
Abstract
The stereochemical activity of lone-pair electrons critically influences lattice anharmonicity and thermal transport in crystals. However, traditional chemical substitution methods lack continuity and reversibility. We propose a strain-engineered bond angle distortion strategy in layered BiCuSeO to continuously modulate lone-pair electrons. Theoretically, tensile strain reduces the O-Bi-O bond angle, expands lone-pair electron spatial distribution, and decreases Bi-O bond charge overlap, intensifying Bi atom anharmonic vibrations. Furthermore, tensile strain induces reverse O atom vibrations and strong lattice dynamic disorder, lowering the phonon band gap and enhancing anharmonic phonon-phonon interactions and Umklapp scattering. Importantly, strain modulates lone-pair electron distribution and interaction strength without uniformly weakening long-range interatomic forces. As a result, 4% tensile strain reduces lattice thermal conductivity of BiCuSeO to 0.53 W/mK (54% decrease) at 300 K. This work establishes a multiscale framework linking strain, lone-pair electron behavior, and phonon dynamics, enabling robust and continuous control of thermal transport properties.
Similar content being viewed by others
Introduction
Dynamic regulation of lattice anharmonicity and thermal transport properties is a central challenge in the design of advanced thermoelectric materials1. Traditional chemical substitution strategies, limited by discontinuous doping and irreversible lattice distortions, struggle to achieve dynamic response at the atomic scale2. Therefore, developing effective regulatory mechanisms is critical to optimizing thermoelectric performance3,4,5,6. Recent studies have shown that stereochemically active lone pair electrons (LPEs) can induce local structural distortions via symmetry breaking, offering an avenue for modulating thermal transport7,8. However, current research primarily focuses on the chemical composition modulated spatial distribution of LPEs9,10, which remains fundamentally limited by the fixed angular restrictions dictated by valence shell electron pair repulsion (VSEPR) theory11,12,13. This intrinsic limitation hinders real-time, atomistic modulation and poses a significant bottleneck to further improvements in thermoelectric performance.
Minimizing lattice thermal conductivity has long been a key objective in thermoelectric materials research14,15,16,17,18,19,20. Typical low thermal conductivity BiCuSeO material features have a unique crystal structure consisting of alternating conducting [Cu2Se2]2− layers and insulating [Bi2O2]2+ layers21. This intrinsic heterointerface provides a unique platform for electron and phonon transport22. The intrinsically low lattice thermal conductivity of BiCuSeO is primarily attributed to the strong lattice anharmonicity induced by the stereochemically active 6 s² LPEs of Bi23. However, its thermoelectric figure of merit (ZT) remains limited by low carrier mobility and the uncontrollable spatial distribution of LPEs. Various chemical doping strategies, including Pb24, Ba21, Sr25, and Ca26, have been employed to enhance performance. Nevertheless, weak interlayer coupling limits charge diffusion and improvements in electrical conductivity. Although Pb doping can reduce lattice thermal conductivity via enhanced phonon scattering27, it inevitably introduces irreversible structural defects and lacks real-time tunability. More advanced approaches, such as Bi/Cu dual-vacancy doping, have been used to increase carrier concentration to some extent28, yet still fail to dynamically regulate the LPE distribution. Moreover, excessive doping or high vacancy concentrations can introduce significant lattice disorder, further degrading thermoelectric performance.
In contrast, strain engineering has recently emerged as a promising non-destructive and reversible approach for tuning material properties. Theoretical studies have investigated the intrinsic properties of unstrained BiCuSeO29, and the effects of strain on thermal conductivity have been systematically explored in other crystal systems30. In BiCuSeO, biaxial strain has been shown to influence electronic structure and phonon behavior31, while uniaxial compressive strain has been found to impact electronic transport and induce structural distortions32. Furthermore, defect-related mechanisms (such as dislocations and grain boundaries) have also been identified as key contributors to phonon scattering and thermal resistance in BiCuSeO33. These efforts have significantly advanced our understanding of structural and transport behavior under strain and defect conditions. However, these studies have yet to explore how strain can be employed to continuously and controllably tune the stereochemical activity of LPEs by directly modulating key geometric parameters such as bond angles and bond lengths. Specifically, previous research has largely focused on macroscopic structural or phononic modifications, without considering the regulation of thermal transport from the perspective of LPE spatial evolution under strain, which is a mechanism fundamentally tied to lattice anharmonicity and phonon scattering processes.
This study proposes and validates an effective mechanism to address the identified gap. Applying tensile strain reduces the O–Bi–O bond angle and increases the Bi–O bond length, resulting in spatial expansion of the Bi³⁺ 6s² LPEs. This expansion enhances lattice anharmonicity and strengthens phonon scattering, enabling continuous, controllable reduction of lattice thermal conductivity. First-principles calculations reveal that increasing tensile strain from 0 to 4% decreases the O–Bi–O bond angle and extends the Bi–O bond length, significantly expanding the stereochemically active LPEs. This expansion leads to reduced orbital overlap, phonon softening, and elevated Grüneisen parameters, indicative of increased anharmonicity and stronger Umklapp scattering. Unlike static chemical or structural modifications, this strain-driven mechanism is dynamic, reversible, and composition-independent, offering superior flexibility and efficiency in phonon engineering.
Results and discussion
The stereochemical activity of LPEs plays a crucial role in determining the lattice dynamics and thermal transport properties of materials. This study proposes a bond-angle distortion regulation strategy based on strain engineering to achieve continuous modulation the activity of the LPEs in layered BiCuSeO crystal. The stereochemical activity of LPEs is conventionally explained using the VSEPR theory. As illustrated in Fig. 1A, the LPEs of Bi atoms in BiCuSeO are identified as the primary contributor to its low lattice thermal conductivity34. By systematically tuning the O-Bi-O bond angle through tensile strain, the degree of distortion in the [BiO2]2+ layer can be significantly enhanced, achieving a continuous transition of the LPE state from delocalized to localized. Specifically, tensile strain provides additional space for LPEs by reducing the bond angle, thereby increasing Coulomb repulsion between the LPEs and the bonding electron pairs. This process significantly enhances lattice anharmonicity and strengthens phonon scattering, leading to effective suppression of lattice thermal conductivity.

A Schematic illustration of strain engineering from 0 to +4% tensile strain and its effect on local structural parameters (denoted as α1 to α5) and the expression strength of lone pair electrons. Increasing tensile strain leads to continuous tuning of lone pair electron activity, with a corresponding enhancement from weak to strong expression. B Structural diagram of BiCuSeO with Bi (purple), Cu (blue), Se (green), and O (red). Insets show the mechanism of lone pair electron tuning: tensile strain modifies the lone pair electrons and affects long-range interactions.
Layered BiCuSeO is a superlattice material with a ZrCuSiAs-type tetragonal crystal structure (space group P4/nmm). Detailed information on the crystal structure is provided in Fig. S1. The crystal structure of BiCuSeO consists of two structural units: the [Bi2O2]2+ layer containing LPEs and the conductive [Cu2Se2]2− layer (Fig. 1B). In the [Bi2O2]2+ layer, the LPEs of Bi occupy the van der Waals (vdW) gaps and form a long-range ordered arrangement along the c-axis, introducing anharmonic characteristics to the crystal. Meanwhile, the conductive [Cu2Se2]2− layer constructs a high carrier mobility pathway through a Cu-Se covalent bond network. These two structural units are connected by weak vdW interactions, forming a three-dimensional electron-phonon decoupled system. Consequently, strain engineering enables effective and sustained modulation of LPE expression within the vdW gaps, thereby regulating lattice thermal conductivity. Furthermore, this study reveals that tensile strain can modify long-range interactions within the material, offering insights into LPEs behavior in crystalline structures.
Figure 2A illustrates the trajectory of BiCuSeO in the x-y plane from molecular dynamics (MD) simulations at 900 K. Despite significant thermal vibrations of all atoms at 900 K, the results indicate that the overall structure of BiCuSeO remains stable (Fig. S2). Notably, O atoms vibrate with significantly larger amplitudes than other atoms, suggesting that they are less constrained around their equilibrium position. In addition, the large vibrations of O atoms may lead to frequent shifts to nearby locations, which further affect their local structural properties and enhance phonon scattering. Figures 2B and S3 present the decay curves of the velocity autocorrelation function (VAF), obtained from ab initio molecular dynamics (AIMD) simulations, revealing differences in atomic dynamics and their impact on lattice thermal conductivity. Typically, a damped oscillatory VAF reflects strong interatomic interactions in solids, whereas in disordered systems, a single valley minimum is observed, arising from the diffusive and incoherent motion of constituent atoms35,36. As shown in Fig. 2B, the VAFs of Bi, Cu, and Se atoms exhibit damped harmonic decay, indicating the presence of strong anharmonic interactions within the lattice. These interactions enhance the Umklapp process scattering, significantly reducing phonon lifetimes and mean-free paths. However, the VAF of O atoms exhibits a single-valley pattern, reflecting incoherent dynamic behavior characterized by the absence of phase correlation in atomic motion and predominantly governed by diffusive dynamics. To quantify the specific impact of these characteristics on lattice thermal conductivity, the Green-Kubo relation is employed, expressed as follows37:
where V is the volume of the system, KB stands for Boltzmann constant, T represents the temperature, and 〈 J(t)⋅J(0)〉 is the autocorrelation function of thermal flow. The VAF of O atoms decays rapidly, leading to a significant reduction in its integral value, which in turn results in a relatively weak contribution of O atoms to the thermal conductivity. Although the VAFs of Bi, Cu, and Se atoms display damped harmonic oscillations with enhanced integral values, their contributions to thermal conductivity remain constrained by their larger atomic masses. To investigate the strain-regulated stereochemical activity of LPEs in the BiCuSeO system, this study established a nonlinear relationship between structural distortions and the spatial distribution of LPEs based on the Continuous Symmetry Measure (CSM) analysis38. As illustrated in Fig. 2C, the [Bi2O2]2+ layer of BiCuSeO undergoes structural distortion under tensile strain, with the O-Bi-O bond angle decreasing from 114.5° (0% strain) to 113.6° (4% strain). This distortion triggers an increase in the CSM value from 3.5 (0% strain) to 3.6 (4% strain), reflecting a reduction in structural centrosymmetry. The increase in CSM value indicates that the structural distortion drives LPEs toward a broader spatial distribution and an outward expansion of the electron cloud morphology39. These changes enhance the anharmonicity of LPEs, thereby reducing lattice thermal conductivity through enhanced phonon scattering.

A Atomic trajectories projected on the x–y plane from molecular dynamics at 900 K, showing displacements of Bi, Cu, Se, and O atoms. B Velocity auto-correlation functions (VAFs) of Bi, Cu, Se, and O atoms, demonstrating element-specific vibrational dynamics. C Correlation between continuous symmetry measure (CSM) values and O-Bi-O bond angle variation under tensile strains. D Temperature dependence of atomic displacement parameter U33(Bi) along the c-axis under varying strains, showing increased Bi vibrations. E Bader charge analysis and electron localization function (ELF) maps for Bi atoms under increasing tensile strain, showing redistribution of electron density and lone pair behavior. F Integrated Crystal Orbital Hamilton Population (ICOHP) and Integrated Crystal Orbital Bond Index (ICOBI) values versus tensile strain, illustrating bonding strength evolution in BiCuSeO.
Figure 2D reveals the influence of tensile strain on the displacement parameter U33(Bi) along the c-axis for Bi atoms carrying LPEs. Here, U33(Bi) refers to the anisotropic displacement parameter of the Bi atom along the crystallographic c-axis, calculated by density functional theory (DFT) to quantify the mean squared thermal vibration amplitude. At 300 K, U33(Bi) increases from 0.0089 (0% tensile strain) to 0.0120 under 4% tensile strain. As the temperature rises to 900 K, U33(Bi) exhibits a more pronounced increase, indicating that strain amplifies the thermal vibrational amplitude of Bi atoms. Notably, the temperature-dependent of U33(Bi) for Bi follows a T 0.9 power law40, highlighting anharmonic motion beyond harmonic model limitations. Moreover, the slope of U33(Bi) versus temperature increases with increasing tensile strain, demonstrating that tensile strain not only intensifies vibrational amplitudes but also enhances the anharmonicity of Bi atoms.
Based on the coupling mechanism of structural distortion and electronic effects in the material, the contraction of the O-Bi-O bond angle and the elongation of the Bi-O bond length under tensile strain synergistically enhance the LPE effect of the Bi atom. Bader charge analysis41 reveals that the charge transferred from Bi atoms to neighboring O atoms gradually decreases under tensile strain, as shown in Fig. 2E. This phenomenon can be attributed to the elongation of the Bi–O bond length, which reduces the charge density overlap. Notably, this charge redistribution significantly enhances the electronic spatial distribution of Bi atoms, resulting in the strengthening of LPEs. To validate the strain effect on LPEs, we establish the relationship between strain and LPE expression by comparing the differences in the Electron Localization Function (ELF)42 between off-center and center cases. The results indicate that as the strain increases from 0 to 4%, the ELF difference of Bi atoms becomes more pronounced. Further quantitative analysis shows a consistent increase in LPE localization with each 1% increment in strain, confirming that tensile strain continuously modulates LPEs. To further quantify the localization behavior of LPEs and their contribution to lattice anharmonicity, we analyzed the Born effective charges (BECs) of Bi atoms under different levels of tensile strain. As shown in Table S1, the average BEC (\({Z}_{{{{\rm{avg}}}}}^{\ast }=\frac{1}{3}({Z}_{xx}^{\ast }+{Z}_{yy}^{\ast }+{Z}_{zz}^{\ast })\)) of Bi increases significantly from 3.59 at 0% strain to 5.14 at 4% strain, representing a 43% increase. This trend indicates a markedly enhanced dynamic charge response and polarizability of Bi atoms under external perturbations, which is closely associated with the increased activity of the LPEs. Together with the increase in Bi–O bond length and the reduction of the O–Bi–O bond angle, these results suggest that the LPEs become more localized and stereochemically active, thereby enhancing the electrostatic response of Bi atoms to lattice distortions. This enhanced polar behavior further highlights the critical role of local bonding environment modulation in strengthening lattice anharmonicity.
As shown in Fig. 2F, the Integrated crystal orbital Hamilton population (COHP) value of Bi–O bonds in the BiCuSeO compound progressively decreases as the tensile strain increases from 0 to 4%, which directly correlates with strain-induced bond length elongation. The increased bond length reduces wavefunction overlap, thereby weakening the chemical bond strength. Furthermore, we quantitatively investigated the bond order of Bi-O atomic pairs under different tensile strains using the integrated crystal orbital bond index (ICOBI). The results demonstrate that tensile strain diminishes electron sharing between Bi and O atoms, leading to a decrease in bond strength. Given the relationship between chemical bond strength and lattice thermal conductivity, expressed as κl ∼ (F/M)1/2, the decrease in chemical bond strength leads to a reduction in lattice thermal conductivity.
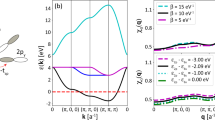
Lattice static strain significantly influences phonon dispersion characteristics by altering the interatomic force field. In the classical one-dimensional atomic chain model, the phonon dispersion is given by \(\omega=2\sqrt{\frac{F}{M}}\,\sin \left(\frac{\pi }{2}\frac{{{{\bf{k}}}}}{{{{{\bf{k}}}}}_{{{{\rm{c}}}}}}\right)\), where F, \({{{\bf{k}}}}\), and \({{{{\bf{k}}}}}_{{{{\rm{c}}}}}\) are the force constant, phonon wavevector, and cutoff wavevector, respectively43,44. The strain induces a broadening of the phonon frequency and enhances anharmonicity. This study achieves continuous modulation of LPEs via strain engineering, revealing a synergistic mechanism between lattice dynamics and electronic orbital interactions. As shown in Fig. 3A, tensile strain continuously decreases the overall phonon vibration frequencies in the BiCuSeO compound. This variation not only enhances the coupling between low-frequency optical phonons and acoustic phonons but also significantly expands the phonon scattering phase space. Due to differences in atomic mass, the 43.0 cm−¹ phonon gap between optical branches blocks three-phonon scattering channels. However, four-phonon scattering processes enable phonon scattering across wide phonon gaps, making it easier to satisfy energy and momentum conservation. Additionally, tensile strain not only reduces the phonon gap (Table S2) but also enhances higher-order phonon scattering processes by modulating interatomic distances and the phonon band structure. Notably, optical branches in the frequency of 188.4 cm−¹ exhibit quasi-flat dispersion (Δω < 10 cm−¹), especially in the Γ-Z direction, which is attributed to reduced elastic force constants under tensile strain (Table S3), confirming strain-induced lattice softening in BiCuSeO. In Fig. 3B, the Γ-point phonon frequency decreases by 39 cm−¹ under 0–4% strain, indicating weakened interlayer interaction ([Bi₂O₂]2+ and [Cu₂Se₂]2− layer) and reduced elastic constants. Furthermore, the effect of tensile strain on the TA’ phonon branch is particularly significant, with a notable decrease in the group velocity of the TA’ phonons compared to the TA/LA phonon branches at a strain of 4% (Table S4). This asymmetric phonon dynamic response provides a crucial entry point for selective regulation of thermal transport. Further analysis reveals that the TA’ phonon branch dominates thermal transport due to its lower inverse participation ratio (IPR), whereas the high IPR values of the TA/LA phonon branches significantly hinder phonon propagation. Therefore, applying tensile strain effectively reduces the contribution of the TA’ phonon branch to thermal transport, thereby lowering the lattice thermal conductivity (Fig. 3C). Additionally, tensile strain decouples Se and Cu atomic vibrations, activates the reverse vibrations of the O atoms, and forms localized scattering centers. Meanwhile, the Bi and O inverse vibrations effectively hinder transverse thermal conduction pathways, further enhancing phonon-phonon anharmonic interactions and significantly impeding thermal flow transmission (Fig. 3D). Furthermore, by constructing the Bi atomic displacement potential well and employing relative regular residual (RRR) curves45 to quantify material anharmonicity (Supporting Information). As shown in Fig. 3E, with increasing tensile strain, the cubic coefficient b of the fitting residuals increases significantly. Combined with the compression of the O–Bi–O bond angle (Fig. 2E), this confirms the enlargement of the LPE space and a significant enhancement of anharmonicity. Moreover, the RRR curves of the Bi atom along the c-axis exhibit a notable “M”-like, suggesting that longitudinal vibrational coupling in Bi–O chains causes avoided crossings in phonon bands (Fig. S4), thereby triggering high-order phonon scattering. Finally, by calculating the Grüneisen parameter of phonon modes under different tensile strains, we confirm that tensile strain enhances the expression of LPEs (Fig. 3F). With increasing strain, the Grüneisen parameter rises from 3.02 (0% strain) to 3.64 (4% strain), confirming that tensile strain effectively expands the occupancy space of LPEs, thereby reinforcing the anharmonic characteristics of the system. Analysis of the two-dimensional potential energy surface at the Z-point reveals that tensile strain significantly reduces the potential well depth, leading to phonon mode softening and enhanced anharmonicity (Fig. S5).

A Phonon dispersion spectra of BiCuSeO at strains from 0% to 4%, showing frequency shifts across the Brillouin zone. B Enlarged view of the low-frequency acoustic branches (TA, TA′, and LA) under varying strain levels, indicating softening of phonon modes. C Phonon spectrum with color-coded inverse participation ratio (IPR) values, revealing the degree of phonon localization. D Representative vibrational modes at the Γ point for different frequencies, showing the atomic displacement patterns and evolution from low to high vibrational frequencies. E Relative regular residuals as a function of atomic displacements, fitted by a fourth-order polynomial. The increasing coefficients indicate enhanced anharmonicity with strain. F Grüneisen parameters (γ) across the first Brillouin zone under different tensile strains, reflecting changes in phonon anharmonicity and thermal transport properties.
As is well known, tensile strain usually leads to lattice expansion, which weakens long-range interatomic interactions that play a key role in phonon-phonon coupling and directly affect thermal conductivity (Fig. 4A). To quantify the effect of strain on interatomic interactions, we normalized the trace of the interatomic force constant (IFC) tensor by using the self-interaction IFC tensor (Supporting Information). Given that the applied tensile strain is along the c-direction, we analyzed the nearest-neighbor force constants along this direction. The second-nearest neighbors (O–Cu, L2) exhibit slightly stronger interactions than the third-nearest neighbors (O–Se, L3), though both are significantly weaker than the first-nearest neighbors (Bi–O, L1) (Fig. 4B). Notably, both second- and third-neighbor pairs demonstrate an “anti-springs” effect characterized by positive force constants. As tensile strain increases, these interactions gradually weaken, not only confirming the strain-induced lattice spacing enlargement but also highlighting the weakening effect of tensile strain on interatomic forces. Figure 4C provides further insight into the trend of force constants among nearest-neighbor atoms. The results indicate that the force constants of Cu–Se and Bi–O bonds decrease with increasing strain, whereas the force constant of the Bi–Se bond anomalously increases. This phenomenon may be attributed to the spatial expansion of the LPE induced by tensile strain and the enhanced localization of the electron cloud, leading to the strengthening of the Bi–Se bond. The radial distribution function (RDF)46 of BiCuSeO is used to reveal the structural ordering of the material (Fig. 4D). The interatomic distances within the material expand significantly under tensile strain, and this change is visualized in the shift of the RDF peak positions towards larger d values, especially for the long-range interatomic interactions. However, the minimal peak alteration suggests that the weakening of interactions is limited, reinforcing our conclusion that tensile strain primarily modifies the spatial distribution and interaction strength of LPEs rather than uniformly reducing the interatomic interactions.

A Schematic diagram of long-range interaction changes under tensile strain; dashed circles and red arrows indicate the phonon interaction range. B Normalized trace of interatomic force constant (IFC) tensors versus interatomic distance at different strains; inset shows strain direction (along the c-axis) and atomic arrangement. C Second-order force constants and interatomic distances for nearest-neighbor pairs (Bi-Se, Bi-O, Se-Cu) under varying tensile strains. D The relationship between radial distribution functions (RDF) and distance under different tensile strains.
To investigate the impact of continuous modulation of LPEs on lattice thermal conductivity, we have performed an iterative BTE solution mixed three-phonon (3 ph) and four-phonon (4 ph) scattering according to the following formula47:
where, κB, T, Ω, N, and ℏ stand for the Boltzmann constant, temperature, volume of the unit cell, regular grid centered at Γ, and the Planck constant, respectively. α and β represent Cartesian coordinates. Additionally, \({\omega }_{\lambda }\), \({\upsilon }_{\lambda }^{\alpha }\), and \({F}_{\lambda }^{\beta }\) denote the phonon frequency, group velocity, and the term associated with the phonon relaxation time for the λ vibrational mode, respectively. As shown in Fig. 5A, the lattice thermal conductivity (κl) of BiCuSeO compound under different tensile strains exhibits strong temperature dependence, following an inverse relationship with temperature. With increasing tensile strain, the κl decreases significantly, which can be attributed to the strain-induced enhancement of anharmonicity and the synergistic effect of additional phonon scattering channels. When considering only three-phonon scattering, the κl of BiCuSeO is 1.38 W/mK (300 K@0% strain) (Fig. S6). However, when four-phonon interactions are taken into account, κl further decreases to 1.17 W/m·K (300 K@0% strain), highlighting the significant impact of four-phonon scattering on thermal transport. At 300 K, as the tensile strain increases from 0 to 4%, the κl decreases from 1.17 W/m·K to 0.53 W/m·K, representing a reduction of ~54%. This indicates that the tensile strain effectively enhances the stereochemically active LPEs of Bi atoms, thereby suppressing the lattice thermal conductivity. Moreover, the material displays pronounced anisotropy (κl(a) = κl(b) > κl(c)), arising from the intrinsic disparity between strong in-plane and weak out-of-plane bonding (Fig. S7). Specifically, the low κl(c) is attributed to suppressed interlayer phonon transport, driven by weak vdW interactions and enhanced anharmonicity associated with stereochemically active LPEs. To support the reliability of our simulations, our calculated κl values at unstrain show good agreement with the experimental measurements reported by Li et al.48, confirming the reliability of our simulation approach. Some deviations are observed when compared to the data from Pei et al.49, which may be attributed to differences in sample preparation, microstructural defects, and measurement conditions. It is noteworthy that experimental data are generally obtained from polycrystalline ceramic samples, where additional phonon scattering from grain boundaries, impurities, and porosity further reduces thermal conductivity compared to the idealized single-crystal models used in our simulations. While our calculations incorporate multiple phonon scattering mechanisms, fully capturing these complex microstructural effects remains challenging. Thus, the focus of this work is on revealing the intrinsic trends and mechanisms of phonon transport under strain rather than achieving exact numerical matches.

A Lattice thermal conductivity (κl) versus temperature at various tensile strains. κl decreases systematically with strain, showing about 54% reduction at 300 K under +4% strain. B Cumulative lattice thermal conductivity as a function of phonon mean free path (MFP) at 300 K. The inset highlights the phonon MFP corresponding to 50% of the cumulative lattice thermal conductivity. C Schematic illustration of anharmonicity changes under tensile strain, showing decreased O–Bi–O bond angle and expanded lone pair electron region. D Four-phonon scattering rate versus phonon frequency at different tensile strains at 300 K. E Umklapp phonon scattering rate versus frequency at 300 K under different strains, with all events satisfying momentum conservation (K₁ = K₂ + K₃ + G). F Total scattering phase space for three- and four-phonon processes under different strains, including three-phonon splitting and combination, and four-phonon splitting, recombination, and redistribution processes.
The cumulative lattice thermal conductivity (\({{\kappa }}_{l}^{C}\)) is significantly affected by the dimensions of the sample, particularly when the sample dimensions surpass the longest phonon mean free path (MFP). Thus, the relationship between the \({{\kappa }}_{l}^{C}\) and the phonon MFP can be described by the following equation:
where \({\Lambda }_{0}\) and \({\Lambda }_{\max }\) represent the eigenvalues of phonon MFP and the maximum phonon MFP, respectively. As shown in Fig. 5B, the cumulative phonon MFPs without any suppression of \({{\kappa }}_{l}^{C}\) are 109 nm and 30 nm under 0% and 4% tensile strain, respectively. When the \({{\kappa }}_{l}^{C}\) is suppressed by 50%, the corresponding effective MFPs are significantly reduced to 2.6 nm (0% strain) and 0.7 nm (4% strain). This result confirms that strain engineering can effectively regulate phonon transport. Compared to conventional defect engineering strategies, this dynamic modulation mechanism enables continuous adjustment of thermal transport properties while maintaining the integrity of the crystal structure, offering an effective approach to designing advanced phonon modulation devices. The physical essence of strain-induced LPE regulation lies in a dual synergistic effect arising from structural distortion (Fig. 5C). On the one hand, the compression of O–Bi–O bond angles shifts the interatomic potential away from its equilibrium position and significantly enhances lattice anharmonicity. On the other hand, the elongation of Bi–O bonds reduces the orbital wavefunction overlap, thereby promoting charge localization on the LPEs of Bi atoms. These two effects together significantly enhance the anharmonicity of the system, creating a powerful phonon scattering center. The enhancement of anharmonicity directly increases phonon scattering rates, particularly leading to a significant rise in the scattering rate of high-frequency optical phonons (Fig. 5D). Under tensile strain, the four-phonon scattering processes undergo significant changes, including splitting, redistribution, and recombination (Fig. S8). Among these, redistribution is the most pronounced due to selection rule constraints. Notably, in the low-frequency region, recombination plays a dominant role, whereas in the high-frequency region, splitting becomes predominant. Additionally, the three-phonon scattering rate exhibits frequency dependence. The scattering rate of the splitting process gradually increases with increasing phonon frequency, while the combining process shows the opposite trend (Fig. S9). This is due to constraints imposed by energy and momentum conservation, where high-frequency phonons primarily scatter with other high-frequency phonons, leading to similar trends in the scattering rates of both high- and low-frequency phonons. Strain-induced asymmetric atomic vibrations disrupt the periodic potential field of the lattice, significantly enhancing lattice vibrational anharmonicity. This anharmonicity induces strong three-phonon coupling effects near the Brillouin zone boundary in reciprocal space. When the phonon momentum approaches a half-integer multiple of the reciprocal lattice vector, the total momentum of the collision process exceeds the first Brillouin zone, requiring momentum conservation correction via a reciprocal lattice vector G (K₁ = K₂ + K₃ + G)50, thereby triggering Umklapp scattering, as illustrated in Fig. 5E. Notably, the optical branches dominated by O atoms exhibit the most significant response to strain, with Umklapp scattering becoming particularly prominent under tensile strain. This phenomenon arises from the enhanced local dynamic disorder of O atoms induced by tensile strain, which manifests in reciprocal space as intensified Umklapp scattering. The underlying physical origin lies in the reconstruction of the LPE distribution in real space, which disrupts the symmetry of lattice vibrations. Figure 5F illustrates the total scattering phase space for three- and four-phonon processes in BiCuSeO compound, including emission (\(\lambda \to {\lambda }^{{\hbox{'}}}+{\lambda }^{{\hbox{'}}{\hbox{'}}}\)), absorption (\(\lambda+{\lambda }^{{\hbox{'}}}\to {\lambda }^{{\hbox{'}}{\hbox{'}}}\)), splitting (\(\lambda \to {\lambda }^{{\hbox{'}}}+{\lambda }^{{\hbox{'}}{\hbox{'}}}+{\lambda }^{{\hbox{'}}{\hbox{'}}{\hbox{'}}}\)), redistribution (\(\lambda+{\lambda }^{{\hbox{'}}}\to {\lambda }^{{\hbox{'}}{\hbox{'}}}+{\lambda }^{{\hbox{'}}{\hbox{'}}{\hbox{'}}}\)), and recombination processes (\(\lambda+{\lambda }^{{\hbox{'}}}+{\lambda }^{{\hbox{'}}{\hbox{'}}}\to {\lambda }^{{\hbox{'}}{\hbox{'}}{\hbox{'}}}\)). The three-phonon scattering exhibits similar emission and absorption phase spaces, and both exceed the scattering phase spaces for the four-phonon scattering, indicating a higher scattering probability for the three-phonon scattering. However, within the four-phonon scattering phase space, the redistribution scattering phase space dominates, while the splitting scattering phase space is the smallest. Furthermore, with increasing tensile strain, the scattering phase space gradually expands, suggesting that lattice distortion-induced anharmonicity enhancement significantly increases phonon-phonon scattering probabilities. This trend indicates that tensile strain enables continuous regulation of phonon transport in BiCuSeO material, thereby offering to optimize their thermoelectric performance. This work breaks through the limitations of traditional chemical substitution strategies by establishing a continuous regulation mechanism for strain-LPEs-thermal transport. It provides theoretical guidance for the design of low thermal conductivity materials and optimization of thermoelectric performance, and is expected to extend to dynamic regulation studies of systems with LPEs, such as SnSe51,52,53 and Bi2Te354.
This study proposes a strain engineering strategy to continuously modulate the stereochemical activity of LPEs in layered BiCuSeO, revealing the dynamic coupling mechanism between LPEs and lattice thermal conductivity. Tensile strain compresses the O–Bi–O bond angles and elongates Bi–O bond lengths, thereby enhancing the structural distortion of the [Bi₂O₂]²⁺ layer and expanding the spatial distribution of LPEs. MD simulations show that local dynamic disorder of O atoms at high temperatures suppresses heat transport, and strain-induced out-of-phase vibrations further hinder transverse phonon transport. Bader charge and ELF analyses reveal that tensile strain reduces the charge overlap density of Bi–O bonds, enhancing the stereochemical activity of LPEs and inducing anharmonic vibrations of Bi atoms. Furthermore, strain softens optical phonon modes, narrows the phonon bandgap, and enlarges the scattering phase space, substantially reducing the contribution of the TA′ phonon mode to thermal conductivity. Crucially, strain primarily modulates the interactions among LPEs, rather than uniformly weakening all interatomic forces. As a result, a 4% tensile strain leads to a 54% reduction in lattice thermal conductivity at 300 K, reaching as low as 0.53 W/mK. This work establishes a quantitative “strain–LPE–phonon” coupling framework, offering theoretical insights and practical strategies for designing low-thermal-conductivity, high-performance thermoelectric materials.
Methods
First-principles calculations
First-principles calculations were performed using the projected augmented wave method in the Vienna ab initio Simulation Package (VASP)55. The exchange-correlation function was described by the Perdew-Burke-Ernzerhof functional within the generalized gradient approximation56. Self-consistent calculations were carried out with an energy convergence criterion of 10−6 and a plane-wave energy cutoff of 520 eV. Bonding interactions within the compound were analyzed using the COHP57 and the ICOBI58 method. Uniaxial tensile strain along the c-axis was simulated by adjusting the lattice constants. The selection of c-axis tensile strain was based on its unique ability to simultaneously reduce the O–Bi–O bond angle and elongate the Bi–O bond, which was crucial for activating LPEs and reducing lattice thermal conductivity. The expression for uniaxial strain is defined as \(\varepsilon=(l-{l}_{0})/{l}_{0}\), where \(l\) and \({l}_{0}\) represent the lattice constants of strained and unstrained, respectively. To visualize the atomic structures, all crystal structure images were generated using the VESTA software59.
Phonon and thermal transport calculations
Phonon dispersion curves were computed using PHONOPY60 based on density-functional perturbation theory within a 3 × 3 × 2 supercell. The atomic displacement parameter (ADP) was calculated from the second-order forces. The phonon mode potential energy surface was constructed using the ModeMap code61. To address higher-order phonon scattering effects, machine-learned interatomic potentials (MLIPs) were trained to obtain moment tensor potentials (MTPs)62 through AIMD simulations. These simulations were performed using 3 × 3 × 2 supercell at the temperatures of 50 K, 300 K, 500 K, 700 K, and 900 K with a time step of 1 fs and a total time of 1 ps. Third- and fourth-order force constants were calculated using MTP, considering interactions up to the 12th and 4th nearest neighbor atoms, respectively. To evaluate the lattice thermal conductivities related to three-phonon (3 ph) and four-phonon (4 ph) scattering, the Boltzmann transport equation was solved using the ShengBTE software63.
Data availability
Data for this study are available in the main text and the Supplementary Information, or can be accessed on Zenodo64. All relevant data supporting the findings of this study are available within the article and its Supplementary Information files. No restrictions apply to data access.
Code availability
Ab-initio density functional theory DFT-based computational packages were used in this study. The VASP package can be purchased from https://www.vasp.at/. The PHONOPY code is available at https://phonopy.github.io/phonopy/. The ModeMap code is available at https://github.com/JMSkelton/ModeMap. The MLIP package is available at https://doi.org/10.17632/fmkvzbk3nt.1. The ShengBTE package is available at https://www.shengbte.org/.
References
Yuan, X. et al. Influence of quartic anharmonicity on lattice dynamics and thermal transport properties of 16 antifluorite structures. Phys. Rev. B 110, 014304 (2024).
Zhao, M. et al. A universal atomic substitution conversion strategy towards synthesis of large-size ultrathin nonlayered two-dimensional materials. Nano-Micro Lett. 13, 1–13 (2021).
Bai, S. et al. Unravelling the thermoelectric properties and suppression of bipolar effect under strain engineering for the asymmetric Janus SnSSe and PbSSe monolayers. Appl. Surf. Sci. 599, 153962 (2022).
Bai, S. et al. Revealing the origin of anisotropic rashba spin-orbital splitting and four-phonon scattering in strontium-tin-selenium thermoelectrics. Adv. Funct. Mater. 35, 2414288 (2025).
Bai, S. et al. Stacking pattern induced high ZTs in monolayer SnSSe and bilayer SnXY (X/Y = S, Se) materials with strong anharmonic phonon scattering. Chem. Eng. J. 455, 140832 (2023).
Wan, D. et al. Unravelling the regulating role of strain engineering on the phonon dispersion, mechanical behavior, and electronic transport properties of pentagonal PtTe2 monolayer. Vacuum 215, 112343 (2023).
Bu, K. et al. Quantifying structural polarization by continuous regulation of lone-pair electron expression in molecular crystals. J. Am. Chem. Soc. 146, 22469–22475 (2024).
Bai, S. et al. Lattice Vibrational Hierarchy and Mean-Free-Path Filtering in Bi6Cu2Se4O6 Superlattice Thermoelectrics. Phys. Rev. X. https://link.aps.org/doi/10.1103/44gy-zmbf (2025).
Dou, Y. et al. Lone-pair engineering: achieving ultralow lattice thermal conductivity and enhanced thermoelectric performance in al-doped gete-based alloys. Mater. Today Phys. 20, 100497 (2021).
Bai, X. et al. Tailoring 5s² lone pair-antibonding orbital interaction by Zr-doping to realize ultrahigh power factor in thermoelectric GeTe. Chem. Eng. J. 461, 142069 (2023).
Gillespie, R. J. & Hargittai, I. The VSEPR Model of Molecular Geometry (Courier Corporation, 2013).
Jenkins, H. D. B. & Waddington, T. C. Lone electron pairs and stereochemistry. Nature 255, 623–625 (1975).
Gillespie, R. J. & Robinson, E. A. Electron domains and the VSEPR model of molecular geometry. Angew. Chem. Int. Ed. 35, 495–514 (1996).
Wan, D. et al. Synergistic effect of bonding heterogeneity and phonon localization in introducing excellent thermoelectric properties in layered heteroanionic NdZnSbO material. J. Energy Chem. 100, 259–272 (2025).
Wan, D. et al. Rattling-like behavior and band convergence induced ultra-low lattice thermal conductivity in MgAl2Te4 monolayer. J. Mater.10, 1004–1016 (2024).
Tang, S. et al. Enhancing phonon thermal transport in 2H-CrX2 (X= S and Se) monolayers through robust bonding interactions. Phys. Chem. Chem. Phys. 25, 22401–22414 (2023).
Bai, S. et al. Rethinking SnSe thermoelectrics from computational materials science. Acc. Chem. Res. 56, 3065–3075 (2023).
Wan, D. et al. Anharmonicity and weak bonding-driven extraordinary thermoelectric performance in wrinkled snse monolayer with low lattice thermal conductivity. Ceram. Int. 50, 9591–9603 (2024).
Bai, S. et al. Theoretical prediction of thermoelectric performance for layered LaAgOX (X = S, Se) materials in consideration of the four-phonon and multiple carrier scattering processes. Small Methods 7, 2201368 (2023).
Zhao, L. D. et al. BiCuSeO oxyselenides: new promising thermoelectric materials. Energy Environ. Sci. 7, 2900–2924 (2014).
Li, J. et al. A high thermoelectric figure of merit ZT > 1 in Ba heavily doped BiCuSeO oxyselenides. Energy Environ. Sci. 5, 8543–8547 (2012).
Liu, Y. et al. Remarkable enhancement in thermoelectric performance of BiCuSeO by Cu deficiencies. J. Am. Chem. Soc. 133, 20112–20115 (2011).
Zhang, X. et al. BiCuSeO thermoelectrics: an update on recent progress and perspective. Materials 10, 198 (2017).
Lan, J. L. et al. Enhanced thermoelectric properties of Pb-doped BiCuSeO ceramics. Adv. Mater. 25, 5086–5090 (2013).
Zhao, L. D. et al. Bi1-XSrXCuSeO oxyselenides as promising thermoelectric materials. Appl. Phys. Lett. 97, 092118 (2010).
Pei, Y. L. et al. High thermoelectric performance of oxyselenides: intrinsically low thermal conductivity of ca-doped BiCuSeO. NPG Asia Mater. 5, e47 (2013).
Ren, G. K. et al. Complex electronic structure and compositing effect in high performance thermoelectric BiCuSeO. Nat. Commun. 10, 2814 (2019).
Zhu, H. et al. Efficient interlayer charge release for high-performance layered thermoelectrics. Natl. Sci. Rev. 8, nwaa085 (2021).
Kumar, S. et al. Lattice thermal conductivity in layered BiCuSeO. Phys. Chem. Chem. Phys. 108, 19158–19164 (2016).
Parrish, K. D. et al. Origins of thermal conductivity changes in strained crystals. Phys. Rev. B 90, 235201 (2014).
Wei, B. et al. Synergistically optimizing electrical and thermal transport in layered BiCuSeO via biaxial strain modulation. Acta Mater. 286, 120699 (2025).
Tan, R. et al. Enhancing thermoelectric properties of BiCuSeO via uniaxial compressive strain: first-principles calculations. J. Alloy. Compd. 743, 610–617 (2018).
Yin, Z. et al. Ultrahigh-pressure structural modification in BiCuSeO ceramics: dense dislocations and exceptional thermoelectric performance. Adv. Energy Mater. 15, 2403174 (2025).
Das, S. et al. Thermoelectric properties of Sn-doped BiCuSeO. Appl. Surf. Sci. 418, 238–245 (2017).
Jellinek, J. et al. Solid–liquid phase changes in simulated isoenergetic Ar13. J. Chem. Phys. 84, 2783–2794 (1986).
Alder, B. J. et al. Studies in molecular dynamics. viii. the transport coefficients for a hard-sphere fluid. J. Chem. Phys. 53, 3813–3826 (1970).
Manjunatha, L. et al. Atomic-level breakdown of green–kubo relations provides new insight into the mechanisms of thermal conduction. Sci. Rep. 11, 5597 (2021).
Waroquiers, D. et al. Statistical analysis of coordination environments in oxides. Chem. Mater. 29, 8346–8360 (2017).
Schmidt, M. et al. Untersuchungen zu Bismutseltenerdoxidhalogeniden der Zusammensetzung Bi2SeO4X (X = Cl, Br, I). Z. Anorg. Allg. Chem. 626, 125–135 (2000).
Safarik, D. J. et al. Localized anharmonic rattling of Al Atoms in Val10.1. Phys. Rev. B 85, 014103 (2012).
Tang, W. et al. A grid-based bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 21, 084204 (2009).
Savin, A. et al. Electron localization in solid-state structures of the elements: the diamond structure. Angew. Chem. Int. Ed. 31, 187–188 (1992).
Wu, Y. et al. Lattice strain advances thermoelectrics. Joule 3, 1276–1288 (2019).
Klemens, P. G. The scattering of low-frequency lattice waves by static imperfections. Proc. Phys. Soc. A 68, 1113–1128 (1955).
Yue, S. Y. et al. Metric for strong intrinsic fourth-order phonon anharmonicity. Phys. Rev. B 95, 195203 (2017).
Li, F. & Lannin, J. S. Radial distribution function of amorphous carbon. Phys. Rev. Lett. 65, 1905–1908 (1990).
Han, Z. et al. FourPhonon: an extension module to ShengBTE for computing four-phonon scattering rates and thermal conductivity. Comput. Phys. Commun. 270, 108179 (2022).
Li, F. et al. Polycrystalline BiCuSeO oxide as a potential thermoelectric material. Energy Environ. Sci. 5, 7188–7195 (2012).
Pei, Y.-L. High thermoelectric performance of oxyselenides: intrinsically low thermal conductivity of Ca-doped BiCuSeO. NPG Asia Mater. 5, e47 (2013).
Peierls, R. Zur kinetischen theorie der wärmeleitung in kristallen. Ann. Phys. 395, 1055–1101 (1929).
Qin, B. et al. The development and impact of tin selenide on thermoelectrics. Science 386, eadp2444 (2024).
Zhao, L. D. et al. Ultralow thermal conductivity and high thermoelectric figure of merit in SnSe crystals. Nature 508, 373–377 (2014).
Zhao, L. D. et al. Ultrahigh power factor and thermoelectric performance in hole-doped single-crystal SnSe. Science 351, 141–144 (2016).
Zheng, Z. et al. Harvesting waste heat with flexible Bi2Te3 thermoelectric thin film. Nat. Sustain. 6, 180–191 (2023).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P. et al. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Deringer, V. L. et al. Crystal orbital hamilton population (COHP) analysis as projected from plane-wave basis sets. J. Phys. Chem. A 115, 5461–5466 (2011).
Müller, P. C. et al. Crystal orbital bond index: covalent bond orders in solids. J. Phys. Chem. C. 125, 7959–7970 (2021).
Momma, K. & Izumi, F. VESTA: a three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 41, 653–658 (2008).
Togo, A. & Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 108, 1–5 (2015).
Skelton, J. M. et al. Anharmonicity in the high-temperature Cmcm phase of SnSe: soft modes and three-phonon interactions. Phys. Rev. Lett. 117, 075502 (2016).
Podryabinkin, E. V. et al. Accelerating crystal structure prediction by machine-learning interatomic potentials with active learning. Phys. Rev. B 99, 064114 (2019).
Li, W. et al. ShengBTE: a solver of the Boltzmann transport equation for phonons. Comput. Phys. Commun. 185, 1747–1758 (2014).
Wan, D. et al. Dataset for publication “Strain-driven lone pair electron expression for thermal transport in BiCuSeO”, Zenodo, https://doi.org/10.5281/zenodo.15618366 (2025).
Acknowledgements
This research is sponsored by the National Science Fund for Distinguished Young Scholars (52525101), the National Natural Science Foundation of China (52450001, 12104370), Tianmushan Laboratory Research Project (TK2024D006, TK2023C021), the International Cooperation and Exchange of the National Natural Science Foundation of China (52411540237), and the Tecent Xplorer Prize. We appreciate the support of high-performance computational resources at Tianmushan Laboratory and Beihang University.
Author information
Authors and Affiliations
Contributions
Da Wan, Investigation, Data curation, Writing—original draft, Writing—review & editing. Shulin Bai: Investigation, Data curation. Sirui Fan: Investigation. Xiao Xiang: Investigation. Zhen Li: Investigation. Yu Liu: Investigation. Peng Kang, Lei Zheng, Li-Dong Zhao, and Huibin Xu: Writing—original draft, Writing—review & editing, Project administration, Investigation, Funding acquisition, Formal analysis.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ankit Kumar, Illia Serhiienko, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wan, D., Bai, S., Fan, S. et al. Strain-driven lone pair electron expression for thermal transport in BiCuSeO. Nat Commun 16, 6284 (2025). https://doi.org/10.1038/s41467-025-61506-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61506-6
