
Abstract
Basidiomycete fungi are the main decomposers of dead wood with an impact on the global carbon cycle. Their degradative mechanisms have been well-studied under aerobic conditions. Here, we study their activity in oxygen-depleted environments. We use metaproteomics in a field study to identify active wood-decomposing fungi and their enzymes at different depths from the wood surface, including in oxygen-depleted conditions. In vitro, we observe that the brown-rot fungus Fomitopsis pinicola can grow on wood in complete anoxia. Using 13C solid-state NMR, we demonstrate the degradation of plant cell-wall polysaccharides and fungal growth in the absence of oxygen. Proteomic analyses reveal that F. pinicola switches from a Fenton chemistry-based process under aerobic conditions to the secretion of plant cell wall-active enzymes in anoxia. Our finding that wood decay fungi can thrive in complete anoxia provides a deeper understanding of lignocellulose degradation mechanisms in nature and raises opportunities for the development of bio-inspired anaerobic processes.
Similar content being viewed by others

Introduction
Forests are a large and persistent carbon sink that stores 45% of the total carbon (C) of terrestrial ecosystems1,2. An equilibrated forest microbiome is required to enable a continuous C cycle and ensure the forest’s resilience towards global changes3. While plants are responsible for the large ecosystem uptake of C through photosynthesis, fungal saprotrophs are the main decomposers of lignocellulose4.
The most efficient fungal wood decayers have been empirically classified into soft-, white-, and brown-rots based on the appearance of the degraded wood following colonisation. Their decaying strategies rely on complex mechanisms combining abiotic chemistry with enzymatic activities5. Differences in the mode of wood decay between brown-rots and white-rots have been supported in recent years by comparing the number of genes encoding plant cell wall-degrading enzymes (PCWDEs) in fungal genomes and machine-learning techniques4,6,7,8. White-rot fungi secrete a complete arsenal of PCWDEs, including oxidative and hydrolytic enzymes, to degrade the different plant cell wall polymers6,9. However, brown-rot fungi, which are predominant decayers of softwood in boreal forests, degrade holocellulose in a staggered mechanism10,11, initiated by the controlled biochemical and enzymatic formation of highly reactive and short-lived hydroxyl radicals through Fenton chemistry, which is believed to be the primary cause of the decay5,12,13,14. For the second stage, brown-rot fungi employ PCWDEs, yet these are a smaller set enzymes compared to white-rot fungi5,8. Lignin and crystalline cellulose remain because they lack of key hydrolytic and oxidative enzymes (e.g. cellobiohydrolases, lytic polysaccharide monooxygenases, multicopper oxidases, and peroxidases)4,15,16. Despite these recent advances in the genomics and biochemistry of fungal saprotrophs, improved understanding of wood decay is needed.
The mechanisms used by white-rot and brown-rot fungi to decay wood were deciphered mostly using cultures made at an ambient O2 concentration of 20.9%, which facilitates the experimental set-up17. While such concentration is relevant to the decay in the surface of wood or in biotechnological set-ups with air supply, it does not correspond to the conditions naturally encountered by fungi inside the wood. Here, the O2 concentration can decrease to a very low percentage as a function of depth in the wood18,19. Despite the physiological relevance of evaluating the influence of O2 concentrations on the ability of white-rot and brown-rot wood-decayers to grow, relatively few studies have addressed this aspect. Cultures under limited O2 concentrations (hypoxia) have shown that some white-rot and brown-rot fungi can grow at low levels of O220 thanks to a metabolic reorganisation allowing them to withstand the hypoxic constraint21,22. However, it remains unclear whether fungal wood decay can take place in the absence of O2 (anoxia).
Here, we explored in situ wood decay with particular emphasis on the constraints imposed by restricted O2 availability towards the central part of the wood. Using metaproteomics on wood stumps and trunks collected in nature, we identified the basidiomycetes fungi at play and their secreted PCWDEs. A targeted in vitro approach undertaken on the brown-rot fungus Fomitopsis pinicola (F. pinicola) uncovered that fungal wood decay can occur in anoxic conditions and provide insights into fungal strategies for lignocellulose degradation in O2-deprived environments.
Results
A restricted microbial community decomposes wood under O2 constraints in nature
To investigate the impact of O2 depletion on microbial wood decay in nature, we collected decaying spruce wood (Picea abies) in a boreal forest at different stages of decay, i.e. trees that were cut down 3, 10, and 15 years before sampling (Fig. 1a, Supplementary Fig. 1a). Using a needle O2 sensor, we could penetrate the two stumps with the highest grade of decay (chopped down 10 and 15 years before sampling), and measured a strong depletion of O2 beneath the surface of dead wood (Fig. 1b, Supplementary Fig. 1b). Macroscopically, we observed fungal mycelium in areas with very restricted O2 concentrations, indicative of the growth of some wood-decaying fungi under minimal O2 concentrations. To get access to the active microbial species and their enzymatic portfolio, we collected samples at different depths in the dead wood, i.e. from the external layer to the centre of wood stumps and trunks (Fig. 1a) and we used a metaproteomic approach because recent breakthroughs in high-resolution tandem mass spectrometry allow to investigate microbiome functionality by identifying and quantifying active proteins. We identified 11,551 proteins of which 93% pertained to fungal genera (see Supplementary Data 1). Only a restricted number of microbial genera, mostly tree-specific, were detected in trunk and stump samples (Fig. 1c, Supplementary Fig. 2). The brown-rot F. pinicola dominated the microbial community of the decaying trunk cut down 3 years before sampling (Supplementary Figs. 2 and 3). The species identity—F. pinicola (Sw.) P. Karst.—was confirmed using molecular tools (Genbank sequence ID PP277039). Heterobasidion sp., a saprotrophic white-rot species, was detected in the centre of the same trunk, and dominated the corresponding stump. In this stump, other species, such as the white-rot Cristinia sp. appeared confined to the centre and intermediary part of the wood. In the 10 years decayed stump, the white-rot Resinicium sp. was the only microorganism detected. Finally, the 15 years decayed stump was colonised by a broader variety of fungal saprotrophs (Meripilus, Trametes) together with mycorrhizal fungi (Russula, Laccaria) and diverse bacteria within Acidipila, Acidisphaera, Acidobacteria, Dyella and Methylovirgula genera (Supplementary Fig. 2). Overall, this in vivo proteotyping approach unveiled distinct colonisation patterns of deadwood samples by a specific subset of fungal species, some being present across the different depths from normoxia (20.9% O2) towards anoxia. To identify the enzymes at play during in vivo wood decay, we assigned putative function to 85% of the proteins (accounting for 91% of the captured signal) (Supplementary Fig. 4). The methodology developed allowed the identification of secreted enzymes, including carbohydrate-active enzymes (CAZymes) involved in the degradation of holocellulose, and lignin, indicating that the detected fungi were actively decaying the wood (Fig. 1d, Supplementary Fig. 4).

a Summary of the experimental design. All protein extractions were carried out in parallel with three independent replicates per sample. Pictures were drawn with Inkscape. b O2 profiles measured in P. abies wood of different decay ages. Individual lines are separate profiles in the same piece of wood, and each line represents an individual profile measured at a distinct spot from the wood surface (see Supplementary Fig. 1b). Measurements stopped before 40 mm depth and could not penetrate further into the wood. Wood of 3 years decay was not soft enough to allow the dioxygen probe to penetrate. c Taxonomic assignation of the fungal proteins extracted from the samples. Genus prevalence, estimated as the cumulative NSAF (normalized spectral abundance factor) percentage of proteins assigned to each genus, is displayed for each sample analysed. d Secreted carbohydrate active enzymes (CAZymes) prevalence in the samples. The abundance of CAZymes acting on key wood substrates is estimated by the cumulative NSAF percentage of the corresponding CAZymes. 3ydT: 3 years decayed trunk, 3ydS: 3 years decayed stump, 10ydS: 10 years decayed stump, 15ydS: 15 years decayed stump, cen. centre, int. intermediate, ext. external part of the wood, near the surface.
Fomitopsis pinicola can grow and decay wood in anoxia
The presence of the brown-rot fungus F. pinicola at the centre of the trunk (Fig. 1c, Supplementary Figs. 2 and 3) attracted our attention since brown-rot fungi are known to rely on Fenton chemistry to decay softwood and these reactions require hydrogen peroxide, and thus O2, which is depleted in the centre of dead wood. We thus hypothesised the existence of an undescribed alternative pathway allowing wood decomposition in a limited O2 environment. To test this hypothesis, we assessed the impact of O2 depletion on the growth of F. pinicola and on wood degradation by designing in vitro set-ups to measure and control this parameter. Of note, we have used milled pine wood (<1.5 mm particle size) in all experimental set-ups. The first system developed was a solid-state culture set-up in a column that mimicked the entire radial profile in the wood and allowed to non-intrusively measure the O2 gradient between the top and the bottom, i.e. from normoxia (proxy of the surface of the tree) to anoxia (proxy of the heart of the tree) (Fig. 2a). Initially, we strived to determine the minimum O2 level required for the saprotroph to colonise the substrate, but surprisingly, we observed the migration of the fungus throughout the entire O2 gradient and into the anoxic zone at the bottom of the column (Fig. 2a).

a Experimental set-up mimicking the O2-gradient during saprotrophic colonisation with non-intrusive measurements of O2 concentrations at the top (blue patch) and the bottom (white patch). The picture was drawn with Inkscape. b O2 concentration measured during the growth of F. pinicola in the set-up at the air-wood interphase (blue) and at the bottom of the column (white). c In vitro saprotrophic solid-state cultures of F. pinicola on softwood in normoxia and anoxia. The experimental set-up ensured constant normoxia (air) and anoxia (N2 with O2 < 2 ppm) (Supplementary Fig. 5). Scanning electron microscopy confirmed the presence of fungal mycelium (framed in dotted lines) in both normoxia and anoxia conditions. Scale bar represents 50 µm. d Box plots depict fungal growth (after 14 days) assessed using quantitative PCR in four biological replicates (n [normoxia] = 34, and n [anoxia] = 33). In each box plot, the lower and upper boundaries of the box represent the first quartile (Q1) and the third quartile (Q3), respectively. The whiskers extend from the quartiles to the minimum and maximum values within 1.5 times the interquartile range. Of note, the water content remained stable between 71.6–75.0% (mass) (73.8% at the start of the cultivation) in both conditions.
To confirm this unexpected observation, we developed a second solid-state culture system in Roux flasks with either air or N2 to provide a stable normoxic or anoxic environment, respectively (Supplementary Fig. 5, Fig. 2c). This experimental set-up demonstrated the ability of F. pinicola to grow under anoxia in two weeks. Indeed, the growth of the mycelium, as assessed qualitatively by microscopy and quantitatively by qPCR, was similar in normoxia and anoxia (Fig. 2d). Of note, we confirmed the growth of F. pinicola on wood in anoxia using alternative culture configurations in an anaerobic jar and an O2-depleted bioreactor (Supplementary Fig. 6).
In the solid-state culture conditions (Fig. 2c), F. pinicola decayed wood to the same extent with 7.4–13.2% mass loss (wood dry weight) in normoxia and 8.0–8.9% in anoxia. Furthermore, we confirmed holocellulose and pectin degradation by the fungus through the identification of glucose and the respective sugar monomers of glucuronoarabinoxylan, galactoglucomannan, and pectin components (Supplementary Fig. 7, Supplementary Fig. 8). Interestingly, holocellulose monomers (i.e. glucose, xylose, mannose) and the main component of pectin (galacturonic acid), were primarily detected in anoxia (Supplementary Figs. 7a, b and 8a). A deeper saccharide analysis of the colonised wood using carbohydrate gel electrophoresis (PACE)23 revealed improved accessibility for the structural polysaccharide backbones independent of the O2 concentration during cultivation compared to the untreated sample (Supplementary Fig. 7c–e). We attribute this improved accessibility to (i) substantial Fenton chemistry-mediated cleavage of glycosidic bonds under normoxic condition, and (ii) Fenton-independent modifications under anoxia. To further investigate this hypothesis, we measured the key reagent in Fenton reactions (i.e. H2O2) as well as oxalate (Supplementary Fig. 8b), its potential precursor24, which we observed microscopically in both culture conditions (Supplementary Fig. 9). While the amount of soluble oxalate was not significantly increased in normoxia, we detected considerably higher concentrations of H2O2 and a lower pH (Fenton-promoting acidic environment) in normoxia compared to the O2-depleted cultures. Therefore, the normoxic condition provided the core prerequisites for promoting the formation of short-lived hydroxyl radicals, which are claimed to be essential to brown-rot wood decay. In contrast, in the O2-depleted conditions, these requirements were not fulfilled.
In anoxia, Fomitopsis pinicola decays wood polysaccharides to build its cell wall
To identify changes in the molecular architecture of pine cell walls induced by F. pinicola growth, we analyzed with solid-state NMR (ssNMR) 13C enriched pine wood25 before and after 28 days of fungal wood decay (solid-state cultures). The quantitative Direct Polarization (q-DP) 1D NMR spectra, normalized to the cellulose C41 peak (89 ppm), showed significant differences in the relative abundance of cell wall components (Supplementary Fig. 10), with the mannan carbon 1 peak at ~101 ppm (M1) being significantly smaller in the decayed wood. Furthermore, the decayed wood showed a decrease in the acetate peak (AcetateCH3, ~21 ppm). In softwoods such as pine, mannan is the only acetylated hemicellulose26. Therefore, the observed decrease in acetylation in the q-DP spectrum can be attributed to the reduction of acetylated mannan. Additionally, two new peaks were observed in the decayed sample at 93 ppm and 97 ppm, corresponding to newly formed unknown oligo/polysaccharide reducing ends27, indicating that F. pinicola degraded cell wall polysaccharides. There was also a significant increase in aliphatics (~30 ppm) and many changes in the aromatic region (110–155 ppm).
Further information was revealed from the 2D 30 ms CP-PDSD 13C NMR spectra (Supplementary Fig. 11). Clear differences were observed in the mannan C4-C6 cross-peak (M4-M6, 80.3, 61.6) ppm confirming the reduction of mannan in the rotting pine wood (Fig. 3A). It was also evident that the xylan threefold (Xn3f) C4-C5 cross-peak (77.8, 63.7) ppm was significantly decreased in rotting pine wood whilst the bound twofold (Xn2f)28 C4-C5 cross-peak (82.0, 64.0) ppm also showed a significant but less marked reduction (Fig. 3A). These changes in mannan and xylan were also clearly visible in the C1-C6 region of the spectrum (Supplementary Fig. 11 and Supplementary Fig. 12). Interestingly, we found evidence that F. pinicola incorporated 13C-labeled pine wood polysaccharides into its own cell wall. Indeed, we identified exclusively in the rotting pine sample C3-C4 cross-peaks at (87.0, 68.3) ppm (Fig. 3B) and C1–C4 (103.9, 68.3) ppm (Supplementary Fig. 11), corresponding to β-1,3-glucan29 and also the C2–C1 peak cross peak at (55.4, 103.9) ppm (Fig. 3B) corresponding to chitin30. It is not possible to know from the NMR spectra whether or not cellulose is degraded because we needed to use cellulose C4(1) as a cell wall internal reference for spectral normalisation. In conclusion, ssNMR revealed that under anoxia F. pinicola can metabolize xylan and mannan from pine cell walls, and uses these hemicellulose polysaccharides as a carbon source to synthesize fungal cell wall polysaccharides, including β-1,3-glucan and chitin.

A An overlay of the 76–91, 58–68 ppm region of the 30 ms CP-PDSD 13C NMR spectra showing the xylan Xn4–Xn5 and mannan M4–M6 cross peaks. There is a significant decrease in the mannan M4–M6 and in both threefold (Xn3f) and twofold (Xn2f) xylan conformations in the decaying pine wood. B Left Hand Side the 82–92, 58–70 ppm region of the 30 ms CP-PDSD 13C NMR spectrum, Right Hand Side the 52–70, 98–110 ppm region. The C3–C4 and C1–C2 cross-peak areas for β−1,3-glucan and chitin respectively are highlighted with dotted yellow circles. These signals are absent in the control pine wood and indicate the emergence of 13C-labeled fungal cell wall polysaccharides in the decaying wood sample. All spectra have been normalized to 89 ppm (C41). A full 30 ms CP PDSD spectrum is shown in Supplementary Fig. 12.
Secretion of a complete set of PCWDEs in anoxia
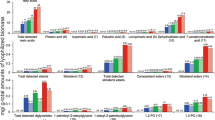
To understand how the degradation could occur without Fenton chemistry, the enzymatic machinery of F. pinicola was explored using mass spectrometry under anoxia and normoxia conditions (experimental set-up from Fig. 2c). Cumulatively, approx. 1200 proteins were identified in the normoxic and anoxic conditions (see Supplementary Data 1 and Supplementary Fig. 13 for the assessment of reproducibility in biological replicates). Most of the proteins were detected in comparable quantities in these two conditions, including key enzymes of energy processing and regulation (Supplementary Fig. 14). Noteworthy, hydrolases (EC 3) accounted for the main difference with >25% increased abundance in anoxia (vide infra). To highlight the main differences between the two conditions, we focused on the most differentially-produced proteins in each condition. Strikingly, a putative oxalate oxidase stood out on the top under normoxia, while being absent in anoxia (Fig. 4a). This enzyme (EC 1.2.3.4) catalyses the formation of H2O2 from oxalic acid in an acidic environment31,32, and hence confirms Fenton-mediated wood decay in the presence of O2. Under anoxia, the top 25 most differentially-produced proteins disclosed a different trend with the striking presence of most of the PCWDEs encoded by the genome of F. pinicola, in particular, glycoside hydrolases (GHs, EC 3.2.1) targeting cellulose, hemicelluloses and pectin (Fig. 4). Two GH10 xylanases, three GH43 arabinofuranosidases, and a single GH115 glucuronidase, which cleave glucuronoarabinoxylan ranked high in abundance in anoxia, but were not detected in normoxia. Consistently, galactoglucomannan-degrading enzymes (one GH5_7 mannanase and three GH27 galactosidases) and a CE15 methyl-glucuronoyl esterase, targeting lignin-xylan ester bonds, were exclusively detected in anoxia. Additional PCWDEs targeting the hemicellulose side chains (GH51 and CE16), cellulose (e.g. GH5_5) and pectin (e.g. GH28) were significantly increased in anoxia (Fig. 4). The endo-xylanase and endo-glucanase activity were confirmed from the secretomes only in the anoxia condition using cyclophellitol-derived activity-based probes (Supplementary Fig. 15). Some proteins of unknown function were also preferentially secreted under anoxia conditions (Fig. 4a). Noteworthy, the protein corresponding to DUF1793 was already identified in Pycnoporus coccineus secretomes induced on wood33, suggesting a potential role related to wood decay. In conclusion, an extensive set of PCWDEs targeting the linkages of glucuronoarabinoxylan, galactoglucomannan, cellulose, and pectin are secreted preferentially or exclusively in anoxic conditions (Fig. 4b).

a Differentially produced fungal proteins in normoxia (left, in blue) and anoxia (right, in black) ranked by fold change and their relative abundance (% NSAF) in the respective conditions. b Differentially secreted CAZyme families under anoxia with their respective target bonds in holocellulose and pectin. “#SC” indicates the total spectral counts for the CAZyme member(s) within a CAZy family. On the right, “#enzymes” indicates the CAZyme(s) member(s) identified/gene(s) encoded by the genome. * indicates that the CAZyme was exclusively found under anoxia. NSAF: normalized spectral abundance factor, GH: glycoside hydrolase, DHase: dehydrogenase, DUF: domain of unknown function. The fold change calculation is defined in the method section.
Discussion
The global approach we initiated on decaying softwood from a boreal forest has uncovered that fungal wood decay can occur in nature under O2-depleted conditions. In vitro cultures of F. pinicola corroborated these initial observations, demonstrating the ability of this brown-rot fungus to grow in anoxic conditions. Using proteomics, we revealed that under anoxia, F. pinicola secretes a full set of glycoside hydrolases and carbohydrate esterases, enabling the degradation of softwood xylan and mannan. A significant decrease of both hemicelluloses during the anoxic culture of wood was confirmed by solid-state NMR spectroscopy, which also demonstrated the fungus synthesized its own cell wall polysaccharides such as β-1,3-glucan and chitin. Thus, our findings provide a functional explanation for the retention of PCWDEs in the genomes of brown-rot fungi8,15,16. Although these fungi have lost most of their oxidative enzymes, such as lytic polysaccharide monooxygenases (LPMOs) and class II peroxidases, critical for crystalline cellulose and lignin degradation, they have preserved a strategically-relevant set of hydrolytic PCWDEs, predominantly targeting hemicelluloses, including recalcitrant xylan.
Our results enable us to propose a mechanism to explain how the fungus penetrates and colonizes regions of wood where O2 is limited or absent (Fig. 5). During the initial phase of decay, in the presence of O2, plant polysaccharide breakdown occurs through a combination of Fenton chemistry and a limited set of PCWDEs5,12,13,14. As decay progresses, fungal growth continues into the substrate until a “critical” O2 concentration is reached, which slows down and eventually stops Fenton chemistry-mediated decay. Deeper within the wood, PCWDEs become the main contributors of plant polysaccharide breakdown, consistent with the second step of the staggered decay mechanism described for brown-rot fungi10,11. In nature, this mechanism allows the fungus to continue its colonization even in the absence of O2.

The data presented in this study suggest a shift from Fenton-mediated wood decay under normoxia towards exclusive PCWDE-driven hydrolytic decomposition in anoxia. In a staggered mechanism, the first stage of decay, in the presence of O2, is characterized by non-specific Fenton chemistry and a small set of PCWDEs. At the later stage of decay, deeper in wood, and hence under O2-restricted conditions, secreted glycoside hydrolases and carbohydrate esterases depolymerize the holocellulose and pectin fraction in the immediate vicinity of the fungal hyphae, which enables the fungus to penetrate the substrate further. F. pinicola is represented by grey filaments, oxidative enzymes by light blue triangles, and hydrolytic CAZymes by black circles. The picture was drawn with Inkscape.
F. pinicola is among the most damaging wood-decaying species in old-growth forests of the Northern hemisphere. Infected dead trees are prone to windthrow and top-breakage, making them high-risk hazard trees34 and compromising timber quality35. Interestingly, empirical observations describe F. pinicola and other wood decayers as heart rot decayers36,37,38. Our findings of fungal growth and decay under anoxia may have implications for heart rot decay, which is not exclusively associated with brown-rot species, or the genus Fomitopsis. For instance, Heterobasidion spp., identified in the center of our dead wood samples, are also described as heart rot decayers39,40. Furthermore, other species like the white-rot fungi Phlebia radiata and Phanerochaete sordida, have been reported to grow on wood under O2-limited conditions21 and resist to hypoxic conditions22. Growth and wood degradation under O2-restricted conditions, including anoxia, may be more widespread among wood-decaying fungi than previously recognized. This raises intriguing questions about the mechanisms of wood decay in white-rot fungi under anoxic conditions, particularly given their extensive repertoire of oxidative enzymes involved in lignocellulose degradation. We believe that a deeper understanding of the biochemical processes of wood decay, particularly the role of O₂, could improve the classification of rot types.
In conclusion, our findings advance the understanding of natural lignocellulose degradation mechanisms, highlighting the critical role of O2 in fungal wood decay. By demonstrating saprotrophic wood decay in an anoxic environment, we reveal a previously unexplored dimension of fungal adaptability. The extent of anaerobic wood depolymerisation by basidiomycetes must be further investigated for the understanding and quantification of carbon dynamics in forest ecosystems, especially in the boreal forest dominated by conifers. This discovery also opens avenues for developing innovative decarbonization strategies through bio-inspired anaerobic processes leveraging the unique capabilities of fungal wood decayers.
Methods
Wood sampling
In May 2022, we sampled wood from Picea abies (L.) Karst (P. abies) stumps in the experimental forest of Stensholt Vang in Denmark (709370, 620263 UTM), one site (site ID 1011) in a larger national tree planting experiment41. At this site, 5-year-old seedlings of native provenance Rye Nørskov F. 300 were planted in 1965 on ploughed cropland. Trees were thinned every 4-6 years leaving behind wood stumps of different, known ages. Wood stumps remaining from thinning events in 2007, 2012 and 2019 were selected for our purpose to represent a gradient of wood degradation. The selected sample trees were located in the same area, just a few metres apart. With a chainsaw, we removed the top of the stump that was exposed to air (approx. 10 cm). From there, we cut off approx. 20 × 20 × 20 cm irregular-shaped pieces from each of the three wood stumps. In addition, a piece of the trunk from the 2019-felled tree with visible carpophores of Fomitopsis sp. was collected (Supplementary Fig. 1a). Pieces of trunk and stumps were wrapped and transported in individual bags.
Strain isolation and authentication
Basidiomes of F. pinicola were collected from the fallen trunk of decayed spruce tree (cut in 2019), at Stensholt Vang forest (Denmark). The isolation process involved transferring tissues from the basidiomes to agar plates containing malt extract supplemented with 0.025% chloramphenicol and 0.04% gentamicin. Successive subcultures without antibiotics were performed to verify the absence of contaminants. To confirm the identity of the isolated strains, the rDNA barcoding region ITS1-5,8S-ITS2 was PCR-amplified and sequenced42. Following molecular authentication (Genbank sequence ID PP277039), the strain corresponding to the species F. pinicola (Swartz: Fr.) Karst. was deposited in the Biological Resource Center CIRM-CF (International Center of Microbial Resources, Marseille, France; https://doi.org/10.15454/KJQW-SJ57) under the accession number CIRM-BRFM 3531.
Dioxygen (O2) profiles in wood
O2 concentrations in the wood from Stenholt Vang were measured from outside going into the wood. Wood O2 profiles were determined from the exterior part (sapwood under the bark) into the inner sapwood with a minimum distance of 50 mm to the air-exposed cutting sections of the stump. Therefore, a needle sensor (OX-NP, Unisense, Århus, Denmark) mounted on a motorised stage (MMS, Unisense, Århus, Denmark, Supplementary Fig. 1b), which moved n steps of 1.0 mm towards the wood centre until a depth of 4-6 cm. The optical sensors were mounted inside a surgical needle for strength. The measured O2 concentrations were calibrated with a two-point procedure: at an atmospheric O2 level through water bubbled with an aquarium pump, and at 0% O2 in a sodium ascorbate solution (pH 9.9, zero oxygen calibration kit, Unisense, Århus, Denmark) and corrected for temperature in the programme SensorTrace Suite (version 3.4.100.15377, Unisense, Århus, Denmark).
Microscopy
Specimens of approx. 2 × 3 × 5 mm were prepared from 3-year-old Norway spruce (P. abies) wood collected in Stensholt Vang forest and from pine (Pinus halepensis) used as substrate in the fungal solid-state cultures. The material was carefully recovered from the wood and immediately fixed in 2% glutaraldehyde in 0.05 M NaPO4 buffer, pH 7.5, and stored at 4 °C. Samples were washed in the buffer, dehydrated in an ascending ethanol series, critical point dried in an EMS 850 CP-drier, mounted onto metal stubs with double-sided tape, sputter-coated with gold in an EM ACE200 automated sputter coater and viewed in Quanta 200 SEM (FEI CompanyTM) at 10 kV.
Protein extraction from wood samples
All protein extractions were carried out in parallel with three independent replicates per sample. The wood samples were sliced into smaller pieces (mm size) using a wood driller and then milled into fine powder, using a cryo-grinder Freezer/Mill 6770 (SPEX SamplePrep, Metuchen, USA) in liquid nitrogen with 15 Hz in 3 cycles of 30 s and an interval cooling time of 1 min. The freeze ground wood was stored at −20 °C. Proteins were extracted using a modified TCA/Acetone method, adapted from Nui et al.43. Briefly, 200 mg of wood powder was suspended with 1 mL of sodium dodecyl sulphate (SDS)-PE buffer (1% SDS (w/v), 0.1 M Tris-HCl at pH 8.0, 2 mM dithiothreitol (DTT), 2 mM phenylmethylsulfonylfluorid (PMSF)) and incubated at 600 rpm at room temperature for 1 h. The insoluble part was discarded after centrifugation at 15,000 × g for 5 min at 4 °C and the supernatant was transferred into Eppendorf tubes (2 mL). Twenty percent (v/v) cold TCA/acetone was added at a 1-to-1 (v/v) ratio to a final concentration of 10% TCA and 40% acetone mixed with the supernatant, and incubated for 5 min on ice for precipitation. The protein pellet was centrifuged at 15,000 × g for 3 min at 4 °C, washed three times with 80% ice-cold acetone followed by centrifugation at 15,000 × g for 3 min at 4 °C. The protein precipitate was air-dried for a maximum of 3 min, resuspended in 25 µL of loading SDS buffer (Laemmli buffer, 10% (w/v) SDS, 40% (w/v) Glycerol, 1% (w/v) DTT, 0.1 M Tris-HCl pH 6.8), heated for 10 min at 95 °C, and stored at 4 °C.
Metaproteomics
Extracted proteins (pooled triplicates from the samples collected in nature) were incubated at 99 °C for 5 min before 25 μL was loaded onto a NuPAGE 4–12% Bis-Tris gel (Thermo Fisher Scientific, Massachusetts, USA), and subjected to 5 min SDS-PAGE migration. Proteins were stained for 5 min with Coomassie SimplyBlue SafeStain (Thermo Fisher Scientific) prior to in-gel trypsin proteolysis with Trypsin Gold (Promega, Wisconsin, USA) using 0.011% ProteaseMAX surfactant (Promega, Wisconsin, USA), as described in Hartmann et al.44. Peptides were quantified using the Pierce Quantitative Fluorometric Peptide Assay (Thermo Fisher Scientific), and 350 ng were injected on an Exploris 480 mass spectrometer (Thermo Fisher Scientific) connected with a Vanquish Neo LC system (Thermo Fisher Scientific) operating in data-dependent mode. The peptides were desalted on an Acclaim PepMap100 C18 precolumn (5 μm, 100 Å, 300 μm id × 5 mm), and resolved on a nanoscale EasySpray PepMap Neo C18 column (2 μm, 100 Å, 75 μm id × 50 cm) with a 95 min gradient at a flow rate of 0.25 μL min−1. The gradient was applied from 5 to 25% of solvent B (100% CH3CN, 0.1% formic acid) over 90 min, and then from 25 to 40% over 5 min, with solvent A being 0.1% formic acid. Peptides were analysed initiated by a full scan of peptide ions in the ultra-high-field Orbitrap analyzer, followed by high-energy collisional dissociation and MS/MS scans on the 20 most abundant precursor ions. Full-scan mass spectra were acquired from m/z 350 to 1500 at a resolution of 120,000 with internal calibration activated on the m/z 445.12002 signal. During ion selection for MS/MS fragmentation and measurement, a 10 s dynamic-exclusion window was applied with an intensity threshold of 5 × 104. Only ions with positive charges 2+ and 3+ were considered. Precursor ions were isolated using a 2.0 m/z isolation window and activated with 30% normalized collision energy.
The Mascot Daemon 2.6.1 search engine (Matrix Science) was employed to match MS/MS spectra to peptides (Peptide-spectrum matches - PSM) and identify taxonomies (Taxon-Spectrum Matches—TSM) in a multi-round search process. An initial Mascot search was performed on a reduced NCBInr‐based database (National Center for Biotechnology Information)45. Genera validated in the first‐round search, along with all their descendants, were extracted from the complete NCBInr database (downloaded in January 2021), supplemented with genome sequences of Agaromycetes from the MycoCosm portal (downloaded in March 2023)7, forming the database for the second Mascot search. Genera validated following this search were used to construct a refined sample-specific database for the final MS/MS spectrum searches. Protein accession numbers were mapped to taxids46. PSMs validated with a Mascot p-value of 0.05 were filtered using a false discovery rate (FDR) <1% and subsequently used to infer peptide and protein identifications. Proteins were grouped if they shared at least one peptide. Label-free quantification was performed based on PSM counts for each protein, applying the principle of parsimony. The count values from taxonomic data (number of TSMs) were scaled relative to their total in the sample.
Protein annotations
The proteins identified though proteomics were annotated as follows: functional annotations of Clusters of Orthologous Groups of proteins (COG), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and Enzyme Commission number (EC) were performed using the eggNOG mapper (eggNOG Database v5.0.2, minimum query and subject coverage 80%, minimum identity 50%)47. Functional domains were detected using InterProScan (Database v5.59-91.0 galaxy3)48. Carbohydrate-active enzymes (CAZymes) were identified using the CAZy database (www.cazy.org, last access June 2023) and subsequent manual curation6. Proteases were identified based on the MEROPS database49 (v.12.4) and Lipases based on the Lipase Engineering Database (v.4.1). Secretomes were predicted using SignalP v650 and deeploc v.251.
Non-intrusive O2 concentration measurements
The O2 concentration was measured during the cultivation non-invasively using flexible, self-adhesive dioxygen sensing patches (FOSPOR RedEye, Ocean Insight, US patent 7,862,770 B2) attached to the inside of i) one-side open glass cylinder at 25 mm, 75 mm, and 125 mm from the opening, or ii) Roux flasks. The O2 sensing patch applied the fluorescence quenching of tris-(4,7-diphenyl-1,10-phenanthroline) ruthenium(II) chloride embedded in a sol-gel. An optical probe sends excitation light (blue LED) to the complex resulting in a quenching of the fluorescence signal in the presence of O2. This signal correlates to O2 partial pressure in the gel matrix as a dynamic equilibrium with O2 in the narrow environment of the patch. The optical signal was detected through a bifurcated optical fibre (RE-BIFBORO-2, Ocean Insight) and a benchtop NeoFox phase fluorometer (NeoFox-GT, Ocean Insight), and the phase shift between the excitation and the emission (excited state lifetime) was determined and transformed into the partial O2 pressure at the patch through the Stern-Volmer equation after calibrating the system in a two-point calibration with air and with N2 according to the manufacturer’s manual.
Fungal solid-state cultures on pine
The fungi were transferred from MA2 agar plates (20 g L−1 malt extract, 20 g L−1 agar) to a liquid medium (10 g L−1 glucose, 2 g L−1 Bacto Peptone, 1 g L−1 yeast extract) and cultivated statically for 14 days at 30 °C passively supplied by air. The mycelium was separated from the liquid medium and resuspended in H2O at 8000 rpm for 1 min (Ultra-Turrax T25, Janke & Kunkel IKA®-Labortechnik). Dried Pinus halepensis (<1.5 mm particle size, untreated mix of heartwood and sapwood without the bark) was rehydrated (1 to 1.778, w(dry pine) w(water)−1) and autoclaved at 110 °C for 30 min, or sterilised by γ-irradiation (10 MeV, 25 kGy) and rehydrated. The resuspended mycelium was inoculated in a ratio of 2.222 mg(fungal dry weight) g(autoclaved pine)−1. In initial experiments, 25 mg g(autoclaved pine)−1 glucose and 2.5 mg g(autoclaved pine)−1 ammonium tartrate were added and mixed carefully to homogenize the mycelium in the substrate. After the method was established, cultures were cultivated without glucose or ammonium tartrate, and similar growth and morphology were observed, which proved the growth of the mycelium purely on wood in the absence of O2.
Solid-state cultures in an O2 gradient
A sterile one-side open glass cylinder (150 mm height, 140 mL volume) with O2 sensing patches was filled at a height of 120 mm with the 96 g fungal-inoculated pine and closed with a gas-permeable membrane. In a similar setup, the column was filled with sterile pine and the fungus was inoculated from the top. Both types of cultures were kept statically at 30 °C for at least 14 days. The dioxygen concentration was determined non-invasively in biological duplicates, which were cultivated from two independent inoculates. After 14 days the pine-mycelium conglomerate was recovered from the cylinder, divided into three parts, and stored at −20 °C for protein extraction.
Solid-state cultures in controlled O2 concentration
Roux flasks (1.1 L) were filled with 30–45 g of the pine-mycelium mixture to form a thin layer (<5 mm) to minimise the gas gradient during the solid-state cultures, which were cultivated statically for 14 days at 30 °C with an active supply of air or N2 (Alphagaz 1 Azote, O2 < 2 ppm, Air Liquide, Paris, France). In one experimental setup, each condition – anoxia and normoxia – was tested as a biological duplicate. Three experimental set-ups were executed in this study. The gas flow was regulated to 9 mL min−1 by digital thermal mass flow controllers (EL-FLOW Select, F-200CV/F-210CV, Bronkhorst, Montigny-Les-Cormeilles, France), passed a sterile filter, and was humidified through a water bottle (300 mL sterile H2O in 500 mL vessel) to maintain the water content of the solid-state cultures. The O2 partial pressure was measured non-intrusively via dioxygen sensing patches at the inside of the Roux flasks during the cultivation. The experimental setup is displayed in Supplementary Fig. 5a. At the end of the solid-state cultures (14 days), the fungus-pine mixtures were harvested from the Roux flasks and stored at −20 °C until cryo-grinding, or used for in vitro assessments of the fungus and sample characteristics (biochemical assays).
Mass loss and water content
Mass loss and water content were determined from two biological replicates, that is two Roux flasks cultivated in normoxia and two Roux flasks cultivated in anoxia. Mass loss was calculated from the weight at the beginning minus the weight at the end of the cultivation. The weight was determined (Sartorius Entris 4202-1S) as the entire weight of the Roux flask and the solid-state cultures therein. The water content [%] was determined at the beginning and the end of the cultivation by measuring wet weight and dry weight using the formula: 1 - (dry weight / wet weight) × 100. The water contents were applied to calculate the dry mass loss from the wet masses of the cultures with the following formula: dry weight mass loss (pine and fungi) = wet mass loss (at the end of the cultivation) × (1 − water content [%]/100). The dry mass loss of the wood (= converted substrate) was calculated taking into account the mass gain of the fungal mycelium calculated from qPCR (see below) The mass loss in the manuscript refers to the dry mass loss of the cultures.
Determination of fungal growth by quantitative PCR (qPCR)
Fungal growth was determined in four biological replicates for each culture condition. From each culture, triplicates were randomly taken. Thereof, fungal genomic DNA was extracted three times from 200 mg cryo-grinded material (mycelium, or mycelium-pine mixture) by the commercial DNA extraction kit NucleoSpin Plant II Kit (Macherey-Nagel, Düren, Germany) according to the manufacturer’s instructions, but with an extended extraction step of 60 min and an elution of the gDNA in two steps with a total volume of 50 µL Milli-Q water at 65 °C. The concentration and the purity were analysed via NanoDrop ND-2000 spectrophotometer (Thermo Fisher Scientific) and confirmed by Qubit™ dsDNA Quantification Assay Kit (Thermo Fisher Scientific) as fluorescence endpoint measurement (excitation: 485 nm, emission: 525 nm) in a Greiner 96 Black Flat Bottom Fluotrac well plate. The qPCR was performed in a final reaction volume of 10 µL including 2 µL sample using the qPCR Master Mix for SYBR Plus (Bio-Rad, California, USA) and 5.8S conserved primer sequences (300 nM in reaction, 5′-TTT CAG CAA CGG ATC TCT TGG C-3′, and 5′-CAA ACA GGC ATG CTC CTC GG-3′) in a modified PCR protocol50: 2 min at 95 °C, 39 cycles of: 15 s at 95 °C and 10 s at 58 °C, and a melting step: 30 s at 65 °C and gradually increase (0.5 °C steps) to 95 °C in a C1000 Touch Thermal Cycler CFX96 real-time system (Bio-Rad), or the AriaMx system (Agilent, California, USA), and quantification cycles (Cq) were determined in regression mode of the Bio-Rad CFX Manager™ software (v 3.0), or Agilent Aria Software v2.0, respectively.
For each condition, 36 qPCRs were performed, and thereof 34 or 33 were considered for normoxia, or anoxia, respectively. The determined gDNA amount correlates linearly with the mycelial fresh weight of the fungus52. The dry weight at the beginning and the end of the cultivation was calculated from the fresh weights taking the water content of the culture into account, and mycelium growth was calculated based on the dry weights.
Anaerobic jar cultures
Standard petri dishes (15 mm × 100 mm) were filled with 25 mL of 2.0% (w/v) agar. When hardening was completed, 2.5 g of rehydrated (1 to 1.778, w(dry pine) w(water)−1), autoclaved (110 °C for 30 min) pine chips (<1.5 mm particle size) were distributed on the agar surface and covered by 20 mL 2.0% (w/v) agar. F. pinicola was transferred from MA2 agar plates (20 g L−1 malt extract, 20 g L−1 agar) to the centre of the plate. The plates were cultivated in an anaerobic jar (Anaerocult™, 2.5 L, Merck, Darmstadt, Germany) with a chemically introduced (Anaerocult™ A, Merck) and controlled (Bandelettes Anaerotest™, Merck) O2-depleted environment, or in normoxia, at 30 °C for 22 days.
Bioreactor
F. pinicola was cultivated from 1.36 g(dry weight) resuspended mycelium (see fungal solid-state cultures on pine) in a stirred tank reactor (3.0 L, 120 rpm) in 1.5 L liquid medium of 15 g L−1 autoclaved pine chips, 3 g L−1 glucose, and 1.8 g L−1 diammonium tartrate at 30 °C for 11 days. Air (normoxic phase) supplied the culture at a flow rate of 0.75 L min−1 (0.5 VVM) for the first 66 h, followed by the removal of O2 by N2 for 6.5 h (identical flow rate). When anoxia was reached, the N2 flow rate was reduced to 0.15 L min−1 (0.1 VVM) until the end of the fermentation. In an identical setup, another bioreactor (identical model) was supplied with air at the same flow rates to cultivate the fungus without O2 restriction. Dissolved oxygen (DO), pH, temperature, and stirring rate were controlled and recorded online during the cultivation.
Proteomics
Extracted proteins from 14-day solid-state cultures in Roux flasks (biological replicates) were dissolved in Laemmli buffer, subjected to SDS-PAGE, and in-gel trypsin proteolyzed with Trypsin Gold (Promega). The resulting peptides (100 ng) were injected in an Exploris 480 mass spectrometer (Thermo Fisher Scientific) connected with a Vanquish Neo LC system (Thermo Fisher Scientific) and operated in data-dependent mode53. MS/MS spectra were assigned with Mascot Daemon 2.6.1 taking into consideration Carbamidomethyl (C) as fixed modification, Deamidated (NQ) and Oxydation (M) as variable options. Peptides with p value below 0.05 were selected. Proteins were validated with at least 2 peptides at FDR 1%.
The fold change (see Fig. 4 of the main manuscript) was calculated the following (1):
with \({{SC}}_{{condition\; A},B}\): spectral counts of a protein from the cultures in normoxia or anoxia.
Quantification of hydrogen peroxide
H2O2 was extracted from 14-day solid-state cultures in Roux flasks from biological duplicates of each condition – anoxia and normoxia – with cold water (1-to-1 ratio) shaking at 450 rpm for 30 min at 4 °C. The supernatant was filtered (0.22 µm) and directly applied (1-to-10 ratio, 10 µL) in a 96-well-plate assay (Invitrogen™ A22188) with 100 µL total volume in 50 mM, pH 7.0 sodium phosphate buffer with 200 µM Amplex Red (10-acetyl-3,7-dihydroxyphenoxazine in DMSO), and 0.1 mU mL−1 Horseradish Peroxidase (HRP) at 23 °C. Resorufin fluorescence (excitation: 563 nm, emission: 587 nm), and absorbance at 575 nm, was monitored over 15 min. H2O2 concentration was determined at the beginning of the assay based on a H2O2 standard curve (0–8.34 µM, H2O2 concentration determined at 240 nm) at the same conditions. Experiments (assay) and controls (no HRP, or 0.834 µM H2O2 added) were performed in triplicates.
pH measurements and glycoside hydrolase activity assay
Secreted proteins were extracted from 2.5 g mycelium-pine mix from 14-days solid-state cultures in Roux flasks from biological duplicates of each condition in 10 mL water shaking at 200 rpm for 6 h at 20 °C. The supernatant was filtered (0.45 µm) and pH was measured with a pH meter (Fisherbrand Accumet AE150, Thermo Fisher Scientific) before storing the sample at −20 °C without cryoprotectants addition. The glycoside activity was determined with cyclophellitol-derived activity-based probes54 added in a 1-to-6 ratio directly in the secretomes, or in NH4OAc-buffered (1 M, 0.1 volume, pH 5.5) supernatants. Samples were incubated at 30 °C for 1 h, mixed with SDS-PAGE loading dye, heated to 96 °C for 5 min, and loaded on a 10% SDS-PAGE gel (Bio-Rad, California, USA). Cy3 and Cy5 filter/laser sets were applied to image enzyme-bound endo-β-glucanase probe (CB664), or endo-β-xylanase probe (SY-F230) directly in the SDS-PAGE gel, respectively.
Monosaccharide detection and quantification
Soluble compounds (released soluble residual monomers of the pine substrate) were extracted from 0.45 g mycelium-wood mix from 14-day solid-state cultures in Roux flasks in 45 mL H2O for 6 h at 80 °C. Solid particles were removed from the supernatant through a 0.22 µm filter, and the liquid phase was analysed by high-performance anion-exchange chromatography (HPAEC) with a CarboPac-PA1 guard column (2 × 50 mm) and a CarboPac-PA1 column (2 × 250 mm), and coupled to a pulsed amperometric detector (PAD) (Dionex ICS6000 system, Thermo Fisher Scientific) at 30 °C. 100 mM NaOH (eluent A) and H2O (eluent C) were used as the solvents at with 25 µL sample volume. Monosaccharides were detected and quantified at 0.25 mL min−1 flow rate starting from 10% eluent A with the following gradient: 0–20 min, 10% A; 20–44 min, 10–100% A; 44–49 min, 100% A; 49–59 min, 10% A. D-galacturonic acid was detected and quantified at a flow rate of 0.25 mL min−1 and 25 µL of the sample was injected. 100 mM NaOH (eluent A) and NaOAc (1 M) in 100 mM NaOH (eluent B) were used as solvents. The initial conditions were set to 100% eluent A, and the following gradient was applied: 0–10 min, 0–10% B; 10–35 min, 10–30% B; 35–40 min, 30–100% B; 40–41 min, 100–0% B; 41–50 min, 100% A. Chromeleon 7.2.10 chromatography data software was used for the integration of the chromatograms. D-glucose was confirmed and quantified by using the glucose oxidase-peroxidase (GOD-POD) method (Glucose Assay Kit, Libios, Vindry-sur-Turdine, France) according to the supplier’s protocol in a 200 µL assay with 10 µL sample at 30 °C.
Liquid chromatography mass spectrometry (LC-MS) analysis
The samples of extracted and filtered soluble compounds (described in the previous section) were mixed with cold ethanol (1-to-9 v/v) and centrifuged at 13,000 × g for 15 min at 4 °C to remove the remaining polysaccharides, and concentrated (Savant SpeedVac SPD1030 Integrated Vacuum Concentrator System, Thermo Fisher Scientific) under reduced pressure at 45 °C for 4 h before Liquid Chromatography Mass Spectrometry (LC-MS) analysis. Samples were filtered (0.2 µm, Chromafil®, Macherey-Nagel). D-galacturonic acid and oxalic acid were confirmed by Ultra High Performance Liquid Chromatography (UHPLC, UltiMate™ 3000 Rapid Separation (RS) HPLC Systems, Thermo Fisher Scientific) coupled to a charged aerosol detector (CAD), and an ISQ-EM mass spectrometer (Thermo Fisher Scientific). Separation was performed on a Kinetex F5 column (Phenomenex, 1.7 µm, 150 × 2.1 mm) at a flow rate of 0.32 mL min−1 at 25 °C. The mobile phases used were solvent A: Ammonium formate 12 mM at pH 2.1, and solvent B: Acetonitrile 100%. The gradient was programmed as follows: solvent A started at 100% for 5 min, then solvent B increased to 80% in 2 min, until the end of running (10 min). Negative-ion ESI–MS spectra (15–800 m/z) were acquired by setting the vaporizer temperature at 145 °C, ion transfer tube temperature at 300 °C, sheath gas pressure at 33 psig, auxiliary gas pressure at 3.4 psig, and sweep gas pressure at 0.5 psig. UHPLC-CAD-ESI-MS data were acquired and analysed with Chromeleon v7.2.10 (Thermo Fisher Scientific), and the quantification was performed by external standard calibrations. The calibration and controls (samples spiked with oxalic acid) were performed with pure oxalic acid (Sigma Aldrich).
Preparation of alcohol Insoluble residue and hemicellulose extraction
Samples were submerged in 96% (v/v) ethanol and boiled at 70 °C for 30 min before homogenization using a ball mixer mill (Glen Creston, now Retsch). The pellet was collected by centrifugation (4000 × g for 15 min) and was washed with 100% (v/v) ethanol, twice with chloroform:methanol (2:1), followed by successive washes with 65% (v/v), 80% (v/v) and 100% (v/v) ethanol. The remaining pellet of AIR was air dried and hemicelluloses were extracted with 4 M NaOH55.
Enzymatic hydrolysis and polysaccharide analysis by carbohydrate gel electrophoresis (PACE)
Enzymes used in this study were GH11 endo-β-1,4-xylanase from Neocallimastix patriciarum56 NpGH11; endo-glucuronoxylanase EcGH30 from Erwinia chrysanthemi57; endo-mannanase AnGH5 from Aspergillus nidulans58; xyloglucanase AaGH12 from Aspergillus aculeatus59. NpGH11 and EcGH30 hydrolyses were carried out at 30 °C, while AnGH5 and AaGH12 were carried out at 37 °C. All hydrolyses were carried out under constant shaking for 24 h. Five hundred micrograms of extracted hemicellulose were used for hydrolysis in 50 mM ammonium acetate buffer pH 6.0. Following hydrolysis, enzymes were heat-deactivated for 10 min and were then taken to dryness under vacuo. Derivatisation of oligosaccharides was performed as described in ref. 55. For carbohydrate electrophoresis, samples were loaded on polyacrylamide gels and electrophoresed at 10 °C at 1000 V for 1 h with 0.1 M TRIS-borate (pH 8.2) solution used as the running buffer. PACE gel scanning was performed using a GBox CCD camera with a transilluminator with long-wave tubes emitting at 365 nm. Images were captured using GeneSnap software.
Solid-state NMR
Pine (Douglas pine, Pseudotsuga menziesii) was labeled with 13C by the labeling facility of IsoLife (Wageningen, The Netherlands), sliced in wood chips of approx. 2 to 5 mm and sterilized by γ-irradiation (10 MeV, 25 kGy). After rehydration of the 13C-enriched pine, fungal mycelium was inoculated (1:30) without adding any additional source of nutrition. The solid-state culture remained for 28 days at 25 °C under anoxic conditions (pure N2) as described above. After freezing in liquid nitrogen, the culture was stored at -80 °C until use.
Solid-state NMR experiments of 13C-enriched pine and of the F. pinicola inoculated pine were acquired on a Bruker 1 GHz AVANCE NEO solid-state NMR spectrometer operating at 1H and 13C Larmor frequencies of 1000.4 and 251.6 MHz, respectively, using a 3.2 mm EFree triple resonance MAS probe. All experiments were conducted at an indicated temperature of 10 °C and an MAS frequency of 14.25 kHz with a recycle delay of 2 s. The 13C chemical shifts were determined using the carbonyl peak at 177.8 ppm of L-alanine as an external reference with respect to tetramethylsilane. The 1H 90° pulse length was 3.25 µs and the 13C 90° was 3.9 µs for pine and 4.3 µs for the pine inoculated by F. pinicola. Cross polarization from 1H to 13C was achieved using ramped (70–100%) 1H radiofrequency amplitude and a contact time of 1 ms60. SPINAL-64 decoupling was applied at a 1H nutation frequency of 70–80 kHz during acquisition60. Sign discrimination in the indirect dimension of the 2D experiments was achieved using the States-TPPI method. Assignments were determined using 2D 13C-13C proton-driven spin diffusion (PDSD) experiment with a mixing time of 30 ms61. The acquisition time in the indirect dimension (t1) of the CP PDSD experiments was 6.4 ms. The spectral width in the indirect dimension was 42.75 kHz with 64 acquisitions per t1 FID. The 2D spectra were processed with Fourier transformation into 8 K (F2) × 2 K (F1) points with exponential line broadening of 20-50 Hz in F2 and cubed sine bell processing in F1 using Bruker Topspin v.3.6. Contour levels are x 1.1 unless otherwise stated. The minimum contour is chosen to show the desired features.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data generated in this study are provided in the Supplementary Information, Supplementary Dataset and Source Data file. Unprocessed NMR data files are available from https://wrap.warwick.ac.uk/id/eprint/191899/. Metaproteomics and proteomics data are available on PRIDE under PXD048319 and PXD048362 accessions, respectively. The accession code for the F. pinicola rDNA sequence is Genbank ID PP277039. Source data are provided with this paper.
References
Bonan, G. B. Forests and climate change: forcings, feedbacks, and the climate benefits of forests. Science 320, 1444–1449 (2008).
Harris, N. L. et al. Global maps of twenty-first century forest carbon fluxes. Nat. Clim. Chang. 11, 234–240 (2021).
Baldrian, P., López-Mondéjar, R. & Kohout, P. Forest microbiome and global change. Nat. Rev. Microbiol. 21, 487–501 (2023).
Floudas, D. et al. The paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 336, 1715–1719 (2012).
Riley, R. et al. Extensive sampling of basidiomycete genomes demonstrates inadequacy of the white-rot/brown-rot paradigm for wood decay fungi. Proc. Natl Acad. Sci. USA 111, 9923–9928 (2014).
Drula, E. et al. The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res. 50, D571–D577 (2022).
Grigoriev, I. V. et al. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 42, D699–D704 (2014).
Hasegawa, N., Sugiyama, M. & Igarashi, K. Random forest machine-learning algorithm classifies white- and brown-rot fungi according to the number of the genes encoding Carbohydrate-Active enZyme families. Appl. Environ. Microbiol. 90, e00482–24 (2024).
Hage, H. & Rosso, M.-N. Evolution of fungal carbohydrate-active enzyme portfolios and adaptation to plant cell-wall polymers. J. Fungi 7, 185 (2021).
Zhang, J. et al. Localizing gene regulation reveals a staggered wood decay mechanism for the brown rot fungus Postia placenta. Proc. Natl. Acad. Sci. USA 113, 10968–10973 (2016).
Anderson, C. E. et al. Capturing an early gene induction event during wood decay by the brown rot fungus Rhodonia placenta. Appl. Environ. Microbiol. 88, e00188–22 (2022).
Eastwood, D. C. et al. The plant cell wall-decomposing machinery underlies the functional diversity of forest fungi. Science 333, 762–765 (2011).
Vanden Wymelenberg, A. et al. Comparative transcriptome and secretome analysis of wood decay Fungi Postia placenta and Phanerochaete chrysosporium. Appl. Environ. Microbiol. 76, 3599–3610 (2010).
Mattila, H. K., Österman-Udd, J., Mali, T. & Lundell, T. Basidiomycota Fungi and ROS: genomic perspective on key enzymes involved in generation and mitigation of reactive oxygen species. Front. Fungal Biol. 3, 837605 (2022).
Martinez, D. et al. Genome, transcriptome, and secretome analysis of wood decay fungus Postia placenta supports unique mechanisms of lignocellulose conversion. Proc. Natl Acad. Sci. USA 106, 1954–1959 (2009).
Kohler, A. et al. Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat Genet 47, 410–415 (2015).
Fukasawa, Y. & Matsukura, K. Decay stages of wood and associated fungal communities characterise diversity–decomposition relationships. Sci. Rep. 11, 8972 (2021).
Thacker, D. G. & Good, H. M. The composition of air in trunk of sugar maple in relation to decay. Can. J. Bot. 30, 475–485 (1952).
Jensen, K. F. Oxygen and carbon dioxide concentrations in sound and decaying red oak trees. Forest Sci 15, 246–251 (1960).
Scheffer, T. C. O2 requirements for growth and survival of wood-decaying and sapwood-staining fungi. Can. J. Bot. 64, 1957–1963 (1986).
Mattila, H. K., Mäkinen, M. & Lundell, T. Hypoxia is regulating enzymatic wood decomposition and intracellular carbohydrate metabolism in filamentous white rot fungus. Biotechnol. Biofuels 13, 26 (2020).
Mori, T., Masuda, A., Kawagishi, H. & Hirai, H. Ethanol fermentation by saprotrophic white-rot fungus Phanerochaete sordida YK-624 during wood decay as a system for short-term resistance to hypoxic conditions. J. Biosci. Bioeng. 133, 64–69 (2022).
Goubet, F., Dupree, P. & Johansen, K. S. Carbohydrate Gel Electrophoresis. (ed Popper, Z.) The Plant Cell Wall. Methods Mol. 715, 81–92, (Humana Press, 2011).
Presley, G. N., Zhang, J. & Schilling, J. S. A genomics-informed study of oxalate and cellulase regulation by brown rot wood-degrading fungi. Fungal Genet. Biol. 112, 64–70 (2018).
Terrett, O. M. et al. Molecular architecture of softwood revealed by solid-state NMR. Nat. Commun. 10, 4978 (2019).
Busse-Wicher, M. et al. Evolution of xylan substitution patterns in gymnosperms and angiosperms: implications for xylan interaction with cellulose. Plant Physiol 171, 4418–2431 (2016).
Klukowski, P. & Schubert, M. Chemical shift-based identification of monosaccharide spin-systems with NMR spectroscopy to complement untargeted glycomics. Bioinformatics 35, 293–300 (2019).
Simmons, T. et al. Folding of xylan onto cellulose fibrils in plant cell walls revealed by solid-state NMR. Nat. Commun. 7, 13902 (2016).
Bourdon, M. et al. Ectopic callose deposition into woody biomass modulates the nano-architecture of macrofibrils. Nat. Plants 9, 1530–1546 (2023).
Jang, M.-K. et al. Physicochemical characterization of α-chitin, β-chitin, and γ-chitin separated from natural resources. J. Polym. Sci. A Polym. Chem. 42, 3423–3432 (2004).
Urzúa, U., Kersten, P. J. & Vicuña, R. Manganese Peroxidase-Dependent Oxidation of Glyoxylic and Oxalic Acids Synthesized by Ceriporiopsis subvermispora Produces Extracellular Hydrogen Peroxide. Appl. Environ. Microbiol. 64, 68–73 (1998).
Berna, A. & Bernier, F. Regulation by biotic and abiotic stress of a wheat germin gene encoding oxalate oxidase, a H2O2-producing enzyme. Plant Mol. Biol. 39, 539–549 (1999).
Couturier, M. et al. Enhanced degradation of softwood versus hardwood by the white-rot fungus Pycnoporus coccineus. Biotechnol. Biofuels 8, 216 (2015).
Runnel, K. & Lõhmus, A. Deadwood-rich managed forests provide insights into the old-forest association of wood-inhabiting fungi. Fungal Ecol 27, 155–167 (2017).
Temnuhin, V. B. Preliminary quantitative estimation of wood decomposition by fungi in a Russian temperate pine forest. For. Ecol. Manag. 81, 249–257 (1996).
Wagener, W. W. & Davidson, R. W. Heart rots in living trees. Bot. Rev 20, 61–134 (1954).
Hennon, P. E. Are heart rot fungi major factors of disturbance in gap-dynamic forests? Northwest Sci 69, 284–293 (1995).
Glaeser, J. A. & Smith, K. T. Wood decay fungi of subalpine conifer forests. In Proc Presented at the 8th Western Hazard Tree Workshop https://www.fs.usda.gov/nrs/pubs/jrnl/2016/nrs_2016_glaeser_001.pdf (2016).
Yuan, Y., Chen, J.-J., Korhonen, K., Martin, F. & Dai, Y.-C. An updated global species diversity and phylogeny in the forest pathogenic genus Heterobasidion (Basidiomycota, Russulales). Front. Microbiol. 11, 596393 (2021).
Garbelotto, M. & Gonthier, P. Biology, epidemiology, and control of heterobasidion species worldwide. Annu. Rev. Phytopathol. 51, 39–59 (2013).
Nord-Larsen, T. & Pretzsch, H. Biomass production dynamics for common forest tree species in Denmark – Evaluation of a common garden experiment after 50 yrs of measurements. For. Ecol. Manag. 400, 645–654 (2017).
Navarro, D. et al. A. large-scale phenotyping of 1,000 fungal strains for the degradation of non-natural, industrial compounds. Commun. Biol. 4, 871 (2021).
Niu, L. et al. Modified TCA/acetone precipitation of plant proteins for proteomic analysis. PLoS ONE 13, e0202238 (2018).
Hartmann, E. M., Allain, F., Gaillard, J.-C., Pible, O. & Armengaud, J. Taking the shortcut for high-throughput shotgun proteomic analysis of bacteria. (eds. Vergunst, A. & O’Callaghan, D.) Host-Bacteria Interactions. Methods Mol. Vol. 1197, 275–285, (Humana Press, 2014).
Ramos-Nascimento, A. et al. Human gut microbiome and metabolite dynamics under simulated microgravity. Gut Microbes 15, 2259033 (2023).
Pible, O. et al. Estimating relative biomasses of organisms in microbiota using “phylopeptidomics. Microbiome 8, 30 (2020).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122 (2017).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Rawlings, N. D. et al. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 46, D624–D632 (2018).
Teufel, F. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 40, 1023–1025 (2022).
Thumuluri, V., Almagro Armenteros, J. J., Johansen, A. R., Nielsen, H. & Winther, O. DeepLoc 2.0: multi-label subcellular localization prediction using protein language models. Nucleic Acids Res. 50, W228–W234 (2022).
Zhou, S., Grisel, S., Herpoël-Gimbert, I. & Rosso, M.-N. A PCR-based method to quantify fungal growth during pretreatment of lignocellulosic biomass. J. Microbiol. Methods 115, 67–70 (2015).
Klein, G. et al. RNA-binding proteins are a major target of silica nanoparticles in cell extracts. Nanotoxicology 10, 1555–1564 (2016).
McGregor, N. G. S. et al. Activity-based protein profiling reveals dynamic substrate-specific cellulase secretion by saprotrophic basidiomycetes. Biotechnol. Biofuels 15, 6 (2022).
Tryfona, T. et al. Grass xylan structural variation suggests functional specialization and distinctive interaction with cellulose and lignin. The Plant Journal 113, 1004–1020 (2023).
Gilbert, H. J., Hazlewood, G. P., Laurie, J. I., Orpin, C. G. & Xue, G. P. Homologous catalytic domains in a rumen fungal xylanase: evidence for gene duplication and prokaryotic origin. Mol. Microbiol. 6, 2065–2072 (1992).
Urbániková, Ľ., Vršanská, M., Krogh, K. B. R., Hoff, T. & Biely, P. Structural basis for substrate recognition by Erwinia chrysanthemi GH30 glucuronoxylanase. FEBS J. 278, 2105–2116 (2011).
von Freiesleben, P. et al. An Aspergillus nidulans GH26 endo-β-mannanase with a novel degradation pattern on highly substituted galactomannans. Enzyme Microb Technol. 83, 68–77 (2016).
Pauly, M. et al. A xyloglucan-specific endo-β-1,4-glucanase from Aspergillus aculeatus: expression cloning in yeast, purification and characterization of the recombinant enzyme. Glycobiology 9, 93–100 (1999).
Fung, B. M., Khitrin, A. K. & Ermolaev, K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 142, 97–101 (2000).
Takegoshi, K., Nakamura, S. & Terao, T. 13C–1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 344, 631–637 (2001).
Acknowledgements
The authors would like to thank the French Institute of Bioinformatics (IFB) for providing computational facilities through their Galaxy interface and the CAZy team for updating the CAZy database. Mireille Haon and Jonas Thomsen are acknowledged for their participation to the collection of samples, Gideon Davies, Hermen Overkleft and Zirui Li for providing chemical probes, and Bastien Bissaro for his help with H2O2 quantification. K.J., P.D. and J.G.B. received funding from the Novo Nordisk Foundation grant NNF20OC0059697 - OxyMiST project. The UK High-Field Solid-State NMR Facility used in this research was funded by EPSRC and BBSRC (EP/T015063/1), as well as, for the 1 GHz instrument, EP/R029946/1. J.G.B. received funding from the European Union’s Horizon 2020 research and innovation programme under Grant Agreement No 101008500.
Author information
Authors and Affiliations
Contributions
R.R., M.N.R., L.T., P.D., B.H., K.J., J.A. and J.G.B. conceived the work. J.G.B coordinated the study. R.R., L.G., R.C., S.L., I.G., T.T., L.T., K.J., R.D. and J.G.B. designed the experiments. R.R., A.L., L.G., R.C., S.L., D.N., J.L., S.G., I.G., H.J.M., G.M., T.T., X.Y. and E.D. performed the experiments. R.R., A.L., L.G., R.C., S.L., I.G., H.J.M., T.T., M.N.R., L.T., P.D., B.H., K.J., R.D., J.A. and J.G.B. analysed the data. R.R., A.L. and J.G.B. wrote the original draft. All authors have approved the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Gry Alfredsen and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Röllig, R., Lebreton, A., Grenga, L. et al. Wood decay under anoxia by the brown-rot fungus Fomitopsis pinicola. Nat Commun 16, 7352 (2025). https://doi.org/10.1038/s41467-025-62567-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62567-3
This article is cited by
-
Arctic driftwood proposal for durable carbon removal
npj Climate Action (2026)
-
Uncovering enzymatic tools promoting lignocellulose breakdown in the anaerobic bacterium Ruminiclostridium cellulolyticum
Communications Biology (2025)
