protein had more high-mannose N-glycans. Similarly, the N-glycan profile of RNA from HeLa cells showed more abundant sialofucosylated glycans. Over 100 glycans were identified and quantified from protein and RNA extracts, and the overlap of the identified glycans between the two populations was more than 70% (76 out of 103) (Fig. 3B). The identified glycans from the glycoproteins were similar to the results from previous studies2, however the GlycanDIA method used only 5 μg of small RNA (5-fold less than used in the initial study) and resulted in deeper and more robust identification of the N-glycans. Remarkably, the most abundant glycans from RNA and protein samples varied greatly (Fig. 3C). A potential explanation is that glycosyltransferases and glycosidases exhibit distinct substrate preferences between proteins and RNAs. As the biosynthetic pathway of glycoRNA remains to be explored, we plan to establish a dedicated glycan reference library for glycoRNA in future work. This resource will enable precise structural analysis of RNA glycosylation.

A The chromatography of N-glycans from HEK 293 T RNA (top) and protein (bottom) samples showed different glycomic profiles. B More than 70% of N-glycans were identified from both RNA and protein. C Examples of the most abundant N-glycans and their relative abundance found from RNA and protein. D Top 16 RNA N-glycans identified from HCT116 cell lines. The fucosylated glycans were not identified due to the GMD protein mutation. E The top glycan subtypes were identified in HCT116 cell lines before and after treatment. Fucosylated glycans were specifically detected following supplementation with exogenous fucose. F N-glycans, such as the Hex(7)HexNAc(6)Neu5Ac(1), were converted into glycans containing fucose after the external fucose feeding. The legend for monosaccharide units and annotations can be found in Supplementary Table 4.
We then investigated the glycans on RNAs in the human colorectal carcinoma cell line, HCT116. This cell line is a glycosylation model that lacks fucosylation due to the mutation of the GDP-mannose-4,6-dehydratase (GMD) enzyme, which blocks the fucose de novo synthesis37. The fucosylation of the cell line’s glycoproteins can be rescued by adding external fucose through the Salvage pathway. Currently, this pathway has not been investigated for the fucosylation of the RNAs. As shown in Fig. 3D and Supplementary Fig. 28B, we detected zero fucosylated glycans from the HCT116 RNA extracts, and the most abundant glycan was a sialylated glycan, Hex(7)HexNAc(6)Fuc(0)Neu5Ac(1) (7601). After treating the cells with 100 mM of fucose, the glycan profile was reprogrammed towards fucosylated and sialofucosylated glycan subtypes (Fig. 3E). For example, over 90% of the sialylated glycan 7601 was converted to the sialofucosylated glycan, Hex(7)HexNAc(6)Fuc(1)Neu5Ac(1) (7611) (Fig. 3F). This result reveals that the N-glycans on HCT116 RNAs can also be modified through the canonical biosynthesis enzymes in carbohydrate metabolism.
Lastly, we applied GlycanDIA to unravel the profile of glycans on RNA from different mouse tissues. As a result, more than 200 N-glycans in total were identified from three replicates of five different tissues (Fig. 4A), and the landscape of glycans from tissue glycoRNAs differed from the profiles of glycoprotein samples38,39. In addition, the relative abundance of different N-glycan subtypes was distinct (Fig. 4B). For example, heart tissue exhibited relatively more abundant high mannose type glycans (30%), while fucosylated glycans predominated, accounting for over 75% of total glycans identified from the brain. Most tissues showed higher ratios of sialic acid-containing glycans, consistent with the total degree of sialylation we assessed using the RNA periodate oxidation and aldehyde labeling (rPAL) method (Supplementary Fig. 29)36. Furthermore, there was variation in the most prevalent glycoforms among different tissues. For example, glycans comprising tetraantennary Hex(7)HexNAc(6) and Hex(7)HexNAc(7) structures were highly abundant in the colon but rarely detected in other samples. Strikingly, although sialylated structures accounted for more than 50% of glycans in most tissues, the sialylation patterns (Neu5Ac vs. Neu5Gc) varied across tissues (Fig. 4C). For example, the colon and the heart had relatively higher Neu5Ac, while the spleen exhibited a higher degree of Neu5Gc. These results were also confirmed with the base peak chromatogram (BPC) of Neu5Ac and Neu5Gc signature ions (Supplementary Fig. 30). The observed heterogeneity in glycan profiles across tissues likely reflects glycoRNA functional specialization. For example, the tissue-specific sialylation patterns might fine-tune immune responses, as sialylated glycoRNA has been found to interact with Siglec receptors2. Moreover, the colon-specific enrichment of glycan features suggests a potential role in mediating host-microbiota interactions, owing to the microbiota-enriched environment of the colon. Overall, the results demonstrate the abundance and tissue-specific distribution of N-glycans in glycoRNA samples, which represent a critical resource for future glycoRNA studies investigating glycan-mediated mechanisms in health and disease.

A Various N-glycans in total were identified from different mouse tissues, and the heatmap showed distinct N-glycan profiles from the different mouse tissues. B The differences in the relative abundance of N-glycan subtypes were also noticed in the mouse tissues. C The high degree of fucosylation was generally noted in all different tissue RNAs, while the degree of sialylation and the level of Neu5Ac and Neu5Gc were tissue-specific. The error bar is the standard deviation obtained from biological replicates of corresponding samples (n = 3, biological replicates of different mouse samples). Three biological replicates from each mouse tissue were analyzed for this study. Data are presented as mean ± SD. The legend for monosaccharide units and annotations can be found in Supplementary Table 4. Source data are provided as a Source Data file. B partially created in BioRender52.
Discussion
DDA-based methods have been prevalent in current glycomic studies. However, the nature of selecting the most abundant ions in DDA often impedes the investigation of subtle glycosylation changes in complex biological samples, especially when the amount of material for analysis is limited. As our showcase in analyzing standard heavy isotope-labeled N-glycans, the detection and quantification from DDA are inconsistent at the attomolar level, while the robust identification and quantitative results from GlycanDIA demonstrate its capability to capture glycan information with lower sample demands. It is noted that the strategy of DIA can always get the best fragmentation at (or near) the apex of the compound peak, while DDA can hardly be promised to trigger at the apex, even with the system optimization or apex-trigger algorithm40. This means the GlycanDIA can generally produce glycan fragmentation with good quality and higher sensitivity. For example, in our analysis of glycans on HEK 293 T RNA, we identified nearly 100 glycans using only 5 μg of small RNAs, while 25 μg of HEK 293 T small RNAs were used in the previous study, finding only 14 N-glycans. This type of increased sensitivity and depth will enable others to more easily study how cells glycosylate various template biopolymers across a wider array of biological conditions. Although the parallel reaction monitoring (PRM) method was recently introduced to glycomic analysis, providing high sensitivity and high resolution, pre-knowledge about the analytes (such as target mass and retention time) is required, and only the ions within the inclusion list can be fragmented41. Meanwhile, GlycanDIA workflow owes the unique advantage that the dataset can continually be re-analyzed since all the precursors in the set mass ranges were fragmented during the data acquisition. This can provide valuable information by re-evaluating glycan modifications and discovering additional glycan structures in future studies. Importantly, GlycanDIA provides a universal tool to monitor the level of glycosylation from different samples, which can be beneficial for identifying novel glycosylated molecules. For a more direct comparison, the general features of different glycomics methods are listed in Supplementary Table 2.
Previously, the quantification of glycans was only investigated at the MS1 level since MS2-based quantification was considered to be ambiguous. This is because glycans often generate common fragments such as hexose (163.06 m/z) and N-acetylhexosamine (204.08 m/z) during the fragmentation event, which may be produced by multiple co-eluting glycans and hardly used for the MS2-based quantification. Meanwhile, unique glycan fragments from CID have been commonly monitored for targeted MRM-based glycomic analysis, which inspired us to develop the GlycanDIA approach11,31. The targeted method essentially is a special DIA-like approach that selects small and discrete mass windows. In this study, we established the pipeline for data analysis using GlycanDIA, which provides essential information on glycan identities and quantities. We first generated predicted fragments for all possible N-glycans using the tool, Glycan Product Generator, which calculates possible glycan fragmentations based on the input structure and outputs the glycans in a format that can be directly imported into Skyline for evaluation. We showed that MS2-based quantification is feasible in glycomic analysis by GlycanDIA after meticulously optimizing the isolation window and picking multiple co-varying transitions. Quantification using the MS2 signals is generally more sensitive because the MS1 signals of a glycan are more likely to have interference from another analyte in the sample with a smaller mass, while the selective fragment ion measurements in the MS2-based quantification approach can be less prone to noise interference. Notably, due to the potential gas phase rearrangement of fucose, having a good separation of glycans and carefully choosing the product ions that represent the parent glycans during the GlycanDIA is crucial42,43.
Glycomics in biological analysis is more often focused on elucidating the glycan composition information, and GlycanDIA can provide superior discovery of such information. Although we have distinguished example glycan isomers from HMO, O-glycan, and N-glycan samples, the current GlycanDIA is still limited in detailing the linkage between each monosaccharide residue and providing better utility for linkage determination due to the major glycosidic bond cleavage (B/Y fragments) during HCD fragmentation. To address these challenges, established workflows already incorporate practical solutions for specific cases. For example, sialic acid and fucose linkages can be inferred by comparing glycan profiles before and after sialidase/fucosidase treatments32. And the integral glycan structure characterization requires other types of fragmentations and applications of the MSn strategy. For example, Wei et al. reported the investigation of the glycan linkages with remarkable details using MS3 and electron-based fragmentations44. In addition, spectral libraries and indexed retention time (iRT) calibration for DIA analysis are critical parameters for cross-sample and cross-batch comparisons to enhance identification confidence. However, limitations in glycan reference standards and inter-instrument variability hinder broader applicability. Ashwood et al. demonstrated the use of glucose oligomers for normalizing retention times during glycomic analysis on a PGC column45. Therefore, the improvement of GlycanDIA in the future can be focused on elucidating glycan structural information by integrating other fragmentation techniques with calibrated glycan standards and the application of the narrow-window DIA strategy, which would improve both the specificity and reproducibility of GlycanDIA46. Regarding software limitations, our software requires prior knowledge of the glycan library and cannot be applied for de novo glycan construction. Notably, we observed promising results in reconstructing spectra for complex glycans like Hex(5)HexNAc(5)Neu5Ac(1) using GlycanDIA data, which aligned well with DDA reference spectra (Supplementary Fig. 31). This suggests potential for a library-free MS2-centric approach, though extensive validation across diverse glycans and algorithm development are still needed. In addition, our current decoy mode—which applies a random mass shift to theoretical fragment ions—helps estimate false discovery rates. However, since glycans often share the same fragment ions, particularly for N-glycans, which share a common chitobiose core, this calculation may lead to an underestimate of the score for distinguishing between similar glycans (the degree of underestimation increases as the similarity of the glycans increases). At the current stage, though its limitations in linkage resolution and software algorithm development, we believe GlycanDIA enables a more comprehensive glycomic analysis compared to the DDA-based methods, offering a unique aspect that is rarely explored in glycomics.
Overall, glycosylation is a prevalent feature of disease progression, with altered patterns observed in diseases such as cancer, diabetes, and Alzheimer’s disease. The different glycosylation significantly influences the biological activity of proteins via alternating glycoprotein structures and potentiating the binding of receptors. We reported an integrative workflow, GlycanDIA, to characterize the glycans using the cutting-edge DIA strategy. Our results exemplify the workflow’s advantages in improved detection and quantification to provide a comprehensive view of different cellular glycosylation, including N-glycans, O-glycans, and HMOs. Meanwhile, information about glycan isomers and low-abundant glycan modifications can also be revealed in many cases. The enhanced capabilities of DIA allow to conduct more in-depth glycomic analysis, providing crucial insights into disease mechanisms and potential biomarkers. In addition, such glycomic information can be used to guide the design of therapeutics with coordinated binding to site-specific glycosylated targets.
Methods
Materials
Research complies with all relevant ethical regulations and was approved by the Animal Care and Use Committee of Boston Children’s Hospital. Details regarding our standard glycoproteins, N-glycans, and human milk oligosaccharides (HMOs) are provided in Supplementary Table 3 for reference. The legend for monosaccharide units and annotations can be found in Supplementary Table 4. The precursor and product ions of heavy isotope-labeled glycan standards are listed in Supplementary Table 5.
Cell membrane extraction
The procedures for cell membrane extraction were described previously47. Briefly, cell pellets were lysed on ice with five alternating on and off pulses in 5 and 10-second intervals using a probe sonicator (Fisher Scientific, NH). Nuclear and cellular debris was removed by centrifugation at 2000 x g for 10 min at 4 °C. The supernatants were then centrifuged at 200,000 x g for 45 min at 4 °C using an Optima Max-XP Ultracentrifuge (Beckman, IN) to extract the plasma membrane. The pellets of the cell membrane were washed with 0.2 M Na2CO3 solution and water, respectively.
RNA Extraction
RNA extraction and processing took place as described36. Specifically, samples were thawed after homogenization, and denaturing was further encouraged by placing the samples at 50 °C and shaking for 5 min. To phase separate the RNA, 0.4X volumes of water was added, vortexed, let to stand for 5 min at 25 °C and lastly spun at 12,000 x g at 4 °C for 15 min. The aqueous phase was transferred to clean tubes, and 1.1X volumes of isopropanol was added. The RNA is then purified over a Zymo-II column (Zymo Research, CA). First, 350 μL of pure water was added to each column and spun at 10,000 x g for 30 s, and the flowthrough was discarded. Next, precipitated RNA from the RNAzol RT extraction (or binding buffer precipitated RNA, below) is added to the columns, spun at 10,000 x g for 10–20 s, and the flowthrough is discarded. This step is repeated until all the precipitated RNA is passed over the column once. Next, the column is washed three times total: once using 400 μL RNA Prep Buffer (3 M GuHCl in 80% EtOH), twice with 400 μL 80% ethanol. The first two spins are at 10,000 x g for 20 s, the last for 30 s. The RNA is then treated with Proteinase K on the column. Proteinase K is diluted 1:19 in water and 50 μL added directly to the Zymo-II column matrix, and then allowed to incubate on the column at 37 °C for 45 min. The column top is sealed with either a cap or parafilm to avoid evaporation. After the digestion, the columns are brought to room temperature for 5 min; lowering the temperature is important before proceeding. Next, eluted RNA is spun out into fresh tubes and a second 50 μL elution with water is performed. To the eluate, 1.5 μg of the mucinase StcE is added for every 50 μL of eluted RNA, and placed at 37 °C for 30 min to digest. The RNA is then cleaned up again using a Zymo-II column. Here, 2X RNA Binding Buffer (Zymo Research, CA) was added and vortexed for 10 s, and then 2X (samples + buffer) of 100% ethanol was added and vortexed for 10 s. This is then bound to the column, cleaned up as described above, and eluted twice with 50 μL water. The final enzymatically digested RNA is quantified using a NanoDrop. To then isolate small RNAs, we followed the Purification of Small and Large RNAs into Separate Fractions protocol in the RNA Clean & Concentrator 5 (Zymo Research, CA) protocol exactly as described.
N-Glycan sample preparation
200 µg of RNase B and serum glycoprotein, 1 mg of cell membrane protein, or 5 μg of RNAs were resuspended with 200 µL of 100 mM HEPES buffer containing 10 mM DTT (pH 7.8), and the mixture was heated using a thermomixer at 100 °C for 3 min. The cleavage of N-glycans was performed by adding 4 µL of PNGase F (500,000 units/mL), followed by incubation in a 37 °C thermomixer overnight. The proteins and RNAs were precipitated by adding ethanol and incubating at − 80 °C for 2 h, followed by centrifuging at 20,000 x g for 15 min at 4 °C. The supernatant containing N-glycans was purified using the porous graphitic carbon (PGC) SPE plate (Thermo Scientific, MA). N-glycans were washed with 0.1% (v/v) TFA in water and were eluted with 60% (v/v) ACN and 0.1% (v/v) TFA in water. The purified glycans were dried using the SpeedVac system (Thermo Scientific, MA) and reconstituted in water for LC-MS/MS analysis.
O-Glycan Sample preparation
The bovine mucin was mixed with 10 μL of 2 M NaOH and 100 μL of 2 M NaBH4 and incubated at 45 °C for 18 h. After the reaction, 10% acetic acid was added to the sample on ice until the pH reached an acidic. Followed by centrifugation at 21,000 x g for 20 min at 4 °C, the supernatant containing free O-glycans was loaded onto the PGC SPE plate and followed as per the above details for purification. The elutes were dried, reconstituted in 89% (v/v) acetonitrile with 1% (v/v) trifluoroacetic acid in water, and further purified by iSPE-HILIC cartridges (HILICON, Sweden).
HMO Sample preparation
For this study, human milk samples were obtained from the Milk Bank at Austin with the informed consent of the donors. The procedures for preparing HMO samples were described previously48. Briefly, the human milk sample (Milk Bank at Austin) was mixed with 90 μL of water and defatted by centrifugation at 3200 x g for 30 min at room temperature. The aqueous layers were transferred into new plates. Then, 2 volumes of ethanol were added to precipitate the proteins at − 80 °C for 2 h. After 30 min of centrifugation at 21,000 x g for 20 min at 4 °C, the supernatant fluids containing mainly oligosaccharides were purified using the PGC SPE plate to remove lactose.
LC-MS/MS Analysis
The glycans were reconstituted using 20 µL water and analyzed using a Vanquish Neo UHPLC System (Thermo Scientific, CA) coupled with an Orbitrap Exploris 240 mass spectrometer (Thermo Scientific, CA). 2 μL of the sample was injected, and the analytes were separated on a self-packed nano PGC column (3 μm, 0.075 × 250 mm). In the comparison experiment, 6520 Accurate Mass Q-TOF LC/MS equipped with a PGC nano-chip (Agilent, CA) was used. A binary gradient using solvent A with 0.1% (v/v) FA in water and solvent B with 0.1% (v/v) FA in ACN was applied to separate N-glycans at a 300 nL/min flow rate. The detailed parameters for MS setup are available in Supplementary Table 1.
Data analysis
For DDA results, the glycans were identified and quantified using GlycoNote and Agilent MassHunter Qualitative Analysis software (v.B08). The settings for GlycoNote were described in the previous publication, and the range of monosaccharide compositions Hex 2-15; HexNAc 2-10; dHex 0-5; Neu5Ac 0-4 was used for general N-glycan analysis27. For DIA results, Glycan Product Generator was used to predict the singly charged glycan fragments, ProteoWizard MSConvert (v3.0) was used for demultiplexing the spectrum, and Thermo Scientific FreeStyle (v1.8) and Skyline (v21.0) software was used for viewing MS1 and MS2 Information49,50,51. GlycanDIA Finder was used to identify and quantify the glycans. For GlycanDIA Finder, we adapted parameters from GlycoNote, including mass shift range, number of decoy sets, FDR, tolerance fragment, and tolerance precursor. We chose a mass shift range of 50% because our precursor mass is 600–1800 m/z. The number of decoy sets was set as 100, which is a balance between simulation time and variability of the decoy set. Smaller tolerances for fragment and precursor ions, 20 ppm and 10 ppm, respectively, were used in our software for resolving the data acquired from high-resolution mass spectrometry instruments. Neu5Gc and other glycan modifications were searched using the add-on mass feature when necessary. An FDR of 2% was chosen as a balance between run time and controlling false-positive identifications. Mass tolerance, intensity threshold, and maximum charge range were set to 10 ppm (MS1), 20 ppm (MS2), 1e4 level, and 3 charges, respectively. Top 5 ions with a signal-to-noise (S/N) ratio above 5 were chosen for quantification (peak area), and the apex of the peak was used as the peak height.
Data processing
The glycan subtypes were annotated in accordance with the previous publication, categorizing them as high mannose, neutral (no sialylation nor fucosylation), fucosylated, sialylated (with Neu5Ac), sialofucosylated (with both Neu5Ac and fucosylation), or Neu5Gc-containing based on the glycan compositions47. Glycan structure was labeled according to the previous studies20,28. To calculate the relative abundance, we employed the label-free approach using the intensity (height or area under the peak) of the specific glycan or subgroup glycans and normalized it to the total abundance of all the identified glycans. Missing values are imputed using MNAR imputation. GraphPad Prism software (Version 9.0.0 or later) was used for data analysis, statistics, graphing and figure assembly. Biorender.com was used to create illustrations.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw mass spectrometry data generated in this study have been deposited in the Mass Spectrometry Interactive Virtual Environment (MassIVE) database under accession code MSV000093677 [https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=9f5160f9b4574db9a9abde9d922fcb5d]. Source data are provided in this paper.
Code availability
The source code of GlycanDIA Finder can be accessed at https://github.com/ChenfengZhao/GlycanDIAFinder.
References
Varki, A. et al. Essentials of Glycobiology. (Cold Spring Harbor Laboratory Press, 2022).
Flynn, R. A. et al. Small RNAs are modified with N-glycans and displayed on the surface of living cells. Cell 184, 3109–3124 (2021).
Reily, C., Stewart, T. J., Renfrow, M. B. & Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 15, 346–366 (2019).
Battistel, M. D., Azurmendi, H. F., Yu, B. & Freedberg, D. I. NMR of glycans: Shedding new light on old problems. Prog. Nucl. Magn. Reson. Spectrosc. 79, 48–68 (2014).
Hirabayashi, J. Lectin-based structural glycomics: Glycoproteomics and glycan profiling. Glycoconj. J. 21, 35–40 (2004).
Ruhaak, L. R., Xu, G., Li, Q., Goonatilleke, E. & Lebrilla, C. B. Mass Spectrometry Approaches to Glycomic and Glycoproteomic Analyses. Chem. Rev. 118, 7886–7930 (2018).
Li, Q., Xie, Y., Wong, M. & Lebrilla, C. B. Characterization of Cell Glycocalyx with Mass Spectrometry Methods. Cells 8, 882 (2019).
Delafield, D. G. & Li, L. Recent advances in analytical approaches for glycan and glycopeptide quantitation. Mol. Cell. Proteomics https://doi.org/10.1074/mcp.r120.002095 (2021).
Kasper, D. M. et al. The N-glycome regulates the endothelial-to-hematopoietic transition. Science 370, 1186–1191 (2020).
Ruhaak, L. R. et al. Glycan labeling strategies and their use in identification and quantification. Anal. Bioanal. Chem. 397, 3457–3481 (2010).
Zhou, S., Hu, Y., DeSantos-Garcia, J. L. & Mechref, Y. Quantitation of Permethylated N-Glycans through Multiple-Reaction Monitoring (MRM) LC-MS/MS. J. Am. Soc. Mass Spectrom. 26, 596–603 (2015).
Venable, J. D., Dong, M.-Q., Wohlschlegel, J., Dillin, A. & Yates, J. R. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods 1, 39–45 (2004).
Doerr, A. DIA mass spectrometry. Nat. Methods 12, 35–35 (2015).
Raetz, M., Bonner, R. & Hopfgartner, G. SWATH-MS for metabolomics and lipidomics: critical aspects of qualitative and quantitative analysis. Metabolomics 16, 71 (2020).
Xie, Y. et al. SWAMNA: a comprehensive platform for analysis of nucleic acid modifications. Chem. Commun. 59, 12499–12502 (2023).
Yang, Y. et al. GproDIA enables data-independent acquisition glycoproteomics with comprehensive statistical control. Nat. Commun. 12, 6073 (2021).
Dong, M. et al. Data-Independent Acquisition-Based Mass Spectrometry (DIA-MS) for Quantitative Analysis of Intact N-Linked Glycopeptides. Anal. Chem. 93, 13774–13782 (2021).
Ye, Z., Mao, Y., Clausen, H. & Vakhrushev, S. Y. Glyco-DIA: a method for quantitative O-glycoproteomics with in silico-boosted glycopeptide libraries. Nat. Methods 16, 902–910 (2019).
Zacchi, L. F. & Schulz, B. L. SWATH-MS Glycoproteomics reveals consequences of defects in the glycosylation machinery. Mol. Cell. Proteom. 15, 2435–2447 (2016).
She, Y.-M., Tam, R. Y., Li, X., Rosu-Myles, M. & Sauvé, S. Resolving isomeric structures of native glycans by nanoflow porous graphitized carbon chromatography–mass spectrometry. Anal. Chem. 92, 14038–14046 (2020).
De Leoz, M. L. A., Simón-Manso, Y., Woods, R. J. & Stein, S. E. Cross-ring fragmentation patterns in the tandem mass spectra of underivatized sialylated oligosaccharides and their special suitability for spectrum library searching. J. Am. Soc. Mass Spectrom. 30, 426–438 (2019).
Wong, M. et al. Metabolic flux analysis of the neural cell glycocalyx reveals differential utilization of monosaccharides. Glycobiology 30, 859–871 (2020).
Xie, Y. et al. Protein oxidation of fucose environments (POFE) reveals fucose–protein interactions. Chem. Sci. 15, 5256–5267 (2024).
Čaval, T., Zhu, J. & Heck, A. J. R. Simply extending the mass range in electron transfer higher energy collisional dissociation increases confidence in N-glycopeptide identification. Anal. Chem. 91, 10401–10406 (2019).
Huber, F. et al. matchms: Processing and similarity evaluation of mass spectrometry data. J. Open Source Softw. 5, 2411 (2020).
Chen, Z. et al. GlycoDeNovo2: An improved MS/MS-based de novo glycan topology reconstruction algorithm. J. Am. Soc. Mass Spectrom. 33, 436–445 (2022).
Liu, M.-Q. et al. GlycoNote with iterative decoy searching and open-search component analysis for high-throughput and reliable glycan spectral interpretation. Anal. Chem. 95, 8223–8231 (2023).
Song, T., Aldredge, D. & Lebrilla, C. B. A method for in-depth structural annotation of human serum glycans that yields biological variations. Anal. Chem. 87, 7754–7762 (2015).
Thomsson, K. A., Bäckström, M., Holmén Larsson, J. M., Hansson, G. C. & Karlsson, H. Enhanced detection of sialylated and sulfated glycans with negative ion mode nanoliquid chromatography/mass spectrometry at high pH. Anal. Chem. 82, 1470–1477 (2010).
Seo, N. et al. Isomer-specific monitoring of sialylated N-glycans reveals association of α2,3-linked sialic acid epitope with Behçet’s disease. Front. Mol. Biosci. 8, https://doi.org/10.3389/fmolb.2021.778851 (2021).
Xu, G. et al. Absolute quantitation of human milk oligosaccharides reveals phenotypic variations during lactation1. J. Nutr. 147, 117–124 (2017).
Xu, G., Goonatilleke, E., Wongkham, S. & Lebrilla, C. B. Deep structural analysis and quantitation of O-linked glycans on cell membrane reveal high abundances and distinct glycomic profiles associated with cell type and stages of differentiation. Anal. Chem. 92, 3758–3768 (2020).
Pett, C. et al. Effective assignment of α2,3/α2,6-Sialic acid isomers by LC-MS/MS-based glycoproteomics. Angew. Chem. Int. Ed. 57, 9320–9324 (2018).
Palmisano, G., Larsen, M. R., Packer, N. H. & Thaysen-Andersen, M. Structural analysis of glycoprotein sialylation – part II: LC-MS based detection. RSC Adv. 3, 22706–22726 (2013).
Muthana, S. M., Campbell, C. T. & Gildersleeve, J. C. Modifications of glycans: Biological significance and therapeutic opportunities. ACS Chem. Biol. 7, 31–43 (2012).
Xie, Y. et al. The modified RNA base acp3U is an attachment site for N-glycans in glycoRNA. Cell 187, 5228–5237 (2024).
Haltiwanger, R. S. Fucose is on the TRAIL of colon cancer. Gastroenterology 137, 36–39 (2009).
Otaki, M. et al. Mouse tissue glycome atlas 2022 highlights inter-organ variation in major N-glycan profiles. Sci. Rep. 12, 17804 (2022).
Helm, J. et al. Non-targeted N-glycome profiling reveals multiple layers of organ-specific diversity in mice. Nat. Commun. 15, 9725 (2024).
Stincone, P. et al. Evaluation of data-dependent MS/MS acquisition parameters for non-targeted metabolomics and molecular networking of environmental samples: Focus on the Q exactive platform. Anal. Chem. 95, 12673–12682 (2023).
Cho, B. G. et al. Targeted N-glycan analysis with parallel reaction monitoring using a quadrupole-orbitrap hybrid mass spectrometer. Anal. Chem. 94, 15215–15222 (2022).
Wuhrer, M., Koeleman, C. A. M., Hokke, C. H. & Deelder, A. M. Mass spectrometry of proton adducts of fucosylated N-glycans: fucose transfer between antennae gives rise to misleading fragments. Rapid Commun. Mass Spectrom. 20, 1747–1754 (2006).
Wuhrer, M., Deelder, A. M. & van der Burgt, Y. E. M. Mass spectrometric glycan rearrangements. Mass Spectrom. Rev. 30, 664–680 (2011).
Wei, J. et al. De novo glycan sequencing by electronic excitation dissociation MS2-guided MS3 analysis on an Omnitrap-Orbitrap hybrid instrument. Chem. Sci. 14, 6695–6704 (2023).
Ashwood, C., Pratt, B., MacLean, B. X., Gundry, R. L. & Packer, N. H. Standardization of PGC-LC-MS-based glycomics for sample specific glycotyping. Analyst 144, 3601–3612 (2019).
Jager, S. et al. In-depth plasma N-glycoproteome profiling using narrow-window data-independent acquisition on the Orbitrap Astral mass spectrometer. Nat. Commun. 16, 2497 (2025).
Li, Q., Xie, Y., Wong, M., Barboza, M. & Lebrilla, C. B. Comprehensive structural glycomic characterization of the glycocalyxes of cells and tissues. Nat. Protoc. 15, 2668–2704 (2020).
Wu, S., Tao, N., German, J. B., Grimm, R. & Lebrilla, C. B. Development of an annotated library of neutral human milk oligosaccharides. J. Proteome Res. 9, 4138–4151 (2010).
Kessner, D., Chambers, M., Burke, R., Agus, D. & Mallick, P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 (2008).
Ceroni, A. et al. GlycoWorkbench: A tool for the computer-assisted annotation of mass spectra of glycans. J. Proteome Res. 7, 1650–1659 (2008).
Adams, K. J. et al. Skyline for small molecules: A unifying software package for quantitative metabolomics. J. Proteome Res. 19, 1447–1458 (2020).
Anthi, A. https://biorender.com/ikhc1he. (2025).
Acknowledgements
The authors appreciate the Skyline and GlycoWorkbench development teams for creating this complementary software for the community. The authors also thank Maurice Wong (University of California, Davis) for the active discussion and Aaron Stacy and Yasmine Bouchibti (University of California, Davis) for preparing HMO samples. Research reported in this publication was supported by grants from Greater Bay Area Institute of Precision Medicine I0036(A) (Y.X.), the Burroughs Wellcome Fund Career Award for Medical Scientists (R.A.F.), the Sontag Foundation Distinguished Scientist Award (R.A.F.), the Rita Allen Foundation (R.A.F.), National Institutes of Health GM151157 (R.A.F.), GM049077 (C.B.L.), AG062240 (C.B.L.), AI118891 (B.A.G.), HD106051 (B.A.G.), and CA196539 (B.A.G.).
Author information
Authors and Affiliations
Contributions
Y.X. conceived the project, designed and performed the experiments, analyzed data, produced figures, and drafted the manuscript. X.L., S.C., Z.L., and F.M.R. performed the MS analysis and edited the manuscript. L.Y., S.W., and C.Z. wrote the code and developed the search engine. Performed the method setup. B.M.G. and R.A.F. performed the glycoRNA experiments and edited the manuscript. C.B.L. planned the overall experimental project and edited the manuscript. B.A.G. supervised the overall experimental project and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
R.A.F. is a stockholder of ORNA Therapeutics. R.A.F. is a board of directors member and stockholder of Chronus Health and Blue Planet Systems. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Frederique Lisacek, Weiping Sun and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xie, Y., Liu, X., Yi, L. et al. Development and application of GlycanDIA workflow for glycomic analysis. Nat Commun 16, 7075 (2025). https://doi.org/10.1038/s41467-025-61473-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61473-y
This article is cited by
-
Functional regulation of GlycoRNA and progress in malignant tumors
Journal of Translational Medicine (2026)
