
Abstract
Facial appearance, one of the most recognizable and heritable human traits, exhibits substantial variation across individuals within and between populations due to its complex genetic underpinning, which remains largely elusive. Here, we report a combined genome-wide association study (C-GWAS) of 946 facial features derived from 44 landmarks obtained from 3D digital facial images of 11,662 individuals of European descent. We identify 253 unlinked single nucleotide polymorphisms (SNPs) across 188 distinct genetic loci significantly associated with facial variation, including 64 SNPs at 62 novel loci and 33 novel SNPs within 29 previously reported face loci that are in very low LD with the previously reported top SNPs. Together, these SNPs account for up to 7.9% of the facial variation per trait, marking an average 2.25-fold increase over previous estimates. Cross-ancestry replication in 9,674 Chinese confirms the effect of 70% of these SNPs. A 382-SNPs prediction model of five nose traits achieves an AUC of 0.67 for individual re-identification from nose images. DNA predicted faces of archaic humans differ more from those of Europeans than from Africans. In genetically modelled Neanderthal faces, 15 of 16 DNA-predicted facial features are in line with skull evidence. Ten DNA-predicted facial features differentiate Neanderthals from Denisovans. Overall, this study substantially enhances our genetic understanding of human facial variation and provides improvements of genetic face prediction in modern and archaic humans.
Similar content being viewed by others
Introduction
Facial appearance, marked by a wide range of similarities and differences across individuals within and between human populations, is shaped largely by genetics1,2,3, and has strong relevance in different fields of science4. Facial variation is a key area of interest in anthropology, where understanding the genetic foundations of facial differences has broad implications5,6. A comprehensive genetic understanding of facial appearance could enable the prediction of faces from DNA, which would be invaluable in studying ancient DNA to ascertain the facial characteristics of archaic human species, as well as reconstructing the faces of historical figures7,8. This is particularly relevant when physical evidence from skull bones has not yet been recovered, as is the case for Denisovans9,10, but also when skulls are available while the skull does not reveal a facial feature of interest, such as the nose. It is also relevant in evolutionary biology, as ancient DNA studies on facial genes could allow unveiling the evolutionary history of facial appearance of modern humans and may identify facial traits of modern humans they received from archaic humans by interbreeding in the distant past11,12,13. Additionally, this knowledge could potentially be used in forensic genetics for DNA-prediction of facial features of unknown perpetrators, who in principle are not identifiable using forensic DNA profiling14. Currently, DNA-based appearance prediction is largely confined to a few categorical traits, most notably eye15, hair16, and skin colour17. Finally, exploring the genetic basis of facial variation is crucial in medical genetics, particularly because numerous medical syndromes manifest distinct facial feature18,19 and facial trait variations are also linked with non-syndromic human diseases20.
Recently, genome-wide association studies (GWASs) on facial features across different human populations21,22 have highlighted the complex genetic underpinnings of facial appearance, marked by multiple genetic loci each contributing small effects to different facial dimensions. Our comprehensive review of the human face genetic literature, as summarized in Supplementary Data 1, identified 440 independent genetic loci (any two DNA variants separated by at least 500 kb) significantly associated with facial features in previous studies. Although no major gene effect is generally observed, and despite the ongoing issue of missing heritability, certain genes have been found to exert significant influences on specific facial traits in particular human populations. For example, PAX3, recognized as a face gene in the first-ever two parallel facial GWASs23,24, is associated with the positioning of the nasion in Europeans, while EDAR is linked to a range of facial traits and other surface ectoderm-derived characteristics, such as hair phenotypes, in East Asians25,26.
There has been a significant transition in facial phenotyping methodologies from traditional anthropological methods used in the past to cutting-edge 3D computational imaging techniques applied today27. In facial GWAS, two primary phenotyping methods using 3D facial images dominate: facial landmarking23 and global-to-local facial segmentation28. Facial landmarking offers several advantages, including avoiding the need to share individual-level data among collaborative partners, which may have privacy implications. It effectively captures facial variation through automatically placed landmarks29,30, providing discrete and replicable results for independent and external datasets, and delivering interpretable findings valuable for applied research, such as genetic prediction studies. The initial limitation of GWASs based on facial traits obtained via facial landmarking was its lower statistical power due to ignoring complex correlations among facial traits influenced by shared genetic and nongenetic factors. This issue has been mitigated by our recently developed combined genome-wide association study (C-GWAS) method31, which has demonstrated increased statistical power in both computer simulations and applications to real data. C-GWAS surpasses the accumulation of single-trait GWAS results and other GWAS-combining methods like MTAG32, especially in the presence of a large number of single-trait GWASs, as is the case in complex morphological traits. Thus far, C-GWAS has proven effective in uncovering novel genetic loci associated with variation of the human face31 and ear33 that remained undetected with standard single-trait GWAS methods. This enhanced power is especially critical for genetic studies of the human face, where increasing sample size to increase statistical power is often unfeasible due to limited funding and other restrictions, and thus increasing the power through methodological advances is essential.
In the present study, we report the results of a C-GWAS on 946 inter-landmark distances between 44 facial landmarks obtained from 3D digital facial images of 11,662 individuals of European descent across five cohorts, followed by cross-ancestry replication analysis in 9674 Chinese individuals. Based on the obtained results, we carried out enrichment and functional annotation analyses. Finally, we applied the generated genetic knowledge to prediction analyses of facial features in both modern and archaic humans. Overall, our study significantly enhances the genetic knowledge underlying human facial variation and provides improvements in genetic face prediction in modern and archaic humans.
Results
C-GWAS and replication highlight 62 novel face loci
The C-GWAS discovery phase included 11,662 individuals of European descent from five cohorts (Supplementary Table 1 and see the “Methods” section): the Rotterdam Study (RS, N = 4242), the TwinsUK study (N = 1080), two US studies from Pennsylvania and Indiana (US-P: N = 1990 and US-I: N = 784), and the Avon Longitudinal Study of Parents and their Children (ALSPAC, N = 3566). From digital 3D facial images, we mapped 44 a-priori defined facial landmarks from which 946 inter-landmark facial distances were obtained (Fig. 1a and Supplementary Table 2) spanning eight regions of the face: forehead, eyes, upper nose (root and bridge), lower nose (wing and tip), mouth, upper cheek, lower cheek, and chin (Fig. 1b). Following General Procrustes analysis and outlier removal, the landmark coordinates were confirmed to be normally distributed across all cohorts (Bonferroni corrected p > 0.05, see the “Methods” section), with specific examples from RS and TwinsUK illustrated in Supplementary Fig. 1. The 946 distances, adjusted for covariate effects, were rank-normalized across all 5 cohorts and used as phenotypes in the subsequent genetic analyses (see the “Methods” section).

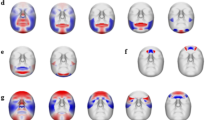
a Localization of 44 landmarks on the face. b Eight facial regions and their corresponding landmarks. c QQ plots for C-GWAS combined p-values (one-sided), MinGWAS adjusted p-values (one-sided), and associated p-values of the most inflated single-trait GWAS (two-sided). Grey dots represent results from the GWAS for the facial trait L21_L23, which had the highest inflation among all single-trait GWASs. LDSC intercepts and \({\lambda }_{{{\rm{G}}}{{\rm{C}}}}\) for the three types of results are indicated. The solid line indicates the expected distribution of p-values under the null hypothesis. d Venn diagram illustrates the overlap between the 188 loci identified by C-GWAS and the 440 loci reported by previous GWASs obtained by our literature survey. e Miami plots display the results of C-GWAS combined p-values (one-sided) and MinGWAS adjusted p-values (one-sided). Solid and dashed lines represent the study-wide significance (\(5\times {10}^{-8}\)) and suggestive significance (\(1\times {10}^{-5}\)) thresholds, respectively. Loci with the study-wide significance are highlighted using three types of marks, including purple dots for overlapping loci between C-GWAS and MinGWAS, green crosses for independent loci from C-GWAS or MinGWAS, and orange triangles for novel loci identified by C-GWAS that were not reported in previous GWASs. The 3D template facial image in this figure is adapted from White et al.82 published under an Open Access license (CC BY 4.0), see http://creativecommons.org/licenses/by/4.0/.
C-GWAS was conducted in two stages. First, 432 pairs of GWAS meta-analyses for symmetrical distances were combined into 432 single-trait GWAS outputs. Next, these were combined with 82 additional GWAS meta-analyses for non-symmetrical distances (total 514) into one C-GWAS output (see the “Methods” section). SNP-based heritability of 514 facial traits GWAS was estimated using LD score regression34 (LDSC) at an average of 0.23 and a range from 0.06 to 0.36 (Supplementary Data 2). Higher heritability was observed for the nose and forehead, whereas lower values for the mouth and cheek (Supplementary Fig. 2). Notably, the mouth and chin exhibited significant heritability divergence between internal variation and distances to other facial regions, highlighting the composite nature of facial variation, driven by cranial structure and soft tissue thickness35.
The C-GWAS method ensures that under the null hypothesis of no association, combined p-values follow a uniform distribution31, allowing the standard genome-wide significance threshold of 5 × 10−8 to be used as the study-wide threshold. Similarly, minimal p-values from 514 single-trait GWASs were adjusted within C-GWAS (see the “Methods” section), referred to as MinGWAS, ensuring they also follow a uniform distribution under the null. This adjustment enables direct comparison between C-GWAS, MinGWAS, and single-trait GWAS results. We assessed statistical inflation using LDSC and the genomic control factor λGC (Fig. 1c). The LDSC intercept quantifies inflation from non-polygenic effects, such as population stratification. 514 single-trait GWASs showed no significant inflation (LDSC intercept < 1.03), although λGC showed slight inflation with an average of 1.06 and a range from 1.03 to 1.08 (Supplementary Data 2), likely due to polygenic effects, consistent with previous GWAS findings for highly polygenic complex traits. Both MinGWAS and C-GWAS showed no LDSC inflation (LDSC intercept < 1.01) but higher lambda values (C-GWAS λGC = 1.25, MinGWAS λGC = 1.21) due to the increased power of multi-trait analysis. After excluding all study-wide significant regions (±500 kbp), λGC decreased but remained elevated (C-GWAS λGC = 1.19, MinGWAS λGC = 1.16, Supplementary Fig. 3), suggesting persistent minor polygenic effects even after removing significant regions.
Compared with our previous face C-GWAS of 78 traits obtained from 13 landmarks in 10,115 individuals31, the current C-GWAS of 946 facial traits from 44 landmarks in 11,662 individuals gained 184% extra statistical power as estimated using the increase in the mean χ2 statistic method described in Turley et al.32. C-GWAS identified 188 distinct genetic loci (separated by >500 kb) at study-wide significance (p < 5 × 10−8), four times more than MinGWAS (Fig. 1e). As the significance of C-GWAS results increases, the proportion of C-GWAS results surpassing MinGWAS rises (Supplementary Fig. 4). Specifically, when the C-GWAS p-value is below 10−5, over 95% of the results are more significant than MinGWAS, and increases to over 99% at study-wide significance. One of the abundant examples, rs970797 at 2q31.1 MTX2 showing p = 2.86 × 10−93 with C-GWAS, while p = 7.9 × 10−21 with MinGWAS. These findings suggest that most face-associated SNPs affect multiple facial dimensions simultaneously, rather than being specific to a single trait.
Of the 188 identified genetic loci, 62 (33%) were novel and had not been reported in previous facial GWASs. Among the remaining 126 loci overlapping with previous face GWASs, 26 (20%) were previously genome-wide but not study-wide significant, confirming the increased power of our approach (Fig. 1d). Among the 188 loci, in addition to the top SNPs at each locus, 41 (21.8%) hosted 65 additional independent study-wide significant SNPs identified through conditional analysis using a modified version of GCTA-COJO36 (Supplementary Data 3, see the “Methods” section), resulting in a total of 253 lead SNPs (Supplementary Data 4). Of these, 97 (38%) were novel SNPs, with 64 located in 62 novel loci and 33 located in 29 previously reported facial loci but in low LD (r2 < 0.1) with the previously reported top SNPs at these loci. The 2q36.1 PAX3 and 14q32.2 BCL11B harboured the largest number of independently associated SNPs, with 6 and 5 SNPs, respectively (Fig. 2). These findings reinforce that multiple independent lead SNPs within the same locus can distinctly influence diverse facial attributes21.

Detailed illustration for two regional Manhattan plots, a for 5 lead SNPs at 14q32.2, and b for 6 lead SNPs at 2q36.1, was presented in three layers. The top layer displayed the distribution and intensity of effects from each lead SNP on the face using the regional percentage of variance explained (regional-PVE, see the “Methods” section) across 8 facial regions. The middle layer is a zoomed view of the C-GWAS combined p-values (one-sided), with multiple lead SNPs and their LD counterparts (p < 1 × 10−5) highlighted in different colours. Each lead SNP is annotated with its rsID and represented by a diamond shape. Protein-coding genes (black texts) and non-coding RNAs (grey texts) in the same region are annotated below the plots according to their chromosomal positions (GRCh37). The bottom layer displays the LD (r2) patterns of multiple lead SNPs and their LD counterparts. LDs are calculated in RS, and hierarchical clustering is applied. Multiple lead SNPs are marked with arrows corresponding to their colours. The 3D template facial image in this figure is adapted from White et al.82 published under an Open Access license (CC BY 4.0), see http://creativecommons.org/licenses/by/4.0/.
Intraclass correlation coefficient (ICC) analysis of allelic effects demonstrated high consistency across the five European cohorts for both, previously known and novel lead SNPs (Supplementary Data 5), with a specific example of effects on nasion width from RS and ALSPAC illustrated in Supplementary Fig. 5. MAMBA37 analysis further assessing posterior probability of replicability (PPR) across all five European cohorts revealed high PPR values approaching 1 (Supplementary Fig. 6). The associations with more significant meta-analysis p-values corresponded to higher PPR values, further corroborating the robustness of our findings. External cross-ancestry replication analyses in 9674 Chinese individuals revealed high replication rates of 69.8% in all lead SNPs and 39.6% in the novel lead SNPs (FDR corrected p < 0.05, Supplementary Data 4). Further details on covariate impact and pan-ethnic effects of lead SNPs are provided in Supplementary Notes 1 and 2.
Biological annotations and pleiotropy of face-associated SNPs
A stratified LDSC38 of C-GWAS summary statistics across various cell and tissue types identified 15 significantly enriched cell types (Bonferroni threshold p = 4.9 × 10−4, Supplementary Fig. 7, Supplementary Data 6). The most substantial enrichments were notably observed in adult dermal fibroblast primary cells (p = 3.3 × 10−11) and cranial neural crest cells (CNCC, p = 2.1 × 10−10). A multi-omics integration analysis (CNCC regulatory network39, multi-tissue eQTLs and chromatin interactions) revealed 365 candidate genes potentially functionally affecting human facial variation (see the “Methods” section, Supplementary Data 4 and 7). GO enrichment analysis of these genes highlighted 177 significant GO-term biological processes (Bonferroni corrected p < 0.001, Supplementary Data 8) clustered into 16 groups (Supplementary Fig. 8 and Supplementary Data 9), covering not only well-established craniofacial morphogenesis processes, such as embryo, skeleton, limb, bone, ear, brain, neuron, eye, differentiation, growth, and the Wnt pathway, but also processes in the key cell types highly relevant to CNCC formation including epithelium, mesenchyme, and mesoderm (Fig. 3a). Additional enrichment in seemingly unrelated processes like vasculum, heart, muscle, urogenital, and digestive systems further emphasize a pleiotropic nature of facial genetic factors. Further details on contributing cell types and candidate gene robustness are provided in the Supplementary Data 10, Supplementary Notes 3 and 4.

a The figure details the enriched biological processes and the specificity of 4 face candidate gene sets annotated by 253 lead SNPs. The outer bar chart contains 177 biological processes with significant enrichment Bonferroni corrected p-values (one-sided hypergeometric test), divided into 16 categories. The dashed line indicates the significance threshold (p = 0.001). The inner radar chart reflects the mean fold enrichment of genes in each gene set across the biological processes in each of the 16 categories. Different colours denote different gene sets. The dashed line represents the mean fold enrichment under the null distribution (no enrichment). The outer ring annotations include the titles of the 16 categories, the top three most significant enriched biological processes in each category, and biological processes in the top 5% of fold enrichment of genes in 4 gene sets across the 177 biological processes, highlighted in the corresponding colour of the gene set. b Hierarchical clustering of 225 face candidate gene sets annotated by 253 lead SNPs, identified 4 clusters based on their effects on facial regions (in terms of regional-PVE across 8 facial regions, see the “Methods” section). Face maps showed the total effects of the annotated genes on the face for gene sets corresponding to four clusters. The 3D template facial image in this figure is adapted from White et al.82 published under an Open Access license (CC BY 4.0), see http://creativecommons.org/licenses/by/4.0/.
Hierarchical clustering analysis revealed four distinct sets of candidate genes predominantly affecting different sets of facial regions (Fig. 3b and Supplementary Fig. 9) and showed specificity in biological processes (Fig. 3a and Supplementary Data 9). For example, genes influencing the nose were more enriched in limb morphogenesis, while those affecting the chin and mouth were more enriched in morphogenesis of branching structures, particularly axons. These findings offer a preliminary map of how embryonic development, driven by shared genetic factors, potentially influences distinct facial features. The extensive pleiotropy was further supported by a GWAS Catalog look-up, where 144 (57%) of the 253 lead SNPs, or their high LD counterparts (r2 > 0.6), were associated with 13 phenotypic categories of non-facial traits (Supplementary Fig. 10). The top-associated categories are anthropometric, brain imaging, metabolic and appearances traits, such as height, waist-to-hip ratio, sulcal depth, cortical surface area, total testosterone levels, contactin-2 levels, male pattern baldness, and hair colour (Supplementary Data 11). These results collectively suggest that the phenotypic spectrum of face-associated SNPs is more extensive than previously may have thought.
Enhanced proportion of genetically explained facial variance and facial PRS profiles
Across the 514 facial traits, the combined proportion of variance explained (PVE) from all 253 lead SNPs together ranged from 2.25% to 7.89% (Supplementary Data 2), a 2.25-fold average increase from our earlier study3, which were based on 31 SNPs and 78 facial traits and ranged from 0.65% to 4.62%. To further explore the explanatory power of the lead SNPs, we extended the analysis to assess PVE across the entire face (face-PVE, including all 514 traits) and within specific facial regions (regional-PVE, including up to 32 traits per region, see the “Methods” section). Combining 253 lead SNPs resulted in a face-PVE of 4.5%, with the highest regional-PVE in the upper nose (6.0%) and lower nose (5.6%), and the lowest in the lower cheek (3.2%) and mouth (3.6%) (Fig. 4a). Notably, the novel SNPs tended to have more widespread effects across multiple facial regions rather than being confined to a specific area (Supplementary Fig. 11). Our comprehensive mapping of genetic effects on distinct facial dimensions highlighting the polygenic and multidimensional nature of facial shape (Supplementary Data 12). Detailed single-trait PVE, face-PVE and regional-PVE analyses are provided in the Supplementary Note 5.

a Illustration of the total effects of 253 lead SNPs on the face, based on regional-PVE across 8 facial regions. The 3D template facial image in a is adapted from White et al.82 published under an Open Access license (CC BY 4.0), see http://creativecommons.org/licenses/by/4.0/. b Enlarged frontal and lateral views of the nose with 14 landmark points highlighted in c and d, and Supplementary Note 6. The scale indicates the p-value of deviation of the landmark from the average in c and d, which is derived from the association test between PRS and landmark coordinates using linear regression. PRS analysis and its effects on the nose are illustrated for: c for L21–L24 and L23–L26, and d for L7–L8 and L8–L9. Each figure is divided into three sections: PRS effects on the nose (left, enlarged nasal view), population profiling (middle, violin plots), and gene contributions (right). In the nasal views, landmarks significantly associated with standardized PRS are marked with orange dots (3 times positive effect offset) or blue dots (3 times negative effect offset). Connecting lines visually represent nasal variation, with scaled distances shown in the top left. Nasal images reconstructed at mean PRS ± 3 SD are framed in orange (+3SD) and blue (−3SD) and generated using a 3D graph auto-encoder (see the “Methods” section). The overall effect of increasing PRS is described in text at the top left. The violin plots show standardized PRS results for four major populations from the 1000 Genomes Project (African, AFR, N = 504; European, EUR, N = 504; East Asian, EAS, N = 503; South Asian, SAS, N = 489), with the band indicating the median, the box representing the first and third quartiles, and whiskers extending 1.5 times the interquartile range. Gene contribution plots on the right list the top regions accounting for 50% of PRS variance. The 3D template facial image in b–d was generated from the average covariate-adjusted facial shape of all RS participants (N = 4242).
Building on these findings, we constructed polygenic risk score (PRS) profiles for 2000 individuals across four major continental populations (African, AFR; European, EUR; East Asian, EAS; South Asian, SAS) using data from the 1000 Genomes Project40, excluding admixed samples from AFR and the American population. The PRS was based on 382 face-associated SNPs, including 253 lead SNPs from our C-GWAS and 129 SNPs from previous facial shape GWASs, for 23 most genetically explainable and independent facial traits (see the “Methods” section, Supplementary Data 13). Facial PRS profiles largely agree with established anthropological knowledge concerning nose shape variation among continental populations (Fig. 4b–d, Supplementary Data 14 and Supplementary Note 6). A comparative analysis of computed tomography scans of 388 adults of AFR, Asian (ASN), and EUR ancestry41 further confirmed significant correlations between mean differences in facial PRS profiles and phenotypic traits between EUR and AFR (Pearson’s r = 0.75), as well as between EUR and ASN (r = 0.81) (Supplementary Note 7). Specifically, considering the average facial PRS profiles of EUR as reference (z = 0), AFR showed notably smaller nose root (z = −1.86), less protruded (z = −1.76) but more upturned (z = 0.74) nose tips, alongside broader nose wings (z = 0.8) and shorter nose bridges (z = −0.82). EAS were characterized by significantly smaller (z = −1.77) and less (z = −2.42) protruded nose tips, along with smaller nose root (z = −1.12). Meanwhile, SAS displayed similar nose shapes to EUR (all |z | <0.5), which is remarkable given their closer geographic proximity to EAS. The latter finding is in line with the study of Zaidi et al.42 finding that the average nose differences between SAS and EUR were smaller than other intercontinental comparisons. Furthermore, our findings are largely consistent with numerous facial photogrammetric studies reviewed by Wen et al.5, reinforcing the correlation between genetic facial profiles and physical facial anthropometry.
Re-identification of individuals from 3D images with facial PRS profiles
Motivated by these findings, we explored the feasibility of re-identification of individuals’ images from their facial PRS profiles, focusing on the nose region, which manifested the highest genetically explainable phenotypic variance (Supplementary Data 14). This was achieved by calculating the cosine similarity scores between PRS profiles of the nose (referred to as nose PRS profile) and 3D image-derived phenotypic nose profiles (referred to as nose 3D profiles) in the RS. When focusing on the top 1% of the nose 3D profiles that most closely mirrored an individual’s nose PRS profile, we observed a 3.4% probability of accurately identifying the correct individual’s nose 3D profile based on its nose PRS profile (cumulative matching characteristic, CMC1% = 3.4%, that is, with a 1% selection threshold, the matching probability was 3.4%). The matching accuracy significantly improved with less stringent selection thresholds, achieving 24% at CMC10% and 73% at CMC50% (Fig. 5a).

a Cumulative Match Characteristic (CMC) curve for the individual re-identification model using the PRS of the five nose traits, as detailed in Fig. 4 and Supplementary Note 6. The curve is based on 50 rounds of 10-fold cross-validation, highlighting CMC1%, CMC10%, and CMC50% in orange crosses and labelled in the top left. b Receiver operating characteristic (ROC) curve for the individual re-identification model using the PRS of the same five nose traits, based on 50 rounds of 10-fold cross-validation. The average AUC is labelled on the top left. c Distribution of AUC values across different prediction validation scenarios in two European cohorts, RS and TwinsUK. The density plot shows the distribution of AUC null values obtained through 10,000 replicates, where each replicate involved PRS constructed from a number and MAF-matched SNPs randomly selected across the genome. AUC values achieved by our PRS models under different validation scenarios were compared with the null distribution using arrowed lines.
Utilizing a binary variable of true and false matches, the similarity score achieved a moderate AUC of 0.67 (Fig. 5b, see the “Methods” section). Because RS was part of the C-GWAS discovery dataset, aiming to establish a benchmark, we generated null AUC values through 10,000 replicates, where each replicate employed PRS constructed from randomly selected SNPs that were matched by number and MAF. The AUC of 0.67 in RS significantly exceeded the null distribution, confirming that our matching accuracy surpasses expectations by random chance (Fig. 5c). Next, we employed the TwinsUK data in this re-identification analysis. Although the TwinsUK dataset was also part of the C-GWAS discovery dataset, we assessed the potential decrease in AUC when applying the PRS to TwinsUK as a separate dataset. By considering 216 SNPs that overlapped between both datasets, the internal validation within the reference sample of RS yielded an AUC of 0.62. When this PRS was then applied to the TwinsUK dataset, we obtained an AUC of 0.60 (Fig. 5c). This small AUC reduction of 0.02 suggests a negligible winner’s curse effect within the training cohort (i.e., RS), as confirmed by additional simulation analyses (Supplementary Note 8). The significant deviation from the null distribution of both AUC outcomes, from RS and TwinsUK, implies that our genetic model captures meaningful effects beyond random noise. While the AUC values obtained here may seem modest, it is important to emphasize that these results were achieved in homogeneous populations and based solely on SNP data for nose traits alone. Expanding the number of genetically predictable facial phenotypes beyond the nose, even if each has limited accuracy, could collectively improve the overall accuracy in individual re-identification.
Face-associated SNPs experienced positive selection in Europeans
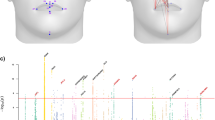
Among the 253 lead SNPs identified in our facial C-GWAS, 69% exhibited a frequency difference > 0.2 among EUR, EAS, and AFR, as per the 1000 Genomes Project data (Supplementary Data 4). This proportion is significantly higher than the average observed in random samples of the same number of SNPs across the genome after 10,000 replicates (p = 0.023, Supplementary Fig. 12). The average of fixation index (FST), which reflects genetic variation between groups and can indicate local positive selection, was significantly higher between EUR and EAS (p = 0.017) and EUR and AFR (p = 0.011) than expected from randomly sampled SNPs (Fig. 6a). However, this pattern was not observed in the EAS-AFR comparison (p = 0.32, Supplementary Fig. 12). Moreover, the mean Population Branch Statistic (PBS) for the 253 lead SNPs compared to randomly selected SNPs was statistically significant in EUR (p < 0.01, Fig. 6a), but not in EAS (p = 0.44) and AFR (p = 0.25). These findings suggest a more pronounced influence of positive selection on face-associated SNPs in Europeans.

a Simulations analyses show the positive selection. Grey bars and curves show the expected distribution density from 10,000 simulations under the null hypothesis. The dashed line indicates the one-sided 0.95 cumulative density threshold. Observed values from 253 lead SNPs are highlighted in orange with one-sided empirical p-values. For illustration of the 2q12.1 region (b) and 9p22.3-p22.2 region c under natural selection, Left: C-GWAS combined p-values within 250 kb of the top SNP plotted against PBS results in EUR. Dashed lines indicate suggestive significance threshold for C-GWAS and the top 1% threshold for PBS, respectively. The top SNP is highlighted with a purple diamond, and LD counterparts are in colour scale. The SNP with both a significant facial effect and a selection signal is annotated. Middle: World map of allele frequency for annotated SNPs based on 1000 Genomes Project sub-populations. Base map is from Natural Earth data v1.4.0 (public domain). Right: Regional-PVE effects of annotated SNP. The 3D template facial image in this figure is adapted from White et al.82 published under an Open Access license (CC BY 4.0), see http://creativecommons.org/licenses/by/4.0/.
Although our targeted SNPs may have higher MAF in EUR because they were identified through a European-based C-GWAS, a recent large-scale face GWAS conducted in East Asians22 identified 244 face-associated SNPs and found evidence of positive selection on these SNPs in Europeans. This suggests that facial differences between Europeans and East Asians, such as more protruded and narrow noses in Europeans, may result from adaptation and positive selection in European populations. The consistency of findings across independent GWAS in different continental populations indicates these results are not merely artefacts of GWAS-based allele frequency biases.
Of the 188 genetic loci where the 253 lead SNPs are located, 22 displayed strong signals of positive selection (top 1% of the genome, Supplementary Data 15), highlighting their importance in the evolutionary history of facial variation in modern humans. Among these, two loci were at the extreme tail of statistical significance: 2q12.1, containing POU3F3 primarily associated with the nose region, and 9p22.3-p22.2 containing BNC2 primarily associated with the chin region (Fig. 6b and c). Notably, BNC2 was previously identified as a skin-colour gene based on association43 and functional evidence44, and is located in a genomic region of Neanderthal ancestry (introgressed segments)45.
Facial PRS profiles of Neanderthals differ more from Europeans than from Africans
Next, we explored whether face-associated SNPs identified via C-GWAS in Europeans could help genetically reconstruct facial traits in archaic humans. Modern human populations outside Africa carry ~1.5–2.1% Neanderthal-derived ancestry46,47. We annotated our 253 lead SNPs with previously identified introgressed segments of Neanderthal in the genomes of EUR and EAS48,49,50. On average, 65 SNPs (25.7%) in EUR and 57 SNPs (22.5%) in EAS overlapped with Neanderthal introgressed segments, including 30 SNPs (11.9%) shared between both populations (Supplementary Data 16). Lead SNPs in EUR-specific segments predominantly influenced the upper nose and forehead, while those in EAS-specific segments primarily affected the chin, and shared lead SNPs were mostly associated with variation in the lower nose (Fig. 7a and Supplementary Fig. 13). This pattern suggests that face-associated SNPs located in Neanderthal introgressed segments have shaped facial variation differently across modern human populations, for instance, prominent brow ridges in EUR51 and flatter chin structures in EAS52. Distinct genetic loci contributed to these population-specific introgression effects (Fig. 7b). Notably, chromosome 6p21.1 contains SUPT3H (EUR-specific) associated with the upper nose, and RUNX2 (EAS-specific) associated with the upper cheek (Supplementary Fig. 14). Additionally, TBX15 on chromosome 1p12, represented by three lead SNPs with broad facial effects, significantly contributed to EAS-specific introgression influence, aligning with previous findings linking TBX15 to Denisovan introgression associated with lip thickness in Latin Americans11. Importantly, our analysis relies solely on C-GWAS lead SNPs overlapping within previously reported introgressed segments, without considering ancestral/derived allelic states. To explore this further, we examined whether the lead SNP in the introgressed segments meets the condition of archaic variants as previously defined53, which would increase confidence that the lead SNP represents a true introgressed variant. However, the presence of such variants in modern populations is not necessarily due to hybridization between Neanderthals and Early Sapiens; it could also be due to incomplete lineage sorting. Furthermore, the limited number of high-confidence introgressed face-associated SNPs identified using this approach restricts our ability to draw robust conclusions (Supplementary Data 17 and Supplementary Note 9).

a Regional-PVE effects of lead SNPs in Neanderthal introgressed segments on the face, divided into EUR-specific, overlapping, and EAS-specific groups. b Top gene regions contributing to total effects for the three SNP groups in a, showing cumulative contributions accounting for 50% of the total. c Violin plots of squared correlations between facial PRS profiles of archaic humans and four major populations in the 1000 Genomes Project (African, AFR, N = 504; European, EUR, N = 504; East Asian, EAS, N = 503; South Asian, SAS, N = 489), grouped by Neanderthals, Neanderthal-Denisovan admixed individual, and Denisovan. The band indicates the median, the box indicates the first and third quartiles, and the whiskers indicate 1.5 times of interquartile range from box. d The mean absolute frequency differences between 10 archaic human samples and 2000 samples from four major populations in the 1000 Genomes Project (same as c) were illustrated for different sets of SNPs, with higher values indicating greater dissimilarity (see the “Methods” section). The dashed line corresponds to the 357 SNPs used to construct the archaic human facial PRS profiles. The violin plots represent replicates of 10,000 random sets of SNPs. The band indicates the median, the box indicates the first and third quartiles, and the whiskers indicate 1.5 times of interquartile range from the box. Labels on the x-axis denote the different SNP sets used: “ALL” represents random 357 genome-wide SNPs, “EUR segments” and “EAS segments” include the SNPs among the 357 overlapping introgression segments specific to Europeans and East Asians, respectively. The 3D template facial image in this figure is adapted from White et al.82 published under an Open Access license (CC BY 4.0), see http://creativecommons.org/licenses/by/4.0/.
Next, we constructed facial PRS profiles based on 357 face-associated SNPs available in 10 archaic humans (8 Neanderthals, 1 Denisovan, and 1 Neanderthal–Denisovan admixed individual) using genomic data obtained from the Allen Ancient DNA Resource54 (see the “Methods” section, Supplementary Table 3 and Supplementary Data 18). When comparing archaic humans to the four major modern human populations in the 1000 Genomes Project, archaic facial PRS profiles are more different from three non-African groups, including EUR, than from AFR (Fig. 7c). This finding aligns with current understanding that Africans are ancestral to all modern humans, making them genetically closer to the common ancestors of both modern and archaic humans. However, this finding may appear as a paradox given that the introgression happened in non-Africans. To explore this further, we calculated the mean absolute differences in allele frequencies for the 357 SNPs between archaic humans and modern human populations, and compared these to 10,000 random sets of number-matched random SNPs. Differences between archaic humans and AFR were consistently smaller than those between archaic humans and all non-Africans from EUR, EAS, and SAS, even when focusing on SNPs within introgressed segments (Fig. 7d). These findings suggest that the similarity in facial PRS profiles between archaic humans and AFR is likely due to shared ancestral alleles. However, we acknowledge a potential bias stemming from the predominantly European GWAS origin of these 357 facial SNPs. Future studies should prioritize facial GWAS in diverse populations, particularly in African cohorts, to reduce such biases and clarify the evolutionary history underlying facial shape genetics.
Facial PRS profiles of Neanderthals align with Neanderthal skulls
Neanderthal skulls have been discovered and show a series of distinct features that are different compared to skulls of modern humans51,55. To validate Neanderthal facial PRS profiles, we mapped them to 16 Neanderthal facial features reflected by skulls, excluding features not reflected in bone structure, such as the nose tip and certain aspects of the nose bridge (Supplementary Table 4, see the “Methods” section). Of the 16 facial features analysed, 15 showed concordances between (a) the direction of the difference predicted by facial PRS profiles in Neanderthals relative to Europeans, and (b) the direction of the reported feature differences for Neanderthals relative to modern humans, examples include a wider face, more protruded brow ridge, flatter cheekbones, and lower palate (Fig. 8a). Permutation tests (see the “Methods” section) indicated that observing 15 concordant features is statistically highly significant, exceeding all 10,000 random permutations (p < 0.0001, Fig. 8b). Only one feature (wider palate in Neanderthals) showed inconsistent classification due to conflicting directions from multiple overlapping PRS (p = 0.0126). No discordant results were observed (p = 0.0012). The statistical significance of these findings indicates that the observed concordance between European-based PRS predictions for Neanderthals and their known facial features relative to modern humans is unlikely to have occurred by chance. To further assess the robustness of these results, we conducted simulations to evaluate how data limitations for archaic humans, such as pseudohaploid data, small sample sizes, and low call rates, might affect PRS accuracy (Supplementary Notes 10 and 11). The results showed that pseudohaploid data and small sample sizes did not bias PRS averages, although they did increase the uncertainty. Additionally, imputing genotypes for low call rate samples yielded PRS averages highly consistent with those from high call rate samples. These findings suggest that, despite moderate PRS r2 values that may not accurately represent individual facial profile and the inherent uncertainty of archaic DNA data, the broader patterns in PRS profiles, such as directionality of averages, remained robust, particularly for extreme values, reinforcing the reliability of archaic PRS profiles for characterizing population-level facial difference.

a Mapping facial PRS profiles of Neanderthals to 16 facial features reflected by skulls. Left panel: PRS (absolute values > 0.4) are sorted and summarized by their facial effects. Right panel: 16 known Neanderthal facial features categorized by different colour according to facial regions, with lines indicating enhancement (solid) or reduction (dashed) predicted by the corresponding PRS. Triangles represent consistent directional predictions, including upward (enhancement) or downward (reduction) those, and circles denote inconsistencies. The diamonds highlight PRS not corresponding to known features. b Comparison of concordance in Neanderthal facial features based on phenotypic and genetic differences. The bar graph assessed the concordance between phenotypic descriptions from archaeological studies and genetic predictions using PRS for 16 facial features of Neanderthals. Each bar represents the number of facial features displaying concordant (orange), discordant (purple), or inconsistent (green) results based on the alignment between skull morphology differences and the direction of mean PRS differences for Neanderthals relative to EUR (see the “Methods” section). The analysis involved 10,000 permutations using 357 MAF-matched SNPs to generate null distributions and assess the statistical significance of the observed patterns with three one-sided significance levels noted at the top. c Same form of illustration as a but displayed the difference between Denisovans and Neanderthals using the PRS difference (Denisovan PRS minus Neanderthal PRS average).
Next, we compared the facial PRS profiles between Neanderthals (8 samples) and EUR from the 1000 Genomes Project using a t-test, which showed significant differences for 18 out of 23 facial traits (FDR < 0.05, Supplementary Data 14). Notably, Neanderthals had a significantly wider and flatter upper cheek region (p = 8.5 × 10−6) than EUR, consistent with fossil evidence showing the broad facial structure and relatively straight cheekbones of Neanderthals relative to modern humans51,56. Additionally, Neanderthals had a more retracted nose tip (p = 1.34 × 10−5) than EUR, which represents a previously unreported finding since no soft tissue evidence exists for Neanderthals. While methods to predict nose shape from skull data have been developed for modern humans57,58, they are trained and validated with head image data of soft and hard tissues of modern humans and are therefore not applicable to archaic humans. More specifically, average Neanderthal PRS profiles predicted shorter (z = –3.14), narrower (z = –1.91), but more protruded (z = 1.35) nose bridges, less protruded (z = –1.99) but more upturned (z = 1.5) nose tips, alongside broader nose wings (z = 1.91), differing more from the nose PRS profiles of EUR (z = 0) than from those of AFR.
Although genomic data are currently only available for a single Denisovan individual, we also performed facial PRS analysis for Denisovans, reflecting preliminary findings. Denisovan PRS fell within the EUR PRS distribution for 17 out of 23 facial traits, with six traits at the extreme tail (p < 0.05, Supplementary Data 14). Notable differences from EUR included a shorter (z = –3.66) nose bridge, less protruded nose tips (z = –3.12), wider orbits (z = 4.19), a higher brow ridge (z = 2.12), a higher chin (z = –2.37), and a smaller tear trough (z = –1.94). Denisovan PRS also differed from Neanderthals for 10 out of 16 mapped traits, including a higher, more protruded brow ridge, a nose with more European characteristics (larger nose root, narrower wings, and lower tip), more protruded cheekbones, a flatter infraorbital region, and a higher, flatter chin compared to Neanderthals (Fig. 8c). Although complete Denisovan skulls have not been discovered yet, a Denisovan partial mandible (Xiahe mandible, >160 kya) found on the Tibetan Plateau10 shows a higher anterior mandible than modern humans but lower than Neanderthals, with a smaller symphyseal angle, indicating a flatter, more retracted chin. Our chin PRS profiles for Denisovans aligned with three of these findings but inconsistently predicted a higher anterior mandible than Neanderthals (Supplementary Data 19). Inspecting previously reported differences in DNA methylated regions among modern humans, Neanderthals and Denisovan8, we found consistent results with Denisovan facial PRS profiles for 9 out of 12 overlapping predictions, including face width, face length, palate height, etc., but inconsistencies were noted for midface prominence and palate width, etc (Supplementary Fig. 15 and Supplementary Data 19).
While these results indicate genetic evidence for facial differences between archaic and modern humans in specific directions, as well as between Neanderthals and Denisovans, we emphasize the limitations of these findings given the small sample size of currently available genome data from archaic humans.
Discussion
This study significantly advances the genetic understanding of human facial variation by expanding previous C-GWAS work31. We increased facial landmarks from 13 to 44 and derived facial distance traits from 78 to 946. Additionally, we enlarged our discovery sample from 10,115 individuals across four cohorts to 11,662 individuals across five cohorts. These enhancements boosted statistical power by 2.84-fold, tripled identified face-associated loci, and doubled genetically explained facial variance, providing a robust foundation for downstream predictive analyses in both modern and archaic human studies. The success of this study empirically exemplified the effectiveness of C-GWAS for combining extraordinarily large numbers of correlated traits across multiple cohorts, setting an important standard for future GWAS in areas like methylation, proteomics, metabolomics, and bio-imaging traits in the brain and other tissues. However, we also note several limitations of the current C-GWAS method in analysing multiple GWASs. First, applying C-GWAS to combine GWASs from distinct genetic backgrounds, such as populations from different continental ancestries, may result in inaccurate effect correlation estimates, potentially reducing statistical power. In such cases, it is advisable to perform a meta-analysis of GWASs from different genetic backgrounds prior to applying C-GWAS for multi-trait combination, thereby improving the reliability of the results33. Second, combining GWAS with varying proportions of missing SNP genotypes using C-GWAS can bias effect estimates since missing SNPs are set to have zero effect by default. When the proportion of missing SNPs is substantial, it is advisable to exclude these variants to minimize bias. Third, to reduce computational burdens and errors from collinearity, it’s advisable to first combine highly correlated GWASs, which we implemented by initially combining symmetric facial traits to improve reliability and efficiency.
We used facial shape GWAS results from East Asians for the replication analysis, despite differences in cohort genetic background (East Asians vs. Europeans) and phenotyping methods (global-to-local segmentation vs. landmark-based). While these differences limit exact replication and reduce replication power due to varying allele frequencies, adjusted replication p-values confirmed a significant proportion of facial lead SNPs, including novel ones, highlighting a robust pan-ethnic genetic influence on facial features when it comes to Europeans and East Asians at least. However, genetic effects specific to non-Europeans were less emphasized, indicating a need for discovery C-GWAS in non-European populations. Furthermore, by integrating multi-omics annotations of facial lead SNPs and their associations with non-facial traits, we have outlined the most comprehensive biological and pleiotropy map of face-associated SNPs. This reinforces known facial shape genetics while providing additional insights into shared genetic links between facial shape and a broader spectrum of human traits, offering a list of candidate traits and corresponding associated SNPs for future research on disease risks and genetic foundations.
The facial lead SNPs we identified significantly enlarged the number of genetic loci involved in facial variation, but collectively explained less than 10% of the phenotypic variation. This limited explanatory power indicates that reconstructing a detailed face solely from DNA, which is akin to drawing an accurate portrait of a person from his/her DNA, is currently beyond any reach. However, re-identification of individuals in facial imagery datasets using facial genetic data may be more achievable. This approach relies on combining multiple predicted traits rather than accurately predicting each individual feature. Previous simulations59 have confirmed that even modestly accurate individual trait predictions can effectively identify individuals when combined. Our model for five nose traits, though not highly accurate on its own, highlights the potential of this approach. Previous research60 reported an AUC of 0.77 for DNA-based facial recognition in diverse populations, such as from the US, primarily driven by distinct genetic ancestry among individuals. In our study, the AUC of 0.67 for nose-based re-identification represents a significant initial demonstration that moderate predictive accuracy can be achieved in a homogeneous population using SNP data, even when only using nose traits alone. Looking ahead, the model’s overall discriminative power could be further enhanced by incorporating more facial features, pigmentation traits61, hair features16, and age estimations14, along with ancestry-informative DNA markers62,63, all of which have previously shown promise for direct DNA-based prediction. Furthermore, novel facial lead SNPs, despite exhibiting lower statistical significance than previously reported ones, demonstrated comparable mean face-PVE and were more likely to influence multiple facial regions. This suggests that SNPs with broader but weaker effects may be missed due to limitations in multi-trait analysis methods. Nonetheless, their overall impact on facial variation may not be weaker than SNPs that strongly affect specific regions. This highlights the importance of identifying more face-associated SNPs, even those with modest statistical significance, to improve model performance. Besides, we were unable to test our nose PRS in external non-European cohorts, but previous studies suggest facial trait PRS can generalize across populations. For instance, Xiong et al.3 showed that PRS derived in one population can capture key facial features in different genetic backgrounds. Specifically, Zhang et al.22 demonstrated that PRS-based facial shapes successfully captured facial shape variations between EUR and EAS populations, closely resembled the true population average shapes across different facial regions both visually and statistically. These suggest that the facial shape effects identified are not confined to a single population, supporting the potential cross-ancestry applicability of our facial PRS profiles. Overall, our findings underscore both the current limitations and the future potential of DNA-based facial recognition as methodologies and datasets expand.
While these findings are preliminary and serve as an initial proof-of-concept rather than providing a next step towards practical implications, we would like to point out that DNA-based facial recognition or face prediction can raise ethical and societal concerns that warrant careful consideration. The ability to recognize or reconstruct a person’s facial features from DNA could lead to privacy violations, unauthorized tracking, and potential discrimination, especially if accuracy varies across genetic backgrounds. There is further the potential of politically or otherwise motivated misuse of such technology. While yet, the scientific and technological knowledge is too premature to know if these concerns will ever become practically relevant, developing ethical guidelines and regulatory frameworks alongside scientific advancements is crucial to managing these potential risks.
A few studies have demonstrated that archaic human introgression influences modern human facial features. For example, Neanderthal introgression has been linked to nose length13, while a 28 kb Denisovan-derived fragment on chromosome 1, near WARS2 andTBX15, affects upper lip thickness and prominence11. Furthermore, research on a wide range of complex traits in the UK Biobank has shown that Neanderthal introgression significantly contributes to the heritability of dermatological traits, such as those related to hair and skin64, suggesting that introgression may similarly affect facial shape due to the link between facial variation and skin development, as demonstrated in our cell type analysis. In our study, genomic regions retaining Neanderthal ancestry were primarily associated with facial variations in population-specific features and showed evidence of natural selection within the corresponding population22, offering notable insights into the genetic basis for facial differences among populations. However, there are limitations to our understanding of introgression’s role in facial traits. Specifically, it’s unclear whether archaic DNA segments recombined with modern human variants shortly after introgression or if the variations emerged later65. Supporting the latter scenario, fewer than 10% of face-associated variants within archaic segments are high-confidence introgressed SNPs in our analysis. This may reflect demographic and evolutionary history: introgression occurred close in time after the out-of-Africa migration46, followed by strong selection pressures on facial shape, particularly on nose shape, in the northern hemisphere due to adaptation to changing climatic conditions such as lower temperature and humidity66. This suggests that a substantial portion of alleles involved in nose shape variation, which substantially contribute to overall facial variation, may have arisen or been selected post-introgression, explaining why archaic alleles have a relatively small effect on facial variation.
Currently, three high-coverage Neanderthal genomes are available: Altai (110 kya, 52x coverage, Siberia)47, Vindija (50 kya, 30x coverage, Croatia)67, and Chagyrskaya (80 kya, 27x coverage, Siberia)68. Genetic evidence indicates that early Neanderthals formed at least two geographic populations: an eastern group (Altai) and a western group (Vindija), which shows genetic continuity with later Neanderthals12. Vindija contributed an additional ~10% of Neanderthal sequences to modern Eurasians69, highlighting differences between these two Neanderthal samples and their historical gene flow with modern humans. However, because of Altai’s higher coverage, most introgression studies have focused on Altai, limiting understanding of Neanderthal diversity. Although our study has integrated various methods48,49,50 to detect introgressed segments, there is still a lack of data on western Neanderthal introgression represented by Vindija. The Vindija sample is expected to be the best representative of the introgression population, as suggested by Reilly et al.12 and recent studies have proposed a narrow time range (~45–49 kya BP) for the introgression period70,71. We anticipate that future research will provide a more comprehensive perspective on the introgression of Neanderthals into modern human populations.
Applying PRS to archaic genomes, such as those of Neanderthals and Denisovans, presents considerable challenges. PRS models are based on modern human populations and rely on assumptions about linkage disequilibrium, allele frequency, and additive genetic effects, which may not apply to archaic lineages that evolved under different environmental conditions and selective pressures72. Moreover, the limited genetic diversity and unique adaptations of archaic populations, evident in traits shaped by now-extinct environments, further complicate attempts to generalize modern PRS to these ancient groups. For instance, Denisovan-specific adaptations, with rare exceptions like high-altitude adaptations in present-day Tibetans73 are not well-captured in modern datasets. Despite these uncertainties, our simulations and fossil skull comparisons indicate that pseudohaploid data and small sample sizes, which characterize current archaic genomes, do not significantly bias overall PRS estimates. This finding suggests that, while absolute values should be interpreted cautiously, PRS still provide reasonably reliable directional insights into phenotypic shifts over evolutionary time.
Until now, our understanding of archaic human facial features has mainly come from a limited number of skull specimens. Neanderthal skulls have offered some insight into their facial structure55, but features like nose shape are not well preserved in bone, making genetic prediction particularly informative. The absence of complete Denisovan skulls makes genetic inference about their facial features especially interesting74. Our archaic facial PRS profiles align well with fossil evidence and previous predictions, predicting detailed nose and cheek features of Denisovan, which had not been characterized in such detail before, and highlighting more comprehensive facial differences between Neanderthals and Denisovan than previously known. Interestingly, our European-calibrated PRS suggests Neanderthal noses may differ more from Europeans than from Africans, which appears inconsistent with cold-climate adaptations in Europeans66. This discrepancy may reflect differences in nasal cavity adaptations in Neanderthals, allowing them to regulate the temperature and humidity of inhaled air within the nasal cavity rather than through external nose shape75. However, it is important to acknowledge that the similarity of facial PRS profiles between AFR and Neanderthal can be due to a discovery cohort bias, i.e., due to a weak portability of the PRS to other populations. A key limitation of our findings is the scarcity of archaic human genomes, especially for Denisovans, with only one high-quality sample available. This may explain inconsistencies between our chin PRS profiles and fossil evidence from the Xiahe mandible, which came from a different individual in a different time and geographic region9,10. Another limitation is that internal craniofacial features, such as those related to the oral cavity and teeth, were overlooked in our study. Expanding the number of archaic samples and improving facial landmark coverage will enhance the accuracy of genetic facial inferences, deepening our understanding of how archaic humans looked and how facial shape evolved in modern humans.
In summary, this study represents the largest, regarding both sample size and phenotype number, facial shape GWAS undertaken to date in Europeans and in general. By integrating genomic and facial imagery datasets that were previously analysed and published separately, allowing to increase sample size, by enlarging the number of facial traits and thus the phenotypic variation considered, and by applying our recently developed C-GWAS method allowing to combine GWASs of almost one thousand facial traits, we have increased by far the number of known genetic loci and candidate genes involved in facial variation. Our findings improve the genetic knowledge underlying human facial variation, provide evidence for the partially shared genetic basis of facial variation in different continental populations, provide insights into facial appearance of archaic humans including Denisovans, for which no complete skull information is available thus far, and underline as well as extend the biological functions and pleiotropy spectrum relating to genes involved in facial shape. The less than 10% of facial variance explained by our improved genetic findings underlines the continuous missing heritability problem of the human face, as well as the limitations in genetic prediction of human faces, which shall motivate more facial GWAS studies in different human populations in the future.
Methods
Ethics statement
The Rotterdam Study has been approved by the Medical Ethics Committee of the Erasmus MC (registration number MEC 02.1015) and by the Dutch Ministry of Health, Welfare and Sport (Population Screening Act WBO, license number 1071272–159521 PG). The Rotterdam Study has been entered into the Netherlands National Trial Register (NTR; www.trialregister.nl) and into the WHO International Clinical Trials Registry Platform (ICTRP; www.who.int/ictrp/network/primary/en/) and under shared catalogue number NTR6831. The TwinsUK Study has been approved by the St. Thomas’ Hospital Local Research Ethics Committee. For the ALSPAC samples, ethical approval for the study was obtained from the ALSPAC Ethics and Law Committee and the Local Research Ethics Committees. For the US Pennsylvania Study samples, the following local ethics approvals were obtained: Urbana-Champaign, IL (PSU IRB 13103); New York, NY (PSU IRB 45727); Cincinnati, OH (UC IRB 2015-3073); Twinsburg, OH (PSU IRB 2503); State College, PA (PSU IRB 44929 and 4320); Austin, TX (PSU IRB 44929); and San Antonio, TX (PSU IRB 1278). For the US Indiana Study samples, the following local ethics approvals were obtained: Indianapolis, IN, and Twinsburg, OH (IUPUI IRB 1409306349).
Study populations
The Rotterdam Study is a population-based cohort study of 14,926 participants’ aged 45 years and older, living in the same suburb of Rotterdam in the Netherlands76. The present study includes 4242 participants of Dutch European ancestry, of whom high-resolution 3dMD digital facial photographs were taken. Genotyping was carried out using the Infinium II HumanHap 550K Genotyping BeadChip version 3 (Illumina, San Diego, CA, USA). Microarray-based genotyping according to the manufacturer’s instructions was performed at Erasmus MC with details provided elsewhere77. All SNPs were imputed using MACH software (www.sph.umich.edu/csg/abecasis/MaCH/) based on the 1000 Genomes Project reference population information78. Quality control procedures and parameters are detailed in Supplementary Table 1. After all quality controls, the current study included a total of 6,729,404 autosomal SNPs. All participants provided written informed consent to participate in the study and to have their information obtained from treating physicians.
The TwinsUK study included 1,574 participants within the TwinsUK adult twin registry based at St. Thomas’ Hospital in London, UK. Participants were photographed using 3dMD (Atlanta, GA, USA) photographic scanning system, and 3D avatars were rendered from two 2D photos taken from the left and right directions. Genotyping of the TwinsUK cohort was done with a combination of Illumina HumanHap300 and HumanHap610Q chips. Intensity data for each array were pooled separately, and genotypes were called with the Illuminus32 calling algorithm, employing a threshold on a maximum posterior probability of 0.95 as suggested in a previous study79. Imputation was performed using the IMPUTE 2.0 software package using haplotype information from the 1000 Genomes Project78. After all quality controls (Supplementary Table 1), the current study included a total of 4,574,672 autosomal SNPs and 1080 individuals (all females of European ancestry) for whom high-resolution 3dMDface digital photographs were taken. All participants provided fully informed consent under a protocol reviewed by the St. Thomas’ Hospital Local Research Ethics Committee.
The ALSPAC is a longitudinal birth cohort in which pregnant women residing in Avon, UK, with expected dates of delivery between 1 April 1991 and 31 December 1992 were recruited80,81. The initial number of pregnancies enroled was 14,541 (14,203 unique mothers). Of the initial pregnancies, there was a total of 14,676 foetuses, resulting in 14,062 live births and 13,988 children who were alive at 1 year of age. As a result of the additional phases of recruitment, the total sample size for analyses using any data collected after the age of seven is 15,447 pregnancies (14,833 unique women), resulting in 15,658 foetuses. Of these 14,901 children were alive at 1 year of age. In addition to this, 4731 ALSPAC children were imaged using a Konica Minolta Vivid 900 laser scanner (Konica Minolta Sensing Europe). Please note that the study website contains details of all the data that is available through a fully searchable data dictionary and variable search tool (http://www.bristol.ac.uk/alspac/researchers/our-data/). The current study sample included 3566 unrelated individuals of European ancestry with quality-controlled images, covariates, and genotype data, which are the same samples analysed in a previous facial GWAS21. Quality control of 3D image, genotyping, and imputation process is the same as in the previous study and has been described elsewhere21. ALSPAC participants were genotyped using the Illumina Human Hap550 quad array, performed by Sample Logistics and Genotyping Facilities at the Wellcome Trust Sanger Institute (Cambridge, UK) and the Laboratory Corporation of America (Burlington, NC), supported by 23andMe. Imputation was performed using the IMPUTE 2.0 software package using haplotype information from the 1000 Genomes Project Phase 1 reference panel (Version 3)78. After quality controls (Supplementary Table 1), a total of 7,417,599 autosomal SNPs were included. Informed consent for the use of data collected via questionnaires and clinics was obtained from participants following the recommendations of the ALSPAC Ethics and Law Committee at the time. Consent for biological samples has been collected in accordance with the Human Tissue Act (2004).
For the two US cohorts of European ancestry from Pennsylvania (US-P) and Indiana (US-I), summary statistics of the 946 facial traits were provided to this study, which were based on samples used in a previous facial GWAS21. As reported by White et al.21, institutional review board approval was obtained at each recruitment site of US-P and US-I, and all participants gave their written informed consent before participation. See White et al.21 for further details.
Facial phenotyping
Participants’ raw 3D facial images were acquired using a 3D photographic scanning system by 3dMD (http://www.3dmd.com/). During scanning, participants maintained a neutral expression with closed mouths. 3D avatars were generated from 2D photos taken from various predefined angles, i.e., from front-top, left-down, and right-down directions. MeshMonk82 software was used to identify 44 predefined facial landmarks (Supplementary Table 2). Quality control on landmarks involved the following iterative process to exclude outliers: (1) Perform scaled General Procrustes analysis on all samples; (2) Calculate average coordinates for each landmark; (3) Compute standardized coordinate differences for each landmark in X, Y, and Z axes; (4) Sum the squared standardized differences and obtain p-values under the χ2 distribution with 3 degrees of freedom; (5) Remove samples with significant deviations after Bonferroni correction. This process iterates until no significant deviations are found. Subsequently, we computed 946 pairwise Euclidean distances between the 44 landmarks, extracting residuals after regressing out covariates (Supplementary Table 1). The residuals, standardized and ranked, represented facial traits and were used in GWAS followed.
GWAS and meta-analysis
We performed GWAS on 946 facial traits using a linear regression model assuming additive allele effects in Plink83. For TwinsUK, which includes related individuals, we used fastGWA84, a mixed linear model that controls for population relatedness. Meta-analysis of GWAS summary statistics from RS, TwinsUK, US-P, US-I, and ALSPAC was conducted using the Inverse Variance Weighting (IVW) method. We retained SNPs present in RS and with a sample size greater than two-thirds of the total (N = 7775), resulting in 6,343,785 SNPs. This ensured consistent effects across populations, as RS, primarily an elderly cohort, may have different genetic structures for facial shape compared to the teenager cohort ALSPAC. Effect-based Inverse Covariance Weighting31 (EbICoW), an extension of IVW, was applied in C-GWAS for each pair of symmetric facial distances, reducing the number of meta-analyses GWASs to 514. SNP-based heritability and inflation factors for these 514 GWASs were estimated using LDSC34 (Supplementary Data 2). Consistency of SNP effects across cohorts was assessed using the Intraclass Correlation Coefficient (ICC) calculated with the R package ‘irr’. Reducing SNPs to trait-specific associated SNPs (meta-analysis GWAS p < 0.05) increased ICC (Supplementary Data 5), indicating heterogeneity in genetic effects on facial shape. We conducted a MAMBA37 analysis on the 253 lead SNPs identified in the C-GWAS. We analysed summary statistics for the 253 lead SNPs across all facial traits, gathering data on effect sizes (beta) and standard errors (se) for each facial trait in each of the five European cohorts. This yielded an input dataset with dimensions 253 (SNPs), 514 (facial traits), and 5 (cohorts). MAMBA then calculated the posterior probability of replicability (PPR) for each SNP-trait association across cohorts, indicating the likelihood that a SNP has a non-zero effect on a facial trait without outliers in multiple cohorts.
C-GWAS
The C-GWAS is an R library that is freely available at https://github.com/Fun-Gene/CGWAS. The summary statistics of the 514 meta-analysis GWASs were used as input for C-GWAS. Details of C-GWAS are provided in the previous study31. In brief, the null hypothesis (H0) of C-GWAS for a SNP under testing is the absence of any allelic effect on all traits, and the alternative hypothesis (H1) is that its allelic effects deviate from 0 for at least one of the multiple traits. C-GWAS incorporates two different tests, each sensitive to either homogeneity effects or heterogeneity effects, to maximize statistical power. All resultant p-values from C-GWAS are adjusted using simulations implemented in C-GWAS to ensure that C-GWAS p-values follow the uniform distribution under the null. A common approach for handling correlated traits in GWAS involves performing separate GWASs for each single trait and selecting the minimum p-value (MinGWAS). However, MinGWAS is inflated under the null hypothesis (no effect on any trait), requiring stricter significance thresholds or adjustments for multiple correlated traits. In this study, we adjusted MinGWAS using the getCoef function in C-GWAS. This function employs simulation analyses to ensure that both C-GWAS and the adjusted MinGWAS follow a uniform distribution under the null. Consequently, our MinGWAS refers to the getCoef-calibrated minimum p-value, allowing direct comparison between the outcomes of C-GWAS, MinGWAS, and any single-trait GWAS. Thus, the traditional genome-wide significance threshold of 5 × 10−8 corresponds to our study-wide significance threshold. Univariate LDSC34 was employed to z-transform C-GWAS p-values to estimate the inflation factor of C-GWAS and MinGWAS results31. The gain of power for C-GWAS was estimated according to the increase in the mean χ2 statistic method described in Turley et al.32. In brief, the power ratio between two GWASs can be derived as \(\frac{{\mathrm{E}}({\chi }_{{\mathrm{GWAS}}1}^{2})-{{\mathrm{E}}({\chi }_{{\mathrm{null}}}^{2})}_{{\mathrm{GWAS}}1}}{{\mathrm{E}}({\chi }_{{\mathrm{GWAS}}2}^{2})-{{\mathrm{E}}({\chi }_{{\mathrm{null}}}^{2})}_{{\mathrm{GWAS}}2}}\). In our current study, \({\mathrm{E}}\left({\chi }_{{\mathrm{Current}}}^{2}\right)=1.459\) and \({{\mathrm{E}}\left({\chi }_{{\mathrm{null}}}^{2}\right)}_{{\mathrm{Current}}}=1.013\). In our previous study31, \({\mathrm{E}}\left({\chi }_{{\mathrm{Previous}}}^{2}\right)=1.142\) and \({{\mathrm{E}}\left({\chi }_{{\mathrm{null}}}^{2}\right)}_{{\mathrm{Previous}}}=0.985\), so that \(\frac{1.459{\mathrm{-}}1.013}{1.142{\mathrm{-}}0.985}=2.84\).
Lead SNP selection and literature integration
COJO implemented in GCTA36,85 is able to identify independently associated SNPs using GWAS summary statistics, which suits our study. However, this approach requires effect size and standardized error to process, thus is not compatible with p-values finally outputted by C-GWAS. We therefore applied a modified version of COJO based on z-statistics distribution of SNPs in the LD region86 to identify SNPs with significant independent effect. In brief, the independent effect of the target SNP can be derived as the difference between the observed effect of the target SNP and the non-independent effect, which can be predicted from other LD SNP87, i.e., zm−rLD\({{{\bf{R}}}}_{{\mathrm{LD}}}^{-1}{{{\bf{z}}}}_{{\mathrm{LD}}}\), where \({z}_{{\mathrm{m}}}\) and \({{{\bf{z}}}}_{{\mathrm{LD}}}\) denote statistics of the target SNP and its LD counterparts, \({{{\bf{r}}}}_{{\mathrm{LD}}}\) and \({{{\bf{R}}}}_{{\mathrm{LD}}}\) denote LD correlations between the target SNP and its LD counterparts and LD counterpart pairs. We used a stepwise selection process identical to COJO to iteratively select lead SNPs in a set of SNPs. This process was applied separately for z-transform C-GWAS p-values of all study-wide significant SNPs in each locus, yielding 41 loci with at least two SNPs with independent effects (Supplementary Data 3). We considered a locus novel if no study-wide significant SNPs of this locus were located within 500 kb of any known face-associated SNPs listed in Supplementary Data 1. We considered a lead SNP novel if it was not in LD (r2 < 0.1) with any known face-associated SNPs in Supplementary Data 1. To determine the study-wide significant threshold of previous facial shape studies, for studies with the threshold reported, we employed the provided thresholds; for studies not mentioning the threshold, we calculated the Bonferroni corrected threshold using the method of effective number of independent tests88.
Genes prioritization and functional annotation
We identified a comprehensive list of 365 candidate genes for subsequent analyses through a structured, multi-step prioritization and functional annotation process. Initially, we determined the closest genes to each of the 253 lead SNPs by utilizing the GRCh37 gene position data from the ‘hsapiens_gene_ensembl’ dataset via the ‘biomaRt’ package in R. To enhance the relevance and functional significance of the identified genes, we proceeded to prioritize functional genes by evaluating associations between SNPs and nearby genes across four key dimensions: (1) regulatory network involvement, (2) cis-expression quantitative trait loci (cis-eQTL) associations, (3) chromatin interactions, and (4) physical proximity. Each of these dimensions contributed to a prioritization score ranging from 0 to 1, which was adjusted for linkage disequilibrium (LD) where applicable. Specifically, we assessed (1) whether lead SNPs or their LD counterparts located in elements regulating the target gene in the CNCC regulatory network from hReg-CNCC39; (2) whether lead SNPs or their LD counterparts are cis-eQTLs of the target gene in FUMA89 results using ‘SNP2GENE’ function; (3) whether lead SNPs or their LD counterparts are in regions with chromatin interactions involving the target gene in FUMA results using ‘SNP2GENE’ function; and (4) whether lead SNPs or their LD counterparts are physically close to the target gene using R package ‘biomaRt’. Genes that met predefined thresholds in any of these four dimensions were retained as functional genes, resulting in a combined total of 581 genes from combining closest genes (Supplementary Data 4) and functional genes (Supplementary Data 7). To refine this gene list further and ensure robustness, we conducted a Gene Ontology (GO) analysis on the 581 genes for biological process (BP) using clusterProfiler90. We retained 365 genes that were significantly involved in at least one BP, applying a Bonferroni-corrected p-value threshold of less than 0.01. This final set of 365 genes constituted our candidate genes for subsequent analyses.
To comprehensively explore the 177 biological processes identified with a stringent Bonferroni-corrected p < 0.001 from GO enrichment analyses with these candidate genes, we performed hierarchical clustering to 177 BPs based on gene set similarity. Utilizing the ‘hclust’ function in R with the ‘ward.D’ method, we clustered the dissimilarity matrix of gene sets involved in 177 BPs, setting the clustering height to 1.5 to achieve an optimal balance between the number and diversity of clusters (Supplementary Fig. 8). For enrichment testing of certain gene set, we employed the ‘phyper’ function in R to assess specific gene sets across 177 BPs from the enrichment analysis of the 356 genes. Fold enrichment was calculated as the ratio of the proportion of genes involved in a certain BP from the gene set to all candidate genes involved in the same BP, divided by the proportion of genes from the gene set to all candidate genes (Supplementary Data 9). Furthermore, all 253 lead SNPs and their high LD counterparts (r2 > 0.6) were annotated for genome-wide significant associations (p < 5 × 10−8) using data sourced from the GWAS Catalog (www.ebi.ac.uk/gwas/) via the ‘SNP2GENE’ function in FUMA. This annotation process allowed us to summarize pleiotropic associations linked to each lead SNP, as detailed in Supplementary Data 11. By searching for manually curated keywords, we classified the associated pleiotropic traits into 13 distinct categories, e.g., “waist-to-hip”, “circumference”, “weight”, “height”, etc., corresponding to anthropometric traits; “cholesterol”, “glucose”, “serum”, “protein”, etc., corresponding to metabolic traits; “myopia”, “orbital”, “cup-disc”, “glaucoma”, etc., corresponding to optical traits; “platelet”, “monocyte”, “neutrophil”, “haemoglobin”, etc., corresponding to haemocytes traits, etc.
Proportion of genetically explained phenotypic facial variance
Summary statistics from 514 facial traits meta-analysis GWASs were employed to estimate the PVE among 253 lead SNPs and 514 facial traits. First, we derived the formula for the PVE of an individual SNP for an individual trait. We then aggregated these PVEs to calculate regional-PVE or face-PVE for multiple SNPs and traits. For the association analysis, a linear model was assumed, ft = bm + e, where ft is the standardized facial trait, b is the effect size of SNP m, and e is the residual. The PVE was derived as \({{{\rm{p}}}{{\rm{v}}}{{\rm{e}}}}_{m}={b}^{2}{\mathrm{Var}}({{\bf{m}}})\). Since the lead SNPs are independent, the PVE for a SNP set M was the sum of \({{{\rm{p}}}{{\rm{v}}}{{\rm{e}}}}_{m}\). To correct for biases from increasing SNP numbers, given that \({{{\rm{p}}}{{\rm{v}}}{{\rm{e}}}}_{m}\) is always greater than 0 under the null, we simulated SNP statistic \({t}_{{\mathrm{null}}}\) under the null and derived the corrected PVE as \((1-{t}_{{\mathrm{null}}}^{2}/{t}_{{\mathrm{m}}}^{2}){b}^{2}{\mathrm{Var}}({{\bf{m}}})\), where \({t}_{{\mathrm{m}}}\) is the statistic of SNP m. We repeated simulations 10,000 times and regarded the average as the PVE. Next, we defined the multi-trait PVE to comprehensively evaluate the explanatory power of individual SNPs across multiple traits. For N independent traits, we directly took the average of \({{{\rm{p}}}{{\rm{v}}}{{\rm{e}}}}_{n}\). For N correlated traits, we transformed the traits into an eigenvector space, where dimensions are uncorrelated, allowing us to use the eigenvalue-weighted average of the PVE in each dimension. Specifically, given the correlation matrix C and its eigenvector matrix V, the multi-trait PVE was pveN = ∑(bTV)2 Var(m)/N. We derived the corrected multi-trait PVE similarly, using a simulated SNP statistics matrix \({{{\bf{T}}}}_{{\mathrm{null}}}\) based on a multivariate normal distribution with correlations C, i.e., \(\sum (1-{{{\bf{T}}}}_{{\mathrm{null}}}^{2}/{{{\bf{T}}}}_{{\mathrm{m}}}^{2}){({b}^{{\mathrm{T}}}{{\bf{V}}})}^{2}{\mathrm{Var}}({{\bf{m}}})/N\), where Tm denotes statistics of SNP m associated with N traits, using ‘mvrnorm’ function in the R package ‘MASS’. We calculated multi-trait PVEs for the entire face, within and between regions, resulting in face-PVE and 36 regional-PVEs, later encompassing 8 facial regions and 28 connection regions in between. We then summarized the regional PVEs of each lead SNP onto candidate genes, including, (1) merging genes with the same PVE patterns as one set and removing redundancy; (2) some candidate genes were prioritized by multiple lead SNPs, summing their regional PVEs accordingly. These yielded a regional PVE matrix for 225 different candidate gene sets (Supplementary Data 12). After excluding 32 candidate gene sets with no significant regional PVEs (Bonferroni corrected p < 0.001), we performed hierarchical clustering on the remaining 193 sets using Spearman’s correlation and the ‘ward.D’ method of ‘hclust’ function in R.
Genetic modelling of facial traits
Among the 946 facial distances, 82 measured symmetric landmarks on both sides of the face or in the mid-axis, leaving 864 distances (432 pairs of symmetric distances). In RS and TwinsUK, the average values for each pair of these symmetric distances were derived, yielding 432 facial traits. Combined with the remaining 82 traits, this resulted in a total of 514 facial traits used in the PRS analysis. To reduce redundancy among the 514 facial traits, we selected 331 traits for genetic modelling based on a threshold of trait correlation r2 < 0.8, prioritizing by their PVEs. Along with 253 lead SNPs, we included 129 previously reported independent SNPs (LD r2 < 0.1) from Supplementary Data 1 with MAF > 0.01 in the RS and p < 0.05 in our discovery phase C-GWAS results, totalling 382 SNPs. A multivariate linear model was constructed for each of 331 traits using these 382 SNPs, and the model performance was evaluated using 10-fold cross-validation r2 calculated in the RS. Due to the genetic heterogeneity of facial shapes, we employed a forward selection strategy to prioritize SNP sets for each model. We refined these models by selecting 23 based on a model fitted values correlation threshold of r2 < 0.1, prioritizing by model cross-validation r2. Thus, these 23 models represent the most genetically explainable and independent traits among the 514 facial traits (Supplementary Data 13). PRSs for these 23 traits were computed using effect sizes from the models, i.e., \({\sum }_{i=1}^{382}{b}_{i}{m}_{i}\), where b is the scaled effect size of SNP m, ensuring the fitted PRS follows a standard normal distribution. Facial PRS profiles for different continental populations in the 1000 Genomes Project (AFR, EUR, EAS, and SAS, totalling 2000 samples) were derived by averaging the PRS of samples within each population (Supplementary Data 14).
We observed that the top 5 cross-validation r2 models were for nose traits. We assessed the identification ability of the top 5 nose PRSs by calculating the cosine similarity in correct matches (a sample’s nose PRSs and traits) and false matches (a sample’s nose PRSs and other samples’ traits). Identification ability was evaluated using 10-fold cross-validation, CMC, and ROC curves in the RS. For CMC calculation, each test set included one correct match and 399 random false matches (N = 400). The proportions of correct matches ranked in the top 4, 40, and 200 were recorded as CMC1%, CMC10%, and CMC50%, respectively. ROC curves and corresponding AUC value were derived with the R package ‘pROC’.
Genetic diversity and positive selection
The effect allele frequency of 253 lead SNPs and their high LD counterparts (r2 > 0.6) in three super-populations, i.e., Europeans (EUR), East Asians (EAS), and Africans (AFR), were derived from the 1000 Genome Project40. We performed two selective statistics for each SNP, including Wright’s fixation index91 (FST) measuring the differences between populations, and the population branch statistic92 (PBS). Both FST and PBS were calculated based on continental populations from EUR, EAS and AFR, which was consistent with the setting of the previous study93. The Weir and Cockerham estimators91 implemented in GCTA85 were used to compute FST. PBS values were subsequently calculated based on the obtained FST values using the method described in Yi et al.92. In brief, for a given SNP among three populations X, Y, and Z, the pair-wise FST are first transformed using a logarithmic conversion as L = −ln(1−FST). The PBS for the target population, for example, X, is then calculated as (LXY + LXZ−LYZ)/2. This represents the amount of allele frequency divergence specific to population X, relative to the other two populations. Empirical significance cut-offs for both FST and PBS were set at the values corresponding to the top 1% values across the genome (Supplementary Data 15). Enrichment analysis94 was conducted for 253 lead SNPs to assess whether these SNPs exhibited significantly higher FST and PBS values than random SNPs. In brief, the average FST and PBS value across 253 lead SNPs was compared to a null distribution, empirically generated by 10,000 times of calculating the average values from a randomly selected 253 SNP set. These random SNP sets were chosen to have matched MAF to 253 lead SNPs. To achieve this, the range of MAFs (0–1) was divided into 50 equal bins separately for each of the three populations (EAS, EUR, and AFR). A matched SNP was then randomly selected from the same bin as the original SNP. The enrichment was considered significant if the observed average value exceeded the 95th percentile of the null distribution, corresponding to a 5% alpha level.
Archaic introgressed regions and facial PRS profiling
We aimed to explore the contribution of the Neanderthal archaic introgression to human facial variation. To achieve this, we gathered the Neanderthal introgressed segments identified in EUR and EAS in three studies48,49,50. In addition to downloading the raw data, we used BEDtools95 to obtain the union of introgressed segments found in the three studies identified in EUR and EAS, respectively. Finally, we acquired 8 segmentation annotation files (three studies and their union in EUR and EAS) for the following analysis. We annotated and attained the overlaps of 253 lead SNPs based on 8 annotations (Supplementary Data 16). We applied the effect sizes estimated from 23 models in the RS to 8 Neanderthals, 1 Denisovan, and 1 Denisova–Neanderthal admixed individual in the Allen Ancient DNA Resource54 (AADR) (Dataverse 8.0) to derive facial PRS profiles of archaic individuals. Due to limited overlap between modern and archaic genotypes for the 382 SNPs used in the 23 models trained in the RS, we replaced non-overlapping SNPs with their highest LD counterparts (at least r2 > 0.6) with available archaic genotypes. This resulted in 357 SNPs (24 had no suitable replacements) being determined and used to re-model and compute PRSs for archaic individuals (Supplementary Data 18). The similarity between population genetic profiles of faces was evaluated by the squared correlation between the facial PRS profiles of the corresponding populations. We compared the mean absolute differences in allele frequencies for 357 SNPs between archaic humans (Archaic, 10 samples) from the AADR and four modern populations from the 1000 Genomes Project (AFR, EUR, EAS, and SAS, totalling 2000 samples). To assess whether these differences were specific to the selected face SNPs, we performed random sampling of 10,000 SNP sets and calculated the mean absolute differences in allele frequencies between archaic and modern human groups for each set.
Validation of archaic facial PRS profiles with previous studies
We selected 16 facial features documented in archaeological and anthropological studies and overlapped with 23 facial traits in our PRS model to establish the direction of phenotypic differences between Neanderthal skulls and modern human skulls. Compared to modern human skulls, Neanderthal cranial fossils exhibited a wide face8,56, long face8,96 and protruded midface51,96, and 13 regional ones, including low51,55 and protruded51,55 brow ridge; large orbital volume56,97; a nose with retracted nose root51,56, protruded nose bridge51,96, tall nostril51 and wide nose wing51,96; barely curved cheekbones51,96 and infraorbital inflation96; wide8,98 and low8,98 palate; high anterior mandible8,52 and flat chin52,55. Moreover, we compared mean PRS obtained from 8 Neanderthals, 1 Denisovan, and 503 modern Europeans in EUR (using the same approach described in the section “Genetic modelling of facial traits” and “Archaic introgressed regions and face PRS profiling”) to each other to determine the direction of genetic difference for each feature. To assess the consistency between facial PRS profiles and skull morphology of Neanderthals, we compared the direction of phenotypic difference from the literature with the direction of mean PRS difference from our study for each feature between Neanderthals and EUR. If the direction of the phenotypic difference matched the direction of the PRS difference (both indicating either an increase and a decrease in Neanderthals relative to EUR), the result was classified as concordant. If the directions were opposite, the result was classified as discordant. If multiple PRS overlapped with a single feature and showed conflicting directions of difference, the result was classified as inconsistent. To assess the statistical significance of our findings, we conducted a permutation test involving 10,000 iterations. In each iteration, 357 MAF-matched SNPs (using the same approach described in the section “Genetic diversity and positive selection”) were randomly selected from the genome. PRSs were recalculated for Neanderthals using these randomly selected SNPs and were reclassified as concordant, discordant, or inconsistent based on the criteria mentioned above. The counts of the three concordance statuses from all iterations were used to construct a null distribution representing the expected distribution of outcomes under the null hypothesis of no true association. The observed results were compared against the null distributions to determine their quantiles. A result was considered statistically significant if its occurrence was rare under the null distribution (e.g., in the top or bottom 1% of the distribution). To confirm the consistency between our Denisovan facial PRS profiles and previous studies, we included a fossil study10 (Xiahe mandible) and a prediction study8 (methylation profiling). We extracted and compared the Xiahe mandible measurements for ‘symphyseal height’ and ‘symphyseal angle’ from SI Table 2 in Chen at al.10, corresponding to the characteristics ‘high anterior mandible’ and ‘flat chin’ in our study. Additionally, we extracted and compared data of six overlapping traits from Denisovan methylation profiles from Table S7 in Gokhman et al.8. Comparisons were made twice: once between Denisovans and modern humans and once between Denisovans and Neanderthals. Details of the consistency are provided in Supplementary Data 19.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Summary statistics data of C-GWAS and MinGWAS, full FUMA results in 253 loci, visualized regional-PVE of 253 lead SNPs, and reconstructed average facial images using 3D graph auto-encoder for 23 traits in face PRS profiles are available at https://doi.org/10.6084/m9.figshare.26360206. Single-trait summary statistics of meta-analyses of GWAS for the European discovery phase data generated in this study are available at https://doi.org/10.5281/zenodo.13730680. GWAS summary statistics for the East Asian replication phase data are available at https://www.biosino.org/node/project/detail/OEP00002283. The ChIP-seq data of CNCC are available in the Gene Expression Omnibus with accession number GSE70751. The regulation region annotation data in embryonic craniofacial tissues are available in the Gene Expression Omnibus with accession number GSE97752. The regulation region annotation data in other general cell types are available in the Roadmap Epigenomics Project (https://egg2.wustl.edu/roadmap/web_portal/chr_state_learning.html#core_15state). The regulatory network of CNCC is available at https://github.com/AMSSwanglab/hReg-CNCC. The 1000 Genomes Project phase 3 data are available at https://www.internationalgenome.org/category/phase-3. Other statistical data for generating figures in this study are provided in the Supplementary Information and Supplementary Data. Individual-level data used in this study have different data accessibility conditions based on the cohorts’ informed consent. For ALSPAC, data access can be granted via an approved application (http://www.bristol.ac.uk/alspac/researchers/access/) according to defined regulations (https://www.bristol.ac.uk/media-library/sites/alspac/documents/researchers/data-access/ALSPAC_Access_Policy.pdf), including on the time frame of response and access, and queries can be sent to the ALSPAC data team (alspac-data@bristol.ac.uk) and the ALSPAC Executive team (alspac-exec@bristol.ac.uk). For TwinsUK, data accessed can be requested by submitting a formal Data Access Application (http://twinsuk.ac.uk/resources-forresearchers/access-our-data/) according to defined regulations (https://twinsuk.ac.uk/resources-for-researchers/our-data/) including on the time frame of response and access, and queries can be sent to the data access and collaboration manager Victoria Vazquez, at King’s College London (Victoria.vazquez@kcl.ac.uk). For RS and the two US datasets (US-P and US-I), informed consent does not allow sharing individual-level data, unless as part of direct collaborations with the cohort owners. For RS, collaboration requests can be directed to Frank J.A. van Rooij at Erasmus MC (f.vanrooij@erasmusmc.nl), for US-P to Mark Shriver at Pennsylvania State University (mds17@psu.edu), and for US-I to Susan Walsh at Indiana University Indianapolis (walshsus@iu.edu); responses to collaboration requests will be answered in a timely fashion, typically one calendar month.
Code availability
MeshMonk is implemented as an open-source toolbox available at https://gitlab.kuleuven.be/mirc/meshmonk. C-GWAS is implemented as an open-source R package available at https://github.com/Fun-Gene/CGWAS. LDSC is implemented as an open-source command line tool available at https://github.com/bulik/ldsc. SpiralNet++ is implemented as an open-source 3D graph auto-encoder available at https://github.com/sw-gong/spiralnet_plus. The latest version of MeshMonk is available at https://doi.org/10.6084/m9.figshare.c.6858271.v199. The latest version of C-GWAS is available at https://doi.org/10.6084/m9.figshare.26360206100. Other softwares used in data analysis includes: Plink (https://www.cog-genomics.org/plink2/), FastGWA (https://yanglab.westlake.edu.cn/software/gcta/#fastGWA), FUMA, GCTA-Fst (https://yanglab.westlake.edu.cn/software/gcta/#Fst), R package ‘shapes’ (version 1.2.7), R package ‘irr’ (version 0.84.1), R package ‘mamba’ (version 1.12), R package ‘clusterProfiler’ (Version 4.6.2), R package ‘org.Hs.eg.db’ (version 3.16.0), R package ‘biomaRt’ (version 2.54.1), R package ‘MASS’ (version 7.3.57), R package ‘pROC’ (version 1.18.4).
References
Johannsdottir, B., Thorarinsson, F., Thordarson, A. & Magnusson, T. E. Heritability of craniofacial characteristics between parents and offspring estimated from lateral cephalograms. Am. J. Orthod. Dentofac. Orthop.127, 200–207 (2005). quiz 260-201.
Djordjevic, J., Zhurov, A. I., Richmond, S. & Visigen, C. Genetic and environmental contributions to facial morphological variation: a 3D Population-Based Twin Study. PLoS ONE 11, e0162250 (2016).
Xiong, Z. et al. Novel genetic loci affecting facial shape variation in humans. Elife 8, e49898 (2019).
Richmond, S., Howe, L. J., Lewis, S., Stergiakouli, E. & Zhurov, A. Facial genetics: a brief overview. Front. Genet. 9, 462 (2018).
Wen, Y. F., Wong, H. M., Lin, R., Yin, G. & McGrath, C. Inter-ethnic/racial facial variations: a systematic review and bayesian meta-analysis of photogrammetric studies. PLoS ONE 10, e0134525 (2015).
Farkas, L. G. et al. International anthropometric study of facial morphology in various ethnic groups/races. J. Craniofac. Surg. 16, 615–646 (2005).
Du, P. et al. Ancient genome of the Chinese Emperor Wu of Northern Zhou. Curr. Biol.34, 1587–1595 e1585 (2024).
Gokhman, D. et al. Reconstructing Denisovan anatomy using DNA methylation maps. Cell 179, 180–192 e110 (2019).
Reich, D. et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 468, 1053–1060 (2010).
Chen, F. et al. A late Middle Pleistocene Denisovan mandible from the Tibetan Plateau. Nature 569, 409–412 (2019).
Bonfante, B. et al. A GWAS in Latin Americans identifies novel face shape loci, implicating VPS13B and a Denisovan introgressed region in facial variation. Sci. Adv. 7, eabc6160 (2021).
Reilly, P. F., Tjahjadi, A., Miller, S. L., Akey, J. M. & Tucci, S. The contribution of Neanderthal introgression to modern human traits. Curr. Biol.32, R970–R983 (2022).
Li, Q. et al. Automatic landmarking identifies new loci associated with face morphology and implicates Neanderthal introgression in human nasal shape. Commun. Biol. 6, 481 (2023).
Kayser, M., Branicki, W., Parson, W. & Phillips, C. Recent advances in Forensic DNA Phenotyping of appearance, ancestry and age. Forensic Sci. Int. Genet. 65, 102870 (2023).
Simcoe, M. et al. Genome-wide association study in almost 195,000 individuals identifies 50 previously unidentified genetic loci for eye color. Sci. Adv. 7, eabd1239 (2021).
Chen, Y. et al. Genetic prediction of male pattern baldness based on large independent datasets. Eur. J. Hum. Genet.31, 321–328 (2023).
Breslin, K. et al. HIrisPlex-S system for eye, hair, and skin color prediction from DNA: massively parallel sequencing solutions for two common forensically used platforms. Forensic Sci. Int. Genet. 43, 102152 (2019).
Gurovich, Y. et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat. Med. 25, 60–64 (2019).
Hsieh, T. C. et al. GestaltMatcher facilitates rare disease matching using facial phenotype descriptors. Nat. Genet. 54, 349–357 (2022).
Dingemans, A. J. M. et al. PhenoScore quantifies phenotypic variation for rare genetic diseases by combining facial analysis with other clinical features using a machine-learning framework. Nat. Genet. 55, 1598–1607 (2023).
White, J. D. et al. Insights into the genetic architecture of the human face. Nat. Genet. 53, 45–53 (2021).
Zhang, M. et al. Genetic variants underlying differences in facial morphology in East Asian and European populations. Nat. Genet. 54, 403–411 (2022).
Liu, F. et al. A genome-wide association study identifies five loci influencing facial morphology in Europeans. PLoS Genet. 8, e1002932 (2012).
Paternoster, L. et al. Genome-wide association study of three-dimensional facial morphology identifies a variant in PAX3 associated with nasion position. Am. J. Hum. Genet. 90, 478–485 (2012).
Tan, J. et al. The adaptive variant EDARV370A is associated with straight hair in East Asians. Hum. Genet. 132, 1187–1191 (2013).
Adhikari, K. et al. A genome-wide association scan implicates DCHS2, RUNX2, GLI3, PAX1 and EDAR in human facial variation. Nat. Commun. 7, 11616 (2016).
Stephan, C. N., Caple, J. M., Guyomarc’h, P. & Claes, P. An overview of the latest developments in facial imaging. Forensic Sci. Res. 4, 10–28 (2019).
Claes, P. et al. Genome-wide mapping of global-to-local genetic effects on human facial shape. Nat. Genet. 50, 414–423 (2018).
de Jong, M. A. et al. Ensemble landmarking of 3D facial surface scans. Sci. Rep. 8, 12 (2018).
Qiao, H. et al. A comprehensive evaluation of the phenotype-first and data-driven approaches in analyzing facial morphological traits. iScience 27, 109325 (2024).
Xiong, Z. et al. Combining genome-wide association studies highlight novel loci involved in human facial variation. Nat. Commun. 13, 7832 (2022).
Turley, P. et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet. 50, 229–237 (2018).
Li, Y. et al. Combined genome-wide association study of 136 quantitative ear morphology traits in multiple populations reveal 8 novel loci. PLoS Genet. 19, e1010786 (2023).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Qian, W. et al. Genetic evidence for facial variation being a composite phenotype of cranial variation and facial soft tissue thickness. J. Genet. Genom.=Yi chuan xue bao 49, 934–942 (2022).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375 (2012). S361-363.
McGuire, D. et al. Model-based assessment of replicability for genome-wide association meta-analysis. Nat. Commun. 12, 1964 (2021).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Feng, Z. et al. hReg-CNCC reconstructs a regulatory network in human cranial neural crest cells and annotates variants in a developmental context. Commun. Biol. 4, 442 (2021).
Genomes Project C et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Simmons-Ehrhardt, T. L., Monson, K. L., Flint, T. & Saunders, C. P. Quantitative accuracy and 3D biometric matching of 388 statistically estimated facial approximations of live subjects. Forensic Imaging 21, 200377 (2020).
Zaidi, A. A., Mattern, B. C., Claes, P., McEvoy, B., Hughes, C. & Shriver, M. D. Investigating the case of human nose shape and climate adaptation. PLoS Genet. 13, e1006616 (2017).
Jacobs, L. C. et al. Comprehensive candidate gene study highlights UGT1A and BNC2 as new genes determining continuous skin color variation in Europeans. Hum. Genet. 132, 147–158 (2013).
Visser, M., Palstra, R. J. & Kayser, M. Human skin color is influenced by an intergenic DNA polymorphism regulating transcription of the nearby BNC2 pigmentation gene. Hum. Mol. Genet. 23, 5750–5762 (2014).
Gittelman, R. M. et al. Archaic hominin admixture facilitated adaptation to out-of-Africa environments. Curr. Biol.26, 3375–3382 (2016).
Green, R. E. et al. A draft sequence of the Neandertal genome. Science 328, 710–722 (2010).
Prufer, K. et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, 43–49 (2014).
Sankararaman, S. et al. The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507, 354–357 (2014).
Vernot, B. & Akey, J. M. Resurrecting surviving Neandertal lineages from modern human genomes. Science 343, 1017–1021 (2014).
Yuan, K. et al. Refining models of archaic admixture in Eurasia with ArchaicSeeker 2.0. Nat. Commun. 12, 6232 (2021).
Weaver, T. D. Out of Africa: modern human origins special feature: the meaning of neandertal skeletal morphology. Proc. Natl Acad. Sci. USA 106, 16028–16033 (2009).
Bergmann, I., Hublin, J. J., Gunz, P. & Freidline, S. E. How did modern morphology evolve in the human mandible? The relationship between static adult allometry and mandibular variability in Homo sapiens. J. Hum. Evol. 157, 103026 (2021).
Zeberg, H. et al. A Neanderthal sodium channel increases pain sensitivity in present-day humans. Curr. Biol.30, 3465–3469 e3464 (2020).
Mallick, S. et al. The Allen Ancient DNA Resource (AADR) a curated compendium of ancient human genomes. Sci. Data 11, 182 (2024).
Schwartz, J. H. & Tattersall, I. Fossil evidence for the origin of Homo sapiens. Am. J. Phys. Anthropol. 143, 94–121 (2010).
Tattersall, I. & Schwartz, J. H. Morphology, paleoanthropology, and Neanderthals. Anat. Rec. 253, 113–117 (1998).
Rynn, C., Wilkinson, C. M. & Peters, H. L. Prediction of nasal morphology from the skull. Forensic Sci. Med Pathol. 6, 20–34 (2010).
Ryu, J. Y. et al. Craniofacial anthropometric investigation of relationships between the nose and nasal aperture using 3D computed tomography of Korean subjects. Sci. Rep. 10, 16077 (2020).
Lippert, C. et al. Identification of individuals by trait prediction using whole-genome sequencing data. Proc. Natl Acad. Sci. USA 114, 10166–10171 (2017).
Sero, D. et al. Facial recognition from DNA using face-to-DNA classifiers. Nat. Commun. 10, 2557 (2019).
Chen, Y. et al. The impact of correlations between pigmentation phenotypes and underlying genotypes on genetic prediction of pigmentation traits. Forensic Sci. Int Genet 50, 102395 (2021).
de la Puente, M. et al. Development and Evaluation of the Ancestry Informative Marker Panel of the VISAGE Basic Tool. Genes (Basel) 12, 1284 (2021).
Ruiz-Ramirez, J. et al. Development and evaluations of the ancestry informative markers of the VISAGE Enhanced Tool for Appearance and Ancestry. Forensic Sci. Int. Genet. 64, 102853 (2023).
McArthur, E., Rinker, D. C. & Capra, J. A. Quantifying the contribution of Neanderthal introgression to the heritability of complex traits. Nat. Commun. 12, 4481 (2021).
Zeberg, H., Jakobsson, M. & Paabo, S. The genetic changes that shaped Neandertals, Denisovans, and modern humans. Cell 187, 1047–1058 (2024).
Noback, M. L., Harvati, K. & Spoor, F. Climate-related variation of the human nasal cavity. Am. J. Phys. Anthropol. 145, 599–614 (2011).
Prufer, K. et al. A high-coverage Neandertal genome from Vindija Cave in Croatia. Science 358, 655–658 (2017).
Mafessoni, F. et al. A high-coverage Neandertal genome from Chagyrskaya Cave. Proc. Natl Acad. Sci. USA 117, 15132–15136 (2020).
Chen, L., Wolf, A. B., Fu, W., Li, L. & Akey, J. M. Identifying and interpreting apparent Neanderthal Ancestry in African individuals. Cell 180, 677–687 e616 (2020).
Iasi, L. N. M. et al. Neanderthal ancestry through time: insights from genomes of ancient and present-day humans. Science 386, eadq3010 (2024).
Sumer, A. P. et al. Earliest modern human genomes constrain timing of Neanderthal admixture. Nature 638, 711–717 (2025).
Brand, C. M., Colbran, L. L. & Capra, J. A. Predicting archaic hominin phenotypes from genomic data. Annu. Rev. Genom. Hum. Genet. 23, 591–612 (2022).
Huerta-Sanchez, E. et al. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 512, 194–197 (2014).
Peyregne, S., Slon, V. & Kelso, J. More than a decade of genetic research on the Denisovans. Nat. Rev. Genet. 25, 83–103 (2024).
de Azevedo, S. et al. Nasal airflow simulations suggest convergent adaptation in Neanderthals and modern humans. Proc. Natl Acad. Sci. USA 114, 12442–12447 (2017).
Hofman, A. et al. The Rotterdam Study: 2014 objectives and design update. Eur. J. Epidemiol. 28, 889–926 (2013).
Kayser, M. et al. Three genome-wide association studies and a linkage analysis identify HERC2 as a human iris color gene. Am. J. Hum. Genet. 82, 411–423 (2008).
Genomes Project C et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
Small, K. S. et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat. Genet. 43, 561–564 (2011).
Boyd, A. et al. Cohort Profile: the ‘children of the 90s’-the index offspring of the Avon Longitudinal Study of Parents and Children. Int. J. Epidemiol. 42, 111–127 (2013).
Fraser, A. et al. Cohort Profile: the Avon Longitudinal Study of Parents and Children: ALSPAC mothers cohort. Int. J. Epidemiol. 42, 97–110 (2013).
White, J. D. et al. MeshMonk: Open-source large-scale intensive 3D phenotyping. Sci. Rep. 9, 6085 (2019).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Jiang, L. et al. A resource-efficient tool for mixed model association analysis of large-scale data. Nat. Genet. 51, 1749–1755 (2019).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Chen, W. et al. Improved analyses of GWAS summary statistics by reducing data heterogeneity and errors. Nat. Commun. 12, 7117 (2021).
Pasaniuc, B. et al. Fast and accurate imputation of summary statistics enhances evidence of functional enrichment. Bioinformatics 30, 2906–2914 (2014).
Li, J. & Ji, L. Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity 95, 221–227 (2005).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Xu, S. et al. Using clusterProfiler to characterize multiomics data. Nat. Protoc. 19, 3292–3320 (2024).
Weir, B. S. & Cockerham, C. C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 38, 1358–1370 (1984).
Yi, X. et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329, 75–78 (2010).
Li, M. J. et al. dbPSHP: a database of recent positive selection across human populations. Nucleic Acids Res. 42, D910–D916 (2014).
Guo, J. et al. Global genetic differentiation of complex traits shaped by natural selection in humans. Nat. Commun. 9, 1865 (2018).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Lacruz, R. S. et al. The evolutionary history of the human face. Nat. Ecol. Evol. 3, 726–736 (2019).
Pearce, E., Stringer, C. & Dunbar, R. I. New insights into differences in brain organization between Neanderthals and anatomically modern humans. Proc. Biol. Sci. 280, 20130168 (2013).
Lacruz, R. S. et al. Ontogeny of the maxilla in Neanderthals and their ancestors. Nat. Commun. 6, 8996 (2015).
Goovaerts, S. Joint multi-ancestry and admixed GWAS reveals the complex genetics behind human cranial vault shape. figshare https://doi.org/10.6084/m9.figshare.c.6858271.v1 (2023).
Xiong, Z. Combined genome-wide association study of facial traits in Europeans increases explained variance and improves prediction. figshare https://doi.org/10.6084/m9.figshare.26360206 (2025).
Acknowledgements
The authors thank all participants and supporting staff from all cohorts used in this study. We are grateful for the dedication, commitment and contribution of the study participants, the general practitioners, pharmacists, and the staff from the Rotterdam Study. We are extremely grateful to all the families who took part in the ALSPAC study, the midwives for their help in recruiting them, and the whole ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists and nurses. We thank Mark Shriver and Susan Walsh for the summary statistics from their Pennsylvania and Indiana datasets, respectively. The Rotterdam Study is supported by the Erasmus MC; the Erasmus University Rotterdam; the Netherlands Organization for Scientific Research (NWO); the Netherlands Organization for Health Research (ZonMw); the Research Institute for Diseases in the Elderly (RIDE); the Netherlands Genomics Initiative (NGI); the Ministry of Education, Culture and Science; the Ministry of Health Welfare and Sport; the European Commission (DG XII); and the Municipality of Rotterdam. The generation and management of GWAS genotype data for the Rotterdam Study were executed by the Human Genotyping Facility of the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC. The UK Medical Research Council and Wellcome (Grant ref: 217065/Z/19/Z) and the University of Bristol provide core support for ALSPAC. This publication is the work of the authors and Manfred Kayser will serve as guarantor for the contents of this paper. A comprehensive list of grant funding is available on the ALSPAC website (http://www.bristol.ac.uk/alspac/external/documents/. grant-acknowledgements.pdf). ALSPAC Genomewide genotyping data was generated by Sample Logistics and Genotyping Facilities at Wellcome Sanger Institute and LabCorp (Laboratory Corporation of America) using support from 23andMe.
Author information
Authors and Affiliations
Contributions
Conceptualization: F.L. and M.K.; Methodology: Z.X.; Formal analysis, Z.X., Y.L., X.L. and H.L.; Data curation: P.G.H., L.M.P., A.G.U., F.R., M.A.I., M.G., E.B.W., G.V.R., S.R., T.N., T.D.S., S.W., F.L. and M.K.; Original draft: Z.X., Y.L., F.L. and M.K.; Review and editing: Z.X., F.L. and M.K.; Visualization: Z.X., Y.L. and X.L.; Supervision: F.L. and M.K.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiong, Z., Li, Y., Liu, X. et al. Combined genome-wide association study of facial traits in Europeans increases explained variance and improves prediction. Nat Commun 16, 6562 (2025). https://doi.org/10.1038/s41467-025-61761-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61761-7
