
Abstract
Sepsis is a severe global health issue with high mortality rates, and sepsis-associated encephalopathy (SAE) further exacerbates this risk. While recent studies have shown the migration of gut immune cells to the lungs after sepsis, their impact on the central nervous system remains unclear. Our research demonstrates that sepsis could induce the migration of IL-7Rhigh CD8low γδ T17 cells from the small intestine to the meninges, where they secrete IL-17A, impairing mitochondrial function in microglia and activating the cGAS-STING-C1q pathway in male mice. This process is accompanied by inhibited ubiquitination of STING at the K150 site, resulting in STING accumulation and increased release of C1q-tagged hippocampal synapses, which are subsequently pruned by activated microglia. Importantly, 4-octyl itaconate mitigates the excessive synaptic pruning by inhibiting γδ T17 cell migration and promoting STING ubiquitination, thereby alleviating SAE. Our findings suggest a potential mechanism of synaptic pruning by microglia via the cGAS-STING-C1q pathway, emphasize the critical role of gut-derived γδ T17 cell migration to the meninges in SAE, and highlight the importance of STING ubiquitination in modulating C1q-mediated excessive synaptic pruning.
Similar content being viewed by others


Introduction
Sepsis-associated encephalopathy (SAE) is a severe complication of sepsis, characterized by diffuse or multifocal neurological dysfunction, and is a leading cause of increased mortality among ICU patients. Although SAE is often considered reversible, approximately 40% of patients experience long-term neurological symptoms, including memory impairment, depression, anxiety, and cognitive dysfunction, especially in cases of severe sepsis1. The lack of in-depth understanding of the mechanisms and effective therapy for SAE remains a big challenge in clinical practice.
The gut is considered the “engine” driving sepsis and multiple organ dysfunction, a concept known as “gut-origin sepsis”2. As a critical immune organ, the gut contains diverse lymphocyte subsets, including γδ T cells and Th17 cells, primarily located in the small intestinal lamina propria (SI-LP). These cells are crucial for maintaining mucosal immune balance and intestinal barrier integrity3. Although γδ T cells comprise a small fraction of total T cells, they are essential for epithelial integrity, tissue repair, host homeostasis, and pathogen defense3. When the intestinal barrier is compromised, leading to bacterial invasion and inflammation, γδ T cells could differentiate into interleukin-17A (IL-17A)-producing γδ T17 cells, which exacerbates the inflammatory response4.
T cells in the meninges can secrete IL-17A, which could affect border-associated macrophages, reduces cerebral blood flow, and impairs cognition5. Similarly, γδ T17 cells can migrate from the SI-LP to the meninges, worsening the acute ischemic brain injury6. IL-17A secreted from γδ T17 cells could activate microglia and contribute to lipopolysaccharide (LPS)-induced neuroinflammation and cognitive deficits7. We recently showed that IL-7Rhigh CD8low γδ T17 cells could migrate from the gut to the lungs and exacerbate cecum ligation and puncture (CLP)-induced acute lung injury and mortality via secreting IL-17A8. However, it remains unclear whether gut-derived IL-7Rhigh CD8low γδ T17 could migrate to the central nervous system (CNS) and exacerbate SAE via IL-17A.
Synaptic pruning, a natural process of synapse elimination, is crucial for proper neural circuit formation, but aberrant pruning can lead to CNS disorders such as schizophrenia, anxiety, and autism9. Aberrant synaptic pruning affects neural signal transmission and network functionality, which is primarily mediated by microglial phagocytosis10. Complement-mediated synaptic pruning by microglia is closely linked to the progressive synaptic loss in LPS-induced depression11. Furthermore, sepsis-activated microglia could release C1q, which tags synapses for abnormal pruning and thereby exacerbates peritoneal contamination and infection-induced SAE12. However, the connection between C1q-mediated synaptic pruning and IL-17A and its potential mechanisms remain to be clarified.
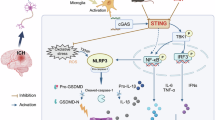
This study demonstrates that the migration of IL-7Rhigh CD8low γδ T17 cells from the small intestine to the meninges during sepsis is a critical factor contributing to SAE. IL-17A mediates mitochondrial damage in microglia and activates the cGAS-STING pathway, which is accompanied by inhibited ubiquitination of STING at the K150 site, resulting in STING accumulation. Increased levels of STING promote the release of C1q and thus enhances C1q-tagged hippocampal synapses, which exacerbates microglia-mediated synaptic pruning and SAE. 4-Octyl itaconate (4-OI), an itaconate derivative with potent anti-inflammatory properties13,14, inhibits IL-7Rhigh CD8low γδ T17 cell migration; increases the ubiquitination of STING at the K150 site in microglia; reduces STING expression; and subsequently decreases C1q-tagged synapses and excessive synaptic pruning, thereby ameliorating SAE.
Results
Gut-derived IL-7Rhigh CD8low γδ T17 cells migrate to the meninges and exacerbate SAE
First, we investigated whether small intestinal γδ T cells could migrate to CNS after sepsis, we performed flow cytometry (FCM) analysis following CLP. We observed significant reduction of γδ T cells in the small intestine, with a concurrent increase in the meninges (Fig. 1a, b), while γδ T cells in the brain parenchyma remained low (Supplementary Fig. 1a). These findings suggest that the meninges serve as the primary CNS destination for migrating small intestinal γδ T cells after sepsis. Notably, CLP induced elevated production of IL-17A by γδ T cells in the meninges, with over 50% of these cells identified as γδ T17 cells (Fig. 1c–e). Building on previous findings that IL-7Rhigh CD8low γδ T17 cells in small intestine could migrate to the lungs after sepsis8, we observed a similar elevation of this subset in the meninges after CLP (Fig. 1f, g). Therefore, we hypothesized that small intestinal IL-7Rhigh CD8low γδ T17 cells could directly migrate to the meninges following sepsis. To investigate this, the small intestine of Kaede-tg mice underwent localized irradiation to track the migration of small intestinal lymphocytes (Fig. 1h; Supplementary Fig. 1b). We found that the most IL-7Rhigh CD8low γδ T17 cells in the meninges originated from the small intestine (Fig. 1i). In contrast, the proportions of natural killer (NK) cells, B cells, T helper 17 (Th17) cells, regulatory T (Treg) cells, neutrophils, monocytes, dendritic cells (DC), and macrophages in the meninges that derived from the small intestine were not significantly altered following CLP (Supplementary Fig. 2a–d).

a, b Flow cytometry (FCM) plots and graphs showing CD45+ γδ T cells in the small intestine and meninges of sham and cecum ligation and puncture (CLP)-treated mice (n = 6–7). c FCM plots and graphs showing IL-17A+ γδ T17 cells (CD45+ CD3+ γδ TCR+ IL-17A+) in the meninges of sham and CLP-treated mice (n = 6–7). d, e FCM plots and graphs showing CD45+ IL-17A+ γδ T cells (CD45+ IL-17A+ CD3+ γδ TCR+) in the small intestine and meninges of sham and CLP-treated mice (n = 6–7). f, g FCM plots and graphs showing IL-7Rhigh CD8low γδ T17 cells (IL-7R+ CD8–) and IL-7Rlow CD8high γδ T17 cells (IL-7R– CD8+) in the small intestine and meninges of sham and CLP-treated mice (n = 6–7). h Treatment schedule and graph of the Kaede red+ γδ T17 cells (IL-17A+ CD3+ γδ TCR+ PE+) in the menings of mice (n = 7). Kaede transgenic mice were treated with a 405 nm laser and analyzed after sham or CLP surgery. i Graph of the Kaede red+ IL-7Rlow CD8high (IL-7R– CD8+ PE+) and IL-7Rhigh CD8low γδ T17 cells (IL-7R+ CD8– PE+) in the meninges of septic mice (n = 6). j, k Golgi staining image of CA1 region and dendritic spine density in septic mice (n = 6). ×6, Scale bar = 200 μm. ×12, Scale bar = 100 μm, Primary and secondary, Scale bar = 10 μm. l, m Transmission electron microscopy (TEM) of synapses and mitochondria in the CA1 region of septic mice (n = 6), ×15, Scale bar = 500 nm. ×20, Scale bar = 2.0 μm. n, o Y-maze tests (n = 12–13). p–r MWM tests (n = 12–13). Behavioral tests data are shown as mean ± SEM, the others are shown as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. ns, not significant. Statistical analyses: unpaired t-tests (b, c, g, i), Welch’s t-test (e), ANOVA with Šídák’s test (k, m, o, q, r). AZ, Active Zone; SC, Synaptic Cleft; SV, Synaptic Vesicles; PSD, Postsynaptic Density. Source data and exact P values are provided as a Source Data file. a, c, d, f–h Include components created in BioRender. Wu, Y. (2025) https://BioRender.com/rin8c1c.
We further investigate the impact of IL-7Rhigh CD8low γδ T17 cells on SAE, γδ T cell-specific knockout mouse models targeting Cd8a and Il7r were used. Specifically, mice were developed by crossing TrdcCreERT2 mice with Il7rflox/flox and Cd8aflox/flox mice, leading to a conditional knockout of Il7r and Cd8a in γδ T cells, respectively. In particular, TrdcCreERT2 Il7rflox/flox mice exhibited a significant reduction in γδ T17 cells in the meninges compared to wild-type (WT) and TrdcCreERT2 Cd8aflox/flox mice (Supplementary Fig. 1c). Consistently, TrdcCreERT2 Il7rflox/flox mice displayed low IL-17A mRNA levels in small intestinal γδ T cells (Supplementary Fig. 1d). Additionally, Golgi staining showed that CLP-induced dendritic spine loss and disorganization were reversed in TrdcCreERT2 Il7rflox/flox mice (Fig. 1j, k), Notably, the reduction in dendritic spine density on both primary and secondary dendrites—an index closely linked to normal neuronal function—was effectively alleviated. Furthermore, transmission electron microscopy (TEM) revealed attenuation of active zone shortening, synaptic vesicle loss, postsynaptic density thinning, and elevated Flameng score (mitochondrial damage) in TrdcCreERT2 Il7rflox/flox mice (Fig. 1l, m).
Behavioral assessments highlighted significant cognitive and memory impairments in CLP-induced SAE, including reduced exploration in Y-maze (Fig. 1n, o) and Morris-water maze (MWM) tests (Fig. 1p–r), increased anxiety-like behavior in open field tests (OFT) (Supplementary Fig. 1g, h), and decreased time spent exploring the novel object in the novel object recognition test (NORT) (Supplementary Fig. 1i). However, these behavioral deficits were markedly alleviated in TrdcCreERT2 Il7rflox/flox mice, suggesting that IL-7Rhigh CD8low γδ T17 cells could exacerbate cognitive dysfunction in SAE.
γδ T cells enhance microglial cGAS-STING expression and exacerbate synaptic pruning via C1q release
Having established that the migration of IL-7Rhigh CD8low γδ T17 cells from the intestine to the meninges could exacerbate SAE, we next explored the molecular mechanisms involved. We found that more than 50% of IL-17A in the meninges was produced by γδ T17 cells (Fig. 1d, e). IL-17A from γδ T17 cells is known to activate microglia, contributing to neuroinflammation and cognitive dysfunction7. Although IL-17A has been implicated in mitochondrial degradation via enhanced mitophagy in bronchial fibroblasts in severe asthma15, its effect on microglial mitochondrial integrity remains unclear. To evaluate the impact of IL-17A, produced by γδ T cells, on microglial function, we treated BV2 cells with recombinant IL-17A (100 ng ml–1) and LPS (1000 ng ml–1). This treatment exhibited decreased mitochondrial membrane potential and mitochondrial swelling, as indicated by JC-1 staining and TEM (Fig. 2a–c), suggesting mitochondrial damage. Mitochondrial dysfunction can release mitochondrial DNA (mtDNA), triggering the cGAS-STING pathway, a key intracellular DNA-sensing mechanism involved in host defense and inflammation16. Activation of the cGAS-STING pathway not only triggers inflammatory responses but also exacerbates various CNS injuries, including traumatic brain injury, spinal cord injury, subarachnoid hemorrhage, and hypoxic-ischemic encephalopathy17. We confirmed elevated cGAS and STING expression in BV2 cells following IL-17A and LPS treatment (Fig. 2d, e). Importantly, we found that mitochondrial damage was necessary for cGAS-STING upregulation, as mitochondria-depleted BV2ρ0 cells did not exhibit increased cGAS and STING expression (Supplementary Fig. 1j–l). To further validate the role of IL-17A secretion by γδ T cells in exacerbating microglial injury (Fig. 2f). Primary γδ T cells were isolated from mouse spleens and primary microglia from fetal mice, with cell purities confirmed by flow cytometry (Supplementary Fig. 1e–f). Following isolation, the two cell types were co-cultured, and IL-17A expression in the γδ T cells was selectively silenced using siRNA. The silenced IL-17A in primary γδ T cells reduced microglial mitochondrial damage and decreased cGAS, STING, and C1q levels in primary microglia (Fig. 2g–l).

a, b JC-1 staining shows mitochondrial membrane potential (FITC+/PE+) in IL-17A and LPS-treated BV2 cells (n = 6). c Transmission electron microscopy (TEM) images of mitochondria and the flameng score of BV2 cells. ×6, Scale bar = 5.0 μm. ×20, Scale bar = 2.0 μm, ×40, Scale bar = 0.5 μm. Western blot (d) and quantification (e) of cGAS and STING in BV2 cells exposed to IL-17A and LPS (n = 6). f Schematic of the transwell co-culture of primary γδ T cells and microglia. g IL-17A mRNA expression levels of primary γδ T cells (n = 8). h TEM images of primary microglia. ×6, Scale bar = 5.0 μm. ×20, Scale bar = 2.0 μm, ×40, Scale bar = 0.5 μm. Western blot (i, k) and quantification (j, l) of cGAS, STING, and C1q in primary microglia (n = 6). m Treatment schedule. Stereotactic injection of IL-17A+-Adv into the hippocampus, followed by behavioral assessments. Western blot (n) and quantification (o) of cGAS and STING in hippocampus (n = 6). Western blot (p) and quantification (q) of C1q, PSD95 and SYN in hippocampal synaptic proteins (n = 6). r, s TEM images of Adv-treated mice (n = 6), ×20, Scale bar = 500 nm. ×15, Scale bar = 2.0 μm. Western blot (t) and quantification (u) of C1q, PSD95 and SYN in hippocampal synaptic proteins (n = 6). Western blot (v) and quantification (w) of cGAS and STING in the hippocampus (n = 6). 3D reconstructed imaging (x) and analysis (y) of microglia engulfing synapses (n = 6). Green, Iba-1; Red, PSD95; Magenta, CD68. Scale bar, 5 μm. Data are shown as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. ns, not significant. Statistical tests: ANOVA with Šídák’s test (b, c, e, u, w, y), unpaired t tests (j, l, o, q, s). AZ active zone, SC synaptic cleft, SV synaptic vesicles, PSD postsynaptic density. Source data and exact P values are provided as a Source Data file. Panel m includes components created in BioRender. Wu, Y. (2025) https://BioRender.com/rin8c1c.
In vivo, hippocampal overexpression of IL-17A increased cGAS and STING expression (Fig. 2m–o) and impaired cognitive function (Supplementary Fig. 3a–g). Conversely, neutralization of IL-17A improved cognition in septic mice (Supplementary Fig. 3h–o). Additionally, reducing IL-17A levels decreased C1q expression in microglia (Fig. 2k, l). Given that C1q can bind to synapses and mediate synaptic pruning in a murine polymicrobial sepsis model12, we examined synaptic proteins in the hippocampus. IL-17A overexpression elevated C1q and reduced the levels of postsynaptic density protein 95 (PSD95) and synaptophysin (SYN), indicating that IL-17A could promote increased synaptic pruning by increasing C1q levels (Fig. 2p–s). However, this synaptic damage was reversed in TrdcCreERT2 Il7rflox/flox mice (Fig. 2t, u), which also exhibited lower expression of cGAS and STING in the hippocampus (Fig. 2v, w). Confocal imaging and 3D reconstruction using Imaris software showed that microglia in the hippocampus of septic mice displayed an activated phenotype, with increased phagocytosis of PSD95, linking microglial activation to synaptic protein loss (Fig. 2x, y).
Considering that STING promotes neuroinflammation18 and contributes to synaptic impairments, we investigated whether pharmacological inhibition of STING could reverse the associated neuroinflammatory responses and synaptic deficits (Fig. 3a–c). Treatment with the STING inhibitor H151 (750 nM) reduced C1q, increased the expression of PSD95 and SYN (Fig. 3d, e), and mitigated synaptic pruning (Fig. 3f, g), which led to improved cognitive outcomes (Fig. 3h, i; Supplementary Fig. 4a–e). H151 also reversed synaptic impairments caused by cGAS overexpression in microglia (Fig. 3j–s; Supplementary Fig. 4f–l). Additionally, direct C1q neutralization reduced microglial phagocytosis of synaptic proteins, resulting in improved cognition and restoring synaptic integrity (Fig. 3t–z; Supplementary Fig. 4m–q). Consistently, microglial depletion with PLX3397 similarly reduced C1q level and synaptic pruning, resulting in improved behavior deficits in septic mice (Supplementary Fig. 5a–k). These findings demonstrate that synaptic pruning after sepsis is closely related to the phagocytosis of C1q-labeled synapses by microglias. IL-17A produced by γδ T cells could drive C1q release via the cGAS-STING pathway, representing a key therapeutic target to reduce cognitive impairment in sepsis.

a Treatment schedule. H151 was administered intraperitoneally before cecum ligation and puncture (CLP), with brain collection 1 day post-CLP and behavioral testing starting 7 days post-CLP. Western blot (b) and quantification (c) of STING in the hippocampus (n = 6). Western blot (d) and quantification (e) of C1q, PSD95 and SYN in hippocampal synaptic proteins (n = 6). 3D imaging (f) and analysis (g) of microglia engulfing synapses (n = 6). Green, Iba-1; Red, PSD95; Magenta, CD68. Scale bar, 5 μm. h, i MWM tests. Western blot (j) and quantification (k) of STING and C1q of primary microglia (n = 6). l Treatment schedule. Hippocampal injection of cGAS+ adeno-associated virus (AAV); H151 administered 21 days later, followed by brain collection and behavioral testing. m Western blot analysis showing cGAS overexpression in the hippocampus. 3D imaging (n) and analysis (o) of microglia engulfing synapses (n = 6). Green, Iba-1; Red, PSD95; Magenta, CD68. Scale bar, 5 μm. Western blot (p) and quantification (q) of cGAS and STING in hippocampus (n = 6). Western blot (r) and quantification (s) of C1q, PSD95 and SYN in hippocampal synaptic proteins (n = 6). t Treatment schedule. C1q neutralizing antibody or IgG injected into hippocampus 1 day before CLP. 3D imaging (u) and analysis (v) of microglia engulfing synapses (n = 6). Green, Iba-1; Red, PSD95; Magenta, CD68. Scale bar, 5 μm. w, x Morris-water maze tests (n = 12). Western blot (y) and quantification (z) of C1q, PSD95 and SYN in hippocampal synaptic proteins (n = 6). Behavioral tests data are shown as mean ± SEM, the others are shown as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. ns, not significant. Statistical tests: unpaired t tests (c, e, g, i, k, o, v, x, z), ANOVA with Šídák’s test (q, s). Source data and exact P values are provided as a Source Data file. Panels a, l and t include components created in BioRender. Wu, Y. (2025) https://BioRender.com/rin8c1c.
K150-mediated STING ubiquitination prevents cognitive dysfunction
Given that sepsis could increase STING expression and contributes to C1q-mediated synaptic pruning in our findings, we investigated whether the ubiquitin-proteasome system (UPS), a key pathway for protein degradation and maintenance of protein homeostasis19, could regulate STING accumulation. We hypothesized that the ubiquitination of STING could play an important role in the pathogenesis of SAE. As expected, we found that exposure of primary microglia to LPS resulted in a significant reduction in STING ubiquitination, which was also observed in the hippocampus of CLP mice (Fig. 4a, b). Mass spectrometry identified several potential ubiquitination sites on STING: lysine 37 (K37), K127, K150, K151, K275, K288, and K235 (Fig. 4c; Supplementary Fig. 6). Subsequent analysis using plasmids harboring point mutations at each of these lysine residues in BV2 cells revealed that mutation of the K150 site markedly decreased STING ubiquitination (Fig. 4d), indicating that K150 is a critical site for STING ubiquitination in microglia. Furthermore, a specific antibody targeting ubiquitinated K150 confirmed a reduction in the hippocampus of septic mice (Fig. 4e, f).

a Western blot showing STING ubiquitination levels in primary microglia. b Western blot showing STING ubiquitination levels in mouse hippocampus. c Mass spectrometry identification of STING ubiquitination site K150. d Western blot analysis of STING ubiquitination levels in primary microglia transfected with mutant plasmids for each of the seven identified ubiquitination sites. Western blot (e) and quantification (f) of K150 in hippocampal of mice (n = 6). g Western blot showing STING ubiquitination levels in hippocampus 21 days after K150R+ adeno-associated virus (AAV) injection. Representative 3D reconstructed imaging (h) and quantitative analysis (i) of microglia engulfing synapses (n = 6). Green, Iba-1; Red, PSD95; Magenta, CD68. Scale bar, 5 μm. Western blot (j) and quantification (k) of STING in hippocampus of mice received K150R+-AAV (n = 6). Western blot (l) and quantification (m) of C1q, PSD95 and SYN in hippocampal synaptic proteins of mice treated with K150R+-AAV (n = 6). n, o Representative transmission electron microscopy (TEM) images of synapse and mitochondria in the CA1 region of mice treated with K150R+-AAV (n = 6), ×15, Scale bar = 500 nm. ×20, Scale bar = 2.0 μm. Representative images (p) and statistical analysis (q) of the Morris water maze (n = 12). Representative images (r) and statistical analysis (s) of the Y-maze tests (n = 12). Representative images (t) and statistical analysis (u) of the open field tests (n = 12). v Statistical analysis of the novel object recognition tests (n = 12). Behavioral tests data are shown as mean ± SEM, the others are shown as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. ns, not significant. Statistical tests: two-sided Student’s unpaired t-tests (f, i, k, m, o, q, s, u) and multiple unpaired t-tests (v). AZ active zone, SC synaptic cleft, SV synaptic vesicles, PSD postsynaptic density. Source data and exact P values are provided as a Source Data file.
To further explore the role of K150, we generated an adeno-associated virus (AAV) vector carrying the K150R mutation and injected it into the hippocampus of Cx3cr1Cre mice to mutate K150 site specifically in microglia. The AAV vector was designed using a double-floxed inverse orientation (DIO) system, which ensures that the mutant STING construct (encoding the K150R mutation) is selectively expressed only in Cre-expressing cells, thereby restricting expression specifically to microglia. This mutation led to reduced STING ubiquitination (Fig. 4g), increased microglial phagocytosis of PSD95 (Fig. 4h, i), and elevated STING protein levels (Fig. 4j, k). Consequently, mice with the K150R mutation in microglia showed enhanced synaptic pruning, evidenced by increased C1q and decreased levels of PSD95 and SYN (Fig. 4l, m). Furthermore, microglial K150R-mutant mice exhibited severe synaptic and mitochondrial damage—with shorter active zones, wider synaptic clefts, fewer synaptic vesicles, thinner postsynaptic densities, and increased Flameng scores indicative of mitochondrial injury (Fig. 4n, o)—along with pronounced learning and memory deficits in behavioral tests (Fig. 4p–v). These findings demonstrate that K150-mediated ubiquitination in microglia is essential for STING degradation and prevention of cognitive dysfunction.
We further explored the ubiquitination process of STING. We used UbiBrowser to predict potential E3 ubiquitin ligases for STING and identified BRCA1, RNF26, RNF5, Trim32, Trim56 as key candidates (Supplementary Fig. 7a). Co-immunoprecipitation (Co-IP) confirmed a direct interaction between RNF5 and STING (Supplementary Fig. 7b–f), which decreased following LPS stimulation or CLP-induced sepsis (Supplementary Fig. 7g, h). Furthermore, RNF5 knockdown in microglia significantly reduced STING ubiquitination (Supplementary Fig. 7i), while RNF5 overexpression increased STING ubiquitination, reduced C1q levels, increased synaptic proteins, and improved cognitive function in septic mice (Supplementary Fig. 7j–v). Importantly, the K150 mutation disrupted the interaction between STING and RNF5, highlighting the dependence of RNF5-mediated STING ubiquitination on K150 (Supplementary Fig. 7w). Together, these findings underscore the importance of K150-mediated STING ubiquitination in microglia in preventing excessive synaptic pruning and cognitive dysfunction, with RNF5 serving as a key regulator of this process.
4-OI reduces γδ T17 cell migration and promotes K150-mediated STING ubiquitination
To explore potential therapeutic strategies for SAE, we considered the role of small intestinal γδ T17 cell migration and the strong correlation between sepsis, gut barrier dysfunction, and systemic inflammatory response20. Proteomic analysis of the small intestine from septic mice revealed significant upregulation of aconitate decarboxylase 1 (ACOD1), which produces itaconate, known for its anti-inflammatory effects13 (Fig. 5a, b). Notably, we also observed elevated ACOD1 expression in the hippocampus following CLP (Fig. 5c). Previous studies have primarily attributed ACOD1 expression to myeloid cells like macrophages, indeed microglia have emerged as the main source of itaconate under neuroinflammatory conditions21. Indeed, microglia-depleted hippocampal tissue exhibits markedly suppressed ACOD1 expression22, and ACOD1 knockout mice display cerebral itaconate deficiency alongside exacerbated ischemic brain injury23. Collectively, these findings confirm microglia as the principal neural source of itaconate22.

a Heatmap of top 20 upregulated differentially expressed genes (DEGs) in small intestine (n = 3). Western blot of ACOD1 in small intestines (b) and hippocampus (c) (n = 6). d Treatment schedule. e Kaplan–Meier survival analysis (n = 7–17). f, g Western blot of ZO-1, MUC2 and Occludin in small intestine (n = 6). h Immunofluorescence images of ZO-1, MUC2, and Occludin of small intestines. i Flow cytometry (FCM) plots of IL-17A+ live γδ T cells (IL-17A+ CD3+ γδ TCR+) in the meninges. j FCM of Kaede red+ IL-7Rhigh CD8low γδ T17 (IL-17A+ CD3+ γδ TCR+ IL-7R+ CD8– PE+) cells in the meninges (n = 6). k Transmission electron microscopy (TEM) images of mice (n = 6), ×15, Scale bar = 500 nm. ×20, Scale bar = 2.0 μm. l, m 3D imaging and analysis of microglia engulfing synapses (n = 6). Green, Iba-1; Red, PSD95; Magenta, CD68. Scale bar, 5 μm. n Golgi staining images of mice (n = 6). ×6, Scale bar = 200 μm. ×12, Scale bar = 100 μm, Primary and secondary, Scale bar = 10 μm. o, p Spontaneous (sEPSCs) and miniature (mEPSCs) excitatory postsynaptic currents of CA1 pyramidal neurons (n = 8). q, r Morris-water maze tests (n = 12). Behavioral tests data are shown as mean ± SEM, the others are shown as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. ns, not significant. Statistical tests: unpaired t-tests (b, c, j, k, m, n, p, r), Log-rank test (e), ANOVA with Šídák’s test (g). AZ, Active Zone; SC, Synaptic Cleft; SV, Synaptic Vesicles; PSD, Postsynaptic Density. Source data and exact P values are provided as a Source Data file. a–d, i, j Include components created in BioRender. Wu, Y. (2025) https://BioRender.com/rin8c1c.
Hence, we further investigated the protective effects of itaconate and 4-octyl-itaconate (4-OI), a blood-brain barrier (BBB)-permeable itaconate derivative, against sepsis-induced systemic inflammation. Initial experiments revealed comparable efficacy between both compounds in attenuating sepsis-induced γδ T cell migration from the small intestine to the meninges (Supplementary Fig. 8a, b). While both agents mitigated hippocampal synaptic damage and mitochondrial dysfunction, 4-OI demonstrated superior neuroprotection, as evidenced by significantly improved mitochondrial ultrastructure (Supplementary Fig. 8c–e), reduced aberrant synaptic phagocytosis (Supplementary Fig. 8f), and more pronounced downregulation of the STING and complement C1q pathways (Supplementary Fig. 8g–j). Both treatments suppressed IL-17A levels equivalently across intestinal, plasma, meningeal, and hippocampal compartments (Supplementary Fig. 8k). The differences in central efficacy likely arise from the limited BBB permeability of unmodified itaconate due to its high polarity, whereas the enhanced lipophilicity of 4-OI permits greater CNS penetration and maintains pharmacological activity through esterase-mediated hydrolysis to active itaconate24.
Dose-response studies using CCK-8 viability assays and survival analyses identified optimal 4-OI concentrations of 250 nM in vitro and 50 mg/kg in vivo (Supplementary Fig. 9a, b). Notably, administration of 4-OI led to a marked reduction in pro-inflammatory cytokines, as demonstrated by multiplex cytokine analysis (Supplementary Fig. 9c, d), with improved survival rates and enhanced intestinal barrier integrity (Fig. 5d–h). Interestingly, 4-OI significantly reduced the number of IL-7Rhigh CD8low γδ T17 cells in the meninges, correlating with decreased migration from the small intestine (Fig. 5i, j). This reduction in migration was accompanied by mitigation of hippocampal synaptic and mitochondrial damage (Fig. 5k), reduced reactive oxygen species (ROS) levels, and enhanced antioxidant activity, as confirmed by dihydroethidium (DHE), malondialdehyde (MDA), and superoxide dismutase (SOD) assays, respectively (Supplementary Fig. 10a–d). We further found that treatment with 4-OI significantly decreased the levels of IL-17A, C1q, and STING in the small intestine, plasma, meninges, and hippocampus, while concurrently increasing itaconate concentrations (Supplementary Fig. 11). Additionally, positron emission tomography-computed tomography (PET-CT) and laser speckle imaging demonstrated improved hippocampal glucose metabolism and restored sepsis-induced decrease in the cerebral blood flow after 4-OI treatment (Supplementary Fig. 12a, b).
Moreover, 4-OI reduced microglial phagocytosis of PSD95 (Fig. 5l, m), and increased dendritic spine density (Fig. 5n). Whole-cell patch-clamp recordings further demonstrated that 4-OI restored synaptic transmission efficacy in CA1 pyramidal neurons, as evidenced by elevated frequencies of both spontaneous and miniature excitatory postsynaptic currents (sEPSCs/mEPSCs) (Fig. 5o, p). In behavioral assays, 4-OI improved cognitive performance in the MWM, OFT, Y-maze and NORT (Fig. 5q, r; Supplementary Fig. 10e–i). Mechanistically, 4-OI reduced the expression of cGAS and STING, leading to decreased C1q and increased levels of PSD95 and SYN, indicating that 4-OI could attenuate C1q-mediated synaptic pruning (Supplementary Fig. 13a–d). To further investigate the molecular basis of 4-OI’s action, we performed molecular docking simulations to assess its binding interactions with cGAS and STING. The predicted binding energy of 4-OI to cGAS was –11.9635 kcal/mol (Supplementary Fig. 14a). Examination of the cGAS-4-OI binding pocket demonstrated that Arg364 of cGAS forms hydrogen bonds with the ester group of 4-OI, anchoring the orientation of the ester moiety and reducing flexibility at the binding site. Similarly, Ser366 engages in hydrogen bonding with the ester group, enhancing the specificity of cGAS for 4-OI. In addition, Phe367 and Glu371 form hydrogen bonds with the carboxylate group of 4-OI, restricting its rotational freedom and further stabilizing the binding conformation. Together, these four residues form an extensive hydrogen bond network with 4-OI, collectively enhancing both binding affinity and selectivity (Supplementary Fig. 14b). In contrast, the docking energy of 4-OI to STING was –8.7913 kcal/mol (Supplementary Fig. 14c), indicating a weaker interaction relative to cGAS. In this complex, the carboxylate group of 4-OI forms a salt bridge with the guanidinium group of Arg237, which likely contributes to local stabilization of STING’s secondary structure (Supplementary Fig. 14d). Nonetheless, the preferential binding of 4-OI to cGAS over STING implies a more selective and potentially functionally relevant interaction with cGAS. To validate these in silico findings, we generated site-directed mutants targeting the predicted 4-OI binding residues. For cGAS, four point mutants (R364A, S366A, F367A, and E371A) were constructed, while for STING, Arg237 was mutated to alanine (R237A). Western blot analysis revealed that mutation of Ser366 (S366A) in cGAS significantly reduced STING protein levels in BV2 microglial cells, along with a concomitant decrease in C1q expression (Supplementary Fig. 14e, f). These results indicate that Ser366 plays a critical role in cGAS-mediated activation of STING and downstream induction of C1q. We speculate that 4-OI binding at the Ser366 site may induce a conformational change or inhibit cGAS enzymatic activity, thereby attenuating STING signaling and ultimately suppressing C1q production (Supplementary Fig. 14g, h). Conversely, mutation Arg237 in STING did not further suppress C1q levels (Supplementary Fig. 14i, j), highlighting the dominant role of cGAS as the functional target of 4-OI in this signaling cascade.
In vitro, 4-OI inhibited IL-17A release and mitigated mitochondrial dysfunction in BV2 cells (Supplementary Fig. 13e, f), which was associated with decreased cGAS-STING-C1q signaling (Supplementary Fig. 13g–j). Additionally, 4-OI also diminished the migration and phagocytic activity of LPS-stimulated microglia (Supplementary Fig. 15a–d), and promoted microglial polarization in vivo, shifting the balance from homeostatic microglias toward disease-associated microglia (DAM) (Supplementary Fig. 15g–j). Furthermore, 4-OI inhibited STING activity in microglia (Supplementary Fig. 15e, f).
Consistent with our findings in the CLP model, we confirmed the therapeutic potential of 4-OI in the colon ascendens stent peritonitis (CASP)-induced sepsis model. As in the CLP model, treatment with 4-OI demonstrated robust neuroprotective effects by reducing synaptic and mitochondrial damage (Supplementary Fig. 16a, b), mitigating synaptic engulfment (Supplementary Fig. 16c, d), suppressing STING and C1q expression, and increasing PSD95 and SYN levels (Supplementary Fig. 16e–h). Additionally, it alleviated sepsis-associated cognitive deficits (Supplementary Fig. 16i–o). Collectively, these results indicate that 4-OI confers significant therapeutic benefits in both CLP- and CASP-induced sepsis models.
Interestingly, transcriptomic analysis of the hippocampus from septic mice also showed upregulation of Acod1 (Supplementary Fig. 13k), which consistent with our findings (Fig. 5c). In the hippocampus, 4-OI enhanced K150-mediated STING ubiquitination and upregulated RNF5 expression (Supplementary Fig. 7x, y; Supplementary Fig. 13l–n). These results confirm that 4-OI could protect against SAE not only by inhibiting γδ T17 cell migration from small intestine into the meninges, but also by promoting K150-mediated STING ubiquitination in the hippocampus.
Silencing Acod1 in microglia aggravates cognitive dysfunction
Given the increased expression of ACOD1 in both the intestine and hippocampus after sepsis, we further investigated its role in microglia and its impact on cognitive impairment in SAE. We utilized Acod1–/– mice and observed that none survived beyond 24 hours post-CLP, necessitating sample collection at 16 hours post-procedure (Supplementary Fig. 17a). While synaptic and mitochondrial structures remained intact in sham-operated Acod1–/– mice, those subjected to CLP exhibited markedly exacerbated synaptic and mitochondrial damage (Supplementary Fig. 17b–d), increased microglial synapse engulfment (Supplementary Fig. 17e, f), elevated STING and C1q expression, and reduced levels of synaptic proteins (Supplementary Fig. 17g–j). Notably, administration of 4-OI mitigated these pathologic changes in Acod1–/– mice.
We silenced Acod1 using Acod1– -AAV in Cx3cr1Cre mice (Fig. 6a). This suppression significantly upregulated cGAS and STING expression in the hippocampus, leading to synaptic and mitochondrial damage (Fig. 6b–e). Silencing Acod1 also elevated C1q levels and reduced the levels of PSD95 and SYN, indicating that Acod1 suppression enhanced complement-mediated synaptic pruning (Fig. 6f, g). Correspondingly, microglial phagocytosis of PSD95 was notably increased following Acod1 suppression, further confirming its involvement in excessive synaptic pruning (Fig. 6h, i). Behavioral assays confirmed that silencing microglial Acod1 exacerbated cognitive impairment (Supplementary Fig. 18a–f).

a Treatment schedule. Hippocampal stereotactic injection of Acod1–-adeno-associated virus (AAV); brain collection 22 days post-infection. Behavioral assessments were conducted 28 days post-infection. Western blot (b) and quantification (c) of cGAS and STING in hippocampus of mice treated with control or Acod1–-AAV (n = 6). d, e Representative transmission electron microscopy (TEM) of synapse and mitochondria in CA1 region in mice treated with control or Acod1–-AAV (n = 6). Western blot (f) and quantification (g) of cGAS and STING in hippocampus of mice received control or Acod1–-AAV (n = 6). Representative 3D reconstructed imaging (h) and quantitative analysis (i) of microglia engulfing synapses (n = 6). Green, Iba-1; Red, PSD95; Magenta, CD68. Scale bar, 5 μm. Western blot (j) and quantification (k) of K150 in the hippocampus of mice treated with control or Acod1–-AAV (n = 6). l Western blot image of STING ubiquitination levels in mouse hippocampus. m Schematic of mechanism. Data are shown as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. ns, not significant. P values were calculated using two-sided Student’s unpaired t-tests (c, e, g, i, k). AZ active zone, SC synaptic cleft, SV synaptic vesicles, PSD postsynaptic density. Source data and exact P values are provided as a Source Data file. a includes components created in BioRender. Wu, Y. (2025) https://BioRender.com/rin8c1c.
Consistent with the promotion of K150-mediated STING ubiquitination by 4-OI, Acod1 suppression led to a significant reduction in K150-mediated STING ubiquitination (Fig. 6j–l), promoting cGAS-STING pathway activation and exacerbating synaptic injury (Fig. 6b–i). Taken together, these findings suggest that ACOD1 could regulate microglial function by promoting K150-mediated STING ubiquitination, thereby limiting cGAS-STING activation and preventing excessive synaptic pruning. Therefore, Acod1 suppression in microglia could worsen hippocampal synaptic and mitochondrial dysfunction by impairing K150-mediated STING ubiquitination, activating the cGAS-STING-C1q pathway, and enhancing synaptic pruning, ultimately leading to cognitive dysfunction after sepsis.
Discussion
Our latest study reveals that IL-7Rhigh CD8low memory γδ T17 cells, originating from the small intestine, could migrate to the lungs and contribute to acute lung injury following sepsis8. Interestingly, this present research further demonstrates that the migration of small intestine-derived IL-7Rhigh CD8low γδ T17 cells to the meninges plays a pivotal role in exacerbating SAE (Fig. 6m), establishing a crucial link between intestinal γδ T17 cells and CNS pathology. Our findings underscore the importance of gut-centric inter-organ interactions.
Prior research investigating the “gut–brain” axis have predominantly focused on gut dysbiosis and intestinal barrier dysfunction in SAE25,26,27. For instance, research have demonstrated that the human commensal Bacteroides fragilis can restore gut permeability, modulate the microbial composition, and alleviate deficits in communicative, stereotypic, anxiety-like, and sensorimotor behaviors25. However, our study provides direct evidence indicating that IL-7Rhigh CD8low γδ T17 cell migration may contribute to the pathogenesis of SAE. Notably, similar mechanisms have been observed in hypertension, where IL-17-secreting T cells in meninges could contribute to CNS injury5. Consistent with these findings, we observed a significant increase in γδ T17 cells in the meninges after CLP, along with an upregulation of IL-7Rhigh CD8low γδ T17 cells. However, the number of γδ T cells in the brain parenchyma remained low in both CLP and sham-operated mice, suggesting a specific migration pattern to the meninges rather than the brain tissue.
IL-17A, a pro-inflammatory cytokine known to exacerbate immune responses under inflammatory conditions28, appears to be a key mediator in this “small intestine-derived γδ T17 cells induced SAE”. We found that over 50% of IL-17A-secreting immune cells in the meninges following sepsis were γδ T cells, significantly higher than that of 30% in the sham group. We further confirmed that IL-7Rhigh CD8low γδ T17 cells are the primary IL-17A source in the meninges after sepsis, as shown using TrdcCreERT2 Il7rflox/flox and TrdcCreERT2 Cd8aflox/flox transgenic mice. The IL-17A secreted by IL-7Rhigh CD8low γδ T17 cells activates microglia, triggering CNS inflammation and exacerbating SAE.
Microglia, as CNS resident immune cells, can be neuroprotective or neurotoxic29. In the CLP model of sepsis, activated microglia could exacerbate SAE by upregulation neuronal NAT10 expression30, induction of hippocampal neuronal ferroptosis via the CXCL2/CXCR2 pathway31, and aberrant synaptic pruning32. In the peritoneal contamination and infection (PCI)-induced SAE model, microglial complement C1q-dependent synaptic pruning is shown to be able to worsen SAE12, although the mechanisms underlying sepsis-induced C1q release remains unclear. Our study reveals that IL-17A-stimulated microglia upregulate C1q-dependent synaptic pruning through activation of the cGAS-STING pathway. Excessive activation of the complement cascade has been implicated in the pathogenesis of a wide range of diseases33. C1q, a pivotal initiator of the classical complement cascade, plays an essential role in synaptic pruning during both normal development and disease states. For instance, in Alzheimer’s disease, both C1q and C3 are involved in microglia-mediated synaptic loss34, while excessive complement C4 activity is associated with reduced synapse numbers in schizophrenia35. Our findings align with previously published datasets that identified significant upregulation of C1q in SAE36, supporting the notion that C1q is critically involved in the pathogenesis of this condition. Furthermore, we found that inhibiting STING with H151 or depleting microglia with PLX3397 significantly reduced C1q levels and synaptic pruning, suggesting that STING-mediated microglial C1q release contributes to the worsening SAE. We primarily focused on excitatory synaptic pruning, as evidenced by increased microglial phagocytosis of PSD95, which may underlie the cognitive dysfunction observed in SAE. By contrast, in conditions such as epilepsy and chronic pain—characterized by heightened excitatory neural activity—augmented inhibitory synaptic pruning has been reported37,38. Importantly, our investigation concentrated on the hippocampal CA1 region, a brain area particularly susceptible to neuroinflammatory insults32,39 and critical for processing diverse inputs essential for learning and memory40.
In addition, our study revealed that IL-17A released from γδ T17 cells exacerbates mitochondrial damage in microglia. Mitochondrial dysfunction, which results in the release of mtDNA, serves as a central trigger for STING activation41. Notably, loss of STING has been shown to alleviate dopaminergic neuron loss in the substantia nigra pars compacta and improve motor function42. Moreover, STING links inflammatory responses with glutamatergic signaling, thereby promoting neuronal cell death and contributing to the pathogenesis of multiple sclerosis43. In this context, demyelination-associated release of DNA fragments activates microglial cGAS-STING signaling, fostering an inflammatory environment that damages both oligodendrocytes and neurons18.
Traditionally associated with antiviral immunity through the detection of cytosolic DNA and activation of type I interferon responses, the cGAS-STING pathway has recently been implicated in neuroinflammatory diseases18. In multiple sclerosis, STING activation enhances autophagy in neurons, increasing their susceptibility to glutamate-induced excitotoxicity, thereby STING inhibitors such as C176 or H151 have been shown to reduce neuroinflammatory damage43. Furthermore, attenuating cGAS-STING signaling has been found to inhibit glial cell activation in aging models44.
In exploring potential therapeutic interventions, we focused on itaconate, an immunomodulatory metabolite produced by the enzyme ACOD1. Elevated itaconate levels, dependent on ACOD1, have been observed in the blood during Plasmodium infection45. Macrophages and myeloid cells produce itaconate under inflammatory stimulation, and microglia also express ACOD1 highly under pro-inflammatory conditions46, which aligns with our findings. Our proteomic and transcriptomic analyses revealed significant upregulation of ACOD1 in both the small intestine and hippocampus after sepsis. Exogenous supplementation with itaconate and its derivative 4-OI has been shown to mitigate systemic inflammation and exert anti-inflammatory effects in the small intestine during CLP-induced sepsis14. Notably, 4-OI attenuates inflammatory responses by targeting glyceraldehyde 3-phosphate dehydrogenase (GAPDH) to reduce aerobic glycolysis in macrophages47, and other studies have reported that itaconate suppresses the production of pro-inflammatory cytokines such as IL-6 and IL-1248. In our study, both itaconate and 4-OI significantly reduced IL-17A levels in the periphery and CNS, while concurrently decreasing the migration of γδ T cells from the small intestine to the meninges. Importantly, 4-OI exhibited a more robust protective effect against synaptic and mitochondrial damage. This enhanced efficacy is likely attributable to its lower polarity, which facilitates efficient BBB penetration and direct neuroprotection. The heightened vulnerability observed in Acod1–/– mice, wherein all subjects succumbed within 24 hours post-CLP, underscores the critical role of normal ACOD1 expression in mounting an effective defense against inflammatory insults. Moreover, treatment with 4-OI downregulated cGAS-STING signaling, an effect that may be attributed to both a reduction in γδ T17 cell migration and the direct attenuation of mitochondrial damage within the brain.
The upregulation of the cGAS-STING pathway has been implicated in exacerbating neuroinflammatory responses and contributing to the pathogenesis of various neurodegenerative diseases, including Parkinson’s disease, Alzheimer’s disease, multiple sclerosis, and amyotrophic lateral sclerosis18. These findings, in conjunction with our data, suggest that 4-OI, by inhibiting STING expression and due to its favorable BBB permeability, holds significant clinical promise. Future investigations should evaluate the therapeutic potential of 4-OI in other neurodegenerative conditions, thereby broadening its clinical application.
To elucidate the mechanism by which 4-OI regulates the cGAS-STING pathway, we performed molecular docking analyses and identified Ser366 of cGAS as a critical binding site for 4-OI. This mechanism complements previous studies reporting that 4-OI inhibits STING activity by alkylating the Cys147 residue, thereby interfering with its post-translational modifications14. However, in our study, mutating the predicted STING binding site (R237A) did not alter C1q expression, suggesting that cGAS—rather than STING—is the primary direct target of 4-OI in this context. Intriguingly, we also observed that 4-OI enhanced STING ubiquitination, suggesting an indirect regulatory mechanism.
Specifically, our data indicate that 4-OI enhances the expression of RNF5, an E3 ubiquitin ligase that promotes ubiquitination and degradation of STING at the K150 residue. RNF5 has been shown to target STING at K150 for ubiquitination and degradation following viral infection49. The K150 site is crucial for recruiting the deubiquitinating enzyme VP1-2 to STING, influencing its ubiquitination by other E3 ligases such as TRIM3250. These findings suggest that STING ubiquitination may differ across diseases. Although its role in CNS disorders remains unclear, our study provides evidence supporting its potential involvement in SAE. By enhancing STING ubiquitination, 4-OI effectively dampens neuroinflammatory processes mediated by the cGAS-STING pathway. Although other E3 ligases such as TRIM32 have been shown to promote STING ubiquitination in different contexts like in herpes simplex virus encephalitis50, the role of these ligases in CNS disorders remains to be elucidated. Future research should explore the involvement of additional ubiquitin ligases and the precise molecular mechanisms governing STING regulation in neuroinflammation, with particular attention to how K150-mediated ubiquitination interacts with other post-translational modifications—such as phosphorylation, acetylation, and SUMOylation.
However, several areas warrant further investigation. We observed that various cell types from the small intestine can migrate to the CNS, and different γδ T cell subsets may have distinct functional roles. Integration of emerging spatial transcriptomics and single-cell technologies could provide a precise map of the spatial distribution and functional heterogeneity of γδ T cells within the meninges and brain, thereby elucidating their specific contributions in sepsis. Additionally, given that sex influences inflammatory responses, future studies should include female subjects to evaluate potential sex-specific differences.
In summary, our study highlights a potential mechanism wherein in the migration of small intestine-derived IL-7Rhigh CD8low γδ T17 cells to the meninges leads to IL-17A secretion, microglia activation, and inhibition of K150-dependent STING ubiquitination. And promotes the cGAS-STING-C1q pathway, playing a key role in cognitive impairment during SAE. By enhancing STING ubiquitination and controlling γδ T17 cell migration, 4-OI emerges as a promising therapeutic agent to alleviate the neurological consequences of sepsis. This study lays the foundation for “gut-brain” axis-targeted therapies in inflammatory CNS diseases, offering new hope for patients with SAE and other neuroinflammatory conditions.
Methods
Animals
Male WT C57BL/6 J mice, TrdcCreERT2 mice, Il7rflox/flox mice, Cd8aflox/flox mice, Cx3cr1Cre, Acod1–/– mice (all on a C57BL/6 background), weighing between 23.0 and 25.0 grams and aged between 8 and 10 weeks, were obtained from Vital River Laboratory Animal Technology Co Ltd., Beijing, China. TrdcCreERT2 mice were crossed with Il7rflox/flox mice and Cd8aflox/flox mice to generate TrdcCreERT2 Il7rflox/flox and TrdcCreERT2 Cd8aflox/flox offspring. To induce Cre activity in γδ T cells, tamoxifen (150 mg kg–1) was administered intraperitoneally to TrdcCreERT2 Il7rflox/flox mice and TrdcCreERT2 Cd8aflox/flox mice for five consecutive days. The CLP model was performed one week after tamoxifen induction. Kaede-transgenic (Kaede-Tg) mice (B6. Cg-Gt (ROSA)26Sor<tm1.1(CAG-kikGR) Kgwa > ) were generously provided by M. Tomura of Kyoto University51. The mice were housed in a controlled, specific pathogen-free environment. They were exposed to a 12:12 light/dark cycle, maintained at a regulated temperature and humidity, and provided with unrestricted access to food and water. Ethical approval for all experiments was obtained from the Experimental Animals Committee of Tongji Medical College (permission number: 4028), in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The study adhered to ARRIVE guidelines (Animals in Research: Reporting In Vivo Experiments).
Animal model
Mice were anesthetized with sodium pentobarbital (0.3% solution) at a dose of 40 mg kg–1 body weight. A midline laparotomy was performed to conduct a central lymphadenectomy, following which the abdominal region was shaved and sterilized.
A 1 cm incision was made along the midline to expose the cecum, which was ligated 1 cm from the distal end using a 4-0 silk suture. A 20-gauge needle was then used to puncture the cecum, allowing a small amount of cecal content to extrude from both openings. After the procedure, the ligated cecum was returned to the abdominal cavity, and the incision was closed in multiple layers. Either 50 mg kg–1 of 4-OI or 10 ml kg–1 of 0.9% saline, or 50 mg kg–1 of itaconate was administered intraperitoneally immediately after CLP or sham-operation. Inhibitor H151 was administered at a concentration of 750 nM via intraperitoneal injection, three times per week for three weeks prior to the CLP procedure. Survival rates were monitored and assessed 7 days post-CLP. Tissue samples were collected 24 hours after CLP, and behavioral assessments commenced from day 7 post-CLP.
In parallel experiments, a separate cohort of mice underwent the colon ascendens stent peritonitis (CASP) procedure to induce polymicrobial sepsis. Briefly, the mice ascending colon was carefully exposed, and an 18-gauge catheter was gently inserted through the colon wall to allow continuous leakage of fecal content into the peritoneal cavity. The stent was secured with a 6-0 silk suture, ensuring that both entry and exit points were firmly sealed around the catheter. The colon was then repositioned, and the abdomen was closed in multiple layers. As in the CLP model, animals received either 4-OI or saline intraperitoneally at the indicated doses. Operators were blind to group assignment.
To increase IL-17A expression in the brain, mice received adenoviruses (Adv) via lateral ventricle injection three weeks before sham surgery or CLP. Each Adv injection contained 1 × 1012 plaque-forming units (PFU) of IL-17A-expressing recombinant AAV (IL-17A+-Adv). Control mice received an equivalent dose of Adv expressing GFP. In a separate experimental group, IL-17A neutralizing antibody was administered intraperitoneally at 100 μg per day for 5 days, with four doses given prior to the CLP challenge and one dose immediately after. Control mice received equivalent doses of normal hamster serum IgG.
To specifically interfere with gene expression in microglia, we injected Cx3cr1Cre mice with DIO-AAV vectors. The AAVs were obtained from Brainvta (Wuhan, China) and included the following constructs: rAAV-SFFV-DIO-cGAS-2a-EGFP-WPREs, rAAV-SFFV-DIO-RNF5-His-2a-EGFP-WPREs, rAAV-CWV-DIO-(EGFP-U6)-shRNA1(Acod1), and rAAV-SFFV-DIO-STING1 (K150R)-2a-EGFP-W.
Kaede photoconversion
To track immune cells in the small intestines in vivo, we performed photoconversion following the method described in a previous study8. Briefly, following CLP or sham surgery, the small intestine of Kaede-transgenic mice was exposed to a 405-nm laser for 10 min, while the surrounding tissue was shielded from light with aluminum foil.
Dose exploration of 4-OI
To identify an optimal therapeutic window for 4-OI with minimal toxicity, we conducted dose–response studies both in vitro and in vivo. In vitro, BV2 microglial cells were stimulated with LPS (1000 ng mL⁻¹) and treated with graded concentrations of 4-OI (0, 10, 50, 125, 250, or 500 µM). After 6 hours of exposure, cell viability was evaluated using the Cell Counting Kit-8 (CCK8) assay (Beyotime, China), measuring absorbance at 450 nm on a microplate reader. For the in vivo dose exploration, C57BL/6 J mice were subjected to CLP and subsequently administered intraperitoneal injections of 4-OI at 5, 10, 20, 50, or 100 mg kg⁻¹ immediately post-surgery. Survival was monitored over a 7-day period.
Brain stereotaxic injection
A previous study52 detailed the stereotaxic injection of viruses and neutralizing antibodies into the hippocampus. Briefly, mice were anesthetized with 1%–2% isoflurane. Openings were made at specific coordinates targeting the unilateral hippocampal CA1 region (x: ± 2.15 mm; y: –2.5 mm; z: –2.25 mm). A volume of 400 nanoliters (nL) was injected into each hippocampus at a rate of 26.67 nL min–1. Following the injection, the needle was left in place for 5 min to ensure proper diffusion before removal. The CLP model was performed 21 days post-viral injection and 1 day after C1q neutralizing antibody administration.
MWM
Training was conducted between days 10 and 13 post-CLP, with testing performed on day 14. Experimental data were collected in a 120 cm circular pool filled with opaque water maintained at 20–22 °C. Mice were trained four times daily for four days to locate a hidden platform submerged 1 cm below the water surface. The average latency to find the platform was calculated from four trials. Each mouse was allowed 60 seconds per trial to search for the platform. Mice remained on the platform for 15 seconds if they found it, or were placed on it for 10 seconds if they failed to locate it. A probe trial without the platform was conducted 24 h after the final hidden platform test. Mice were tested for 60 seconds to locate the platform. The number of times the mice crossed the platform target and the total time spent in the target quadrant were recorded. Mice were monitored via video cameras throughout training and probe trials. Data were analyzed using TopScan Lite (Clever Sys. Inc.).
Y-maze test
Y-maze test was conducted on day 7 post-CLP. Three identical arms (30 cm long, 5 cm wide, 20 cm high) were positioned at 120° angles in the Y-maze. In the initial 10-minute training session, mice explored two arms while the third arm was blocked. One hour later, mice were given free access to all arms during the retention test. Mice were recorded exploring the novel arm for 5 min. Each trial was separated by cleaning the Y-maze arms with 75% ethanol. Arm entries and time spent in the novel arm were recorded, and short-term memory was calculated as the ratio of novel arm time to total exploration time. Data were examined using TopScan Lite (Clever Sys. Inc.).
Open field test
OFT was conducted on day 8 post-CLP. The testing area was an acrylic box measuring 40 cm long, 40 cm wide, and 30 cm high. The center of the base contained a 20 × 20 cm square core region. Each mouse was gently placed in the center of a dimly lit open-field arena and allowed to explore for 5 min. A mobile camera automatically recorded and tracked their movement. The time spent in the central region was used to assess exploratory behavior. To eliminate odors, the chamber floor was wiped with 75% ethanol after each session. Data were examined using TopScan Lite (Clever Sys. Inc.).
NORT
NORT was conducted on day 9 post-CLP. Two similar objects were placed at the edges of a 40 × 40 × 30 cm box. Mice were then gently placed in the box and allowed to explore the objects freely for 5 min, with the time spent on each object recorded. Exploration was defined as sniffing or touching an object within 0–2 cm with the nose. One hour later, one object was replaced with a novel one, and mice were given another 5 min to explore both objects, with the time spent on each recorded. The novel object recognition rate was calculated as (time spent on the novel object/total time spent on both objects) × 100%.
Tissue preparation
Cold PBS was perfused transcardially into sedated mice. Brains and small intestines were then collected. For immunofluorescent labeling, tissues were fixed overnight at 4 °C in 4% paraformaldehyde and cryoprotected in 30% sucrose for at least 2 days. Brains were sectioned at 30 μm thickness using a Leica CM1950 cryostat. Small intestines were paraffin-embedded and sectioned at 4 μm thickness. Blood was collected from sedated mice via cardiac puncture into anticoagulant tubes for biochemical analysis. Plasma was obtained and stored at –80 °C. Small intestines were stored at –80 °C after a gentle flush with cold PBS. After dissociation, the hippocampus was snap-frozen in liquid nitrogen and stored at –80 °C for protein extraction.
Immunofluorescence
Brain slices were washed in PBS and blocked for 2 h at room temperature with 5% BSA (Biofroxx, Germany) and 0.3% Triton X-100 in PBS. The sections were incubated overnight at 4 °C with mouse anti-Iba1 (1:100, Cat# ab283319, Abcam), rabbit anti-PSD95 (1:250, Cat# 37657, CST) and rat anti-CD68 (1:100, Cat#137001, Biolegend) primary antibodies. After PBS washing, the sections were incubated with Alexa Fluor 488-conjugated goat anti-rabbit (1:1000, Cat# ab150077, Abcam), Alexa Fluor 549-conjugated goat anti-mouse (1:1000, Cat# ab150116, Abcam) and Alexa Fluor 647-conjugated donkey anti-rat (1:1000, Cat# ab150155, Abcam) for 1 h at room temperature the following day. The slices were mounted with SouthernBiotech DAPI Fluoromount-G after a final wash.
After deparaffinization in xylene, intestinal sections were rehydrated through a graded ethanol series to water. Sections were heated in citrate buffer (pH 6.0) for 20 min for antigen retrieval. After cooling to room temperature, the sections were blocked for 1 h with 5% BSA and 0.3% Triton X-100 in PBS. Sections were incubated overnight at 4 °C with rabbit anti-ZO-1 (1:100, Cat# 21773-1-AP, Proteintech), rabbit anti-MUC2 (1:100, Cat# 27675-1-AP, Proteintech), and rabbit anti-Occludin (1:100, Cat# 27260-1-AP, Proteintech) primary antibodies. The following day, sections were washed in PBS and incubated with Alexa Fluor 488-conjugated goat anti-rabbit (1:1000, Cat# ab150077, Abcam) for 1 h at room temperature. The slices were mounted with SouthernBiotech DAPI Fluoromount-G after a final wash.
Microscopy and analysis
Immunofluorescent-stained brain slices were imaged using a Dragonfly spinning disk confocal microscope (Andor Technology) equipped with 405, 488, 561 and 640 nm laser lines. Z-stack images were captured using a 60x oil immersion objective. Image processing and analysis were performed using Imaris 10.2 (Bitplane). Z-stack images were generated using Imaris 10.2 for 3D reconstruction and quantification of Iba1, PSD95 and CD68 expression. Immunofluorescent signal intensity in ROIs was compared across experimental groups.
Immunofluorescent-stained small intestine sections were scanned at ×20 magnification using an Olympus VS200 slide scanner. High-resolution images were analyzed using Image J (1.8.0, NIH, USA). The fluorescence intensity of ZO-1, MUC2, and Occludin was quantified in randomly selected fields of view.
Primary microglia culture
Based on a recent study53, we made slight modifications to the primary microglia culture procedure. Cortical brain tissues from 1-day-old C57BL/6 J mice were dissected in cold HBSS (Servicebio, China, cat. no. G4203). After removing the meninges, the brain tissues were minced and rinsed three times with HBSS. The tissues were then digested in 0.25% trypsin-EDTA (ThermoFisher, cat. no. 25200056) for 20 min at 37 °C and triturated into a single-cell suspension. The primary cells were plated on poly-L-lysine-coated plates in Neurobasal medium supplemented with 10% FBS (Gibco, USA. cat. no. 10099-141), 1% GlutaMA× (Gibco, USA. cat. no. 35050061), and 1% penicillin-streptomycin (P.S.) (Gibco, USA. cat. no. 15140122). After 10 days of culture, the cell culture flasks were shaken at 250 rpm at 37 °C for 2 h. The collected culture media were centrifuged at 1000 rpm for 10 min, and the cells were resuspended in DMEM (Gibco, USA. cat. no. 11995073) with 10% FBS for inoculation.
Primary γδ T culture
Primary γδ T cells were isolated from mouse spleens. Briefly, T25 cell culture flasks were coated with 5 μg mL–1 TCR γδ antibody one day prior. After anesthesia, mouse spleens were extracted under sterile conditions, homogenized with 2 mL of precooled mouse lymphatic separation solution, filtered through a 70-mesh filter, gently layered over 5 mL of precooled DMEM medium, and centrifuged at 2000 × g for 20 min. The intermediate layer was carefully aspirated, washed with PBS, and centrifuged at 420 × g for 6 min at 4 °C. Cells were resuspended in inoculation medium (DMEM + 10% FBS + 1% P.S + 0.1 mM β-mercaptoethanol + 5 μM Zoledronic acid monohydrate + 1000 IU IL-2 + 20 ng mL–1 IL-7) at 1 × 105 cells mL–1 and seeded into six-well plates for growth. The culture medium was replaced every 3 days with DMEM containing 10% FBS, 1% P.S, 0.1 mM β-mercaptoethanol, 1000 IU IL-2, and 20 ng mL–1 IL-7. On day 12, primary cells were transfected and stimulated.
Primary γδ T cells-microglia co-culture
Primary γδ T cells were isolated from mouse spleens, and microglia were obtained from the brains of postnatal day 1 (P1) mice. For co-culture experiments, microglia were seeded in the bottom chamber of a transwell device with 0.4 μm pore-size polycarbonate membrane inserts, while γδ T cells were seeded in the top chamber. To investigate the impact of IL-17A, γδ T cells were transfected with siIL-17A or siCtrl using Lipofectamine RNAiMAX. Additionally, γδ T cells were treated with recombinant mouse IL-17A (100 ng mL–1) and LPS (1000 ng mL–1) for 6 h, followed by a medium change. The transwell inserts containing γδ T cells and microglia were co-cultured for 24 h post-treatment. After co-culture, microglia were harvested for analysis.
Quantitative PCR (qPCR)
qPCR analyses were conducted in three distinct contexts: (1) primary γδ T cells transfected with siRNA targeting IL-17A; (2) γδ T cells isolated from the small intestine of WT, TrdcCreERT2 Il7rflox/flox, and TrdcCreERT2 Cd8aflox/flox mice; and (3) hippocampal tissue harvested from mice injected intraperitoneally with an IL-17A–neutralizing antibody. Total RNA was extracted from each sample (Vazyme, China), and cDNA was synthesized using a reverse transcription kit (Vazyme, China) following the manufacturer’s protocol. qPCR was performed on a real-time PCR detection system (Bio-Rad, US) under cycling parameters of 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s and 60 °C for 30 s. IL-17A transcripts were detected using the following primers: Forward 5’-CAGACTACCTCAACCGTTCCAC-3’ and Reverse 5’-TCCAGCTTTCCCTCCGCATTGA-3’.
Cell culture and treatment
BV2 murine microglial cells were obtained from Punosai Life Science and Technology Co., Ltd. and cultured in RPMI-1640 medium (Gibco, USA. cat. no. 11875119) supplemented with 10% FBS and 1% P.S. at 37 °C in 5% CO2. To model SAE, BV2 cells were treated with 1000 ng mL–1 LPS for 6 h. After incubation, Cells were collected and processed for further assays. To block the cGAS-STING pathway, BV2 cells were treated with H151 (0.75 μM) for 2 h prior to LPS stimulation. To generate BV2ρ0 cells, which are devoid of mitochondria, BV2 cells were treated with ethidium bromide (50 ng mL–1) for 4 weeks54.
Cell transfection
For gene silencing, primary γδ T cells were transfected with IL-17A siRNA, and BV2 microglial cells transfected with RNF5 siRNA. Additionally, BV2 cells were transfected with cGAS and STING protein plasmids containing site-directed mutations for site analysis. All siRNAs and plasmids were obtained from Obio Technology Co., Ltd., Shanghai. Following the manufacturer’s instructions. Lipofectamine RNAiMA× was used for siRNA transfections, and Lipofectamine 3000 was used for plasmid transfections. Transfection efficiency was confirmed by Western blot analysis.
Western blot
Total proteins were extracted from cells and tissues, and protein concentrations were measured using the BCA assay. Equal amounts of protein were separated by SDS-PAGE and transferred to PVDF membranes (Millipore). Membranes were incubated overnight at 4 °C with primary antibodies against ZO-1 (1:1000, Cat# 21773-1-AP, Proteintech), MUC-2 (1:1000, Cat# 27675-1-AP, Proteintech), Occludin (1:1000, Cat# 27260-1-AP, Proteintech), cGAS (1:1000, Cat# 26416-1-AP, Proteintech), STING (1:1000, Cat# 19851-1-AP, Proteintech), C1q (1:1000, Cat# 11602-1-AP, Proteintech), PSD95 (1:1000, Cat# ab18258, Abcam), SYN (1:1000, Cat# 17785-1-AP, Proteintech), ACOD1 (1:1000, Cat#17805, CST), RNF5 (1:1000, Cat#A8351, Abclonal), Ubiquitin (UB) (1:1000, Cat# 10201-2-AP, Proteintech), β-actin (1:1000, Cat# 66009-1-Ig Proteintech),GAPDH (1:1000, Cat# 60004-1-Ig, Proteintech) and a custom-made antibody for the K150 ubiquitination site on STING (1:1000, Cat# RPM0002, Abclonal). After incubation, membranes were treated with HRP-conjugated anti-mouse or anti-rabbit IgG. Protein bands were visualized using ECL Western blot Detection Reagents (Beyotime) and captured with a UVP gel documentation system (UVP, LLC, Phoenix). Band intensity was quantified using ImageJ (1.8.0, NIH, USA).
Synaptosomal proteins were extracted from mouse hippocampi using the Syn-PER Synaptic Protein Extraction Reagent (Thermofisher) following the manufacturer’s instructions. Western blot analysis was then performed on the isolated synaptosomal proteins.
FCM analysis
As previously dicribed55, cell suspensions from the meninges and small intestine were prepared. After counting 1 × 106 cells, they were resuspended in 100 µL of 1% BSA in PBS, blocked with 5 ng µL–1 anti-CD16/CD32 antibody for 5 min at 4 °C, and then stained with the desired antibodies. Antibodies used for extracellular staining (2 ng µL–1) included fixable viability dye, CD45, CD3, CD4, CD19, CD25, CD49b, TCR γδ, IL7R, CD8, F4/80, CD11b, CD11c, P2RT12, CLEC7A, Ly6G, Ly6c and CD103. For intracellular staining, cells were first labeled with surface markers, then frozen and permeabilized before being labeled with Foxp3 and IL-17A antibody (4 ng µL–1). Samples were analyzed using a Beckman CytoFLE× or BD LSRFortessa X-20 cytometer (BD Biosciences, San Jose, CA). Data were analyzed using FlowJo 10.0 (FlowJo, Oregon, USA).
JC-1 assay
Mitochondrial membrane potential in BV2 microglial cells was measured using the JC-1 detection kit (Elabscience, China) according to the manufacturer’s instructions. BV2 cells were seeded in 6-well plates following experimental criteria. After treatment, cells were stained with JC-1 at 37 °C for 20 min. The cells were rinsed with buffer solution and analyzed using a fluorescence microscope or flow cytometer after incubation. The mitochondrial membrane potential was assessed by the red-to-green fluorescence ratio.
Co-IP
BV2 cells, primary microglia, and mouse hippocampal tissues were lysed on ice for 30 minutes in an NP-40-based lysis buffer (1% NP-40, 150 mM NaCl, 50 mM Tris-HCl, pH 7.4) supplemented with protease inhibitors. After centrifugation to remove cellular debris, the clarified lysates were incubated overnight at 4 °C with an anti-STING antibody (1:1000, Cat# 19851-1-AP, Proteintech) to immunoprecipitate STING-containing complexes. Protein A/G magnetic beads were then introduced to capture the antibody-protein complexes, followed by rigorous washing to eliminate non-specific interactions. The immunoprecipitated proteins were eluted in sample buffer and separated by SDS-PAGE. Western blot analysis was performed to assess the interactions between STING and proteins of interest, including BRCA1 (1:1000, Cat# 22362-1-AP, Proteintech), RNF26 (1:200, Cat# 16802-1-AP, Proteintech), RNF5 (1:1000, Cat# A8351, Abclonal), TRIM32 (1:1000, Cat# 10326-1-AP, Proteintech), TRIM56 (1:500, Cat# 25509-1-AP, Proteintech), as well as to evaluate STING ubiquitination.
Ubiquitination level detection
MG132 (100 µM) was added to cells 6 h before collection to inhibit proteasome activity and accumulate ubiquitinated proteins in cell culture studies. Hippocampus tissues were lysed in IP lysis buffer containing protease and phosphatase inhibitors to extract proteins. The lysates were incubated overnight at 4 °C with an anti-STING antibody (1:1000, Cat# 19851-1-AP, Proteintech) and protein A/G magnetic beads for Co-IP. After washing, the co-immunoprecipitated proteins were eluted from the beads. Eluted proteins were analyzed by Western blot using an anti-UB antibody (1:1000, Cat# 10201-2-AP, Proteintech) to measure ubiquitination.
Detection of ubiquitination modification sites
Immunoprecipitation (IP) was performed to pull down STING from BV2 microglial cells for the identification of ubiquitination modification sites. Ubiquitination sites were identified by mass spectrometry following STING isolation. The identified modification sites were used to create mutant plasmids for each ubiquitination site using site-directed mutagenesis. These mutant plasmids were then transfected into BV2 cells to specifically alter STING ubiquitination sites. Ubiquitination levels of WT and mutant STING proteins were compared in subsequent assays to identify key ubiquitination sites.
Microglia depletion
PLX3397 (Selleck, China) was used to deplete brain microglia. As previously reported55, PLX3397 was incorporated into AIN-76A chow at a concentration of 300 mg kg–1. The PLX3397-supplemented chow was prepared by Jiangsu Xietong, Inc., Nanjing. Mice were fed AIN-76A chow or PLX3397-supplemented chow (300 mg kg–1 in AIN-76A) for 21 days. Microglia depletion was evaluated by quantifying microglia numbers and assessing Iba1 expression after the 21-day feeding period.
Golgi staining
After anesthesia, mouse brains were immediately placed in Golgi stain fixing solution and immersed in dye solution for 14 days at room temperature in the dark. The treatment solution was changed after 1 h and then stored at 4 °C in the dark for 3 days. Brain samples were sectioned into 60 μm coronal slices using a vibrating microtome. Dendritic spine morphology was examined under a microscope.
ROS detection by DHE staining
Oxidative stress in the hippocampal CA1 region was measured using dihydroethidium (DHE) staining. Brain slices from treated animals were processed as previously described. Sections were treated with Beyotime DHE staining reagent according to the manufacturer’s instructions. F Slices were incubated with DHE (10 μM) for 30 min in a light-protected, humidified chamber at 37 °C. Superoxide anions oxidize DHE to ethidium, which intercalates with DNA and fluoresces red, indicating tissue ROS levels.
After incubation, sections were washed in PBS and mounted with anti-fade media. Fluorescent signals were captured using an Olympus fluorescence microscope (Olympus, Japan) with DHE filters. Red fluorescence intensity in the hippocampal CA1 region was quantified using ImageJ (NIH, USA) to assess oxidative stress. Oxidative stress levels between experimental groups were assessed by statistical analysis of mean fluorescence intensity.
TEM
Mice were perfused transcardially with 2.5% glutaraldehyde. 1-mm coronal brain slices were collected immediately after perfusion. The CA1 region of the hippocampus was meticulously microdissected and post-fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer at 4 °C for 24 h. After treatment, BV2 and primary microglia were centrifuged and fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer at 4 °C for 24 h.
Both tissue samples and cells were processed similarly after fixation. After three washes with 0.1 M sodium cacodylate buffer, the samples were post-fixed in 1% OsO₄ in the same buffer at 4 °C for 1.5 h. After dehydration in graded ethanol (50%, 70%, 90%, 100%), the samples were embedded in epoxy resin. Thin sections (60–70 nm) were cut using a Leica EM UC7 ultramicrotome and placed on copper grids. For contrast enhancement, sections were stained with 2% uranyl acetate for 15 min and lead citrate for 10 min.
For assessing mitochondrial damage, Flameng score was employed56. This ordinal scale reflects progressive ultrastructural degeneration, with higher scores indicating increased severity of mitochondrial pathology. 0: Intact mitochondrial ultrastructure with preserved electron-dense matrix granules and normal membrane integrity. 1: Mild structural preservation with partial loss of matrix granule density and early signs of matrix rarefaction. 2: Moderate swelling characterized by discernible mitochondrial enlargement accompanied by matrix clearing (electron-lucent appearance) and cristae separation. 3: Advanced structural compromise featuring cristae fragmentation/dissolution, matrix condensation, and partial membrane discontinuity. 4: Severe organelle degeneration demonstrating complete cristae disintegration, loss of inner/outer membrane integrity, and vacuolar transformation of mitochondrial architecture. Electron micrographs were captured using an 80 kV Hitachi HT7800 transmission electron microscope (HITACHI, Japan). Ultrastructural characteristics of hippocampal CA1 synapses, BV2 cells, and primary microglia were identified and analyzed.
Patch-clamp electrophysiology
Twenty-four hours post-CLP, mice were deeply anesthetized with isoflurane and decapitated, and brains were quickly removed into ice-cold, carbogen-saturated (95% O2 and 5% CO2) artificial cerebrospinal fluid (ACSF) containing (in mM): 125 NaCl, 2.5 KCl, 1.25 NaH₂PO₄, 25 NaHCO₃, 25 glucose, 2 CaCl₂, and 1 MgCl₂. Coronal sections (300 µm) of the hippocampus were prepared using a vibrating microtome (VT1200S, Leica) and allowed to recover at 32 °C in oxygenated ACSF for at least 30 min. For electrophysiological recordings, individual slices were transferred to a perfusion chamber maintained at 32 ± 1 °C, where the hippocampal CA1 region was visualized through an upright microscope (BX51WI, Olympus) equipped with a ×40 water-immersion objective and differential interference contrast optics. Whole-cell patch-clamp recordings of spontaneous (sEPSCs) and miniature (mEPSCs) excitatory postsynaptic currents were performed on visually identified CA1 pyramidal neurons using borosilicate glass electrodes (3–5 MΩ) filled with a K-gluconate-based internal solution (135 mM K-gluconate, 10 mM HEPES, 2 mM MgCl₂, 4 mM Na₂ATP, adjusted to pH 7.2). For mEPSC recordings, tetrodotoxin (1 µM; Alomone Labs) was included in the superfusate to block action potentials. Signals were amplified with a MultiClamp 700B amplifier (Molecular Devices), digitized at 10 kHz via a Digidata 1550 (Molecular Devices), and analyzed offline using pClamp software (Molecular Devices), where sEPSC and mEPSC frequency and amplitude were measured over stable 2–3 min epochs.
MDA and SOD activity determination
MDA and SOD activity were measured using kits (Beyotime, China) according to the manufacturer’s instructions. The hippocampus was excised, weighed, and homogenized in MDA and SOD assay solutions. Plasma and homogenate supernatants were added to the reaction system. MDA activity was measured at 532 nm by absorbance and reported as μmol mg–1 or μM. SOD activity in the samples was measured by absorbance at 450 nm and expressed as U mg–1 or U mL–1 of total protein.
Determination of cytokine levels
Cytokine levels in mouse plasma and hippocampus were measured using the ABplex Mouse Cytokine 8-Plex Assay Kit (Abclonal). The analyzed cytokines included IL-2, IL-4, IL-12p70, IL-1β, IL-6, IFN-γ, TNF-α, and IL-17A. The samples were clarified by centrifugation and analyzed by FCM.
Scratch assay
BV2 microglial cell migration was assessed using a scratch assay. BV2 cells were seeded into 12-well plates and cultured until 90% confluence. A linear scratch was made in the cell monolayer using a sterile 200 µL pipette tip. After scratching, wells were gently rinsed with PBS to remove floating cells and debris before fresh media was added. Cell migration into the scratch region was tracked for 24 h using the Opera Phenix Plus high-content imaging system (Revvity). Images were frequently captured to monitor cell migration. The migration rate was calculated as the proportion of the scratch area covered by migrating cells relative to the control group. ImageJ (NIH, USA) was used for image analysis to compare cell migration rates between experimental conditions.
Phagocytosis assay
The phagocytic capability of primary microglial cells was assessed using the Cell Meter™ Fluorimetric Phagocytosis Assay Kit (Red Fluorescence, Cat#21225). Primary microglial cells were allowed to adhere overnight in a 96-well confocal cell culture plate. After experimental treatments, cells were incubated with Protonex™ 600-labeled beads according to the manufacturer’s instructions. After incubation, non-internalized beads were washed away, and fluorescence from ingested beads was detected using a Zeiss LSM 880 confocal laser scanning microscope. Phagocytic activity was quantified as bead engulfment (%), calculated as the ratio of red fluorescent area (engulfed beads) to total cell area. ImageJ software (NIH, USA) was used to calculate this ratio and statistically compare microglial phagocytic capability between experimental groups.
PET-CT imaging for brain metabolism
Brain metabolic activity was assessed via PET-CT imaging on mice 24 h after CLP. To minimize glucose fluctuations that could affect 18F-fluorodeoxyglucose (18F-FDG) uptake, mice were fasted for 4–6 h with access to water before imaging. After fasting, mice were intravenously administered 18F-FDG at a dose of 15.6 ± 1.7 MBq in 0.5 mL saline. Following injection, mice were housed in a warm, calm environment for 30–45 min to enhance tracer uptake and minimize stress and muscular activity that could affect glucose distribution.
Small-animal PET-CT scanners with high resolution and sensitivity were utilized under isoflurane anesthesia. Optimized scanning covered the entire brain, with a focus on the hippocampi. CT scans provided anatomical reference, followed by PET scans to assess glucose metabolism.
The SUVmean, which represents the average 18F-FDG uptake adjusted for body weight and injected dose, was used to measure brain metabolic activity, particularly in the hippocampal region. SUVmean data were statistically compared between experimental groups to evaluate post-CLP brain metabolism.
Laser speckle contrast imaging (LSCI) for cerebral blood flow
Cerebral blood flow dynamics in mice were assessed by LSCI 24 h post-CLP. After anesthesia, the scalp was gently removed to expose the skull. LSCI uses coherent laser light to illuminate cortical tissue. Red blood cells in cerebral vessels alter the speckle pattern captured by a high-resolution camera. These oscillations are used to map blood flow across the cortical surface in real time. Data were collected using a laser-synchronized high-sensitivity CCD camera. Relative blood flow changes were computed based on speckle contrast. To evaluate the impact of CLP on cerebral perfusion, blood flow was quantified and statistically compared between experimental groups.
TMT-based quantitative proteomics analysis
Sample preparation
Proteomic analysis was conducted on CLP and sham-operated mice (n = 3 per group). After anesthesia, intestinal tissues were promptly removed and snap-frozen in liquid nitrogen. Weighed protein samples were added to SDS L3-EDTA lysis buffer. The samples were homogenized (25,000 × g, 4 °C, 5 min). The homogenate was then treated with 10 mM DTT and 45 mM IAM and incubated in the dark for 45 min. Proteins were precipitated with cold acetone, followed by centrifugation (25,000 × g, 4 °C, 15 min). After air-drying the pellet, SDS L3-free lysis solution was added to fully solubilize the proteins. Following another centrifugation (25,000 × g, 4 °C, 15 min), protein concentration was measured using the Bradford method. Proteins were desalted after trypsin digestion. The freeze-dried peptides were separated using a Shimadzu LC-20AB liquid chromatography system.
High-performance liquid chromatography and mass spectrometry
Peptides were separated using gradient elution on a self-packed C18 column on an Easy nLC 1200 system. The isolated peptides were analyzed using Data Dependent Acquisition (DDA) tandem mass spectrometry. TMT-proteomic analysis was performed by BGI-Shenzhen, China.
Enzyme-linked immunosorbent assay (ELISA)
Small intestine, plasma, meninges, and hippocampus samples from mice 24 hours after CLP for ELISA. The supernatants were then assayed for STING, C1q, IL-17A and itaconate levels using commercially available ELISA kits (MEIMIAN, China), following the manufacturer’s instructions.
Bioinformatics analysis
The UniProtKB database (Release 2016_10) was used to obtain FASTA protein sequences of differentially expressed proteins for Gene Ontology (GO) mapping and annotation. KEGG Orthology (KO) identities and pathways were determined by aligning the FASTA sequences of significantly altered proteins with the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://geneontology.org/). Protein expression data from relevant KEGG pathways were visualized using a hierarchical clustering heat map.
Protein modeling and molecular docking
The structural formula of 4-octyl itaconate was downloaded from the PubChem database. The protein crystal structure of STING and cGAS was obtained from the pdb database (PDB id: 4LOK, 4LEZ). Molecular docking was performed using the software Discovery Studio (version 19.1.0). The resulting STING and cGAS models were visualized, and figures were generated using the PyMOL Molecular Graphic System (version 4.6).
Statistical analysis
Data reported as mean ± standard deviation (Mean ± SD) were analyzed using GraphPad Prism 10.0 software. For behavioral experiments, data were expressed as mean ± standard error of the mean (Mean ± SEM) to more accurately capture population variance. Data normality was assessed using the D’Agostino & Pearson test or Shapiro–Wilk test. One-way analysis of variance (ANOVA) and two way ANOVA with Tukey’s or Šídák’s multiple comparisons test was used for multiple-group comparisons. Unpaired Student’s t-test or Mann-Whitney test was used for group comparisons, and Log-rank (Mantel–Cox) test was performed for survival rate analysis. P values < 0.05 were considered statistically significant. A supplementary table detailing the specific statistical tests, P values, and post hoc analyses used for each experiment is provided in Source Data. All data analyses and statistical figures were generated using GraphPad Prism 10.0.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Mouse hippocampal transcriptome data were obtained from public databases [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE128925]. The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the iPro× partner repository with the dataset identifier PXD051763. Additionally, the mass spectrometry raw data for the STING protein can be accessed through the same repository under the identifier PXD057241. Uncropped western blots for data in the main and Supplementary Figs. are provided in the Source data file. Source data are provided with this paper.
References
Robba, C., Crippa, I. A. & Taccone, F. S. Septic encephalopathy. Curr. Neurol. Neurosci. Rep. 18, 82 (2018).
Assimakopoulos, S. F. et al. Gut-origin sepsis in the critically ill patient: pathophysiology and treatment. Infection 46, 751–760 (2018).
Nielsen, M. M., Witherden, D. A. & Havran, W. L. γδ T cells in homeostasis and host defence of epithelial barrier tissues. Nat. Rev. Immunol. 17, 733–745 (2017).
Wu, P. et al. γδT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity 40, 785–800 (2014).
Santisteban, M. M. et al. Meningeal interleukin-17-producing T cells mediate cognitive impairment in a mouse model of salt-sensitive hypertension. Nat. Neurosci. 27, 63–77 (2024).
Benakis, C. et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat. Med. 22, 516–523 (2016).
Sun, J. et al. IL-17A is implicated in lipopolysaccharide-induced neuroinflammation and cognitive impairment in aged rats via microglial activation. J. Neuroinflamm. 12, 165 (2015).
Xie, B. et al. Gut-derived memory γδ T17 cells exacerbate sepsis-induced acute lung injury in mice. Nat. Commun. 15, 6737 (2024).
Zhang, Y. Q. et al. Dopamine D2 receptor regulates cortical synaptic pruning in rodents. Nat. Commun. 12, 6444 (2021).
Dejanovic, B. et al. Complement C1q-dependent excitatory and inhibitory synapse elimination by astrocytes and microglia in Alzheimer’s disease mouse models. Nat. Aging 2, 837–850 (2022).
Hao, W. et al. Gut dysbiosis induces the development of depression-like behavior through abnormal synapse pruning in microglia-mediated by complement C3. Microbiome 12, 34 (2024).
Chung, H. Y. et al. Microglia mediate neurocognitive deficits by eliminating C1q-tagged synapses in sepsis-associated encephalopathy. Sci. Adv. 9, eabq7806 (2023).
Mills, E. L. et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556, 113–117 (2018).
Li, W. et al. 4-octyl itaconate as a metabolite derivative inhibits inflammation via alkylation of STING. Cell Rep. 42, 112145 (2023).
Ramakrishnan, R. K. et al. IL-17 induced autophagy regulates mitochondrial dysfunction and fibrosis in severe asthmatic bronchial fibroblasts. Front. Immunol. 11, 1002 (2020).
Ghosh, M., Saha, S., Li, J., Montrose, D. C. & Martinez, L. A. p53 engages the cGAS/STING cytosolic DNA sensing pathway for tumor suppression. Mol. Cell 83, 266–280.e266 (2023).
Hu, X. et al. Emerging role of STING signalling in CNS injury: inflammation, autophagy, necroptosis, ferroptosis and pyroptosis. J. Neuroinflamm. 19, 242 (2022).
Ferecskó, A. S. et al. STING-triggered CNS inflammation in human neurodegenerative diseases. Biomedicines 11, e20191422 (2023).
Meyer-Schwesinger, C. The ubiquitin-proteasome system in kidney physiology and disease. Nat. Rev. Nephrol. 15, 393–411 (2019).
Chancharoenthana, W., Kamolratanakul, S., Schultz, M. J. & Leelahavanichkul, A. The leaky gut and the gut microbiome in sepsis - targets in research and treatment. Clin. Sci. 137, 645–662 (2023).
He, Y. et al. A metabolite attenuates neuroinflammation, synaptic loss and cognitive deficits induced by chronic infection of Toxoplasma gondii. Front. Immunol. 13, 1043572 (2022).
Chausse, B., Lewen, A., Poschet, G. & Kann, O. Selective inhibition of mitochondrial respiratory complexes controls the transition of microglia into a neurotoxic phenotype in situ. Brain Behav. Immun. 88, 802–814 (2020).
Vigil, T. M., Frieler, R. A., Kilpatrick, K. L., Wang, M. M. & Mortensen, R. M. Aconitate decarboxylase 1 suppresses cerebral ischemia-reperfusion injury in mice. Exp. Neurol. 347, 113902 (2022).
Liu, C. et al. One-step synthesis of 4-octyl itaconate through the structure control of lipase. J. Org. Chem. 86, 7895–7903 (2021).
Dinan, T. G. & Cryan, J. F. Gut-brain axis in 2016: Brain-gut-microbiota axis - mood, metabolism and behaviour. Nat. Rev. Gastroenterol. Hepatol. 14, 69–70 (2017).
Xu, K. et al. Phosphatidylserine improves aging sepsis survival, modulates gut microbiome, and prevents sepsis-associated encephalopathy. Biomed. Pharmacother. 178, 117200 (2024).
Xu, Y. et al. Palmatine ameliorated lipopolysaccharide-induced sepsis-associated encephalopathy mice by regulating the microbiota-gut-brain axis. Phytomedicine 124, 155307 (2024).
Mills, K. H. G. IL-17 and IL-17-producing cells in protection versus pathology. Nat. Rev. Immunol. 23, 38–54 (2023).
Li, J. et al. Microglial phenotypic transition: signaling pathways and influencing modulators involved in regulation in central nervous system diseases. Front Cell Neurosci. 15, 736310 (2021).
Gao, S. et al. N-acetyltransferase 10 mediates cognitive dysfunction through the acetylation of GABA(B)R1 mRNA in sepsis-associated encephalopathy. Proc. Natl. Acad. Sci. USA 121, e2410564121 (2024).
Yang, Y. S. et al. Quercetin protects against sepsis-associated encephalopathy by inhibiting microglia-neuron crosstalk via the CXCL2/CXCR2 signaling pathway. Phytomedicine 134, 155987 (2024).
Yin, X. Y. et al. HMGB1 mediates synaptic loss and cognitive impairment in an animal model of sepsis-associated encephalopathy. J. Neuroinflamm. 20, 69 (2023).
He, Q., Wei, Y., Qian, Y. & Zhong, M. Pathophysiological dynamics in the contact, coagulation, and complement systems during sepsis: potential targets for nafamostat mesilate. J. Intensive Med. 4, 453–467 (2024).
Hong, S. et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016).
Sekar, A. et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016).
Zhang, Q., Lu, C., Fan, W. & Yin, Y. Exploring the molecular mechanism of sepsis-associated encephalopathy by integrated analysis of multiple datasets. Cytokine 180, 156609 (2024).
Fan, J. et al. Preferential pruning of inhibitory synapses by microglia contributes to alteration of the balance between excitatory and inhibitory synapses in the hippocampus in temporal lobe epilepsy. CNS Neurosci. Ther. 29, 2884–2900 (2023).
Watson, Z. & Tang, S. J. Aberrant synaptic pruning in CNS diseases: a critical player in hiv-associated neurological dysfunction? Cells 11, e20191422 (2022).
Li, H. R. et al. β-Nicotinamide mononucleotide activates NAD+/SIRT1 pathway and attenuates inflammatory and oxidative responses in the hippocampus regions of septic mice. Redox Biol. 63, 102745 (2023).
Ben-Simon, Y. et al. A direct excitatory projection from entorhinal layer 6b neurons to the hippocampus contributes to spatial coding and memory. Nat. Commun. 13, 4826 (2022).
Wang, C. et al. Ultrasound-responsive low-dose doxorubicin liposomes trigger mitochondrial DNA release and activate cGAS-STING-mediated antitumour immunity. Nat. Commun. 14, 3877 (2023).
Sliter, D. A. et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262 (2018).
Woo, M. S. et al. STING orchestrates the neuronal inflammatory stress response in multiple sclerosis. Cell 187, 4043–4060.e4030 (2024).
Jiménez-Loygorri, J. I. et al. Mitophagy curtails cytosolic mtDNA-dependent activation of cGAS/STING inflammation during aging. Nat. Commun. 15, 830 (2024).
Ramalho, T. et al. Itaconate impairs immune control of Plasmodium by enhancing mtDNA-mediated PD-L1 expression in monocyte-derived dendritic cells. Cell Metab. 36, 484–497.e486 (2024).
Chen, Y. J. et al. Targeting IRG1 reverses the immunosuppressive function of tumor-associated macrophages and enhances cancer immunotherapy. Sci. Adv. 9, eadg0654 (2023).
Liao, S. T. et al. 4-Octyl itaconate inhibits aerobic glycolysis by targeting GAPDH to exert anti-inflammatory effects. Nat. Commun. 10, 5091 (2019).
Bambouskova, M. et al. Electrophilic properties of itaconate and derivatives regulate the IκBζ-ATF3 inflammatory axis. Nature 556, 501–504 (2018).
Zhong, B. et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity 30, 397–407 (2009).
Bodda, C. et al. HSV1 VP1-2 deubiquitinates STING to block type I interferon expression and promote brain infection. J. Exp. Med. 217, e20191422 (2020).
Tomura, M. & Kabashima, K. Analysis of cell movement between skin and other anatomical sites in vivo using photoconvertible fluorescent protein “Kaede”-transgenic mice. Methods Mol. Biol. 961, 279–286 (2013).
Wu, X. et al. Foxq1 activates CB2R with oleamide to alleviate POCD. Brain Pathol. 35, e13289 (2025).
Liu, Y. et al. NG2 glia protect against prion neurotoxicity by inhibiting microglia-to-neuron prostaglandin E2 signaling. Nat. Neurosci. 27, 1534–1544 (2024).
Zecchini, V. et al. Fumarate induces vesicular release of mtDNA to drive innate immunity. Nature 615, 499–506 (2023).
Shi, Y. et al. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J. Exp. Med 216, 2546–2561 (2019).
Li, J. et al. Mechanism of the hypoxia inducible factor 1/hypoxic response element pathway in rat myocardial ischemia/diazoxide post‑conditioning. Mol. Med. Rep. 21, 1527–1536 (2020).
Acknowledgements
The study was funded by Noncommunicable Chronic Diseases-National Science and Technology Major Project (Grant No. 2023ZD0506504 to [Y.S.]); National Natural Science Foundation of China (Grant No. 82472223 and 82071480 [to J.C.Z.]; Grant No. 82272231 [to S.Y.Y.]; Grant No. 82302471 [to Y.J.Z.]; Grant No. 82402568 [to B.X.]); National Key Research and Development program from Ministry of Science and Technology of the People’s Republic of China (Grant No. 2021YFC2500801 [to S.Y.Y.]); Science foundation of Union Hospital, Tongji Medical College, Huazhong University of Science and Technology (2022xhyn054 [to Y.Y.]); and Research Grant of Key Laboratory of Anesthesiology and Resuscitation (Huazhong University of Science and Technology), Ministry of Education (Grant No. 2024MZFS 007 [to Y.M.W.]).
Author information
Authors and Affiliations
Contributions
J.-c.Z., K.H., and Y.-m.W. conceived and designed experiments. Y.-m.W. and Y.-j.Z. carried out most of the experiments, analyzed the data, and prepared figures. Y.Yu, Z.-f.Z., and X.W. carried out the TMT-based quantitative proteomics analysis. B.X. and M.-q.H. conducted CLP surgery. X.-y.Z., X.-q.S., and M.-y.W. carried out the Kaede photoconversion. X.-y.W. and Y.Y. conducted cell culture. J.-c.Z., S.-y.Y., and Y.S.g contributed to experimental design, data analysis, discussions, and advice. Y.-m.W., J.-c.Z., and Y.-j.Z. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Fabio Grassi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, Y., Zhang, Y., Yu, Y. et al. Small intestinal γδ T17 cells promote SAE through STING/C1q-induced microglial synaptic pruning in male mice. Nat Commun 16, 6779 (2025). https://doi.org/10.1038/s41467-025-62181-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62181-3
