
Abstract
Direct conversion of CO2 into valuable organic products is probably the most important but challenging issue for global sustainability efforts. Metal carbides are promising as vital catalytic components in achieving this goal. Understanding the evolution of chemical orbitals and the corresponding energy levels on their interfaces are essential for targeted product synthesis. In this study, we discover that a highly active FeCo alloy carbide has a distinctive oxygen-bonding ability to regulate the evolution of oxygen-containing reaction intermediates. Combining with the copper/zinc/aluminum catalytic component, the designed tandem catalyst allows for the extremely high C2+ alcohols selectivity (49.1 percent) and space-time yield (245.7 milligram per gram catalyst per hour) at a CO2 conversion of 51.1 percent. The excellent catalyst stability (>1000 hours) and potential economic viability make this process promising in eliminating carbon emissions at industrial application scale.
Similar content being viewed by others
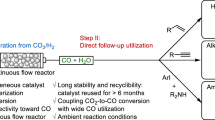


Introduction
C2+ alcohols (C2+OH), widely used as green fuel additives and organic solvents in industry, are primarily produced through sugar fermentation or fossil fuel refining. Given the depletion of fossil fuel reserves and the increasing concerns over global food shortages, it is crucial to develop alternative methods for C2+OH synthesis1. CO2, an abundant and sustainable carbon source, presents a promising feedstock for producing value-added chemicals2,3,4,5. In particular, thermal-catalytic hydrogenation of CO2 coupled with sustainable water photo/electrolysis H2 is attracting more and more attention due to its integrated advantages of industrial applicability, greenhouse effect elimination, and non-fossil fuel-based product synthesis6,7,8,9,10,11. However, discovering efficient heterogeneous catalysts that can effectively convert CO2 and H2 into C2+OH remains a significant challenge.
Metal carbides, particularly iron carbides of χ-Fe5C2, ε-Fe2C, and θ-Fe3C, have been recognized as a kind of active compounds for Fischer–Tropsch synthesis (FTS) to produce various chemicals12,13,14,15,16,17,18,19,20,21, including the promising alcohols (C2+OH)22,23,24. Nevertheless, the reported carbides are difficult to apply to the CO2 hydrogenation to produce C2+OH due to the distinct water-rich atmosphere relative to the conventional FTS and the stronger oxidation capability of CO2 compared with that of CO. Catalysts with strong hydrogenation capacity are highly needed to activate the inert gas molecule CO2. But unfortunately, this requirement makes the catalysts unsuitable for binding the oxygen (O)-containing species, resulting in inferior performance for C2+OH synthesis from CO2 hydrogenation6,25,26,27. Therefore, even though great progress has been made in FTS, no reported catalysts show a clear advantage for the CO2 transformation into C2+OH at a practical level.
To enhance the C2+OH synthesis performance, appropriate O-bonding ability, that is neither very weak to no activate the C–O bond of CO2 nor so strong as to oxidize the catalytic interface and break the C–O bond in C2+OH intermediates, is required for the Fe-based carbide. Modulation of the oxygen affinity of the catalyst through a rational design is thus necessary to realize the efficient C2+OH synthesis of CO2 hydrogenation. The alloy carbide is the optional measure to regulate the electronic structure of Fe. Cobalt (Co) is well-known for its excellent catalytic activity in CO/CO2 hydrogenation reactions22,24,28,29. Unlike iron (Fe), which tends to partially convert to Fe3O4 during CO2 hydrogenation, cobalt and cobalt carbide (e.g., Co2C) remain predominantly, having a lower oxygen affinity for Co compared to Fe18,30,31,32,33,34. This makes cobalt and cobalt carbide active for non-dissociative CO adsorption and subsequent insertion processes in the Co/Co2C catalysts25,35,36. Lee et al. reported that a FeCo alloy carbide phase could be formed from a FeCo alloy during the CO2 hydrogenation reaction at 340 °C37. Ma et al. developed CoFe bimetallic carbide catalysts derived from Co/α-Fe2O3 nanorods through interdiffusion of Co and Fe species during the reduction process, where Co atoms replace Fe atoms to form FeCo alloy carbides under reaction conditions22. Furthermore, they synthesized a monodisperse ε’-(CoxFe1−x)2.2C alloy carbide catalyst, which is obtained from CoxFe3−xO4 spinel oxide nanoparticles as precursors38. Zhong et al. demonstrated that Co atoms can be uniformly distributed and doped into Fe2C, leading to the formation of (FexCoy)2C alloy carbide24. Guo et al. further reported that Co atoms readily incorporate into the iron carbide when the precursors contain Co and Fe species in close proximity. By tuning the carburization time and temperature, specific CoFe alloy carbides with controlled composition and structure were successfully obtained39. In addition, both the elements of Co and Fe belong to the family of transition metals, with adjacent atomic numbers and similar chemical properties. Co (3d74s2) has one more 3d electron than Fe (3d64s2) allows for electronic structure modulation. Therefore, the presence of Co offers a unique advantage by promoting the formation of Fe-based alloy carbides, thereby enhancing its catalytic performance in CO2 hydrogenation to C2+OH products.
We propose, by controlling the component and composition of bimetal FeX catalysts (X = Cu, Co, Mn, and Zn) in the in-situ fabrication conditions (Supplementary Table 1), a stable FeCo alloy carbide can be developed to achieve appropriate O-bonding ability. Through the introduction of CuZnAl (CZA) catalytic component to supply abundant O-containing intermediates6,23,25,26,27, the multifunctional tandem catalyst (FeCo alloy carbide/CZA) can realize the efficient synthesis of C2+OH with recorded space–time yield up to 245.7 mg gcat−1 h−1. Other bimetal FeX catalysts are not satisfactory. The phase evolution of bimetal FeX catalysts during reduction and reaction processes was monitored by in situ XRD. The phases of Cu, MnCO3, ZnO were detected on spent bimetal FeX catalysts, indicating the alloy carbide formed difficultly by these metals (Supplementary Figs. 1 and 2a). As illustrated in Fe2p XPS spectra, the binding energy of the Fe–C bond in FeCo alloy carbide was higher than that of other FeX catalysts, suggesting the bonding ability of Fe and C was stronger than those of other FeX catalysts (Supplementary Fig. 2b). The formation of FeCo alloy carbides led to lattice stress effect, which affected the electronic structure of Fe atoms. Therefore, the elements Fe and Co readily form alloy carbide, which can be attributed to their adjacent atomic numbers and similar chemical properties. Further evidence and analysis will be provided in the following discussion. Unlike previous studies on FeCo alloy carbides28,39, to the best of our knowledge, this is the first report of Co-stabilized alloy carbides that enable efficient CO2 hydrogenation to C2+OH.
Here, we show the exceptional long-term stability of the designed Fe-based tandem catalyst over 1000 h from CO₂ hydrogenation to C2+OH, as well as its distinct oxygen-binding capability. More importantly, we elucidate the underlying mechanism of CO2 conversion to C2+OH products in a clear and intuitive manner.
Results
Catalytic performance from CO2 hydrogenation to C2+OH
The bimetallic FeCo catalyst (molar ratio of Fe/Co = 3) was prepared by the co-precipitation method. Then we physically mixed it with the CZA catalytic component in powder form to catalyze the CO2 hydrogenation reaction under the given reaction conditions (320 °C, 5.0 MPa, H2/CO2 = 3, 4500 ml gcat−1 h−1). Surprisingly, the multifunctional catalyst (FeCo&CZA) delivered extremely high C2+OH selectivity of 49.1% at a CO2 conversion of 51.1%, which is far superior to the counterpart Fe&CZA without Co introduction (CO2 conversion and C2+OH selectivity of 44.9% and 21.2%, respectively; Fig. 1a and Supplementary Table 2). Moreover, the C2+OH space–time yield (STY, 245.7 mg gcat−1 h−1; Fig. 1b and Supplementary Table 2) obtained from FeCo&CZA outperformed most of the Fe-based catalysts reported so far (Fig. 1c and Supplementary Table 3)26,27,40,41,42,43,44,45,46,47,48,49,50. Regarding to the crucial industrial application index of catalyst stability, the CO2 conversion from FeCo&CZA catalyst was well maintained at ca. 56.0% even after long-term stability test of 120 h, and C2+OH selectivity was more than 40% (Fig. 1d). The C2+OH performance at 1000 h fluctuated in the range of 36–45% with the yield of C2+OH above 17%. That was even superior to almost all the reported catalysts that usually collected data within 8–48 h, confirming its good durability and application potential properties (Supplementary Fig. 3).

a CO2 conversion and product selectivity of four catalysts. b Alcohol distribution and STY of C2+OH from four catalysts. c Comparison of CO2 conversion, CO selectivity, and STY of C2+OH from FeCo&CZA with the results reported in other literature. d Stability of FeCo&CZA catalyst in the CO2 hydrogenation to C2+OH. Reaction conditions: 320 °C, 5.0 MPa, H2/CO2 = 3, 4500 ml gcat−1 h−1.
We then conducted a series of control experiments to further clarify the difference between the FeCo&CZA and the other catalysts. The sole Fe and FeCo catalysts exhibited inferior C2+OH selectivity (12.5% and 12.0%, respectively, Fig. 1a and Supplementary Table 2), and no obvious C2+OH products were found on the Co, CZA and Co&CZA counterparts (Supplementary Table 4). We also tested the FeCo&CZA catalyst for the FTS process using CO + H2 and found it is not ideal (Supplementary Table 4). These findings suggest that both FeCo carbide and CZA compounds play essential roles in C2+OH production, and that CO2 hydrogenation and the FTS could follow different reaction mechanisms.
It is generally believed that the introduction of CZA helps to generate abundant O-containing intermediates (e.g., CHxO*, x = 0, 1, and 2) on the catalytic interface, thereby boosting the C2+OH synthesis through C–C couplings between CHx* and CHxO*26. Compared to the Fe&CZA catalyst, we observed a significant performance enhancement by adjusting the FeCo compound to CZA catalyst ratio from 16:1 to 2:1, while the Fe&CZA catalyst itself remained insensitive to such changes. (Supplementary Fig. 4 and Supplementary Table 4). The significant increase in C2+OH selectivity, from 21.2% with Fe&CZA to 49.1% with FeCo&CZA, confirms that the introduction of Co governed the C2+OH synthesis performance.
Notably, the proportion of Co in the FeCo component is decisive to the C2+OH synthesis performance of the multifunctional FeCo&CZA catalyst. We varied the Fe/Co molar ratio from 1 to 5, including FeCo(1:1), FeCo(3:1), and FeCo(5:1), but only FeCo(3:1)&CZA displayed excellent C2+OH selectivity of >40.0% (Supplementary Table 2), indicating that the FeCo(3:1) has unique active sites to modulate the evolution of reaction intermediates. Besides the Fe/Co ratio, the mixing manner of FeCo and CZA components influenced the catalytic performance significantly. As shown in Supplementary Fig. 5, the particle mixing and dual-bed modes with a prolonged distance between FeCo and CZA were detrimental to C2+OH synthesis. Both CO2 conversion and C2+OH selectivity were suppressed to below 44.0% and 27.5%, respectively. As the distance between the components decreased, CO selectivity dropped in the FeCo&CZA catalyst, suggesting that closer proximity facilitated the transformation and consumption of the key intermediate CO*, thereby accelerating C2+OH synthesis through C–C coupling steps. In addition, we investigated the influence of reaction conditions, such as temperature and pressure, on the catalytic performance of FeCo&CZA (Supplementary Table 5). The CO2 conversion increased with the increase of temperature from 300 to 340 °C, and C2+OH selectivity increased first and then decreased. As the pressure decreased from 5 to 3 MPa, both CO2 conversion and C2+OH selectivity were suppressed, suggesting that high pressure promotes the generation of C2+OH.
Catalysts characterization and structural evolution during the reaction
Confirmation of FeCo alloy carbide
To determine the active phases for CO2 activation and C2+OH synthesis, the structure and composition of the optimal FeCo&CZA catalyst (Fe/Co molar ratio of 3) before and after the reaction were investigated in detail. FeCo alloy oxide (Supplementary Figs. 6 and 7) was confirmed in the fresh FeCo catalyst by X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and high-resolution transmission electron microscopy (HRTEM). After the reaction, carbide phases were formed in the spent FeCo&CZA catalyst (FeCo&CZA-spent) due to the carburization effect, as confirmed by the ex-situ XRD patterns (Fig. 2a), XPS, TEM, and 57Fe Mössbauer spectra (Supplementary Figs. 8, 9 and Supplementary Table 6). In particular, compared to the spent Fe&CZA (Fe&CZA-spent) catalyst, where χ-Fe5C2 is the dominant carbide phase, the FeCo&CZA-spent catalyst retained prominent diffraction peaks corresponding to the carbide phase. Notably, these peaks shifted to higher 2θ angles without the appearance of any new diffraction peaks (Fig. 2a), indicating a lattice contraction effect, likely caused by Co incorporation into the Fe carbide structure28,37,51.

a XRD patterns of the Fe&CZA-spent and FeCo&CZA-spent catalysts. b Fourier transforms (FT) of the k3-weighted EXAFS spectra of the Fe&CZA-spent and FeCo&CZA-spent catalysts at the Fe K-edge. c AC-HAADF-STEM and EDX-mapping images of FeCo&CZA-spent catalyst. d Schematic illustration of the phase evolution of the FeCo component in FeCo&CZA catalyst during reduction and reaction processes. e TPO-MS profiles of the FeCo alloy carbide and monometallic Fe carbide with ca. 10% water/He feeding. f Surface configurations of bonded O atoms on Fe5C2(510) and (Fe3/4Co1/4)5C2(510) models and the corresponding binding energies under high O coverage. g Crystal orbital Hamilton populations (COHP) analysis of O* adsorption on Fe5C2(510) and (Fe3/4Co1/4)5C2(510) surfaces.
Additionally, the carbide diffraction peaks shifted to even higher 2θ angles as the Fe/Co molar ratio decreased (Supplementary Fig. 10a) in FeCo&CZA-spent catalysts51,52, further confirming the enhanced lattice contraction effect. The sole FoCo-spent catalysts with varying Fe/Co ratios were investigated for the process of alloy carbide formation (Supplementary Fig. 10b). For the Fe/Co ratios of 5:1 and 3:1, only the FeCo alloy carbide phase was detected, consistent with the observations for the FeCo&CZA-spent catalysts. However, when the Co content was increased to 50% (Fe/Co = 1:1), both the alloy carbide and FeCo alloy phases were observed in the FeCo(1:1)-spent catalyst. This indicates that an excessive amount of Co is unfavorable for the carburization process. In contrast, the FeCo(1:1)&CZA-spent catalyst exhibited complete carburization of the metallic phase. This suggests that the presence of CZA could significantly promote the carburization of the FeCo alloy26, likely due to the generation of carbon-containing species such as CO* via the reverse water-gas shift (RWGS) reaction (CO2 + H2 → CO + H2O) on the CZA component, which can be employed as a carburization species.
In addition, the H2-temperature programmed reduction (TPR), CO-temperature-programmed desorption (TPD) (Supplementary Fig. 11) and N2 adsorption–desorption isotherm (Supplementary Fig. 12, Supplementary Table 7) further verified the structural evolution of the catalysts. In H2-TPR, the Fe catalyst underwent stepwise reduction from the initial α-Fe2O3 through Fe3O4, FeO, and finally to metallic Fe. In contrast, the FeCo catalyst exhibited a distinct reduction peak corresponding to the transformation of the FeCo alloy oxide phase into the FeCo alloy. Notably, the reduction peaks shifted toward lower temperatures upon Co addition28, suggesting that the Co facilitates the reduction of the FeCo catalysts. The carburization behaviors of catalyst were monitored by CO-TPD, the desorption peaks obviously shifted toward the higher temperature22, indicating that the carburization of FeCo metallic phase is difficult than that of Fe catalyst, corresponding to the results of XRD (Supplementary Fig. 10b). Nitrogen physisorption analysis revealed that the FeCo catalyst possessed a larger specific surface area and a predominantly mesoporous structure compared to the Fe catalyst. After the reaction, the surface area of FeCo-spent and Fe-spent became comparable. Taken together, it is reasonable to speculate the formation of the FeCo alloy carbide phase.
To verify the above speculation, we next used the (Fe3/4Co1/4)5C2 with an Fe/Co molar ratio of 3 as a model system and performed density functional theory (DFT) computations to identify the most thermodynamically stable configuration from the broad compositional space of FeCo alloy carbide (Supplementary Fig. 13). Radical distribution function (RDF) analysis confirmed that the lattice contraction observed in the FeCo alloy carbide originated from a shortened Fe–C bond length upon Co incorporation. The simulated XRD pattern of (Fe3/4Co1/4)5C2 showed diffraction peaks shifted to higher angles compared to Fe5C2 (Supplementary Fig. 14), consistent with the experimental XRD results of Fe&CZA-spent and FeCo&CZA-spent catalysts (Fig. 2a). Further extended X-ray absorption fine structure (EXAFS) analysis provided coordination information for Fe and Co species. The Fe–C bond length in FeCo&CZA-spent (1.98 Å) was shorter than that in Fe&CZA-spent (2.08 Å) (Fig. 2b; Supplementary Fig. 15; Supplementary Tables 8 and 9), in good agreement with the RDF results. Additionally, aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) and the corresponding energy-dispersive X-ray (EDX) mapping revealed a uniform distribution of Fe, Co, and C elements in the spent catalyst (Fig. 2c).
The phase evolution of the FeCo&CZA catalyst was further investigated using in situ X-ray diffraction (XRD) (Supplementary Fig. 16). During the H₂ reduction process, the FeCo alloy oxide was first converted into a metallic FeCo alloy. Subsequently, FeCo alloy carbide formed during the CO₂ hydrogenation step, driven by the carburization of the FeCo alloy through CO generated via the reverse water-gas shift (RWGS) reaction (Fig. 2d). Therefore, based on the combined results from multiple characterization techniques and DFT simulations, it is reasonable to conclude that the FeCo&CZA-spent catalyst contains a FeCo alloy carbide phase, rather than separate Fe- or Co-based carbides.
Exploration of O-bonding ability
An intriguing observation is that while the Fe&CZA-spent catalyst exhibits co-existing oxide (primarily Fe3O4) and carbide phases (mainly χ-Fe5C2 and θ-Fe3C), the FeCo&CZA-spent catalyst does not show any oxide phases, as confirmed by the 57Fe Mössbauer, XRD, and EXAFS techniques (Supplementary Figs. 9, 10, and 15). The absence of oxides in the FeCo&CZA-spent catalyst implies that the FeCo carbide could have a different oxygen affinity compared to the Fe carbide, which influences its response to water produced mainly from the RWGS reaction31. This hypothesis was then supported by temperature-programmed oxidation-mass spectrometry (TPO-MS, Fig. 2e) and in-situ water oxidation XRD results (Supplementary Fig. 17), where FeCo alloy carbide showed robust stability than the monometallic Fe carbide (Fe5C2) under the water oxidation conditions53,54,55. Meanwhile, the water contact angles test demonstrated an enhanced hydrophobicity of the FeCo alloy carbide compared to the Fe carbide17,56 (Supplementary Fig. 17), which re-confirmed its strong resistance to water oxidation. As a result, though a significant Fe3O4 phase was detected in Fe&CZA-spent after a long-time reaction, the Fe3O4 peak was unobvious in the FeCo&CZA-spent (Supplementary Fig. 18 and Supplementary Table 10). Therefore, the formation of FeCo carbide effectively suppressed the oxidation and avoided the in-situ generation of Fe3O4 phase.
To gain a deeper insight into the oxygen affinity as well as the water binding in terms of atomic scale, we performed DFT computations on both FeCo and Fe carbides. We employed the (510) surface of (Fe3/4Co1/4)5C2 and Fe5C2 model as the object based on the results of XRD simulation (Supplementary Fig. 14a and b), Wulff construction (Supplementary Fig. 14c and d), and the experimental results (Supplementary Figs. 8f and 10). The rationality of the (510) crystal face was also verified in conjunction with the catalytic performance (Supplementary Fig. 19). Taking monolayer water adsorption as an example, we found that the chemisorption of water on the Co sites of the (Fe3/4Co1/4)5C2(510) surface was prohibited. The average adsorption energy of water decreased from −0.820 eV on the Fe5C2(510) to −0.817 eV on the (Fe3/4Co1/4)5C2(510) (Supplementary Fig. 20). RDF analysis further revealed the H-originated adsorption behavior on the Co sites, whereas Fe sites preferred to interact with O in water molecules, implying the stronger O-bonding ability of Fe sites (Supplementary Fig. 20). Upon water dissociation, the O-containing groups (e.g., O* and OH*) also displayed weaker binding with the (Fe3/4Co1/4)5C2(510) than the Fe5C2(510). Specifically, even under the high coverage of O*, the binding strength of O* on the (Fe3/4Co1/4)5C2(510) remains to be weaker by 0.15 eV (Fig. 2f). This offers the reduction of O* and OH* into water molecules (the reverse reaction of water dissociation) to be accelerated on the (Fe3/4Co1/4)5C2(510) surface (Supplementary Fig. 21), which helps to prevent undesirable oxidation of the carbide active sites from water dissociation and ensure the long-term stability of the catalyst (Fig. 1d).
Crystal orbital Hamilton populations (COHP) analysis revealed a clear difference in the O-bonding ability between Fe5C2(510) and (Fe3/4Co1/4)5C2(510) (Fig. 2g). The more negative value of the integrated COHP (ICOHP) for Fe5C2(510) (−7.75 eV) compared to (Fe3/4Co1/4)5C2(510) (−7.61 eV) indicated that Fe5C2 forms more bonding states below the Fermi level, thus resulting in a stronger oxophilicity property than the (Fe3/4Co1/4)5C2. It is noteworthy that CO2 hydrogenation involves numerous CHxO* reaction species. Such strong O-bonding ability may serve as an additional driving force to facilitate C–O bond cleavage in CHxO* species (e.g., COH* and CH2O*), potentially influencing the catalytic selectivity toward the desired C2+OH products. This aspect will be discussed in further detail in the following sections.
Reaction mechanism of FeCo alloy carbide catalyst for C2+OH synthesis
To decipher the reaction mechanism and the nature of active sites toward C2+OH, the reaction kinetic simulations and the in situ diffuse reflectance infrared Fourier transform spectra (DRIFTS) were carried out. First, DFT computations identified two CO2 adsorption sites on both (Fe3/4Co1/4)5C2(510) and Fe5C2(510) surfaces. Site 1 involves the interaction of the adsorbates with three Fe/Co atoms, while in Site 2, the adsorbed species form four bonds with four neighboring Fe/Co atoms (Supplementary Fig. 22). Then, we focused on these two active sites and studied the detailed reaction pathways of CO2 hydrogenation to the C1 products to identify the potential reaction intermediates for C–C couplings.
For the (Fe3/4Co1/4)5C2(510), we found that the Site 1 and Site 2 displayed different product preferences (Fig. 3a and b, Supplementary Tables 11 and 12). Site 1 tended to generate CH3OH via continuous hydrogenation of CHxO* species, whereas Site 2 readily formed the CHx* (x = 2 and 3) species by dissociating the key reaction species of CH2O* and finally produced CH4. This distinctive product's preference imparts the (Fe3/4Co1/4)5C2(510) with a unique capability to produce both CHxO* and CHx* for C–C coupling, explaining why the sole FeCo alloy carbide, devoid of an oxide phase, can directly produce C2+OH products (Supplementary Table 2). In contrast, the generation of CH4 was always energy-favorable at both Site 1 and Site 2 of Fe5C2(510) (Fig. 3c and d, Supplementary Tables 13 and 14). Specifically, once CH2O* and CHO* were formed on the Site 1 and Site 2 of Fe5C2(510), respectively, the cleavage of the C–O bonds in these species was preferential, leading to the generation of abundant CHx* species to produce hydrocarbons, while the stabilization of CHxO* on the (Fe3/4Co1/4)5C2(510) favors the alcohols formation.

a–d Reaction pathways of CO2 hydrogenation for C1 intermediates synthesis on Sites 1 and 2 of (Fe3/4Co1/4)5C2(510) and Fe5C2(510) surfaces. The transition state (TS) in each elementary step is shown as the inset. e, f Possible C-C coupling pathways and the corresponding energy barriers on (Fe3/4Co1/4)5C2(510) and Fe5C2(510) surfaces.
Nevertheless, even though the sole FeCo alloy carbide component could produce C2+OH alcohols due to the co-existence of active sites for the formation of both CHxO* and CHx*, the large formation barriers for the CHxO* (ca. 1.39 eV, Fig. 3) prohibited its overall performance. However, once the CZA (K2O/Cu(111)/ZnO(101)) component was introduced, the catalytic performance was significantly enhanced, underscoring the critical role of CZA in promoting alcohol production. To further unveil the role of CZA in C2+OH synthesis from CO2 hydrogenation, the reaction mechanism of CO2 hydrogenation into C1 products on the interface of CZA was finally studied (Fig. 4a, Supplementary Table 15). Our computations indicated that the adsorption of CO2 on the interface was energetically favorable with a downhill energy of −0.80 eV. Once the CO2* was formed, CO2* was readily protonated by H* to form HCOO* with an energy barrier of 0.63 eV. Subsequently, the CZA preferred to catalyze the CO2 hydrogenation via the following steps: CHOO* → CH2OO* → CH2O* + O* → CH2O* + OH* → CH2O* + H2O* → CH2O* + H2O (l) → CH3O* → CH3OH* → CH3OH. The rate-limiting step was identified as the CH2O* + O* → CH2O* + OH* with a smaller energy barrier (1.03 eV) than that of (Fe3/4Co1/4)5C2(510). In addition, by comparison with CH2O* hydrogenation (0.27 eV) and CH2O* spillover (ca. 0.20–0.24 eV), the CZA itself was less inclined to generate C2+OH via the coupling of CH2* and CH2O* (0.53 eV), or the self-coupling between two CH2O* species (1.22 eV), as shown in Supplementary Figs. 23 and 24. Therefore, the critical role of CZA in the designed tandem catalysts should be an important reaction intermediate supplier to provide abundant CHxO* species via a fast spillover process. What’s more, the formed CO from the RWGS process at the CZA surface could also be directly used as the reactant to participate in the C2+OH synthesis, and contribute to the improvement of the catalytic performance (Fig. 3a and b). In-situ DRIFTS of the CZA catalyst was performed (Fig. 4b), the signal of CO was dominant, corresponding to the catalytic performance via the RWGS reaction. The HCOO*, CHO* and CH2O* signals were also detected, which reconfirmed the supplement of abundant CHxO* species.

a Reaction paths of CO2 hydrogenation on the interface of K2O/Cu(111)/ZnO(101) and b In-situ DRIFTS of CZA catalyst.
To prove our conjecture, different C–C coupling pathways on the (Fe3/4Co1/4)5C2(510) and Fe5C2(510) were studied based on the identified reaction intermediates during the C1 pathways (Fig. 3e and f, Supplementary Tables 16 and 17). Among them, the coupling between the CH2O* and CH2* was found to be the most favorable step with a small energy barrier of 0.52 eV. Subsequently, the generated CH2CH2O* will then be hydrogenated into CH3CH2O* by overcoming an energy barrier of 0.75 eV, whereas the dissociation of CH2CH2O* into C2H4 was suppressed due to a larger barrier of 1.07 eV. Then, the final hydrogenation of CH3CH2O* led to the formation of desirable ethanol. Yet, the larger coupling barrier between CH2CH2O* and CH2* to C3+OH (1.14 eV) relative to that of CH2CH2O* hydrogenation (0.75 eV) explained why the (Fe3/4Co1/4)5C2(510) to be highly selective for ethanol instead of C3+OH (Supplementary Fig. 25). In contrast, Fe5C2(510) showed clear preference to produce C2H4 with a lower barrier of 0.51 eV for the CH2CH2O* dissociation, whereas the further hydrogenation of CH2CH2O* into CH3CH2O* involved a higher barrier of 0.62 eV (Fig. 3f and Supplementary Table 17). Therefore, hydrocarbons were identified to be major products at the Fe5C2(510) surface.
In-situ DRIFTS was performed to monitor the important intermediates during the CO2 hydrogenation reaction under reaction conditions of 320 °C and 5 MPa. For the FeCo&CZA catalyst (Fig. 5a), carbonate (CO3*) and bicarbonate (HCO3*) with typical bands at 1455, 1510, and 1547 cm−1 were observed. The bands appeared at 1357 and 1392 cm−1 evidenced the formation of formate species (HCOO*). The bands at 1303, 1689, and 1697 cm−1 can be attributed to the stretching vibration of C = O and the deformation vibration of C–H in CH2O*. CHO* was also detected at 1750 cm−1. The stretching vibrations of C = C (1649 cm−1) and C-H (3050–3150 cm−1) can be corresponded to the unsaturated alkenes. The RWGS reaction was confirmed with the appearance of gas phase CO (2108 and 2180 cm−1). The stretching vibration of C–O in alcohols was observed at 1044 cm−1, and the bands between 2897 and 2966 cm−1 can be attributed to the stretching vibration of C–H in C2H5O*57,58,59,60,61,62. In comparison, there was almost no presence of C = C band and C2H5O* on CZA only, implying that pure CZA catalyst did not catalyze multi-carbon products synthesis (Fig. 4b and Supplementary Fig. 26). The peak intensity of gas phase CO (2108 and 2180 cm−1) delivered from CZA catalyst dominated, which stemmed from RWGS reaction. The peak intensity of gas phase CO delivered from the Fe&CZA catalyst strengthened, whereas the typical bonds for C–O in alcohols weakened, which can be attributed to the cleavage of the C–O bond in O-containing intermediates due to the strong O-bonding ability. It comprehensively verifies that the absence of Co in the catalyst is detrimental for C2+OH synthesis due to the reduced coverage of O-containing intermediates on the catalytic interface (Supplementary Fig. 26).

a In-situ DRIFTS of the FeCo&CZA-spent catalyst. b The optimal reaction pathway for ethanol synthesis on (Fe3/4Co1/4)5C2(510) surface. Color: C: Gray, H: White, O: Red.
In the fingerprint region (1200–1900 cm−1), the FeCo catalyst exhibited more abundant characteristic peaks than the Fe catalyst (Supplementary Fig. 27), indicating a richer variety of surface intermediates. The peaks corresponding to carbonate species (CO3* and HCO3*) with typical bands at 1510 cm−1 appeared as early as 150 °C, and their intensities increased progressively with temperature on both catalysts. However, the C–H stretching peak of the CH2O* (~1303 cm−1) emerged at 250 °C for the FeCo catalyst, while it appeared later, at 290 °C, for the Fe catalyst. The FeCo catalyst also exhibited stronger signal intensity. A similar trend was observed for the C–H vibration of the C2H5O* species (~2966 cm−1), further confirming the enhanced activity. Therefore, the introduction of Co could promote the formation of O-containing intermediates, which can be attributed to the effect of Site 1 (Fig. 3a) on the surface of FeCo alloy carbide. Theoretical computations indicated that the addition of Co can effectively tailor the O-bonding ability of iron carbide, which is important to control the cleavage of the C–O bond in O-containing intermediates and produce C2+ alcohols (Fig. 3). In contrast, once CHxO* adsorbed on the surface of Fe5C2, the cleavage of the C–O bonds in CHxO* was preferential, leading to the formation of hydrocarbons by coupling of CHx* species (Fig. 3c and d). Therefore, the absence of Co in the catalyst is detrimental for C2+OH synthesis due to the reduced coverage of O-containing intermediates on the catalytic interface. This is the reason why the intensity of CHxO* for FeCo&CZA is stronger than that of Fe&CZA as confirmed by in-situ DRIFTS.
In general, we conclude that the high performance of the FeCo&CZA-spent catalyst can be attributed to two key factors: the tailored O-bonding ability of the (Fe3/4Co1/4)5C2(510) surface, which serves as a highly active center for C–C coupling, and the critical spillover of CH2O* intermediates from the CZA component, which provides sufficient reactive species to participate in the coupling process. In a way, the key characteristic of CZA could also make it active to enhance the C2+OH selectivity of Fe5C2, but the strong oxygen affinity of the monometallic Fe5C2 led CHxO* to dissociate easily (e.g., CH2O* → CH2* + O*), which disturbed the reaction network of C2+OH synthesis. These analyses offered a theoretical explanation for why FeCo alloy carbide showed a high sensitivity to the CZA ratio while Fe carbide was less affected by it (Supplementary Fig. 4 and Supplementary Table 4). This tandem effect finally optimized the overall reaction pathway in two individual ways and leads to the great improvement of C2+OH selectivity on (Fe3/4Co1/4)5C2(510) surface as drawn in Fig. 5b.
Analysis of industrial application prospects
Overall, our work demonstrated a great promise of using FeCo&CZA for CO2 hydronation to produce value-added C2+OH. In terms of the less expensive and easy batch preparation of the FeCo&CZA catalyst (Fig. 6a), the success in a hundred-gram level fixed-bed reaction operation (Fig. 6b), and excellent C2+OH synthesis performance (Fig. 6c, Supplementary Table 4), the designed catalytic system possesses tremendous potential for industrial application. To assess its viability, we finally conducted professional technical and economic analyses (TEA) on an ethanol production plant with an annual capacity of 200,000 tons of CO2 (process details illustrated in Fig. 6d, and Supplementary Tables 18–24). The estimated annual output of ethanol and olefins is approximately 30,000 and 10,000 tons, respectively. According to Aspen Plus simulations performed by Wison Company, the total product costs amount to 61.61 million US$, while the annual sales revenue reaches 76.76 million US$ (see Supplementary Table 23 for details) based on 8000 h of operation per year. This yields a total after-tax profit of 3.99 million US$ per year (based on Chinese policy).

a–c The picture of the pressed FeCo&CZA catalyst with cylindrical morphology (a), hundred-gram level fixed-bed reactor (b), and the corresponding reaction products (c). d The process flow diagram of the ethanol synthesis plant with CO2 and H2 as feedstocks. e The cost contribution of different annualized processes. f The sensitivity analysis of the main raw materials (H2 and CO2) cost. g The effect of carbon tax on CO2 price.
Though H2 supply remains the most expensive cost factor (Fig. 6e), the overall process can be economically viable if the H₂ price is reduced to below 1.6 US$ kg−1, assuming a CO₂ price of 27.7 US$ ton−1 (Fig. 6f). Alternatively, with the current H₂ price fixed at 1.4 US$ kg−1, a CO₂ price of at least 54.2 US$ ton−1 is required to achieve positive revenue (Fig. 6f). We note that the cost of H2 production from renewable energy (green hydrogen) is expected to drop to 1 US$ kg−1 by 205063,64. The implementation of carbon tax policies is anticipated to further reduce the cost of CO₂ as a raw material (Fig. 6g)65. Therefore, with decreasing reactant costs and advancements in energy-saving technologies, the CO₂-to-C₂ + OH route is likely to become increasingly feasible in the near future66.
Methods
Synthesis of FeCo alloy oxide
The FeCo alloy oxide was synthesized by a co-precipitation method. In a typical procedure, Fe(NO3)3·9H2O of 60.6 g (Sinopharm Chemical Reagent Co., Ltd.) and Co(NO3)2·6H2O of 14.6 g (Sinopharm Chemical Reagent Co., Ltd.) were dissolved in deionized water (50 mL) to obtain solution A (molar ratio of Fe/Co is 3/1). Meanwhile, Na2CO3 of 127.2 g (Sinopharm Chemical Reagent Co., Ltd.) was dissolved into deionized water (600 mL) to obtain solution B. Then solution A and solution B were dropwise added into an empty beaker under vigorous stirring at 80 °C, and the pH value was maintained at ca. 9 by adjusting the adding rate of the two solutions. The obtained suspension was aged at 80 °C for 12 h. The FeCo alloy oxide with an Fe/Co molar ratio of 3 was obtained after washing the precipitate with deionized water, drying at 80 °C for 12 h, and calcining at 350 °C for 4 h in the air. The Na content (ca. 3 wt%) in FeCo alloy oxide can be controlled by quantifying the amount of deionized water for washing. As reference samples, FeCo alloy oxides with Fe/Co molar ratios of 1 and 5 were prepared by changing the Co salt dosage [Co(NO3)2·6H2O of 43.7 and 8.7 g, respectively]. And the other preparation processes were the same as those of the FeCo (3:1) catalyst. Unless specially noted, FeCo represents the FeCo alloy oxide with a Fe/Co molar ratio of 3 in this work.
Other bimetallic oxides were prepared with the same methods with Cu(NO3)2·3H2O of 12.1 g (Sinopharm Chemical Reagent Co., Ltd.), Mn(NO3)2·6H2O of 14.4 g (Sinopharm Chemical Reagent Co., Ltd.), Zn(NO3)2·6H2O of 14.9 g (Sinopharm Chemical Reagent Co., Ltd.) instead of Co salt.
Synthesis of monometallic oxide
The monometallic Fe and Co oxide catalysts were synthesized by a precipitation method. The preparation procedure was the same as that of FeCo alloy oxide, except for the absence of Co salt or Fe salt. The element content in the catalysts was characterized by inductively coupled plasma-atomic emission spectroscopy (ICP-AES), and the results are shown in Supplementary Table 25.
Synthesis of CuZnAl catalyst
The CuZnAl (CZA) catalyst was synthesized by a co-precipitation method. Cu(NO3)2·3H2O of 26.6 g (Sinopharm Chemical Reagent Co., Ltd.), Zn(NO3)2·6H2O of 11.9 g (Sinopharm Chemical Reagent Co., Ltd.), and Al(NO3)3·9H2O of 3.8 g (Sinopharm Chemical Reagent Co., Ltd.) were dissolved into deionized water (50 mL) to obtain solution A. Meanwhile, K2CO3 of 82.9 g (Sinopharm Chemical Reagent Co., Ltd.) was dissolved into deionized water (300 mL) to obtain solution B. Then solution A and solution B were dropwise added into an empty beaker under vigorous stirring at 80 °C, and the pH value was maintained at ca. 9 by adjusting the adding rate of the two solutions. The obtained suspension was aged for 12 h at 80 °C. The CZA catalyst was obtained after washing the precipitate with deionized water, drying at 80 °C for 12 h, and calcining at 350 °C for 4 h in air. The K content (ca. 5 wt%) in the CZA catalyst can be controlled by quantifying the amount of deionized water for washing.
Fabrication of multifunctional catalysts composed of FeCo and CZA
The multifunctional catalysts composed of FeCo and CZA with different intimacy modes were prepared. FeCo&CZA with the closest proximity was prepared by physically mixing two catalyst powders in an agate mortar for 10 min, and then pressing, crushing and sieving to 20–40 mesh. FeCo/CZA was prepared by mixing the granules (20-40 mesh) of two catalytic components in a vessel. In the dual-bed modes (FeCo||CZA and CZA||FeCo), two catalytic components with a 20–40 mesh size distribution were separated by quartz wool.
Synthesis of bimetallic FeCo alloy carbide and monometallic Fe carbide via CO carburization
The bimetallic FeCo alloy carbide and monometallic Fe carbide were also synthesized by CO carbonization for the characterization of temperature-programmed water oxidation-mass spectrometry (TPO-MS), in-situ water oxidation X-ray diffraction (XRD), and water contact angle test. Briefly, the bimetallic FeCo oxide and the monometallic Fe oxide were exposed to pure CO atmosphere at 300 °C for 3 h.
Catalyst characterization
The XRD patterns of the catalysts were recorded by a PANalytical X’Pert PRO MPD X-ray diffractometer (Netherlands) with Cu Kα radiation. The morphologies of the catalysts were observed by Hitachi S-4800 field emission scanning electron microscopy (FE-SEM, Japan) and JEM-2010 transmission electron microscopy (TEM, 220 kV, Japan). An aberration-corrected high-angle annular dark-field scanning TEM (AC-HAADF-STEM, FEI Titan Themis 80-200) was employed to observe the elemental distribution of the catalysts. The elemental composition and chemical bonding were analyzed by a Thermo Scientific Escalab 250XI X-ray photoelectron spectroscopy (XPS, America) with Al Kα radiation. The metal content of the catalysts was determined by ICP-AES (Perkin-Elmer Optima 3300DV). The water contact angle test was performed by DSA25 drop shape analyzer (KRÜSS, Germany). The H2-temperature-programmed reduction (TPR), CO-temperature-programmed desorption (TPD) were performed by a chemical adsorption/reduction apparatus (Beijing Builder, PCA-1200) equipped with a thermal conductivity detector. The N2 adsorption–desorption isotherm was tested on a Micromeritics ASAP 2020 analyzer.
In-situ XRD patterns were recorded using the Rigaku SmartLab instrument with Mo Kα radiation (λ = 0.7 nm, 40 kV, 60 mA). In order to allow the X-ray to penetrate the in-situ high-pressure reaction cell, a Mo target was used for the test. The reduction (H2) or reactant gas (H2/CO2 = 3/1) was fed into the in-situ high-pressure reaction cell, and the heating rate of 5 °C min−1 was employed to reach the reduction and reaction temperatures. The patterns were recorded with a scanning rate of 10° min−1 after reaching the specified temperature and pressure (3 MPa) and maintaining for 2 min.
The 57Fe Mössbauer spectra of the FeCo&CZA-spent, FeCo-spent, Fe&CZA-spent, and Fe-spent catalysts were recorded at room temperature using a Topologic 500 A spectrometer. The radioactive source was supplied by 57Co (Rh) moving in a constant acceleration mode. The velocity was calibrated by a standard α-Fe foil.
The X-ray absorption spectra (XAS) of Fe K-edge and Co K-edge were collected on the 1W1B beamline at the Beijing Synchrotron Radiation Facility (BSRF), China. The radiation was monochromatized by a Si (111) double-crystal monochromator. X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) data reduction and analysis were processed by Athena software.
The temperature-programmed water oxidation-mass spectrometry (TPO-MS) was carried out on a fixed-bed micro-reactor connected to a Hiden QGA Quantitative Gas Analysis System (Hiden Analytical). In brief, the home-made FeCo alloy carbide or monometallic Fe carbide of 0.1 g was placed into the micro-reactor and pretreated at 150 °C for 1 h under flowing of He gas (30 mL min−1), then the sample was cooled down to 30 °C, and a saturated vapor of water was fed into the quartz tube using He as carrier gas (ca. 10% water/He) for 30 min. Then, the temperature-programmed heating from 30 to 800 °C with a rate of 5 °C min−1, simultaneously with mass spectrometer analysis of the product, was performed. The water and H2 were analyzed with the corresponding mass-to-charge (m/z) signals: water of 18 and H2 of 2.
In-situ water oxidation XRD patterns were recorded using the Rigaku SmartLab instrument with Cu Kα radiation (λ = 1.5406 nm, 40 kV, 60 mA). The He flow containing water vapor was fed into the in-situ reaction cell at atmospheric pressure. The heating rate of 5 °C min−1 was employed to reach the specified temperature. The patterns were recorded with a scanning rate of 10° min−1 after reaching the specified temperature and maintaining for 2 min.
In-situ diffuse reflectance infrared Fourier transform spectra (DRIFTS) were recorded using a Thermo Nicolet NEXUS 470 Fourier transform infrared spectrometer (America). Prior to the measurements, the FeCo&CZA-spent, FeCo-spent, and Fe&CZA-spent catalysts were pretreated at 150 °C for 1 h under He flow to remove the products being adsorbed on the catalysts. After increasing the temperature to 320 °C under He flow, a mixture gas of CO2 and H2 (CO2/H2 = 1/3) was fed into the chamber. The spectra were recorded by collecting 64 scans at 2 cm−1 resolution with the catalysts under 320 °C and 5 MPa.
Catalytic activity test
The catalytic performance was analyzed in a stainless steel fixed-bed reactor (ZXBLUE CO., Ltd., Beijing) with an inner diameter of 6 mm. Typically, the Fe-based catalyst of 0.1 g or the multifunctional catalyst of 0.2 g (weight ratio of Fe-based component to CZA was maintained at 1) with a granule size of 20–40 mesh was fixed in the middle of the reactor by quartz wool for the catalytic performance test. Prior to the reaction test, the catalyst was reduced at 400 °C for 8 h in a pure H2 flow (flow rate of 60 mL/min) at atmospheric pressure. After reduction, the reactor was cooled to room temperature, and the feed gas (gas composition, 5% Ar, 23.75% CO2, and 71.25% H2) was introduced into the reactor. The reaction pressure and temperature were 5.0 MPa and 320 °C, respectively. The gaseous products from the reactor were analyzed by an online gas chromatograph (FuLi-9790II) equipped with two detectors [one is the thermal conductivity detector (TCD) connected to the TDX-01 (60–80 mesh, 3 mm × 2 m) and Porapak-Q (60–80 mesh, 3 mm × 1 m) packed columns for the analysis of Ar, CO, CH4, and CO2, the other one is the flame ionization detector (FID) connected to the HP-Plot/Q capillary column (0.53 mm × 30 m) for the analysis of light hydrocarbons]. The aqueous and oil products collected by an ice trap (0 °C) from the reactor were analyzed by an off-line gas chromatograph (FuLi-9790II) equipped with an FID detector and an InertCap-5 (GL Sciences, 0.25 mm × 30 m) capillary column. Sec-butyl alcohol and n-decane were employed as internal standards for the analysis of aqueous and oil products, respectively. The carbon balances in all catalytic runs were within 93-105%. CO2 conversion and product selectivity were calculated by the following equations. It should be noted that the selectivity of hydrocarbons and oxygenates in this work was discussed, with the exception of CO.
CO2 conversion was calculated according to Eq. (1):
where \({{{{\rm{C}}}}{{{\rm{O}}}}}_{2 \, {{{\rm{i}}}}{{{\rm{n}}}}{{{\rm{l}}}}{{{\rm{e}}}}{{{\rm{t}}}}}\) and \({{{{\rm{C}}}}{{{\rm{O}}}}}_{2 \, {{{\rm{o}}}}{{{\rm{u}}}}{{{\rm{t}}}}{{{\rm{l}}}}{{{\rm{e}}}}{{{\rm{t}}}}}\) represent moles of CO2 at the inlet and outlet, respectively.
CO selectivity was calculated according to Eq. (2):
where \({{{{\rm{C}}}}{{{\rm{O}}}}}_{{{{\rm{o}}}}{{{\rm{u}}}}{{{\rm{t}}}}{{{\rm{l}}}}{{{\rm{e}}}}{{{\rm{t}}}}}\) represents moles of CO at the outlet.
Product selectivity was according to Eq. (3):
where \({N}_{i}\) and \({n}_{i}\) represent the mole percentage and carbon number of product i.
Product yield was calculated according to Eq. (4):
The large-scale catalytic operation with FeCo&CZA catalyst of 50 g (6 mm*4 mm) loading was performed on a stainless steel hundred-gram level fixed-bed reactor (KLYT CO., Ltd., Beijing) as shown in Fig. 6b with an inner diameter of 30 mm. The product analysis method was similar to that described above.
Computational details
All the computations were carried out by spin-polarized DFT method with van der Waals (vdW) corrections, as implemented in Vienna ab initio Simulation Package (VASP)67,68. The exchange correlation energy was modeled by the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA)69. An energy cutoff of 400 eV was adopted for the plane-wave basis. Single gamma-point sampling was used for Brillouin zone integration. The geometry optimization and self-consistent field (SCF) convergence criterion were set to 0.02 eV Å−1 and 10−5 eV, respectively. The VTST package with combined climbing image nudged elastic band (CI-NEB)70 and dimer71 methods was used to obtain the reaction pathway and transition states. Fe5C2(510) and (Fe3/4Co1/4)5C2(510) were modeled by 2 × 1 supercell with the thickness of four atomic layers. Two of them (corresponding to one unit cell) are fixed at the atomic positions. CZA catalyst was modeled by a Cu–ZnO interface following previous work2. To imitate the experimentally observed ZnO(101) surface instead of a ZnO cluster, a large supercell of 4 × 4 Cu(111) with a lattice over 2.1 nm was constructed as a support to build Cu(111)–ZnO(101) interface. To avoid interactions between periodic images, a vacuum space of 15 Å was used in the perpendicular direction of the material's surface. We added the alkali near the active sites to include the effect of alkali on the reaction mechanism. Crystal orbital hamilton populations (COHP) analysis was computed by the LOBSTER program72.
Economic and technical analysis
The proposed scenario from CO2 hydrogenation to ethanol was simulated by Aspen Plus version 11 software to obtain energy and mass balances. The non-random two-liquid model and Redlich–Kwong equation (NRTL-RK) were selected as the global property method. The flow of the system displayed the main converting steps to produce ethanol via CO2 hydrogenation reaction, impurity gas removal, oil-water separation, and extractive distillation (Fig. 6d). H2, CO2, H2O, CH4, ethanol, octane, etc., were taken as conventional components. Gas and oil phase by-products were simulated with CH4, C2H4, and C8H18. Steam tables (STEAM-TA) were used for steam and water process simulations. The Stoichiometric reactor (Rstoic) block, RadFrac block, and Compressor (Compr) block were used to simulate the reactor, columns, and steam turbine, respectively. In order to separate the ethanol from the water, glycerol was used as the extractant, breaking the azeotrope system by extractive distillation. After the simulation, the economic analysis was also performed by the Aspen Capital Cost Estimator version 11.
Data availability
The data that support the findings of this study are available on request from the corresponding author. Source data are provided with this paper.
References
Turnhout, E. et al. Do we need a new science-policy interface for food systems?. Science 373, 1093–1095 (2021).
Kattel, S., Ramírez, P. J., Chen, J. G., Rodriguez, J. A. & Liu, P. Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 355, 1296–1299 (2017).
Zhong, M. et al. Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 581, 178–183 (2020).
Yang, Y. et al. Operando studies reveal active Cu nanograins for CO2 electroreduction. Nature 614, 262–269 (2023).
Zuo, J. et al. Selective methylation of toluene using CO2 and H2 to para-xylene. Sci. Adv. 6, eaba5433 (2020).
Xu, D. et al. Advances in higher alcohol synthesis from CO2 hydrogenation. Chem 7, 849–881 (2021).
Parastaev, A. et al. Breaking structure sensitivity in CO2 hydrogenation by tuning metal–oxide interfaces in supported cobalt nanoparticles. Nat. Catal. 5, 1051–1060 (2022).
Jin, S. et al. Atom-by-atom design of Cu/ZrOx clusters on MgO for CO2 hydrogenation using liquid-phase atomic layer deposition. Nat. Catal. 7, 1199–1212 (2024).
Hu, J. et al. Sulfur vacancy-rich MoS2 as a catalyst for the hydrogenation of CO2 to methanol. Nat. Catal. 4, 242–250 (2021).
Garg, S., Xie, Z. & Chen, J. G. Tandem reactors and reactions for CO2 conversion. Nat. Chem. Eng. 1, 139–148 (2024).
Ye, J. et al. Hydrogenation of CO2 for sustainable fuel and chemical production. Science 387, eadn9388 (2025).
De Smit, E. et al. Stability and reactivity of ε–χ–θ iron carbide catalyst phases in Fischer–Tropsch synthesis: controlling µC. J. Am. Chem. Soc. 132, 14928–14941 (2010).
Wang, P. et al. Synthesis of stable and low-CO2 selective ε-iron carbide Fischer–Tropsch catalysts. Sci. Adv. 4, eaau2947 (2018).
Xu, K. et al. ε-Iron carbide as a low-temperature Fischer–Tropsch synthesis catalyst. Nat. Commun. 5, 5783 (2014).
Yang, C., Zhao, H., Hou, Y. & Ma, D. Fe5C2 nanoparticles: a facile bromide-induced synthesis and as an active phase for Fischer–Tropsch synthesis. J. Am. Chem. Soc. 134, 15814–15821 (2012).
Santos, V. P. et al. Metal organic framework-mediated synthesis of highly active and stable Fischer–Tropsch catalysts. Nat. Commun. 6, 6451 (2015).
Fang, W. et al. Physical mixing of a catalyst and a hydrophobic polymer promotes CO hydrogenation through dehydration. Science 377, 406–410 (2022).
Zhong, L. et al. Cobalt carbide nanoprisms for direct production of lower olefins from syngas. Nature 538, 84–87 (2016).
Rommens, K. T. & Saeys, M. Molecular views on Fischer–Tropsch synthesis. Chem. Rev. 123, 5798–5858 (2023).
Xu, Y. et al. A hydrophobic FeMn@Si catalyst increases olefins from syngas by suppressing C1 by-products. Science 371, 610–613 (2021).
Lyu, S. et al. Stabilization of ε-iron carbide as high-temperature catalyst under realistic Fischer–Tropsch synthesis conditions. Nat. Commun. 11, 6219 (2020).
Zeng, Z. et al. CoFe alloy carbide catalysts for higher alcohols synthesis from syngas: evolution of active sites and Na promoting effect. J. Catal. 405, 430–444 (2022).
Lin, T. et al. Direct production of higher oxygenates by syngas conversion over a multifunctional catalyst. Angew. Chem. Int. Ed. 58, 4627–4631 (2019).
Gong, K. et al. Maximizing the interface of dual active sites to enhance higher oxygenate synthesis from syngas with high activity. ACS Catal. 13, 4533–4543 (2023).
Zhang, S. et al. Revealing and regulating the complex reaction mechanism of CO2 hydrogenation to higher alcohols on multifunctional tandem catalysts. ACS Catal. 13, 3055–3065 (2023).
Wang, Y. et al. Direct conversion of CO2 to ethanol boosted by intimacy-sensitive multifunctional catalysts. ACS Catal. 11, 11742–11753 (2021).
Xu, D., Yang, H., Hong, X., Liu, G. & Tsang, S. C. E. Tandem catalysis of direct CO2 hydrogenation to higher alcohols. ACS Catal. 11, 8978–8984 (2021).
Liu, N. et al. Elucidating the structural evolution of highly efficient Co-Fe bimetallic catalysts for the hydrogenation of CO2 into olefins. Appl. Catal. B: Environ. 328, 122476 (2023).
Zhang, L. et al. Direct conversion of CO2 to a jet fuel over CoFe alloy catalysts. Innovation 2, 100170 (2021).
Zhang, S., Liu, X., Shao, Z., Wang, H. & Sun, Y. Direct CO2 hydrogenation to ethanol over supported Co2C catalysts: Studies on support effects and mechanism. J. Catal. 382, 86–96 (2020).
Zhu, J. et al. Dynamic structural evolution of iron catalysts involving competitive oxidation and carburization during CO2 hydrogenation. Sci. Adv. 8, eabm3629 (2022).
Wang, M. et al. Stabilizing Co2C with H2O and K promoter for CO2 hydrogenation to C2+ hydrocarbons. Sci. Adv. 9, eadg0167 (2023).
Sun, F. et al. Effects of cobalt carbide on Fischer–Tropsch synthesis with MnO supported Co-based catalysts. J. Energy Chem. 42, 227–232 (2020).
Chai, B. et al. Highly selective syngas conversion to higher oxygenates over CuZnAl|CoFe multifunctional catalysts. Int. J. Hydrog. Energy 110, 168–180 (2024).
Zhang, R. et al. C2 oxygenate synthesis via Fischer–Tropsch synthesis on Co2C and Co/Co2C interface catalysts: how to control the catalyst crystal facet for optimal selectivity. ACS Catal. 7, 8285–8295 (2017).
Zhao, Z. et al. Insight into the formation of Co@Co2C catalysts for direct synthesis of higher alcohols and olefins from syngas. ACS Catal. 8, 228–241 (2018).
Kim, K. Y. et al. Cobalt ferrite nanoparticles to form a catalytic Co–Fe alloy carbide phase for selective CO2 hydrogenation to light olefins. ACS Catal. 10, 8660–8671 (2020).
Zeng, Z. et al. A monodisperse ε’-(CoxFe1–x)2.2C bimetallic carbide catalyst for direct conversion of syngas to higher alcohols. ACS Catal. 12, 6016–6028 (2022).
Liu, N. et al. Fine-tuning the active phases of CoFe alloy carbides for boosting olefin synthesis from CO2 hydrogenation. ACS Catal. 15, 179–192 (2025).
Kusama, H., Okabe, K., Sayama, K. & Arakawa, H. Ethanol synthesis by catalytic hydrogenation of CO2 over Rh–FeSiO2 catalysts. Energy 22, 343–348 (1997).
Gogate, M. R. & Davis, R. J. Comparative study of CO and CO2 hydrogenation over supported Rh–Fe catalysts. Catal. Commun. 11, 901–906 (2010).
Yang, C. et al. Hydroxyl-mediated ethanol selectivity of CO2 hydrogenation. Chem. Sci. 10, 3161–3167 (2019).
Xu, D., Ding, M., Hong, X. & Liu, G. Mechanistic aspects of the role of K promotion on Cu–Fe-based catalysts for higher alcohol synthesis from CO2 hydrogenation. ACS Catal. 10, 14516–14526 (2020).
Guo, H. et al. Roles investigation of promoters in K/Cu–Zn catalyst and higher alcohols synthesis from CO2 hydrogenation over a novel two-stage bed catalyst combination system. Catal. Lett. 145, 620–630 (2014).
Xu, D., Ding, M., Hong, X., Liu, G. & Tsang, S. C. E. Selective C2+ alcohol synthesis from direct CO2 hydrogenation over a Cs-promoted Cu–Fe–Zn catalyst. ACS Catal. 10, 5250–5260 (2020).
Yao, R. et al. Monometallic iron catalysts with synergistic Na and S for higher alcohols synthesis via CO2 hydrogenation. Appl. Catal. B: Environ. 298, 120556 (2021).
Takagawa, M., Okamoto, A., Fujimura, H., Izawa, Y. & Arakawa, H. Ethanol synthesis from carbon dioxide and hydrogen. Stud. Surf. Sci. Catal. 114, 525–528 (1998).
Yang, H. et al. Tuning the selectivity of CO2 hydrogenation to alcohols by crystal structure engineering. Chem 10, 2245–2265 (2024).
Zhang, Q. et al. Hydrogenation of CO2 to higher alcohols on an efficient Cr-modified CuFe catalyst. Appl. Catal. B: Environ. 337, 123013 (2023).
Wang, Y. et al. PdFe alloy–Fe5C2 interfaces for efficient CO2 hydrogenation to higher alcohols. Appl. Catal. B: Environ. 345, 123691 (2024).
Gnanamani, M. K. et al. Hydrogenation of carbon dioxide over Co–Fe bimetallic catalysts. ACS Catal. 6, 913–927 (2016).
Xu, F. et al. MOFs-derived Fe–Co bimetallic catalyst for selective CO2 hydrogenation to light olefins. Ind. Eng. Chem. Res. 63, 20800–20811 (2024).
Jia, Y. et al. Mechanism of stability and deactivation of N-doped CuFeZn catalysts for C2+ alcohols synthesis by hydrogenation of CO2. Fuel Process. Technol. 250, 107901 (2023).
Yao, B. et al. Transforming carbon dioxide into jet fuel using an organic combustion-synthesized Fe–Mn–K catalyst. Nat. Commun. 11, 6395 (2020).
Yang, C. et al. The Interplay between structure and product selectivity of CO2 hydrogenation. Angew. Chem. Int. Ed. 58, 11242–11247 (2019).
Zhao, C. et al. Solid-diffusion synthesis of single-atom catalysts directly from bulk metal for efficient CO2 reduction. Joule 3, 584–594 (2019).
Khan, M. K. et al. Selective conversion of carbon dioxide into liquid hydrocarbons and long-chain α-olefins over Fe-amorphous AlOx bifunctional catalysts. ACS Catal. 10, 10325–10338 (2020).
Yang, R., Fu, Y., Zhang, Y. & Tsubaki, N. In situ DRIFT study of low-temperature methanol synthesis mechanism on Cu/ZnO catalysts from CO-containing syngas using ethanol promoter. J. Catal. 228, 23–35 (2004).
Frei, M. S. et al. Role of zirconia in indium oxide-catalyzed CO2 hydrogenation to methanol. ACS Catal. 10, 1133–1145 (2019).
Pokrovski, K., Jung, K. T. & Bell, A. T. Investigation of CO and CO2 adsorption on tetragonal and monoclinic zirconia. Langmuir 17, 4297–4303 (2001).
Deng, W. et al. Crucial role of surface hydroxyls on the activity and stability in electrochemical CO2 reduction. J. Am. Chem. Soc. 141, 2911–2915 (2019).
Wang, L. et al. Incorporating nitrogen atoms into cobalt nanosheets as a strategy to boost catalytic activity toward CO2 hydrogenation. Nat. Energy 2, 869–876 (2017).
Dhabi, A. Green Hydrogen Cost Reduction: Scaling up Electrolysers to Meet the 1.5 °C Climate Goal (International Renewable Energy Agency, International Renewable Energy Agency, 2020).
Council, H. & Company, M. Hydrogen Insights—A Perspective On Hydrogen Investment, Market Development and Cost Competitiveness (Hydrogen Council, 2021).
Stiglitz, J. E. et al. Report of the High-level Commission on Carbon Prices (World Bank, 2017).
Dagdougui, H., Sacile, R., Bersani, C. & Ouammi, A. Hydrogen Infrastructure for Energy Applications: Production, Storage, Distribution and Safety (Academic Press, 2018).
Blöchl, P. E., Jepsen, O. & Andersen, O. K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 49, 16223–16233 (1994).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Sheppard, D., Xiao, P., Chemelewski, W., Johnson, D. D. & Henkelman, G. A generalized solid-state nudged elastic band method. J. Chem. Phys. 136, 074103 (2012).
Xiao, P., Sheppard, D., Rogal, J. & Henkelman, G. Solid-state dimer method for calculating solid-solid phase transitions. J. Chem. Phys. 140, 174104 (2014).
Nelson, R. et al. LOBSTER: local orbital projections, atomic charges, and chemical-bonding analysis from projector-augmented-wave-based density-functional theory. J. Comput. Chem. 41, 1931–1940 (2020).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2023YFB4104500 (M.B.W.), 2023YFB4104502 (Y.W.)), the National Natural Science Foundation of China (22478436 (Y.W.), 22108310 (Y.W.), 52072409 (M.B.W.)), the Key Research and Development Program of Shandong Province (2024ZLGX08 (M.B.W.)), the Science and Technology Innovation Project of the Shandong Energy Group Co., Ltd. (SNKJ2023A03 (M.B.W.), SNKJ2021BJ04 (Y.W.)), the China Scholarship Council (CSC, 202108050125 (W.W.)) and the Research Projects of Liaocheng University (318052407 (W.W.)). Japan Society for the Promotion of Science (JSPS, 23K23132 and 23H05404 (N.T.)) and Japan Science and Technology Agency (JST, JPMJSA1605 (N.T.)) supported this work. K.S.N. and X.Y.G. are grateful to the Ministry of Education, Singapore (Research Centre of Excellence award to the Institute for Functional Intelligent Materials, I-FIM, project No. EDUNC-33-18-279-V12 (K.S.N.)) and to the Royal Society (UK, grant number RSRP\R\190000 (K.S.N.)) for support. X.Y.G. gratefully acknowledges the support of the Humboldt Research Fellowship (X.Y.G.), Germany. A portion of the calculations used the computational resource from Centre of Advanced 2D Materials, funded by the National Research Foundation, Prime Ministers Office, Singapore, under its Medium-Sized Centre Program, as well as the computing time granted by the Resource Allocation Board and provided on the supercomputer Lise and Emmy at NHR@ZIB and NHR@Göttingen as part of the NHR infrastructure. The calculations for this research were conducted with computing resources under the project hbc00064 (X.Y.G.).
Author information
Authors and Affiliations
Contributions
W.W., X.Y.G., and Y.W. designed the study, analyzed the data and wrote the original paper. W.W., S.L. and J.X. performed the catalyst preparation, characterization, and catalytic tests. X.Y.G., K.S.N. and T.F. performed the theoretical simulations. X.H.G. and J.L. performed the in-situ XRD tests. J.Z. characterized the catalysts by AC-HAADF-STEM. H.J. evaluated this catalytic system by TEA. F.C. analyzed the XAFS data. Y.C., M.Q.W. and T.X. operated the hundred-gram level fix-bed reaction. G.Y., Y.L. and Q.L. provided helpful discussion. Y.W., K.S.N., N.T. and M.B.W. supervised the whole project and polished the paper. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jae Sung Lee and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, W., Guo, X., Wang, Y. et al. Transformation of CO2 to C2+ alcohols by tailoring the oxygen bonding via Fe-based tandem catalyst. Nat Commun 16, 7265 (2025). https://doi.org/10.1038/s41467-025-62727-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62727-5
