
Abstract
Lattice oxygen-mediated photocatalytic ethane dehydrogenation represents a sustainable strategy for ethylene production, yet achieving a balance between high productivity, selectivity, and durability remains challenging. Here, we report a defective NiO-300 catalyst, where precisely engineered Ni vacancies activate lattice oxygen by weakening Ni–O bond and improving lattice oxygen mobility. This promotes efficient ethane activation and C–H bonds cleavage through photoinduced hole capture, intensifying ethane dehydrogenation via a light-boosted Mars-van Krevelen mechanism. The NiO-300 catalyst manifests a high ethylene yield of 604.5 μmol g−1 h−1 with 100% selectivity and stability over 200 cycles. In situ spectroscopic and theoretical studies elucidate the generation of active oxygen species, the evolution of Ni coordination, the formation of key intermediates, and the underlying photocatalytic mechanism. Our findings highlight cation vacancy engineering as a powerful tactic to fully activate lattice oxygen for solar-driven alkene production from alkane dehydrogenation over oxide photocatalysts.
Similar content being viewed by others

Introduction
Ethylene (C2H4), a valuable feedstock in the modern chemical industry, is produced mainly from steam cracking of naphtha or hydrocarbons at high temperatures (>800 °C), which consumes intensive energy (i.e., fossil fuel) with huge CO2 emissions1,2. Alternatively, the discovery of shale gas containing abundant ethane (C2H6) has motivated growing interest in ethane dehydrogenation (EDH) for ethylene production3. Thermal catalytic EDH with molecular O2 is thermodynamically favorable and typically endows high ethane conversion at reduced temperatures (ca. 500 °C); however, it usually encounters low ethylene selectivity, due to easy overoxidation of ethylene by O24,5. Moreover, the co-feeding of C2H6 and O2 poses safety risks due to the flammability of the gas mixture, necessitating on-site O2 supply through costly air separation units6,7. In comparison, the thermocatalytic nonoxidative EDH approach is advantageous in preventing overoxidation, yet it suffers from severe catalyst deactivation induced by coking and/or sintering8,9. It is therefore extremely desirable to explore improved tactics to promote selective EDH to ethylene, especially those driven by sustainable energy.
Semiconductor photocatalysis has recently shown enormous prospects in alkane dehydrogenation for alkene production, enabling alkene production under milder conditions by overcoming thermodynamic limitations10,11,12. In this process, the lattice oxygen (OL) of oxide catalysts, acting as a solid oxygen carrier, captures photo-generated holes and converts into reactive oxygen species (O•–), facilitating C–H bond activation/cleavage and catalyzes EDH eventually via a light-enhanced Mars-van Krevelen (MvK) mechanism13,14,15. Such an OL-mediated pathway circumvents direct O2 participation, ensuring high ethylene selectivity16. However, the insufficient availability and poor refilling ability of active OL severely hinder the practical application of photocatalytic ethane-to-ethylene conversion17. Given that the catalytic function of OL is closely linked to its coordination environment, modifying the atomic arrangement around OL may optimize its activity18,19,20. Bearing this in mind, we propose that introducing metal vacancies to reduce the coordination number of OL (i.e., effectively weakening the strength of metal-oxygen bond) can increase the proportion of active OL, reinforcing the OL-mediated EDH process. Coupled with a rationally designed reaction system, this approach is expected to achieve selective and durable ethylene photosynthesis. Notably, recent progress in thermocatalytic anaerobic methane oxidation supports this concept, demonstrating that introducing cation vacancies in perovskite ferrites enhances oxygen mobility, lowers electron density over oxygen, and reduces the barrier for C–H bond cleavage, thereby promoting carbon oxidation21.
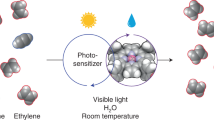
Herein, we demonstrate that utilizing nickel oxide (NiO) as a model catalyst, strategically introducing Ni vacancies to modulate OL coordination significantly enhances photocatalytic EDH, achieving high ethylene selectivity and recyclability. The NiO catalysts with varied concentrations of Ni defects were prepared via a precipitation-calcination method. It is validated that the formation of Ni vacancies endows NiO preferred physicochemical properties and rich active sites for ethane adsorption/activation by modifying the localized electronic configuration. Furthermore, the cation vacancies give OL a larger degree of freedom to escape from lattice constraints for operating the EDH reaction. Photocatalytic EDH experiments show that the optimal NiO-300 catalyst with dense Ni vacancies delivers a C2H4 production of 105.6 μmol g–1 h–1 in a batch reactor, about 3.5 times that of NiO-500 counterpart with sterile Ni vacancies. Remarkably, when catalyzing EDH in a continuous flow system, NiO-300 manifests a C2H4 yield of 604.5 μmol g–1 h–1 with 100% selectivity. After facile regeneration, NiO-300 exhibits long-term durability, presenting a self-healing ability in 200-round reaction cycles without any coke formation. Both theoretical and experimental studies confirm that after reaction the partially reduced NiO-300 catalyst revives naturally in the air by refilling the consumed OL with molecular O2, regaining the original activity, which indeed promises the potential of industrial application after optimizing the reaction system.
Results
Catalyst synthesis and characterizations
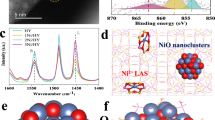
Three NiO-x catalysts (x = 300, 400, and 500) with different concentrations of Ni defects were synthesized. Temperature-dependent operando X-ray diffraction (XRD) confirms the formation of cubic NiO (PDF#47-1049) at temperatures above 250 °C (Fig. 1a), which is reaffirmed by powder XRD (Supplementary Fig. 1)22. The electron paramagnetic resonance (EPR) spectra of NiO-x show distinct signals (g = 2.24, Fig. 1b), confirming the existence of Ni vacancies23, whose amount decreases with the increase in calcination temperature. X-ray photoelectron spectroscopy (XPS) analysis validates the surface chemical composition (i.e., Ni and O) and the presence of Ni3+ and Ni2+ cations (Supplementary Fig. 2). A detailed analysis reveals that NiO-300 possesses a higher Ni3+/Ni2+ ratio compared to NiO-400 and NiO-500, indicating a denser concentration of cation vacancies in NiO-300, while the oxygen vacancy (Ov) concentrations across the NiO-x samples remain nearly identical. This reveals that the calcination temperature primarily influences the formation of Ni vacancies rather than Ov in NiO-x19,24.

a Temperature-dependent operando XRD. b The EPR spectra. c PDF. d Normalized Ni K-edge XANES spectra. e FT k2χ(k) Ni K-edge EXAFS spectra. f The WT for the k2-weighted Ni K-edge EXAFS. g HAADF-STEM image of NiO-300. h The two-dimensional distribution of atomic contrast intensity in the HAADF-STEM image. i The enlarged dotted rhombic area in Fig. 1h. Source data are provided as a Source Data file.
UV-vis-NIR diffuse reflectance spectroscopy (DRS) spectra present that NiO-x catalysts possess strong and broad optical absorption from the UV to the near-IR region (Supplementary Fig. 3a). This wide-range light absorption endows NiO-x with unique photothermal conversion ability, enabling efficient utilization of full-spectrum solar energy to promote the EDH reaction25. Combined with the Tauc plots (Supplementary Fig. 3b) and the Mott-Schottky curves (Supplementary Fig. 4), the band structures of NiO-x are determined (Supplementary Fig. 5). Moreover, Raman analysis exposes that Ni defects increase the interatomic deformation of NiO-x (Supplementary Fig. 6). The pair distribution function (PDF) of NiO-x displays the interactions between Ni–O and Ni–Ni at 2.08 Å and 2.95 Å, respectively (Fig. 1c), whilst the peak intensities of NiO-300 are drastically lower, indicating the weaker bond strength between Ni and neighboring O atoms26.
X-ray absorption spectroscopy (XAS) unveils the edge positions of NiO-x change to the higher energies, compared with that of NiO reference (NiO-ref.), indicating that the formation of cation vacancies in NiO-x leads to the higher oxidation states of Ni (Fig. 1d and Supplementary Fig. 7). In the Fourier-transformed EXAFS (FT-EXAFS) spectra (Fig. 1e), the peaks at 1.64 Å and 2.58 Å are attributed to the first shell Ni–O and the second shell Ni–Ni scattering, respectively27,28. Comparatively, the weaker intensity of Ni–O coordination in NiO-300 suggests that it should hold a reduced constraint of OL and hence the enhanced OL mobility to participate in oxidative reactions (Fig. 1e and Supplementary Fig. 8). Also, the wavelet transformation (WT) proves the weakened Ni–Ni and Ni–O strength in NiO-300, compared to those in the counterparts (Fig. 1f and Supplementary Fig. 9)29.
Transmission electron microscopy (TEM) determines the morphologies and microstructures of NiO-x, among with NiO-300 exhibits the smaller particle size. This makes the coordination of the surface atoms unsaturated, reducing the average coordination number30,31, as verified in the EXAFS fitting parameters at the Ni K-edge for various samples (Supplementary Table 1), which is conducive to exposing more active sites on the surface of NiO-30032. The high-resolution TEM (HRTEM) image presents apparent lattice deformation of NiO-300 (Supplementary Fig. 10), owing to the presence of rich Ni vacancies33. The visualization of Ni defects is confirmed by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). The atomic scatterings of Ni vacancies (dotted red circles) can be discerned clearly (Fig. 1g and Supplementary Fig. 11), which are also exposed by the two-dimensional distribution of atomic contrast intensity (Fig. 1h)23,34. A detailed analysis of the two-dimensional distribution plot shows obvious lattice distortion near the Ni vacancy (Fig. 1i). However, these phenomena are quite moderate over NiO-500 with fewer cation defects (Supplementary Fig. 12). The above results visibly reveal the construction of profuse cation vacancies and the thus-induced changes in the local coordination of OL in NiO-300.
Density functional theory (DFT) calculations disclose that cation vacancy is thermodynamically more stable than anion vacancy in NiO (Supplementary Fig. 13). The local atomic configurations show that Ni defect decreases the coordination number of neighboring OL and induces a lattice distortion (Fig. 2a and Supplementary Fig. 14), forming coordination-unsaturated OL that may possess a higher degree of freedom to escape from the lattice35. Further, the lower Ov formation energy of NiO with Ni vacancy (v-NiO) (0.14 eV) compared to pure NiO (0.65 eV) confirms that the presence of Ni defects facilitates the formation and mobility of OL, thus endowing OL with a higher degree of freedom to escape from the lattice (Supplementary Fig. 15). The pair-correlation function (PCF) derived from ab initio molecular dynamics (AIMD) simulations were performed to explore the structural stabilities of v-NiO and pure NiO. It is revealed that v-NiO exhibits a lower PCF intensity compared to that of pure NiO at the same temperature (Fig. 2b and Supplementary Fig. 16) indicating weaker metal-oxygen interaction and increased structural disorder in v-NiO, although the OL atoms are mainly stabilized around their positions36.

a Local atomic configuration of NiO (110) model without (top) and with (bottom) a Ni vacancy. b PCF of v-NiO at different temperature ranges. c and (d) The phonon spectra of (c) v-NiO and (d) pure NiO. e Calculated band structure of v-NiO. f The PDOS of v-NiO. Source data are provided as a Source Data file.
In the phonon spectra (Fig. 2c, d), v-NiO gives several imaginary frequencies of non-harmonic vibrations, suggesting the metastable property of v-NiO relative to pure NiO, which may arouse its OL deviating from the equilibrium position37. The calculated band structure describes a reduced band gap of v-NiO, due to the impurity level (Fig. 2e and Supplementary Fig. 17), which will enhance light absorption and charge separation for photocatalysis38. The projected density of states (PDOS) indicates that the O 2p-orbital and Ni 3d-orbital mainly contribute to the valence band (VB) maximum (Fig. 2f and Supplementary Fig. 18), while the Ni d-orbital donates the conduction band (CB) minimum39. This means the photogenerated holes in VB will gather predominately around O, preferably generating the active O•– species for adsorbing ethane and activating its C–H bond, and the photoexcited electrons in CB tend to migrate to Ni sites. The charge density difference analysis reveals a stronger interaction between v-NiO and adsorbed C2H6 (Supplementary Fig. 19), revealing the desired feature of v-NiO for ethane conversion reaction. Such theoretical findings highlight the modified coordination of OL after forming Ni vacancies, which could ultimately supply more active OL for the photocatalytic EDH reaction.
Photocatalytic alkane dehydrogenation performance
Photocatalytic EDH performance of the catalysts was first examined in a batch reactor, mainly aiming to gain insights into the reaction mechanism. The as-prepared NiO-x catalysts are all active to the EDH reaction to produce C2H4, without generating other carbonaceous products except CH4. Specifically, NiO-300 shows a C2H4 evolution of 105.6 μmol g–1 h–1 (Fig. 3a), which is about 3.5 and 50 times that of NiO-500 and commercial NiO (NiO-c), respectively. A high ethylene selectivity of 93% is achieved over NiO-300, together with an ethane conversion of up to 45.7%. The NiO-300 catalyst displays deactivation in continuously operating the EDH reaction for 3 cycles (Supplementary Fig. 20), due to the consumption of OL in the photo-reinforced MvK process40. However, the used NiO-300 regains its original activity after regeneration via contact with the air to refill the consumed OL by O2; and it exhibits no deactivation in successive 20 cycles (Supplementary Fig. 21), with its color restored almost identical to the fresh sample (Supplementary Fig. 22). Meanwhile, XRD, XPS and XAS analyses confirm that the regenerated NiO-300 experiences no changes in the crystal, surface and coordinating structures (Supplementary Fig. 23). These results emphasize the fine reusability of NiO-300 and the refillable characteristic of its OL toward the EDH reaction.

a Photocatalytic EDH performance of NiO-x in a batch reactor. b EDH performance of NiO-300 under varied conditions. c GC-MS spectra of ethylene produced from 13CH312CH3. d GC-MS spectra of water produced from the spent NiO-300 regenerated by 18O2. e Photocatalytic EDH abilities of NiO-300 under different alkane concentrations. f Photocatalytic EDH performance of NiO-300 at different GHSV in a continuous flow system. g Stability tests of NiO-300 in a continuous flow system (the durations for EDH reaction, Ar purge, and air regeneration in each cycle are all 5 min, respectively, GHSV = 3000 mL g–1 h–1). Error bars represent standard deviations obtained from three independent measurements. Source data are provided as a Source Data file.
Wavelength-dependent experiments show that the C2H4 yield decreases with the wavelength from full-spectrum to 760 nm, which emphasizes that light is the essential driving force for the EDH reaction (Supplementary Fig. 24). Moreover, no C2H4 generates when the system is absent of NiO-300, C2H6, or photoirradiation (Fig. 3b, columns 1-3), confirming that the EDH reaction proceeds photocatalytically by light excitation of the catalyst. The surface temperature of NiO-300 reaches about 280 °C under light irradiation (Supplementary Fig. 25); however, the EDH reaction is terminated completely when operated in the dark at 300 °C (Fig. 3b, column 4). This authorizes that the EDH reaction is driven by the photocatalysis effect of the NiO-300 catalyst. Despite this, the relatively high temperature should facilitate mass transfer and modulate the catalyst’s lattice dynamics, thus contributing to the reaction41. In addition, engaging O2 into the reaction system leads to substantial decreases in both ethylene yield and selectivity (Fig. 3b, column 5), underlining the advantages of OL in mediating the preferential EDH reaction.
Gas chromatography-mass spectrometer (GC-MS) analysis on ethylene generated using 13CH312CH3 validates that the C2H4 product stems from C2H6 (Fig. 3c). After the reaction, the used NiO-300 catalyst was exposed to 18O2 and then re-applied to catalyze the EDH reaction, with the formed water analyzed by GC-MS. The water product presents definite fragments of H218O (Fig. 3d), proving directly the participation of OL and the refilling of consumed OL by molecular O2.
As revealed above, NiO-300 photocatalyst is efficient to EDH in the batch reactor. Further, to demonstrate the industrial promise of the light-driven EDH strategy for ethylene production, the performance of NiO-300 was examined in a continuous flow system (Supplementary Fig. 26). Strikingly, NiO-300 shows much enhanced EDH performance with 100% C2H4 selectivity at different feedstock concentrations (Fig. 3e, columns 1-3). To be specific, it delivers a high C2H4 yield of 604.5 μmol g–1 h–1 and a reaction conversion of ca. 28.0% using 1 vol.% C2H6 at a gas hourly space velocity (GHSV) of 6000 mL g–1 h–1, corresponding to a carbon balance of nearly 100%. When using the concentrated feedstocks, the C2H4 productivity further upgrades, achieving the highest value of 2129.3 μmol g–1 h–1 in 30 vol.% C2H6. The high EDH efficiency and selectivity are attributed to the promoted reaction kinetics that powerfully blocks ethylene overoxidation. Also, NiO-300 can dehydrogenate propane efficiently, affording 100% propylene in a yield of 1098.5 μmol g–1 h–1 (Fig. 3e, column 4). Moreover, NiO-300 exhibits 100% ethylene selectivity persistently at varied GHSVs (Fig. 3f), affording a maximum ethane conversion of ca. 50.1%. However, in a continuous O2 and C2H6 co-intake system, CO2 and CH4 become the predominant products (Supplementary Fig. 27), which suggests that the chemical looping mode that utilizes OL for selective C–H activation circumvents overoxidation by the decoupling oxidation and dehydrogenation steps, thus ensuring high C2H4 selectivity. The NiO-300 catalyst, operating without direct O2 engagement and without the need for cocatalysts, demonstrates the state-of-the-art performance for photocatalytic EDH—achieving high ethylene yield and unity selectivity, along with exceptional durability over 200 cycles (Supplementary Table 2). These results stress the vast promise of NiO-300 catalyst for converting the low-carbon alkanes to olefines by light.
The NiO-300 catalyst can keep its intrinsic performance over 200 cycles (Fig. 3g), under the condition of simplistic and quick renewal by exposure to the air after each cycle, manifesting its robust durability for EDH in the continuous flow system. The accumulation of C2H4 in this durability test realizes ca. 1747.6 μmol, delivering a turnover number (TON) of 92 relative to the catalyst. After the reaction, Raman spectroscopy approves no carbon formation on the catalyst (Supplementary Fig. 28), revealing the supremacy of the photocatalytic EDH technique in resisting coke deposition, which is hard to accomplish in thermocatalytic anaerobic alkane conversion42. Moreover, TEM and AC-STEM analyses find no obvious changes in the particle size or morphology of NiO-300 after multiple regeneration cycles (Supplementary Fig. 29), proving its structural robustness during the “reaction-regeneration” cycles.
Definitely, the OL of NiO-300 has proved to be refillable for durably mediating the EDH reaction. Nevertheless, such a refillable property of OL is dependent on its appropriate participation in the reaction process. That is, if the NiO-300 catalyst is employed excessively for running the EDH reaction, but without timely regeneration, the OL will become unrecoverable. For instance, after catalyzing the EDH reaction for 10 cycles using the “reaction-regeneration” method, the used NiO-300 catalyst was directly applied to the reaction without exposure to air between cycles. This leads to significantly deteriorated performance, with CO2 and CH4 becoming the dominant products, accompanied by the reduction of the catalyst to form Ni particles. This phase transformation is irreversible, even after re-calcination in air, indicating permanent catalyst degradation (Supplementary Fig. 30). These findings however reflect that OL of NiO-300 is the active site for mediating the selective EDH reaction, rather than the metallic Ni species.
Mechanistic investigations
Temperature-programmed reduction of hydrogen (H2-TPR) indicates that NiO-300 presents the reduction peak at a lower temperature than NiO-400 and NiO-500 (Supplementary Fig. 31a), reflecting its weaker strength of Ni–O bonds that should permit higher mobility of OL43. Consistently, in the profiles of temperature-programmed desorption of oxygen (O2-TPD) (Supplementary Fig. 31b), NiO-300 appears a larger desorption peak than those of NiO-400 and NiO-500, which mirrors that the former possesses much more portable OL to work the oxidative EDH reaction44. Further, C2H6-TPD verifies the markedly higher ethane adsorption of NiO-300 than those of the counterparts (Supplementary Fig. 31c)36,45. Moreover, the NiO-300 affords a higher photocurrent response than NiO-400 and NiO-500 (Supplementary Fig. 32a), mirroring the quicker charge transport of NiO-300, which matches with its lower electronic resistance revealed by electrochemical impedance spectra (EIS) (Supplementary Fig. 32b). The steady-state photoluminescence (PL) and the time-resolved PL (TRPL) spectra show that NiO-300 manifests the reduced PL intensity and a longer lifetime compared with the counterparts (Supplementary Fig. 32c, d). These outcomes underline that constructing Ni vacancies exactly in NiO facilitates the mobility and enriches the numbers of active OL as well as accelerates the migration and separation of charge carriers to boost photocatalytic EDH reaction39,46.
In situ EPR experiments were conducted to verify the formation of reactive oxygen (O•–) species by OL upon capturing photogenerated holes, as well as to assess their reactivity. No signal attributable to O•– species are detected for NiO-300 under dark conditions in an Ar atmosphere (Fig. 4a and Supplementary Fig. 33). Upon light irradiation, new paramagnetic signals emerged at g = 2.027, 2.019, and 2.002, which are characteristic of O•– species generated by photoexcited holes trapped by OL of NiO12,47. Notably, when the reaction atmosphere was switched from Ar to C2H6 under illumination, the intensity of these O•– signals decreases significantly. This observation suggests that the O•⁻ species actively react with ethane molecules, confirming their critical role in C–H bond activation48,49. These findings demonstrate that surface O•– species, derived from OL, are essential for light-driven ethane activation and conversion.

a In situ EPR spectra of NiO-300 in the dark field with an Ar atmosphere and under light illumination with an Ar or C2H6 atmosphere. b and (c) In situ XAFS of NiO-300 in ethane under light irradiation (b) the normalized Ni K-edge spectra and (c) the FT k2χ(k) Ni K-edge EXAFS spectra. d Operando DRIFTS spectra of NiO-300 in ethane under light irradiation for different durations. e The calculated potential free-energy diagrams of NiO in EDH reaction. f The corresponding intermediate configurations (side views) of potential free-energy diagrams of v-NiO in Fig. 4e. Source data are provided as a Source Data file.
In situ XAS measurements were manipulated to monitor the coordination environment of Ni during EDH using a custom-designed in situ photocatalytic cell, which allows for the real-time detection of the evolution of Ni species under illumination and in the presence of a C2H6 atmosphere50. The normalized XANES spectra of NiO-300 disclose that the Ni K-edge moves to the lower binding energies with the prolongation of light irradiation (Fig. 4b), which indicates the decreased Ni valence along with the consumption of OL in the EDH reaction. In the FT-EXAFS spectra (Fig. 4c), the intensity of the Ni–O (1.64 Å) peak decreases gradually with light irradiation, which correlates to the consumption of OL. To be specific, the photoinduced holes being captured by OL promotes the formation of active O•– species that reinforce ethane activation and transformation; as the reaction proceeds, the active OL is consumed progressively, leading to the diminution in Ni–O coordination number. Meanwhile, the strength of the Ni–Ni (2.58 Å) peak increases consistently according to the irradiation, which substantiates the rise in the coordination number of Ni–Ni, owing to the vanishing of OL51. In contrast, the Ni K-edge of NiO-500 also shifts to the lower binding energies upon light irradiation (Supplementary Fig. 34a). However, the intensities of Ni–O and Ni–Ni signals remain almost unchanged after photoirradiation (Supplementary Fig. 34b), due to much less Ni vacancies that makes its OL with too low mobility to contribute the activation and conversion of ethane.
Operando diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was employed to identify the key intermediates engaged in the photocatalytic EDH reaction (Supplementary Table 3). Being illuminated with light (Fig. 4d), the detection of vibrations attributable to C–H (2975 and 2879 cm–1) and CH2 (2970 cm–1) bonds suggests the generation of *CH3–CH2 and *CH2–CH2 moieties, which are created from the dissociation of activated ethane molecules52. Besides, the involvements of these species are also supported by the definite signals corresponding to CH3 (1330 cm–1) and C–C (1085 and 1045 cm–1) moieties53. Particularly, the bands at 1650, 1630 and 1540 cm–1 are indexed to the featured vibrations of C = C bonds, which illustrates the formation of CH2 = CH2 in an adsorbed state12. On the other hand, the gradually enhanced band (3665−3360 cm–1) and vibration (1506 cm–1) point to the generation of OH species, designating the accumulation of H2O during the EDH reaction.
DFT calculations were carried out to reveal the thermodynamic property of the EDH reaction (Fig. 4e, f and Supplementary Fig. 35). The adsorption energy of *C2H6 on v-NiO is slightly superior to that of NiO (I to II), due to the existence of Ni vacancies. Then, both v-NiO and NiO can extract α–H of adsorbed ethane through spontaneous downhill processes by using OL to form hydroxyl (II to III). Strikingly, the extraction of β–H over v-NiO is a thermodynamically favorable process (III to IV), while that over pure NiO requires energy of 0.26 eV, depicting the significance of Ni vacancies-triggered OL in activating the C–H bond of ethane. Afterward, v-NiO and NiO show minor differences in energy requirement for desorbing C2H4 (IV to V) and adsorbing the second ethane (V to VI). However, for abstracting α–H of the second ethane (VI to VII), v-NiO requires a much lower energy (0.21 eV) than NiO (1.21 eV), further exposing the merit of OL in v-NiO for the EDH reaction. In the next steps (VII-XI), OL of the catalysts will be consumed with the release of H2O and CH2 = CH2, due to the strong hydrogen adsorption ability of OL. Significantly, the recovery and replenishment of consumed OL with molecular O2 over v-NiO and NiO are extremely favorable processes in thermodynamics (XI to XIII), consistent with the excellent regeneration ability of used NiO-300 catalyst after exposure to O2 in the air.
Based on the findings of in situ EPR, in situ XAS, operando DRIFTS and DFT calculations, the photocatalytic EDH mechanism over NiO-300 was proposed (Fig. 5). Initially, C2H6 is adsorbed on the surface of NiO-300 catalyst (step I). Upon light irradiation, the photoexcited holes are trapped by OL to produce active O•– species, which empowers H abstraction from adsorbed ethane, yielding the C2H5/C2H4 fractions and OH species. Concurrently, the photoinduced electrons transfer to Ni species, resulting in partial reduction of the catalyst (steps II and III). Afterward, the adsorbed C2H4 desorb from the catalyst surface to generate gaseous ethylene (step IV), and the remaining OH species combine to form H2O, accompanied by spending of OL and creation of Ov (step V). Lastly, by contacting the air, the molecular O2 fulfills the Ov to render the recovery of OL, thus regenerating the catalyst and finishing the catalytic loop (step VI)54,55,56.

The proposed photocatalytic EDH mechanism over NiO-300.
Discussion
In summary, the activity of lattice oxygen (OL) in NiO is triggered by precisely constructing Ni vacancies to tune the coordination environment, resulting in high activity, unity selectivity and sustained durability for photocatalytic ethylene production from ethane in a continuous flow system. The creation of Ni vacancies reinforces light harvesting and promotes charge separation, and importantly, enhances the mobility of OL to escape from the lattice constraint for reaction. Upon photoexcitation, OL accepts the photoinduced holes to form active O•– species, which mediates ethane dehydrogenation via a light-enhanced Mars-van Krevelen mechanism. The consumed OL is refilled spontaneously by molecular O2 from the air, allowing for easy catalyst regeneration and ensuring durability over 200 cycles. The mechanism of OL-mediated EDH is exposed through in situ EPR, in situ XAS, operando DRIFTS and DFT calculations. This study may inspire great interest in solar-driven dehydrogenation of low-carbon alkanes for alkene production over oxide catalysts by cation vacancy engineering to maximally trigger the reactivity of OL.
Methods
Materials
Nickel(II) chloride hexahydrate (NiCl2·6H2O, ≥99.9%), ammonium bicarbonate (NH4HCO3, ≥99.0%) and commercial nickel(II) oxide (NiO, ≥99.9% metals basis) were purchased from were purchased from Aladdin Ltd. Ethane gas with different concentrations (x vol.%C2H6/Ar), propane (5 vol.% C3H8/Ar) and oxygen (20 vol.% O2/Ar) were supplied by Dalian Special Gases Co., Ltd. 13CH312CH3 (98% enriched) gas was provided by Cambridge Isotope Laboratories, Inc. 18O2 (98% enriched) gas was purchased from Wuhan Isotope Technology Co., Ltd. All materials were used without further purification. Deionized water was used in the synthesis of all catalysts.
Synthesis of NiO-300
The NiO-300 catalyst was synthesized through aqueous-phase co-precipitation. Stoichiometric amounts of NiCl2·6H2O (0.2377 g, 1 mmol) and NH4HCO3 (0.1976 g, 2.5 mmol) were separately dissolved in 50 mL of deionized H2O (Caution: NH4HCO3 decomposition releases NH3 gas—perform this procedure in a fume hood). The two solutions were combined dropwise under vigorous stirring and aged for 2 h. The resulting light green precipitate was washed several times with H2O and dried under vacuum at 60 °C overnight. Thermal treatment was then conducted in a muffle furnace by heating the dried powder in an alumina crucible to 300 °C at a rate of 2 °C min−1 under static air, followed by holding at the target temperature for 60 min. Catalyst yields are around 54%.
Synthesis of NiO-400
The NiO-400 catalyst was synthesized via the same co-precipitation method using NiCl2·6H2O (0.2377 g) and NH4HCO3 (0.1976 g), following identical procedures. The resulting precursor was calcined at 400 °C (2 °C min⁻¹ ramp, 1 h dwell), affording the NiO-400 catalyst with a yield of around 55%.
Synthesis of NiO-500
For the NiO-500 catalyst, an identical co-precipitation protocol was employed using NiCl2·6H₂O (0.2377 g) and NH4HCO3 (0.1976 g). The dried precursor was calcined at 500 °C (2 °C min−1 ramp, 1 h dwell) to obtain the NiO-500 catalyst (Catalyst yield is around 50%).
Batch-reactor photocatalytic EDH reaction
A mixture of 100 mg of catalyst and 1 mL of H2O was dispersed evenly on a quartz disk, followed by heating at 100 °C for 20 min to get a catalyst film. Then, the catalyst film-loaded quartz disk was fixed in a gas-closed glass reactor (120 mL in volume) with a quartz window on the top to harvest light irradiation. Afterward, the reactor was vacuumed to get an anaerobic environment, followed by introducing the feedstock gas (i.e., 1 vol.% C2H6/Ar) with a partial pressure of 1 atm. A 300 W Xe lamp with a light intensity of 1.2 W cm−2 was used as the light source. After 30 min, the gas products were analyzed and quantified by a gas chromatograph (GC) equipped with a methanizer and flame ionization detector.
Continues flow system photocatalytic EDH reaction
For the typical reaction cycle, 100 mg of catalyst was mixed with 250 mg of quartz sand (AR, 50-80 mesh) and loaded into the quartz reactor of the continuous flow system. Then, an Ar flow was used as purge gas to remove air in the reaction system at a gas hourly space velocity (GHSV) of 9000 mL g−1 h−1. After that, the purge gas was turned off and the feedstock gas (i.e., 1 vol.% C2H6/Ar) was introduced at certain GHSVs (i.e., 3000, 6000 or 9000 mL g−1 h−1) to start photocatalytic EDH reaction for 5 min, using a 300 W Xe lamp (light intensity: 1.2 W cm−2) as the light source. The generated products were analyzed and quantified by a GC equipped with a thermal conductivity detector and flame ionization detector. After the EDH reaction, the feedstock gas was turned off and an air flow (i.e., 9000 mL g−1 h−1) was introduced to the quartz reactor for 5 min to regenerate the catalyst.
Data availability
The data that support the findings of this study are all available within the paper and its Supplementary Information. Atomic coordinates of the optimized structure and molecular dynamics trajectories of the initial and final configurations are provided in Supplementary Data. Source data are provided with this paper.
References
McFarland, E. Unconventional chemistry for unconventional natural gas. Science 338, 340–342 (2012).
Gärtner, C. A., Veen, A. C. V. & Lercher, J. A. Oxidative dehydrogenation of ethane on dynamically rearranging supported chloride catalysts. J. Am. Chem. Soc. 136, 12691–12701 (2014).
Li, X., Pei, C. & Gong, J. Shale gas revolution: catalytic conversion of C1-C3 light alkanes to value-added chemicals. Chem 7, 1755–1801 (2021).
Pan, Y. et al. Titanium silicalite-1 nanosheet-supported platinum for non-oxidative ethane dehydrogenation. ACS Catal. 11, 9970–9985 (2021).
Yao, R., Herrera, J. E., Chen, L. & Chin, Y. H. C. Generalized mechanistic framework for ethane dehydrogenation and oxidative dehydrogenation on molybdenum oxide catalysts. ACS Catal. 10, 6952–6968 (2020).
Jin, R. et al. Low temperature oxidation of ethane to oxygenates by oxygen over Iridium-cluster catalysts. J. Am. Chem. Soc. 141, 18921–18925 (2019).
Wang, W. et al. Tandem propane dehydrogenation and surface oxidation catalysts for selective propylene synthesis. Science 381, 886–890 (2023).
Lian, Z. et al. Revealing the janus character of the coke precursor in the propane direct dehydrogenation on Pt catalysts from a kMC simulation. ACS Catal. 8, 4694–4704 (2018).
Ping, L. et al. Unraveling the surface state evolution of IrO2 in ethane chemical looping oxidative dehydrogenation. ACS Catal. 13, 1381–1399 (2023).
Hu, Z. et al. High-rate and selective C2H6-to-C2H4 photodehydrogenation enabled by partially oxidized Pdδ+ species anchored on ZnO nanosheets under mild conditions. J. Am. Chem. Soc. 146, 16490–16498 (2024).
Song, R. et al. Ethylene production via photocatalytic dehydrogenation of ethane using LaMn1−xCuxO3. Nat. Energy 9, 750–760 (2024).
Wang, P. et al. Photocatalytic ethylene production by oxidative dehydrogenation of ethane with dioxygen on ZnO-supported PdZn intermetallic nanoparticles. Nat. Commun. 15, 789 (2024).
Wang, X. et al. High-throughput oxygen chemical potential engineering of perovskite oxides for chemical looping applications. Energy Environ. Sci. 15, 1512–1528 (2022).
Li, Z. et al. Lattice oxygen in photocatalytic gas-solid reactions: participator vs. dominator. Angew. Chem. Int. Ed. 63, e202409876 (2024).
Wang, C. et al. Near 100% ethene selectivity achieved by tailoring dual active sites to isolate dehydrogenation and oxidation. Nat. Commun. 12, 5447 (2021).
Luongo, G. et al. Highly selective oxidative dehydrogenation of ethane to ethylene via chemical looping with oxygen uncoupling through structural engineering of the oxygen carrier. Adv. Energy Mater. 12, 2200405 (2022).
Gan, T. et al. “Atomic topping” of MnOx on Al2O3 to create electron-rich, aperiodic, lattice oxygens that resemble noble metals for catalytic oxidation. J. Am. Chem. Soc. 146, 16549–16557 (2024).
Han, W. K. et al. Activating lattice oxygen in layered lithium oxides through cation vacancies for enhanced urea electrolysis. Angew. Chem. Int. Ed. 61, e202206050 (2022).
Mochizuki, C. et al. Defective NiO as a stabilizer for Au single-atom catalysts. ACS Catal. 12, 6149–6158 (2022).
Liu, X. et al. Tunable cationic vacancies of cobalt oxides for efficient electrocatalysis in Li-O2 batteries. Adv. Energy Mater. 10, 2001415 (2020).
He, J. et al. Subsurface A-site vacancy activates lattice oxygen in perovskite ferrites for methane anaerobic oxidation to syngas. Nat. Commun. 15, 5422 (2024).
Kou, T. et al. Carbon doping switching on the hydrogen adsorption activity of NiO for hydrogen evolution reaction. Nat. Commun. 11, 590 (2020).
Zhou, Z. et al. Cation-vacancy-enriched nickel phosphide for efficient electrosynthesis of hydrogen peroxides. Adv. Mater. 34, 2106541 (2022).
Wang, Z. et al. Copper-nickel nitride nanosheets as efficient bifunctional catalysts for hydrazine-assisted electrolytic hydrogen production. Adv. Energy Mater. 9, 1900390 (2019).
Yuan, J.-P. et al. Modeling the enzyme specificity by molecular cages through regulating reactive oxygen species evolution. Angew. Chem. Int. Ed. 62, e202303896 (2023).
Yan, Y. et al. Atomic-level platinum filling into Ni-vacancies of dual-deficient NiO for boosting electrocatalytic hydrogen evolution. Adv. Energy Mater. 12, 2270099 (2022).
Li, H. et al. Highly dispersed NiO clusters induced electron delocalization of Ni-N-C catalysts for enhanced CO2 electroreduction. Adv. Funct. Mater. 33, 2208622 (2023).
Sun, H. et al. Atomic metal-support interaction enables reconstruction-free dual-site electrocatalyst. J. Am. Chem. Soc. 144, 1174–1186 (2022).
Pei, Z. et al. Atomically dispersed Ni activates adjacent Ce sites for enhanced electrocatalytic oxygen evolution activity. Sci. Adv. 9, eadh1320 (2023).
Xue, S. et al. Enriching surface-ordered defects on WO3 for photocatalytic CO2-to-CH4 conversion by water. Proc. Natl Acad. Sci. Usa. 121, e2319751121 (2024).
Huang, Y. et al. Size-dependent selectivity of electrochemical CO2 reduction on converted In2O3 nanocrystals. Angew. Chem. Int. Ed. 60, 15844–15848 (2021).
Wu, T., Han, M. Y. & Xu, Z. J. Size effects of electrocatalysts: more than a variation of surface area. ACS Nano 16, 8531–8539 (2022).
Chen, W. et al. Vacancy-induced catalytic mechanism for alcohol electrooxidation on nickel-based electrocatalyst. Angew. Chem. Int. Ed. 63, e202316449 (2024).
Roy, C. et al. Impact of nanoparticle size and lattice oxygen on water oxidation on NiFeOxHy. Nat. Catal. 1, 820–829 (2018).
Pan, L. et al. Undoped ZnO abundant with metal vacancies. Nano Energy 9, 71–79 (2014).
Boyn, J. N. & Carter, E. A. Probing pH-dependent dehydration dynamics of Mg and Ca cations in aqueous solutions with multi-level quantum mechanics/molecular dynamics simulations. J. Am. Chem. Soc. 145, 20462–20472 (2023).
Volgmann, K. et al. Local determination of the amount of integration of an atom into a crystal surface. Nat. Commun. 5, 5089 (2014).
Wei, F. et al. Dynamic cooperations between lattice oxygen and oxygen vacancies for photocatalytic ethane dehydrogenation by a self-restoring LaVO4 catalyst. Chin. Chem. Lett. 35, 108313 (2024).
Zhou, K. L. et al. Platinum single-atom catalyst coupled with transition metal/metal oxide heterostructure for accelerating alkaline hydrogen evolution reaction. Nat. Commun. 12, 3783 (2021).
Wang, X. et al. Exploring the dynamic evolution of lattice oxygen on exsolved-Mn2O3@SmMn2O5 interfaces for NO oxidation. Nat. Commun. 15, 7613 (2024).
Su, B. et al. Hydroxyl-bonded Ru on metallic TiN surface catalyzing CO2 reduction with H2O by infrared light. J. Am. Chem. Soc. 145, 27415–27423 (2023).
Liu, L. et al. Rivet of cobalt in siliceous zeolite for catalytic ethane dehydrogenation. Chem 9, 637–649 (2023).
Ye, R. et al. Boosting low-temperature CO2 hydrogenation over Ni-based catalysts by tuning strong metal-support interactions. Angew. Chem. Int. Ed. 63, e202317669 (2024).
Xu, X. et al. Engineering Ni3+ cations in NiO lattice at the atomic level by Li+ doping: the roles of Ni3+ and oxygen species for CO oxidation. ACS Catal. 8, 8033–8045 (2018).
Liu, Y. et al. Ultrafine Pt nanoparticles on defective tungsten oxide for photocatalytic ethylene synthesis. Small 20, 2402004 (2024).
Luo, Z. et al. Unveiling the charge transfer dynamics steered by built-in electric fields in BiOBr photocatalysts. Nat. Commun. 13, 2230 (2022).
Wang, P. et al. Selective photocatalytic oxidative coupling of methane via regulating methyl intermediates over metal/ZnO nanoparticles. Angew. Chem. Int. Ed. 62, e202304301 (2023).
Sun, M. et al. Electronic asymmetry of lattice oxygen sites in ZnO promotes the photocatalytic oxidative coupling of methane. Nat. Commun. 15, 9900 (2024).
Kang, L. et al. Photo-thermo catalytic oxidation over a TiO2-WO3-supported platinum catalyst. Angew. Chem. Int. Ed. 59, 12909–12916 (2020).
Yuan, L. et al. Dynamic evolution of atomically dispersed Cu species for CO2 photoreduction to solar fuels. ACS Catal. 9, 4824–4833 (2019).
Li, S. et al. Oxygen-evolving catalytic atoms on metal carbides. Nat. Mater. 20, 1240–1247 (2021).
Yang, C.-C., Yu, Y.-H., Linden, V. D. B., Wu, J. C. S. & Mul, G. Artificial photosynthesis over crystalline TiO2-based catalysts: fact or fiction?. J. Am. Chem. Soc. 132, 8398–8406 (2010).
Zhang, R. et al. Photocatalytic oxidative dehydrogenation of ethane using CO2 as a soft oxidant over Pd/TiO2 catalysts to C2H4 and syngas. ACS Catal. 8, 9280–9286 (2018).
Duan, X. et al. Simultaneously constructing active sites and regulating Mn-O strength of Ru-substituted perovskite for efficient oxidation and hydrolysis oxidation of chlorobenzene. Adv. Sci. 10, 2205054 (2023).
Liu, X. et al. In situ modulation of A-site vacancies in LaMnO3.15 perovskite for surface lattice oxygen activation and boosted redox reactions. Angew. Chem. Int. Ed. 60, 26747–26754 (2021).
Li, J. et al. Activating lattice oxygen in perovskite ferrite for efficient and stable photothermal dry reforming of methane. J. Am. Chem. Soc. 147, 14705–14714 (2025).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22372035, 22432003, 22302039, 52222102 and 22272024), the 111 Project (D16008), and King Abdullah University of Science and Technology (KAUST), Center of Excellence for Renewable Energy and Storage Technologies under award number 5937 and the KAUST Supercomputing Laboratory under project k10175. The authors acknowledge the National Synchrotron Radiation Research Center (NSRRC) in Taiwan for access to the BL32A beamline of the Taiwan Photon Source (TPS) for in-situ XAS measurements, the Electron Microscopy Center of Fuzhou University for access to electron microscopy facilities, and Anhui Absorption Spectroscopy Analysis Instrument Co., Ltd. for assistance with XAFS measurements.
Author information
Authors and Affiliations
Contributions
S.W. and H.Z. conceived and directed the project. F.W. designed the experiments and analyzed the results. J.Z. and W.L. conducted theoretical studies. Y.C.L., Y. H. H. and S. F. H. performed and analyzed the in situ XAS tests. J.F. and Z.Y. analyzed the HAADF-STEM data. K.L., L.T. and X.F.L. analyzed and discussed the reaction mechanism. F.W. and S.W. prepared the manuscript. C.F. and H.Z. reviewed and edited the manuscript. All the authors contributed to the analysis and interpretation of the data and commented on the final draft of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xiaohong Zhang, Zhen Zhao and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wei, F., Zhao, J., Liu, YC. et al. Photocatalytic ethylene production over defective NiO through lattice oxygen participation. Nat Commun 16, 6586 (2025). https://doi.org/10.1038/s41467-025-61634-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61634-z
This article is cited by
-
Engineering Fluorine Doping and Ru–Schottky Interfaces in TiO2 for Efficient Photocatalytic CO2 Reduction with H2O
Transactions of Tianjin University (2026)
-
Eco-Friendly Synthesis of Nickel Oxide Nanoparticles: Multifunctional Dielectric, Antibacterial, and Photocatalytic Studies
Transactions on Electrical and Electronic Materials (2026)
-
Efficient photothermal CO2 methanation over Ni nanoparticles on oxygen-deficient V2O3 via strong metal-support interaction
Science China Chemistry (2025)
