
Abstract
Atomically precise construction of ultra-small electronic devices meets the urgent need for further device miniaturisation and enables numerous electronic applications. In particular, single-molecule junctions are attractive because they serve as platforms for studying fundamental scientific laws at the single-molecule level and can be used to build functional devices. Here, we present a robust methodology using anisotropic hydrogen plasma etching of graphene and in situ Friedel-Crafts acylation reaction to construct, with atomic precision, uniform covalently bonded graphene-molecule-graphene (GMG) single-molecule junctions with clear zigzag graphene edges. Applying the methodology to an azulene-type molecule, stable GMG single-molecule junctions are constructed with high yield (~82%) and high uniformity (~1.56% conductance variance over 60 devices). The reliability of the platform is shown via real-time and direct electrical monitoring of the three-level conductance fluctuation of an individual azulene molecule. This work demonstrates a universal single-molecule platform that offers countless opportunities to reveal intrinsic molecular properties and build high-performance functional molecular nanocircuits.
Similar content being viewed by others
Introduction
Advances in materials preparation and manufacturing technologies have fuelled the unprecedented flourishing of electronic devices1,2,3,4,5. The development of ultra-small electronic devices not only enables further miniaturisation in the semiconductor industry6,7,8, but also enables diverse important applications in quantum information9,10, high-precision detection11,12, and other fields13,14. Various micro/nano processing technologies and low-dimensional materials, such as two-dimensional materials15, nanowires16, and quantum dots17,18, are used to construct increasingly miniaturised devices. Electronic devices made of a single molecule, generally named single-molecule devices, are currently considered to be the physical limit of device minimisation.
Single-molecule devices are the crucial building blocks of miniaturised functional devices for information processing and to explore fundamental physical, chemical, and biological mechanisms from a single-molecule perspective. To date, the approaches to the fabrication of single-molecule devices mainly include mechanically controllable break junctions19,20,21, lithography-defined junctions22,23, electromigration break junctions24,25,26, scanning tunnelling microscopy break junctions (STM-BJs)27,28,29, and others1. When used as electronic components, ideal single-molecule junctions should possess several critical characteristics, including high-yield manufacturing, high stability, and high uniformity. Carbon nanomaterials have the advantages of high electrical and thermal conductivity, mechanical stability, and good compatibility with molecules, making them suitable as electrodes in molecular junction-based devices30,31. Carbon nanotubes have been first used as electrodes to build covalently-bonded single-molecule junctions, successfully addressing the issue of device stability22,32. Taking advantage of the easy processing of two-dimensional graphene, graphene-based single-molecule junctions have been fabricated in batches, thus overcoming the challenges of device fabrication and performance variations caused by the strong dependence on chirality and diameter of the conductivity of carbon nanotubes23,33,34,35,36. However, even using graphene electrodes, high yield, stability, and uniformity are significantly limited by the difficulty in controlling the electrode gap size, uneven electrode configurations, and the variability of the electrode-molecule interface from device to device. To advance the field and truly revolutionise electronics, we need to reliably construct homogeneous graphene-based single-molecule junctions with atomic scale-precise electrodes and interfaces.
Graphene is a two-dimensional atomic crystal with anisotropic processability and chemical tunability. Due to its anisotropic lattice structure, few-layer graphene can be controllably tailored along the lattice direction with a specific edge configuration37,38. Furthermore, graphene can be easily modified with specific functional groups thanks to its rich carbon chemistry39,40,41. Here, we demonstrate a strategy for the construction of single-molecule junctions with atomically precise graphene electrodes and interfacial covalent bonding (Fig. 1), achieved by implementing anisotropic etching and selective functionalisation of graphene edges. Specifically, remote hydrogen plasma etching37,42 is exploited for precise anisotropic etching of three-layer graphene, generating triangular electrodes with atomically precise zigzag edge configuration and controllable nanogap size. Subsequently, through edge-selective functionalisation, specific carboxyl groups are modified in situ on the edges of the graphene electrodes using a Friedel-Crafts acylation reaction39,40,41. The edge carboxyl groups can further react with single molecules anchored via amino groups to form robust and specific interfacial connections, resulting in stable single-molecule junctions. This approach enables atomic-level precise control over various components, including graphene electrodes, the graphene/molecule interface, and bridging by a single molecule. In the following, we also detail the robustness of the experimental approach and the consistency between the results of different experiments, as required for viable molecular electronics.

Bottom: AFM illustration of graphene-based triangular point electrodes. Top: Schematic of the device centre with an azulene-type molecule in the graphene gap. O, N, and C atoms are shown in brownish-red, light blue, and grey, respectively; carbon atoms within the azulene units are highlighted in red and blue. A pair of artificial holes is introduced into the channel region at a distance of 300 nm, and the other epitaxial holes are spaced by 280 nm to ensure that there is only one pair of electrodes with a nanogap that matches the molecular length.
Results
Precise shaping of graphene electrodes by anisotropic hydrogen etching
We present an atomically precise procedure for the production of nanogap graphene electrodes in which etching is monitored in real time by current measurement. As a first step in the device fabrication process (Supplementary Section 1.1), graphene sheets are mechanically exfoliated on silicon wafers with a 300 nm SiO2 layer and pre-labelled metal marks (Supplementary Figs. 1 and 2), which are prepared by photolithography and thermal evaporation. The number of graphene layers is distinguished by the contrast in the grey values of the corresponding optical micrographs43, and the three-layer graphene is chosen for further use. Metallic Cr/Au electrodes with 8/80 nm thicknesses are patterned on graphene by electron beam lithography (EBL) and thermal evaporation (Supplementary Fig. 3). To determine the graphene lattice orientation, circular pattern arrays are inserted into the outer channel region by EBL and oxygen-reactive ion etching. Then, through remote hydrogen plasma etching (Supplementary Figs. 4 and 5), the circular pattern is turned into a regular hexagonal hole with a zigzag edge configuration, thus obtaining the lattice orientation of the entire graphene sheet (Supplementary Section 1.2 and Supplementary Fig. 6).
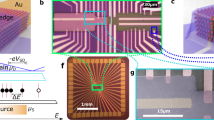
By adopting the atomic-level controllable etching rate in anisotropic remote hydrogen plasma, the pre-prepared holes are developed into regular hexagonal holes due to hydrogen atoms attacking the edge carbon atoms along the orientation of the graphene lattice. Eventually, the adjacent hexagonal vertex contacts form a pair of triangular graphene point electrodes with a molecular-scale gap as the etching time increases (Fig. 2a). The graphene sheets with external metal electrodes serve as the basis of the device (Fig. 2b) and allow for easy monitoring of the current through the device by connection of the external circuitry to the metal electrodes. To prepare triangular graphene electrodes, two artificial holes with diameters of 75 nm and spaced by 300 nm are inserted into the channel region along the graphene lattice direction, and the other epitaxial holes are spaced by 280 nm, which promises the construction of only a single pair of matching nanogap electrodes (Supplementary Fig. 7). During the anisotropic etching (500 °C, 30 W RF power, Hydrogen 9.7 sccm, producing an etching rate of ~1.9 nm/min), the degree of tailored graphene is monitored in real time by measuring the device current, which decreases as the etching progresses. When the conductance is lower than the measured noise (~10 pA), it means that the graphene channel is completely cut off, forming a pair of electrodes with a nanoscale gap (Fig. 2c and Supplementary Fig. 8). In addition, the etching process was evaluated by continuously monitoring the current and visualising the evolution of the conductance using heat maps (Supplementary Fig. 9a). The conductance distribution at the final stage (Supplementary Fig. 9b) and the etching time distribution of graphene nanometric regions (Supplementary Fig. 9c) were also analysed. Statistical analysis of 75 devices reveals a high degree of consistency in both conductance and etching time, with variation coefficients as low as 9.5% and 2%, respectively. The length of the graphene nanogap needed to insert a desired molecule of a given size is determined by precisely controlling the etching time after the cut point. The successful preparation of a smooth triangular graphene electrode pair with a gap size that matches the molecular scale is confirmed by atomic force microscopy (AFM) imaging as exemplified by Fig. 2d.

a Schematic representation of the formation process of graphene triangular point electrodes with zigzag edges. C atoms are shown in blue; H atoms are partially omitted and the displayed ones are shown in yellow. b Optical image of the device pattern. c SEM image of graphene electrodes. d AFM image of graphene electrodes. e HRTEM image of graphene edges after etching. The enlarged graphic is shown in the upper right. The FFT of the enlarged graphic is shown in the lower right, where [10\(\bar{1}\)0] marked with white circles indicates the zigzag orientation. Atomically resolved STEM images of the graphene electrode basement (f) and zigzag edge (g).
The lattice structure and edge configuration of graphene electrodes play a critical role in device performance. To determine these characteristics of the electrodes, the prepared graphene samples are transferred onto transmission electron microscope (TEM) grids by wet preparation for characterisation (Methods)44. The clean straight edges of the graphene hexagonal topography are observed by high-resolution TEM (HRTEM) (Fig. 2e and Supplementary Fig. 10). Fast Fourier transform from a selected area near the straight edge reveals the crystal orientation of graphene. The six spots shown in the inset of Fig. 2e correspond to the zigzag ([\(10{\bar{1}}0\)]) direction45. Since the line connecting the centre to the diffraction point ([\(10{\bar{1}}0\)]) is perpendicular to the zigzag direction in real space, the zigzag direction of the graphene edge can be determined. For a more intuitive characterisation, atomic-resolved scanning transmission electron microscopy (STEM) is used, with a low accelerating voltage of 60 kV to reduce the damage to the graphene sample. From the STEM image, the graphene hexagonal network with a C–C bond length of ~0.14 nm is clearly visible (Fig. 2f). Adjacent graphene layers exhibit an exact AB-stacking structure. The atomically regular zigzag configuration of the graphene edge is also observed in the STEM image (Fig. 2g), confirming the atomically precise control of etching. From the regular hexagonal configuration of the edge, it can be inferred that the edge benzene rings maintain intact aromatic characteristics. The regular lattice structure and atomically precise zigzag configuration of graphene electrodes provide the basis for the construction of single-molecule junctions accurate at an atomical level.
In situ functionalisation of graphene electrodes
To realise atomically precise electrode-single molecule interfaces, we first perform carboxyl modification of the triangular graphene point electrodes via a solvent-controlled Friedel-Crafts acylation reaction (Fig. 3a). Amide groups are then formed by covalent bonding to amino-anchored molecules. In detail, the mechanisms of Friedel-Crafts acylation reaction are regulated by solvents. The graphene edges are attacked by the highly reactive acyl cations (COCl+) or the less reactive complexes (COCl2‧AlCl3), corresponding to ionic and nucleophilic mechanisms, respectively46. In a 1,2-dichloroethane solution, the graphene edges are attacked by COCl+ through an ionic mechanism via a kinetically controlled process, and graphene tailoring occurs as confirmed by both experimental and theoretical analyses46. In a tetrachloroethane (TTCE) solution containing acyl chloride and aluminium chloride, the graphene edges are attacked by COCl2‧AlCl3 through an electrophilic substitution mechanism, generating acyl chloride groups that are further hydrolysed to form carboxyl groups. Furthermore, because of the high reaction barrier to further tearing of graphene46, the reaction terminates at the edge, thus guaranteeing precise edge functionalisation.

a Schematic representation of the process for the in situ graphene edge functionalisation by a Friedel-Crafts acylation reaction. C atoms are shown in blue; O atoms are shown in red; Cl atoms are shown in green; and H atoms are omitted. b XPS spectrum of the functionalised graphene. The peaks of sp2 C, C–O, C=O, and π–π* are centred at ∼284.5, ∼286.4, ∼288.9, and ∼290.7 eV, respectively. c TGA-FTIR spectra of the functionalised graphene at different temperatures. d XRD patterns of pristine graphene (Gr) and edge-selectively functionalised graphene (EFG). The [002] peak at 26.5° means a d-spacing of 0.34 nm for graphene. e HRTEM image of the basement of the functionalised graphene. f Raman spectra of the edge and centre of the functionalised graphene, whose D/G ratio is 0.63 at the graphene edge. g Raman mapping of the peak intensity ratio ID/IG of the functionalised graphene.
To evaluate the edge modification, equivalent graphene samples are characterised (Fig. 3 and Supplementary Section 2). The carboxyl functionalisation of graphene is confirmed by different features: the characteristic C=O peak (~288.9 eV) in the high-resolution C1s spectrum obtained by X-ray photoelectron spectroscopy (XPS) (Fig. 3b and Supplementary Fig. 11a); an increase in the O1s/C1s atomic ratio (from 0.21 to 0.43 after functionalisation) in full XPS surveys spectra (Supplementary Fig. 11b and Supplementary Fig. 12); and the appearance of a C=O stretching vibration peak at 1684 cm–1 in thermogravimetric analyser coupled with Fourier transform infrared analysis (TGA-FTIR) (Supplementary Figs. 13–16 and Fig. 3c). From the TGA-FTIR characterisation, it is also observed that the functional groups remain stable up to ~250 °C.
The π-π* peak (~290.7 eV) from XPS (Fig. 3b and Supplementary Fig. 11a) and invariant characteristic peaks of X-ray diffraction (XRD) (Fig. 3d) indicate that the basal plane of graphene has not been destroyed after the reaction. Peaks at ∼1047, ∼1650, and ∼3420 cm–1 assigned to C–O, C=O, and O–H species, respectively in FTIR spectra of pristine graphene and functionalised graphene also confirm the successful modification (Supplementary Fig. 16). The HRTEM (Fig. 3e) and AFM images (Supplementary Fig. 17) also confirm that the modified graphene maintains an intact lattice structure without basal plane reactions. Moreover, in the Raman spectra of the equivalent graphene samples on the silicon wafer (Supplementary Fig. 18), the D peak at the edge is significantly enhanced after the reaction (the D/G ratio is 0.63), while the D peak at the basal plane is negligible, indicating that the reaction occurs selectively at the edge (Supplementary Fig. 19 and Fig. 3f). Spatial Raman mapping of the D/G peak ratio (Fig. 3g) more intuitively shows the functional groups located at the edges of graphene, further demonstrating the edge-selective functionalisation of graphene.
Molecule connection and characterisation
Single-molecule junctions are built by inserting an amino-terminated azulene-type molecule (Supplementary Section 3 and Supplementary Figs. 20–22) into the graphene nanogap, where it engages in covalent amide bonding with graphene’s carboxyl edges (clearly, the same procedure can be adopted for other amino-terminated molecules). The assembly reaction is carried out using pyridine as the solvent and 1-(3-dimethylaminopropyl)−3-ethylcarbodiimide hydrochloride as a dehydrating/activating agent. Through electrical characterisation, it is observed that the device conductance changes from ~0 nS (i.e., a non-measurable current) in the open circuit condition to ~1 nS after assembly (Fig. 4a), indicating that the molecule is successfully connected. From the inelastic electron tunnelling spectroscopy (IETS) of the device at 2 K (Fig. 4b), we observe the characteristic peaks of amide bonding, namely υ(C=O) (~226 mV) and υ(N–H) (~452 mV), and of the molecular backbone, such as δ(C–H) (~178 mV) and υ(C=C) (~206 mV), confirmed by theoretical simulations. This observation indicates that the azulene-type molecule is fixed (with a stable structure that preserves the molecular frequencies) into the nanogap between the graphene electrodes and therefore works as a molecular bridge for electrical conduction purposes. The azulene-type molecule is fluorescent, with excitation and emission bands at 300–430 nm and 500–600 nm, respectively (Supplementary Fig. 23). From stochastic optical reconstruction microscopy using a 405 nm laser as the excitation source, the fluorescence emission from a single azulene-type molecule in nanogap electrodes is observed, further validating the effective assembly of a single molecule into the device (Fig. 4c and Supplementary Fig. 24). Incidentally, it is worth emphasising that the possibility of observing optical properties of the molecular bridge is granted by the structural stability of the molecular junction. This fact indicates opportunities for using the present approach to build functional molecular junctions in which conductive, optical and combined conductive-optical properties of molecules are exploited.

a I–V curves before and after single-molecule connection. The inset shows the corresponding structure of the single-molecule junction. b Reproducible IETS spectra (Top) were measured three times by a lock-in second-harmonic technique at 2.0 K with an AC amplitude of 10.0 mV (Vpk) and a frequency of 661 Hz. Calculated infrared and Raman spectra of the single-molecule junction are also reported (bottom) at the B3LYP/6-31 G(d,p) level of computational accuracy. All the peaks are marked in the IETS spectra (VD = hω/e). The peaks corresponding to ~226 and ~452 mV are attributed to the vibrational frequencies of υ(C=O) and υ(N–H), respectively, in the amide bonding groups, and δ(C–H) (~178 mV) and υ(C=C) (~206 mV) denote the vibrational mode of the molecular backbone. c Fluorescent super-resolution imaging of the single-molecule junction. d I–V characteristics for 60 individual devices measured at room temperature. e Plots of the slope of ln(ID)‒VD0.5 vs. 1/T for a device. The inset shows a schematic of the barrier model. f Comparison of yield and coefficient variability of the single-molecule junctions obtained using our atomically precise technique with those reported in the literature using electromigration (EM) for gold-based junctions, oxidative plasma ion etching (OPIE) for SWCNT-based and Gr-based junctions, and electrical breakdown (EB) technique for covalently bonded and π-π stacking Gr-based junctions22,23,25,49,50,51,52,53,54,55.
To determine the reproducibility of the prepared single-molecule devices, a total of 73 devices are fabricated (the remaining two used for device characterisations). 60 of them produce valid electrical signals. Therefore, the yield of the prepared devices is as high as ∼82%. Furthermore, the I–V characteristics of all 60 devices measured at room temperature are highly consistent with each other (Fig. 4d). Statistical analysis of the results from the 60 devices provides a conductance of (1.16 ± 0.12) × 10–5 G0 (G0 = 2e2/h = 77.5 μS), with a conductance variance of ∼1.56% and a coefficient variability of 10.73% (Supplementary Table 1 and Supplementary Fig. 25). During 100 measurements of the same device, the I–V curves highly overlap, showing a conductance variance of ∼0.93% (Supplementary Fig. 26). In comparison, azulene-type single-molecule junctions fabricated by a STM-BJ technique with gold electrodes and Au-N interfacial connections have a similar size conductance of ~2.8 × 10–5 G0 (Supplementary Fig. 27).
The conductance of naphthalene-type single-molecule devices (Supplementary Figs. 28 and 29), used as a control system, also shows high consistency with a variation of ~0.48% (Supplementary Table 2). These electrical characterisations consistently demonstrate that single-molecule junctions with high stability, uniformity, and reproducibility are successfully fabricated by the atomic precision construction technique.
Charge transport and protonation/deprotonation dynamics
Based on these reliable devices, the charge transport characteristics of a single azulene-type molecule are further examined. Temperature-dependent I–V characteristics show that the device conductance increases monotonically as temperature increases from 80 to 300 K (Supplementary Fig. 30), indicating a thermally activated charge transport mechanism. Furthermore, the slope of (ln(I) vs. V0.5) vs. 1/T is linear (Fig. 4e and Supplementary Fig. 31), which is consistent with the thermionic emission mechanism47,48. To further assess the quality of the present atomic precision (AP) technique to construct static single-molecule junctions, the achieved coefficient variability and yield are compared (our comparison does not claim to be exhaustive, given the vast amount of studies in the field) with those of different types of techniques, such as electromigration (EM) for gold-based junctions25,49, oxidative plasma ion etching (OPIE) for SWCNT-based junctions22,50, OPIE23,51 and electrical breakdown (EB) technique52,53,54,55 for graphene-based junctions (Fig. 4f). The static single-molecule junctions fabricated by the present approach show the lowest coefficient variability and the highest yield.
The constructed single-molecule junctions also provide a useful platform to monitor the chemical reactions of active groups on individual molecules at the single-event level. Since the azulene group is liable to protonation, protonation and deprotonation processes are investigated using the azulene-type single-molecule devices. Azulene protonation is induced by treating the device with trifluoroacetic acid in propylene carbonate solvent (0.1 M). Tetrabutylammonium hydroxide in propylene carbonate solvent (0.1 M) is instead used to remove the active hydrogen and therefore restore azulene to its initial state (deprotonation). According to the I–V characteristics of the molecular device (Fig. 5a and Supplementary Fig. 32), the conductance of the protonated molecule is lower than that of the deprotonated molecule. During repeated cycles, the device exhibits high repeatability (Fig. 5b), indicating the reversible reactions of the azulene-type molecule in the device. Control experiments on similarly treated naphthalene-type single-molecule devices show practically no change in conductance (Supplementary Fig. 33), thus indicating that the conductance change originates from the reaction of the azulene-type molecule (Fig. 5c).

a I–V curves of the single-molecule junction after protonation (blue) and deprotonation (red) of the molecular bridge. b Switching cycles of the alternately protonated and deprotonated device. VD = 0.1 V. c Reversible reaction processes and transition rates between species A, HA, and H2A at 295 K. The conversion rate constant is k = 1/τ. d Representative (Left) and enlarged (Middle) I−t trajectories, and corresponding statistical histograms (Right) of the device with VD = 0.4 V at 295 K. e Proportions of the A, HA, H2A species at different temperatures. f Theoretical energy profile of the reversible protonation process. g Plots of the temperature-dependent thermodynamic parameters in van’t Hoff equation fitting (lnK vs. 1000/T) and the kinetic parameters in Arrhenius equation fitting (lnk vs. 1000/T). In all figures, error bars are calculated using data obtained from three independent devices.
The in situ protonation dynamics of a single azulene-type molecule is monitored in real time in a solution of propylene carbonate solvent with 10–5 M trifluoroacetic acid. Molecular transitions between three conduction states are observed by means of the real-time electrical spectroscopy (Fig. 5d). Our observation is consistent with the fact that the azulene molecule has two proton binding sites, and therefore the states of deprotonation (A), monoprotonation (HA), and diprotonation (H2A) are expected to co-exist during protonation reactions (Fig. 5c). Simple theoretical calculations of molecular orbitals (Supplementary Fig. 34) and transmission spectra (Supplementary Figs. 35–37) help us assign the three conduction states to three differently protonated molecular structures. We see that, in all cases, the HOMO is much closer to the expected Fermi level of graphene56 than the LUMO, enabling the current at zero or very small bias voltage (this can be argued considering the broadening effect of the molecular reorganisation energy on the I–V characteristics)57,58. The HOMO is most spread over the molecule in the A species. Since the tails of its spatial distribution determine the electronic coupling strengths to the electrodes, we expect that the highest conduction state corresponds to the molecular bridge in form A. Based on the same argument, the intermediate conductive state is assigned to the HA molecular bridge and the lowest conduction state to H2A.
Traces of the current as a function of time give us information on the occupation of the three conductive states over time, and therefore on the proportion of occurrence of the three molecular species in the junction. From the temperature-dependent proportions of the three states (Supplementary Fig. 38), we see that the A population decreases while HA and H2A populations increase with temperature (Fig. 5e). Therefore, the two-step reversible protonation path exhibits a significant temperature dependence. On the contrary, the temperature-dependent current measurement on the control naphthalene-type device does not show meaningful changes in conductive state, thus back-confirming the observation of protonation dynamics in the azulene molecule (Supplementary Fig. 39).
The kinetic and thermodynamic parameters are also obtained for the individual protonation/deprotonation reactions. Specifically, the hidden Markov model is applied to further analyse the real-time electrical spectroscopy and gain information on the single-event processes. From the single-exponential fitting of the time intervals between conductive switches in idealised I–t curves (Supplementary Fig. 40), the lifetime (τ) of each species is identified and the conversion rate constant is obtained as k = 1/τ. For the reactions at 295 K, the protonation rates of the first and second steps (A → HA and HA → H2A) are ~16 and ~901 s–1, respectively; the reverse processes HA → A and H2A → HA have rates of ~21 and ~86 s–1, respectively (Fig. 5c). It is necessary to notice that the transition from HA to H2A is too fast to detect, which can be attributed to the potential kinetic factors perhaps. Based on the temperature-dependent reaction rates and the Arrhenius equation (k = Aexp(–Ea/RT)), the activation energy for the A → HA process is calculated to be 0.32 eV approximately. The activation energy is in excellent agreement with the theoretical reaction barrier (Fig. 5f), despite the approximations inherent in the calculation of the latter which make the match partly fortuitous (Supplementary Fig. 41 and Supplementary Table 3). Furthermore, the transformation rate and extent of protonation (A ↔ HA ↔ H2A) are calculated in Fig. 5g to quantify the protonation dynamics of the azulene ring. In addition, using the temperature-dependent proportions of the initial (A) and final (H2A) states and the van’t Hoff equation (–RTln(K) = ΔH–TΔS), the thermodynamic parameters for the entire reaction are ΔH = ~ 0.18 eV and ΔS = ~ 0.44 meV/K (Fig. 5g). Thus, the single-molecule reaction kinetic and thermodynamic behaviours hidden in the averages of macroscopic reactions can be derived from quantitative analysis of single events in single-molecule junctions.
Furthermore, our modular single-molecule device platform is compatible with various architectures, including the graphene field-effect transistor with back-gate control via doped silicon and 300 nm SiO2 dielectrics, achieving effective gating at 2.4 V for graphene (Supplementary Fig. 42). The gate performance can be further enhanced by computer-aided design (CAD)-defined geometries, electron-beam lithography, and high-κ dielectric deposition (e.g., HfO2/Al2O3/Al stacks). A streamlined four-step nanofabrication process enables ionic liquid gating for precise modulation (Supplementary Fig. 43). At the same time, hydrogen-terminated graphene edges allow versatile molecular bridge functionalisations through phenolic, sulfonation, and one-step coupling reactions, thus facilitating low-barrier molecular junctions and bioconjugation for potential applications in chemistry and life science.
Discussion
We develop an atomically precise procedure for the construction of single-molecule junctions by appropriate combined use of anisotropic hydrogen plasma processing and in situ edge-selective graphene functionalisation. Remote hydrogen plasma etching of three-layer graphene tailors pre-patterned circular holes into hexagonal holes to form graphene-based triangular point electrodes with atomically precise zigzag edges. Then, carboxyl groups are selectively grafted at the edges of graphene electrodes, thus enabling linkage to amino-terminated molecules to form stable single-molecule junctions. The above fabrication process of atomically precise and controllable single-molecule devices involves several meticulously executed steps to ensure atomic precision at each stage. Initially, high-quality graphene with a defined number of layers is obtained through mechanical exfoliation, ensuring atomic-level structural integrity. Subsequent etching processes are performed with atomic precision, as verified by atomically resolved scanning transmission electron microscopy. The atomic precision of graphene edge-selective functionalisation is further validated through a combination of theoretical analysis and multiple characterisation techniques. Furthermore, the molecules themselves are clearly synthesised with atomic precision. Collectively, these meticulously controlled procedures result in the successful fabrication of atomically precise single-molecule devices. Owing to the atomic accuracy of each component, the single-molecule junctions are prepared with high yield (∼82%) and exhibit an unprecedented homogeneity (∼1.56% conductance variance over 60 devices). Based on the azulene-type single-molecule junction, here considered as a case study, the charge transport characteristics and protonation dynamics of the molecular bridge are thoroughly investigated and explained, demonstrating the functionalisation flexibility and diversity. We believe that these atomically accurate single-molecule junctions offer a technical platform for conducting high-reliability research on basic scientific laws at the single-molecule level and for building functional devices in next-generation integrated nanocircuits. The structural stability and reproducibility of the produced devices also contribute to form a robust basis for future scenarios where molecular electronics may be utilised for quantum computing and artificial intelligence purposes59.
Methods
Materials
Graphene nanoplatelets are bought from Cheap Tubes Inc. Aluminium chloride (99%) is bought from Alfa Aesar. Oxalyl chloride, 2.0 M solution in dichloromethane, is bought from Acros. 1,1,2,2-Tetrachloroethane (TTCE, >97%) is bought from TCI. The detailed synthetic procedure of azulene-type molecules is described in the Supplementary Section 3. A control naphthalene-type molecule is synthesised as in a previous work60.
Optical determination of the graphene structure
A standard optical microscope (Olympus BX51TRF with a 50×, 0.8 numerical aperture (NA) objective) with a charge-coupled device camera (Olympus DP71) is used to image graphene samples. A mark pattern (6 nm of Cr followed by 60 nm of Au) is deposited onto silicon wafers by thermal evaporation (Thermal Evaporator ZHD-300) using a photolithographic process.
Device fabrication
High-quality graphene flakes are directly obtained on SiO2/Si substrates (with a 300 nm SiO2 layer) via mechanical exfoliation of Kish graphite. To facilitate the positioning of exfoliated graphene and its alignment for subsequent EBL, gold marks are deposited on the silicon substrate via thermal evaporation through a photolithographically-patterned resist mask. Before the exfoliation process, the silicon substrate is ultrasonically cleaned in isopropanol, acetone, deionised water, and ethanol, followed by oxygen plasma cleaning to remove ambient adsorbates (48 sccm, 50 W radiofrequency (RF) power, 5 min exposure). After folding in half several times to fully exfoliate the Kish graphite, the adhesive tape is pressed with appropriate force onto the SiO2/Si substrate. Graphene flakes with different numbers of layers are prepared after peeling the tape away from the wafer. The number of layers in graphene flakes is rapidly identified by a simple and efficient method based on a standard optical microscope with a charge-coupled device camera and image contrast calculation from graphene optical images. Few-layer graphene is chosen for device fabrication, as it shows due to suppressed over-etching and evident anisotropic etching effect. Source and drain electrodes separated by 3 µm are produced via a typical lift-off approach. EBL is employed to pattern the poly(methyl) methacrylate (PMMA) mask, followed by thermal evaporation and electron beam evaporation to deposit metal (8 nm Cr, 80 nm Au) and SiO2 (40 nm), respectively. A SiO2 layer is used to protect the metal electrodes during the following high-temperature annealing.
The first step in preparing triangular point graphene electrodes with uniform edge configuration is to determine the crystallographic orientation of graphene. The crystallographic orientation of graphene can be identified by pre-etching. A PMMA layer (950 K A6) is spin-coated at 4000 rpm on graphene and then baked at 180 °C for 2 min. An image of the graphene is captured via an optical microscope and imported into MATLAB to eliminate angular deviation for field alignment. Pre-etched patterns (hole radius 0.1 μm, spacing 0.5 μm) are designed with Auto-CAD and DY2000 according to the pre-aligned image. The pre-etched patterns are then exposed on PMMA via EBL. After exposure, the resist is developed in a mixed solution (isopropanol: methyl isobutyl ketone, 3:1), followed by rinsing with ethanol and drying with N2. The patterns are finally transferred onto graphene through the patterned resist mask via reactive ion etching (RIE) (48 sccm, 50 W RF power, 20 s exposure). After etching, the device is soaked in acetone overnight to remove PMMA. After anisotropic hydrogen plasma etching in the plasma-enhanced hydrogen etching (PEHE) system (500 °C, 30 W RF power, Hydrogen 9.7 sccm), an image of the resulting hexagonal holes is captured via SEM (Hitachi S4800) and then imported into Auto-CAD to calculate the zigzag orientation. Then, by using the same procedure, another cycle of precise anisotropic etching is performed again within the graphene channel region along the predefined zigzag direction determined by pre-etching, resulting in the formation of triangular point electrodes. The triangular point electrodes are characterised using SEM and AFM (Bruker dimension icon, scansyst mode, scansyst air tip).
Graphene transfer, HRTEM, and STEM characterisation
A polymer-free transfer method44 is used to prepare graphene films for atomically resolved STEM characterisation. Quantifoil carbon film TEM grids (Quantifoil GIG-1010-3A) are used as target substrates. The graphene sample on SiO2/Si substrates is prepared via mechanical exfoliation. A hexagonal hole array (hole radius 0.1 μm, spacing 1 μm) is created on the graphene sample via EBL and remote hydrogen etching. After cleaning the sample with N2 gas flow, a TEM grid is placed on top of the graphene flakes. A drop of isopropanol is added to the sample. After completing isopropanol evaporation, the carbon film is brought into close contact with the graphene sample via surface tension. Contact is further improved by baking the sample on a hot plate at 100 °C for 2 min. After cooling the sample, hydrogen fluoride is used to selectively etch the SiO2 layer, and the grid together with the graphene sample is separated from the Si substrate in a few seconds. The sample is taken with tweezers and rinsed carefully several times with deionised water. Finally, the sample is dried in air for further characterisation. TEM and STEM experiments are performed in FEI Tecnai F20 TEM and JEM-2100F TEM at 200 kV, and Nion U-HERMES200 at 60 kV to characterise the atomic structures of the samples. Real-space amplitude images are reconstructed from the inverse FFT.
Edge-selective functionalisation of graphene nanoplatelets
After completing the construction of the graphene-based triangular point electrodes, it is necessary to chemically functionalise the hydrogen-terminated graphene electrodes for further covalent connection. However, it is difficult to characterise the tiny number of functional groups in these nanodevices. To find the most suitable reaction conditions and characterise the functional groups, graphene nanoplatelets are used to simulate the graphene electrodes in chemical reactions on silicon wafers. 12.56 mmol aluminium chloride, 6.4 mmol oxalyl chloride, and 80 mg graphene nanoplatelets are stirred in 80 mL TTCE under argon atmosphere protection. 3.2 mL of oxalyl chloride is added dropwise into the ice water bath for 1 h. Then, the reaction equipment is placed into an oil bath, and the reaction is carried out at 100 °C for 18 h. The reaction is quenched by glacial hydrochloric acid. The liquid is filtered by a Buchner funnel. The products are washed with ethyl chloride, methanol, and water several times to obtain functionalised graphene nanoplatelets. The functionalised graphene nanoplatelets are dried in a vacuum oven at 170 °C for 5 days to obtain edge-selectively functionalised graphene (EFG), which is also used for characterisation to confirm the successful modification of functional groups.
Edge-selective functionalisation of graphene
Dense hole arrays are fabricated and etched by PEHE to mimic triangular graphene point electrodes. 1.57 mmol aluminium chloride and 0.8 mmol (0.4 mL) oxalyl chloride are mixed in 40 mL TTCE in an ice bath for 1 h. The graphene with an array of hexagonal holes, on silicon wafers, is rinsed in such solvent, in argon atmosphere. The reaction is carried out at 100 °C for 12 h. Finally, the wafer is immersed in glacial hydrochloric acid to quench the reaction.
Characterisation of functionalised graphene
X-ray diffraction is performed on X-Pert3 Powder (PANalytical). TGA is recorded on a TA Q600 SDT (TA Instrument). TGA-FTIR is used on the TG/FTIR/GCMS (PerkinElmer). XPS is conducted on AXIS Supra. The SEM is carried out with a Hitachi S4800 field emission scanning electron microscope. Raman mapping is obtained by confocal Raman microscopy (WITEC, alpha300R). All G peaks within the region are selected and subjected to peak analysis. The statistical results determine that the G peaks with values below ~7 are considered as the substrate.
Electrical characterisation
A Keysight B1500A semiconductor characterisation system and a Karl Suss (PM5) manual probe station are used for electrical characterization at room temperature in the ambient atmosphere. The time resolution is 4 ms. Specially, the constant voltage is determined by the DC auxiliary output of the UHFLI lock-in amplifier, and a DL1211 amplifier is used to amplify the current signal of the control molecule with the high-speed acquisition card (NIDAQ) at a rate of ~250 Sa/s. The temperature-dependent electrical characterisation of the devices is carried out on Lakeshore TTPX cryogenic probe station featuring the Model 336 temperature controller with the nitrogen cooling system. The temperature-dependent measurement is performed from 80 to 300 K in vacuum. The proton dynamics experiment is carried out from 300 to 320 K. The I–V tests were performed on Keysight B1500A semiconductor characterisation system or UHFLI lock-in amplifier (with DL1211 amplifier).
Single-molecule connection and characterisation
The azulene and control naphthalene molecules are separately linked to the ends of the graphene electrodes by soaking the as-prepared graphene device into the pyridine solution of the target molecules (∼10‒4 M). A well-known carbodiimide dehydrating/activating agent EDCI is added to promote the dehydration reaction. The reaction is performed in an inert atmosphere at room temperature for 48 hours. After the reaction is completed, the device is carefully rinsed with acetone and deionised water and dried with N2 gas flow.
Protonation and deprotonation of single-molecule devices
The protonation and deprotonation of azulene-type and naphthalene-type single-molecule junctions are performed in acid/alkaline droplets. The acid/alkaline solvents are a mixture of trifluoroacetic acid/tetrabutylammonium hydroxide: H2O = 1: 50 (v/v). 1 mL (0.1 M) trifluoroacetic acid is used to trigger the protonation, while 1 mL (0.1 M) tetrabutylammonium hydroxide is used to deprotonate. After every acid/alkaline treatment, the single-molecule junctions are rinsed with abundant water.
Theoretical calculation
The geometries of all molecular structures are optimised using the B3LYP61 hybrid exchange-correlation functional method and the empirical Grimme’s D3 dispersion correction with Becke-Johnson damping62,63,64. A propylene carbonate solvation environment is considered using the SMD implicit solvation model65 by defining the dielectric constant. The electron density is expanded on a 6-31 G(d) basis set.
Frequency calculations are performed to verify that the intermediates have no imaginary frequency (and therefore they correspond to a local minimum in the energy landscape), while the transition structures have only one imaginary frequency (so as to correspond to saddle points). All calculations are performed with the Gaussian 16 software66.
Data availability
The data that support the findings of this study are available within the paper and Supplementary Information. The datasets used in the Supplementary Information are available online from the Zenodo repository at https://zenodo.org/records/16728895. Source data are provided with this paper.
References
Xiang, D., Wang, X., Jia, C., Lee, T. & Guo, X. Molecular-scale electronics: From concept to function. Chem. Rev. 116, 4318–4440 (2016).
Hensleigh, R. et al. Charge-programmed three-dimensional printing for multi-material electronic devices. Nat. Electron. 3, 216–224 (2020).
Tan, C. et al. 2D fin field-effect transistors integrated with epitaxial high-k gate oxide. Nature 616, 66–72 (2023).
Jang, S., Shim, H. & Yu, C. Fully rubbery Schottky diode and integrated devices. Sci. Adv. 8, eade4284 (2022).
Kim, Y. et al. Chip-less wireless electronic skins by remote epitaxial freestanding compound semiconductors. Science 377, 859–864 (2022).
Meier, D. & Selbach, S. M. Ferroelectric domain walls for nanotechnology. Nat. Rev. Mater. 7, 157–173 (2022).
Robinson, A. L. Problems with ultraminiaturized transistors. Science 208, 1246–1249 (1980).
Moore, G. E. Cramming more components onto integrated circuits. Proc. IEEE 86, 82–85 (1998).
Scappucci, G. et al. The germanium quantum information route. Nat. Rev. Mater. 6, 926–943 (2021).
Kagan, C. R., Lifshitz, E., Sargent, E. H. & Talapin, D. V. Building devices from colloidal quantum dots. Science 353, aac5523 (2016).
Aragonès, A. C. et al. Electrostatic catalysis of a Diels–Alder reaction. Nature 531, 88–91 (2016).
Hu, W. et al. Direct flipping dynamics and quantized enrichment of chirality at single-molecule resolution. Sci. Adv. 10, eado1125 (2024).
Choi, J.-H. et al. Exploiting the colloidal nanocrystal library to construct electronic devices. Science 352, 205–208 (2016).
Baets, R. An ultra-small silicon laser. Nature 498, 447–448 (2013).
Kang, K. et al. Layer-by-layer assembly of two-dimensional materials into wafer-scale heterostructures. Nature 550, 229–233 (2017).
Jia, C., Lin, Z., Huang, Y. & Duan, X. Nanowire electronics: From nanoscale to macroscale. Chem. Rev. 119, 9074–9135 (2019).
Borsoi, F. et al. Shared control of a 16 semiconductor quantum dot crossbar array. Nat. Nanotechnol. 19, 21–27 (2024).
van Dam, J. A., Nazarov, Y. V., Bakkers, E. P. A. M., De Franceschi, S. & Kouwenhoven, L. P. Supercurrent reversal in quantum dots. Nature 442, 667–670 (2006).
Xiang, D., Jeong, H., Lee, T. & Mayer, D. Mechanically controllable break junctions for molecular electronics. Adv. Mater. 25, 4845–4867 (2013).
Huang, C., Rudnev, A. V., Hong, W. & Wandlowski, T. Break junction under electrochemical gating: Testbed for single-molecule electronics. Chem. Soc. Rev. 44, 889–901 (2015).
Reed, M. A., Zhou, C., Muller, C. J., Burgin, T. P. & Tour, J. M. Conductance of a molecular junction. Science 278, 252–254 (1997).
Guo, X. F. et al. Covalently bridging gaps in single-walled carbon nanotubes with conducting molecules. Science 311, 356–359 (2006).
Jia, C. et al. Covalently bonded single-molecule junctions with stable and reversible photoswitched conductivity. Science 352, 1443–1445 (2016).
Yuan, L., Jiang, L. & Nijhuis, C. A. The drive force of electrical breakdown of large-area molecular tunnel junctions. Adv. Funct. Mater. 28, 1801710 (2018).
Gehring, P. et al. Complete mapping of the thermoelectric properties of a single molecule. Nat. Nanotechnol. 16, 426–430 (2021).
Park, H. et al. Nanomechanical oscillations in a single-C60 transistor. Nature 407, 57–60 (2000).
Xu, B. & Tao, N. J. Measurement of single-molecule resistance by repeated formation of molecular junctions. Science 301, 1221–1223 (2003).
Reddy, P., Jang, S.-Y., Segalman, R. A. & Majumdar, A. Thermoelectricity in molecular junctions. Science 315, 1568–1571 (2007).
Zhao, C. et al. Electrical monitoring of single-event protonation dynamics at the solid-liquid interface and its regulation by external mechanical forces. Nat. Commun. 15, 8835 (2024).
Novoselov, K. S. et al. A roadmap for graphene. Nature 490, 192–200 (2012).
Jariwala, D., Sangwan, V. K., Lauhon, L. J., Marks, T. J. & Hersam, M. C. Carbon nanomaterials for electronics, optoelectronics, photovoltaics, and sensing. Chem. Soc. Rev. 42, 2824–2860 (2013).
Zhang, J. et al. Contacting individual graphene nanoribbons using carbon nanotube electrodes. Nat. Electron. 6, 572–581 (2023).
Niu, W. et al. Exceptionally clean single-electron transistors from solutions of molecular graphene nanoribbons. Nat. Mater. 22, 180–185 (2023).
Dief, M. E., Low, J. P., Díez-Pérez, I. & Darwish, N. Advances in single-molecule junctions as tools for chemical and biochemical analysis. Nat. Chem. 15, 600–614 (2023).
Xin, N. et al. Concepts in the design and engineering of single-molecule electronic devices. Nat. Rev. Phys. 1, 211–230 (2019).
Chen, Z. et al. Quantum interference enhances the performance of single-molecule transistors. Nat. Nanotechnol. 19, 986–992 (2024).
Yang, R. et al. An anisotropic etching effect in the graphene basal plane. Adv. Mater. 22, 4014–4019 (2010).
Luo, D. et al. Anisotropic etching of graphite flakes with water vapor to produce armchair-edged graphene. Small 10, 2809–2814 (2014).
Baek, J.-B., Lyons, C. B. & Tan, L.-S. Covalent modification of vapour-grown carbon nanofibers via direct Friedel–Crafts acylation in polyphosphoric acid. J. Mater. Chem. 14, 2052–2056 (2004).
Seo, J.-M., Tan, L.-S. & Baek, J.-B. Defect/edge-selective functionalization of carbon materials by “direct” Friedel-Crafts acylation reaction. Adv. Mater. 29, 1606317 (2017).
Li, M. et al. Graphene: Preparation, tailoring, and modification. Exploration 3, 20210233 (2023).
Ma, B., Ren, S., Wang, P., Jia, C. & Guo, X. Precise control of graphene etching by remote hydrogen plasma. Nano Res 12, 137–142 (2019).
Ma, B., Wang, P., Ren, S., Jia, C. & Guo, X. Versatile optical determination of two-dimensional atomic crystal layers. Carbon 109, 384–389 (2016).
Meyer, J. C., Girit, C. O., Crommie, M. F. & Zettl, A. Hydrocarbon lithography on graphene membranes. Appl. Phys. Lett. 92, 123110 (2008).
Bachmatiuk, A. et al. Low voltage transmission electron microscopy of graphene. Small 11, 515–542 (2015).
Li, M. et al. Precise edge functionalization and tailoring of graphene via solvent-controlled reactions. Carbon 197, 519–525 (2022).
Li, M. et al. Dipole-modulated charge transport through PNP-type single-molecule junctions. J. Am. Chem. Soc. 144, 20797–20803 (2022).
Wang, W., Lee, T. & Reed, M. A. Mechanism of electron conduction in self-assembled alkanethiol monolayer devices. Phys. Rev. B 68, 035416 (2003).
Park, J. et al. Coulomb blockade and the Kondo effect in single-atom transistors. Nature 417, 722–725 (2002).
Liu, S. et al. Single-molecule detection of proteins using aptamer-functionalized molecular electronic devices. Angew. Chem. Int. Ed. 50, 2496–2502 (2011).
Cao, Y. et al. Building high-throughput molecular junctions using indented graphene point contacts. Angew. Chem. Int. Ed. 51, 12228–12232 (2012).
Xu, Q. et al. Single electron transistor with single aromatic ring molecule covalently connected to graphene nanogaps. Nano Lett. 17, 5335–5341 (2017).
Ullmann, K. et al. Single-molecule junctions with epitaxial graphene nanoelectrodes. Nano Lett. 15, 3512–3518 (2015).
Sun, H. et al. Efficient fabrication of stable graphene-molecule-graphene single-molecule junctions at room temperature. ChemPhysChem 19, 2258–2265 (2018).
Mol, J. A. et al. Graphene-porphyrin single-molecule transistors. Nanoscale 7, 13181–13185 (2015).
Yu, Y.-J. et al. Tuning the graphene work function by electric field effect. Nano Lett. 9, 3430–3434 (2009).
Migliore, A. & Nitzan, A. Nonlinear charge transport in redox molecular junctions: A marcus perspective. ACS Nano 5, 6669–6685 (2011).
Migliore, A. & Nitzan, A. Irreversibility and hysteresis in redox molecular conduction junctions. J. Am. Chem. Soc. 135, 9420–9432 (2013).
Yan, C. et al. From molecular electronics to molecular intelligence. ACS Nano 18, 28531–28556 (2024).
Fu, H. et al. Dipole-improved gating of azulene-based single-molecule transistors. J. Mater. Chem. C. 10, 7803–7809 (2022).
Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Johnson, E. R. & Becke, A. D. A post-Hartree-Fock model of intermolecular interactions: Inclusion of higher-order corrections. J. Chem. Phys. 124, 174104 (2006).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Marenich, A. V., Cramer, C. J. & Truhlar, D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396 (2009).
Frisch, M. J. et al. Gaussian 16, Revision A.0.16 (Gaussian, Inc., Wallingford CT, 2016).
Acknowledgements
We thank Abraham Nitzan and Ruixi Qiao for their fruitful suggestions. We acknowledge Electron Microscopy Laboratory of Peking University, China for the use of Cs corrected Nion U-HERMES200 scanning transmission electron microscopy and FEI Tecnai F20 transmission electron microscopy. We acknowledge primary financial support from the National Key R&D Programme of China (2022YFE0128700 (to X.G.), 2021YFA1200101 (to Z.Y.), 2021YFA1200102 (to C.J.), 2023YFF1205803 (to X.G.), and 2024YFA1208100 (to C.J.)), the National Natural Science Foundation of China (22403004 (to M.L.), 22173050 (to C.J.), and 22172003 (to J.Z.)), and the Beijing National Laboratory for Molecular Sciences (BNLMS-CXXM-202407 (to X.G.)).
Author information
Authors and Affiliations
Contributions
X.G. and C.J. conceived the main idea of the paper. M.L., B.Y., and J.G. fabricated the devices. W.H. and M.L. performed the protonation dynamic experiment. X.K.G. and J.C. carried out the molecular synthesis. J.Z., M.L., Z.L. and B.Y. obtained the HRTEM/STEM characteristics. C.G. and C.Z. conducted the STM-BJ experiment. W.H. and C.Y. conducted the IETS experiments. X.X. obtained the fluorescent super-resolution imaging. B.W., J.W. and A.M. built and analysed the theoretical models. X.G., C.J., M.L., J.G., C.Z., Y.Z., Y.X., Z.Y. and A.M. analysed the data and wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, M., Yin, B., Wang, B. et al. Atomically precise construction of uniform single-molecule junctions for molecular electronics. Nat Commun 16, 7657 (2025). https://doi.org/10.1038/s41467-025-63113-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63113-x
