
Abstract
TRPA1 is an essential calcium (Ca2+)-permeable channel involved in nociception and inflammation. It exhibits complex and mechanistically elusive Ca2+ regulation with initial potentiation then rapid desensitization. We find that the universal Ca2+ sensor Calmodulin (CaM) binds TRPA1 in cells at rest and suppresses channel activity. Combining biochemical, biophysical, modeling, NMR spectroscopy, and functional approaches, we identify an evolutionarily conserved, high-affinity Ca2+/CaM binding element in the TRPA1 distal C-terminus. Genetic or biochemical perturbation of Ca2+/CaM binding to this site yields hyperactive channels that exhibit drastic slowing of desensitization with minor effect on potentiation. Higher extracellular Ca2+ partially rescues slowed desensitization. Our results identify a critical regulatory element in an unstructured TRPA1 region highlighting the importance of these domains, they reveal Ca2+/CaM is an essential TRPA1 auxiliary subunit required for proper channel function, and they suggest that Ca2+/CaM binding at this distal site stabilizes a long-range allosteric mechanism to drive rapid desensitization.
Similar content being viewed by others


Introduction
Calcium (Ca2+) is a unique ion since it regulates cell excitability and serves as a second messenger in signal transduction cascades involved in almost all aspects of cellular life1. Excessive Ca2+ entry is cytotoxic, and cells dedicate substantial resources to maintain a 20,000-fold lower cytoplasmic concentration than extracellular or endoplasmic reticulum stores1. Extracellular Ca2+ influx is facilitated by Ca2+-permeable ion channels whose activity must be tightly controlled to prevent spurious initiation of Ca2+ signaling pathways and apoptosis. In the peptidergic C fiber subset of peripheral sensory neurons, extracellular Ca2+ influx triggers pain signals and the exocytotic release of neuropeptides that initiate neurogenic inflammation and neuronal hypersensitivity2,3,4,5,6. These processes are believed to play a role in the transition from acute to chronic pain7,8,9. Identifying the Ca2+-permeable ion channels involved in initiating and maintaining neurogenic inflammation and determining their regulatory mechanisms could provide the basis for rational drug design to alleviate these symptoms.
A direct role for the wasabi receptor, TRPA1 (Transient Receptor Potential Ankyrin subtype 1), in human pain has been illustrated by the discovery and characterization of genetic variants that are associated with painful disorders10,11,12. TRPA1 is a homotetrameric Ca2+-permeable non-selective cation channel that is expressed at the plasma membrane in a subset of peripheral sensory neurons originating from dorsal root, trigeminal, and nodose ganglia as well as non-neuronal tissues including airway epithelia, enterochromaffin cells, and cardiac tissue (Fig. 1a, b)13,14,15,16,17,18. Mammalian TRPA1 is a ligand-gated chemosensor that is activated by covalent modification of conserved cytoplasmic cysteine residues by environmental and endogenous electrophiles, including allyl isothiocyanate (AITC), cinnamaldehyde, and acrolein (Fig. 1a, black circles)19,20,21,22. In sensory neurons, active TRPA1 channels then facilitate extracellular Ca2+ entry to initiate pain signals and neuropeptide release13,23,24,25,26,27,28. Neuropeptide-evoked immune responses release pro-inflammatory mediators that sensitize neurons to subsequent painful stimuli by activating or priming receptors, including TRPA113,29,30,31. The ability of TRPA1 to initiate pain and neurogenic inflammation, as well as be sensitized by these signals, places it in the center of a regulatory pathway that could go awry13. Accordingly, animal knockout studies show that TRPA1 plays a key role in the establishment of neurogenic inflammation and neuronal hypersensitivity, making it a prime drug target for managing chronic pain and inflammation15,32,33,34.

a Cartoon schematic of a full-length hTRPA1 monomeric subunit with relevant structural features, a previously identified Calmodulin binding domain (CaMBD, pink), and the CaM binding element discovered here (DCTCaMBE, yellow) denoted. Dashes and transparencies indicate structurally unresolved regions. Arrows indicate truncations used in this study. b Ribbon diagram of hTRPA1 atomic model for residues S448-T1078 (PDB: 6V9W). Each subunit colored differently for clarity. Features denoted as in (a). c WT hTRPA1 interacts with CaM in a Ca2+-dependent manner. Immunoblotting analysis of 3xFLAG-WT hTRPA1 after CaM-agarose pulldown at the indicated Ca2+ concentrations from lysates of HEK293T cells transfected with 3xFLAG-WT hTRPA1 or empty vector (mock). Tubulin from whole cell lysates (10%, inputs) was the loading control. d Quantification of CaM-agarose binding assays as in (c) at the indicated Ca2+ concentrations relative to the maximum enrichment within each replicate. n = 11. e Ratiometric Ca2+ imaging of HEK293T cells transfected with empty vector (mock) or WT hTRPA1 and empty vector (mock), WT CaM, or CaM1234. Cells were stimulated with 100 µM AITC. Scale bars indicate 50 µm. f Quantification of Fura-2 ratio data as from (e). n = 3 independent replicates. g Immunoblotting analysis of 3xFLAG-WT hTRPA1, WT CaM or CaM1234 protein expression in biotin-labeled plasma membranes from transfected cells. CaM was the negative control for plasma membrane localization. h Quantification of the plasma membrane localization of WT hTRPA1 relative to Tubulin inputs from data as in (g). n = 4, two-tailed Student’s t-test. i Co-expression with a functional CaM C-lobe suppresses WT TRPA1 activity. Ratiometric Ca2+ imaging of HEK293T cells transfected with WT hTRPA1 and WT or the indicated CaM mutants. Cells were stimulated with 100 µM AITC. Scale bars indicate 50 µm. j Quantification of 100 µM AITC-evoked change in Fura-2 ratio of data from (i). n = 3 (WT CaM, CaM12, CaM34, and CaM1234) or 4 (CaM C-lobe and CaM34 C-lobe) independent replicates. d, f, h, j Data represent mean ± SEM. P-values indicated in figure panels. d, f, j One-way ANOVA with Tukey’s post hoc analysis.
TRPA1 exhibits complex Ca2+ regulation; Ca2+ entry initially enhances channel activity (i.e., potentiation), but then causes rapid inactivation (i.e., desensitization) as cytoplasmic Ca2+ rises21,35,36. TRPA1 electrophile agonist modifications can persist for at least 10 min, yet TRPA1 desensitizes on a millisecond timescale35,37. Thus, Ca2+ regulation is critical to limit the TRPA1 functional window, however, the mechanism underlying this regulation is poorly understood. Previous work suggests that TRPA1 potentiation and desensitization are independent regulatory events that may engage distinct channel machinery36,38. Direct Ca2+ binding sites have been proposed in TRPA1 cytoplasmic and transmembrane domains to control potentiation and/or desensitization, however, deep mechanistic insight is lacking to explain whether and how these sites affect Ca2+ regulation and many of these sites remain controversial38,39,40. Moreover, most of these elements reside within or near key TRPA1 structural domains that contribute to channel gating. Genetic perturbation in these elements might compromise intrinsic channel structure or function, further complicating interpretation of their effects on Ca2+ regulation.
Ion channels can also be regulated by the universal Ca2+ sensor Calmodulin (CaM), which was recently shown to bind TRPA1 in a Ca2+-dependent manner and affect its Ca2+ regulation21,36,39,40,41. CaM was proposed to mediate these effects in part through a CaM binding domain (CaMBD) located adjacent to the TRP helix in the membrane proximal TRPA1 cytoplasmic C-terminus (Fig. 1a, b, CaMBD in pink and TRP helix in purple)41. Genetic perturbation of the CaMBD had modest effects on TRPA1 Ca2+ regulation, perhaps due to incomplete loss of CaM binding, which raised the intriguing possibility that TRPA1 contains another CaM binding site41. Here, we identify a previously unreported, highly conserved, high-affinity CaM binding element in the distal structurally unresolved TRPA1 cytoplasmic C-terminus (the DCTCaMBE) that we propose to be the main site for a TRPA1:Ca2+/CaM interaction (Fig. 1a, yellow). We show that TRPA1 binds CaM best at basal Ca2+ concentration through its DCTCaMBE and that this interaction can be ablated with short truncations, single point mutations, or a TRPA1 C-terminal peptide without affecting Ca2+-independent channel properties, allowing us to decouple Ca2+/CaM binding and TRPA1 function. TRPA1 DCTCaMBE mutants revealed that Ca2+/CaM binding to this site is dispensable for proper potentiation, but it is critically required for rapid desensitization, providing further support for mechanistic independence of these two regulatory events. To date, most CaM-regulated ion channels engage both the N- and C-lobes, wherein Ca2+-mediated conformational changes in CaM confer Ca2+ sensing to the regulated channel42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59. In contrast, we find that the TRPA1 DCTCaMBE exclusively binds the Ca2+/CaM C-lobe and that only the CaM C-lobe was necessary to suppress TRPA1 function. Thus, we propose that CaM binding per se likely does not directly trigger desensitization. Instead, we found desensitization-resistant TRPA1 channels could be partially rescued by increasing the extracellular Ca2+ concentration available to permeate through open channels. Thus, we present a model that Ca2+/CaM acts as a long-range allosteric regulator to prime an intrinsic TRPA1 Ca2+ binding site that is the true desensitization gate. In this way, we propose that Ca2+/CaM serves as a regulatory binding partner for TRPA1 akin to the auxiliary subunits of voltage-gated ion channels to establish the proper TRPA1 functional window in cells60,61,62,63,64.
Results
Ca2+/calmodulin regulates TRPA1 in cells via its C-lobe
To initially ask whether CaM can directly regulate TRPA1, we assayed wild type (WT) human TRPA1 (hTRPA1) for its ability to bind CaM-agarose at distinct Ca2+ concentrations. Consistent with literature precedence41, hTRPA1 failed to bind CaM in the absence of Ca2+ (Fig. 1c, d, 0 µM Ca2+). Instead, we observed a robust interaction with hTRPA1 at resting, cytosolic Ca2+ concentration (0.1 µM) with a stepwise decrease in binding at intermediate (500 µM) and extracellular (2 mM) Ca2+ concentrations (Fig. 1c, d). These results are consistent with prior results that TRPA1 binds Ca2+/CaM, but not apo CaM41.
We next probed whether CaM regulates TRPA1 function by co-expressing WT hTRPA1 in HEK293T cells with WT CaM or a CaM variant that has a point mutation in each EF hand that prevents Ca2+ binding (CaM1234), which was previously shown to be unable to bind to TRPA141, and monitoring channel activity by ratiometric Ca2+ imaging. Cells co-expressing WT CaM exhibited reduced AITC-evoked hTRPA1 activity compared to cells co-transfected with empty vector or cells co-expressing CaM1234 (Fig. 1e, f, solid black versus grey or striped). These differences in channel activity were not due to WT or mutant CaM-induced changes to hTRPA1 expression or subcellular localization (Fig. 1g, h and Supplementary Fig. 1a).
Previous work showed that co-expression with WT CaM or a CaM mutant with only a functional C-lobe (CaM12) enhanced mouse TRPA1 channel desensitization, demonstrating that the CaM C-lobe is critical to this regulation41. In our ratiometric Ca2+ imaging assay, enhanced desensitization would appear as reduced channel activity. To ask which CaM lobes are important to our observed activity suppression, we co-expressed WT hTRPA1 with different CaM constructs. Consistent with prior electrophysiological recordings41, we found that CaM12 significantly suppressed hTRPA1 activity, while a CaM mutant that only retained a functional N-lobe (CaM34) did not (Fig. 1i, j, black boxed versus carat bars). These results hint that the CaM C-lobe is sufficient to mediate hTRPA1 regulation; however, it is possible that the CaM N-lobe, independent of Ca2+ binding competency, is also required. Thus, we co-expressed the isolated CaM C-lobe or a Ca2+-binding deficient mutant (CaM34 C-lobe) with WT hTRPA1 and assayed for channel activity. Such experiments revealed that the CaM C-lobe suppressed hTRPA1 activity akin to WT CaM and CaM12, while the CaM34 C-lobe mutant did not (Fig. 1i, j, gray versus white bars). Importantly, none of the CaM constructs affected hTRPA1 expression (Supplementary Fig. 1b).
Collectively, these results suggest that CaM suppresses WT hTRPA1 activity predominantly through its C-lobe. This recapitulates and expands on prior electrophysiological data showing that the CaM C-lobe is necessary for TRPA1 regulation41. While HEK293T cells endogenously express WT CaM, a large majority is likely pre-bound to other effector proteins, leaving low levels of free CaM65. hTRPA1 overexpression seemingly overwhelms this free CaM pool, yielding enhanced channel activity, which can be rescued by concurrent overexpression of WT CaM (Fig. 1e, i). Moving forward, we sought to identify the site and mode of Ca2+/CaM binding as well as the role for Ca2+/CaM binding in TRPA1 regulation.
TRPA1 distal C-terminus contains a critical Ca2+/CaM binding element
A Ca2+/CaM binding domain (CaMBD) was previously identified near the TRP helix, which is involved in channel gating and resides within the structurally resolved core of hTRPA1 (Fig. 1a, b, pink)41. While the CaMBD was proposed to contribute to TRPA1 Ca2+ regulation, mutagenesis of this site did not ablate Ca2+/CaM binding and suggested that TRPA1 contains another Ca2+/CaM binding element41. To ask whether and to what extent the CaMBD governs the hTRPA1-Ca2+/CaM interaction, we tested a minimal hTRPA1 construct that was previously used for structural determination66 that retains the CaMBD (hTRPA1S488-T1078) for CaM binding (Fig. 1a, b and Supplementary Fig. 2). However, this minimal hTRPA1 construct failed to bind CaM in vitro, indicating that the CaMBD is not sufficient for mediating this interaction (Fig. 2a).

a–c, f Representative immunoblotting analysis of 3xFLAG-tagged WT or truncated hTRPA1 constructs after CaM-agarose pulldown at the indicated Ca2+ concentrations from lysates of transfected HEK293T cells. Samples were probed as in Fig. 1c. Data are representative of three (a, b, f) or six (c) independent experiments. Truncations in (a–c) are diagramed in Fig. 1a. d Quantification of CaM-agarose enrichment of 3xFLAG-WT (grey) or Δ1–61 hTRPA1 (black) at the indicated Ca2+ concentrations relative to the maximum enrichment within each replicate. Data represent mean ± SEM. n = 6, two-tailed Student’s t-test. P-values indicated in figure. e Simplified cartoon schematic from Fig. 1a denoting sites of truncations used in (a–d) and a suite of hTRPA1 unresolved C-terminus truncations tested in (f).
The hTRPA1S488–T1078 construct truncates the disordered N-terminus (amino acids 1–61), the structurally unresolved ankyrin repeats (ARs, amino acids 61-447 containing AR1-11), and the structurally unresolved distal C-terminus (amino acids 1079–1119), suggesting one or more of these domains contain critical CaM binding sites (Fig. 1a, dashed and transparent regions). TRPA1 is unique among the mammalian TRP channels in that it has the largest N-terminal AR domain (ARD) consisting of 16 ARs (Fig. 1a)17,67. Though a functional or regulatory role for the large TRPA1 ARD is unclear, ARs are known to mediate protein-protein interactions and human-rattlesnake TRPA1 chimeras with swaps in the ARD affected Ca2+ regulation raising the possibility that the TRPA1 ARD contains a CaM binding site68,69. Surprisingly, we found that a hTRPA1 construct containing the full ARD but lacking the structurally unresolved N- and C-termini (hTRPA1M62–T1078) failed to bind CaM indicating that a critical CaM binding element resides in one or both of those regions (Fig. 2b). Thus, we made hTRPA1 truncations lacking only the unresolved N- or C-termini (hTRPA1Δ1–61 and hTRPA1Δ1079–1119, respectively) and found that while loss of the unresolved N-terminus did not affect Ca2+/CaM binding compared to WT hTRPA1, loss of the unresolved C-terminus ablated Ca2+/CaM binding at each Ca2+ concentration (Fig. 2c, d).
The hTRPA1 unresolved C-terminus includes the terminal 41 amino acids, which accounts for only 3.7% of the channel sequence. An acidic cluster in this domain proximal to the coiled coil was previously proposed to be involved in Ca2+ regulation, indicating a role for the unresolved C-terminus in channel modulation38. To initially map the distal C-terminal CaM binding element (DCTCaMBE), we built a suite of truncations lacking sequential blocks of 10 residues (Fig. 2e) and assayed them for CaM binding. We found that even the smallest of these truncations lacking only the terminal 11 amino acids (hTRPA1Δ1109–1119) failed to bind CaM at basal Ca2+ concentration (Fig. 2f) indicating that at least part of the DCTCaMBE resides within the extreme end of the hTRPA1 C-terminus.
Loss of Ca2+/CaM binding yields hyperactive TRPA1 channels
Identification of the DCTCaMBE in the distal C-terminus far from the hTRPA1 core structure raises the intriguing possibility that we could selectively ablate Ca2+/CaM binding without compromising intrinsic channel function. Such a construct would facilitate a detailed characterization of whether and how CaM regulates hTRPA1 without relying on CaM mutants or inhibitors that may disrupt other aspects of CaM biology or that could indirectly impact channel function. Excitingly, the hTRPA1Δ1109–1119 construct yielded functional channels with increased basal and agonist-evoked activity compared to WT hTRPA1 with both the electrophile agonist AITC and the non-electrophile agonist carvacrol (Fig. 3a, b and Supplementary Fig. 3). This increased activity was not due to enhanced mutant protein production or surface localization (Fig. 3c, d and Supplementary Fig. 3c) and instead suggests that loss of Ca2+/CaM binding yields hyperactive hTRPA1 channels.

a Ratiometric Ca2+ imaging of HEK293T cells transfected with 3xFLAG-WT or Δ1109–1119 hTRPA1. Cells were stimulated with AITC (10 or 100 µM). Scale bars indicate 50 µm. b Quantification of baseline, 10 µM or 100 µM AITC-evoked changes in Fura-2 ratio from cells as in (a) transfected with WT (black) or Δ1109–1119 hTRPA1 (red). n = 4 independent experiments, n ≥ 90 cells per transfection condition per experiment, two-tailed Student’s t-test. c Surface biotinylation. Immunoblotting analysis of 3xFLAG-Δ1109–1119 hTRPA1, WT CaM or CaM1234 protein expression in biotin-labeled plasma membranes as in Fig. 1g. CaM was the negative control for plasma membrane localization. d Quantification of the plasma membrane localization of 3xFLAG-WT (grey) or Δ1109–1119 hTRPA1 (red) relative to Tubulin. WT hTRPA1 quantification is repeated from Fig. 1h. n = 4 independent experiments, one-way ANOVA with Bonferroni’s post hoc analysis. e Ratiometric Ca2+ imaging of HEK293T cells co-transfected with 3xFLAG-Δ1109–1119 hTRPA1 and 3xFLAG-WT CaM or CaM1234. Cells were stimulated with 100 µM AITC. Scale bars indicate 50 µm. f Quantification of 100 µM AITC-evoked change in Fura-2 ratio of data as in (e) of cells co-expressing Δ1109–1119 hTRPA1 and WT CaM (red bars) or CaM1234 (striped bars). n = 4 independent experiments, n ≥ 90 cells per transfection condition per experiment, two-tailed Student’s t-test. b, d, f Data represent mean ± SEM. P-values indicated in figure panels.
To directly test whether the hTRPA1Δ1109–1119 construct relieves Ca2+/CaM-mediated channel suppression, hTRPA1Δ1109–1119 was co-expressed in HEK293T cells with WT CaM or CaM1234. Unlike WT hTRPA1 (Fig. 1e, f, i, j), we found that hTRPA1Δ1109–1119 channels were unaffected by CaM co-expression (Fig. 3e, f) consistent with a loss of Ca2+/CaM binding capability (Fig. 2f). Akin to WT channels, WT CaM and CaM1234 had no effect on hTRPA1Δ1109–1119 expression or surface localization (Fig. 3c, and Supplementary Fig. 1c).
The canonical hTRPA1 Ca2+ regulation profile can be observed in Xenopus laevis oocytes by two-electrode voltage clamp (TEVC), as previously reported, providing a system in which to study how loss of CaM binding affects TRPA1 Ca2+ regulation (Fig. 4a)10,69. First, we characterized the Δ1109–1119 hTRPA1 mutant compared to WT channels for its opening and closing kinetics in the absence of Ca2+, its peak current amplitudes, as well its AITC sensitivity to ensure that this mutation did not compromise channel integrity (Fig. 4a–c and Supplementary Fig. 4). These control experiments revealed that the Δ1109–1119 hTRPA1 mutant exhibits gating kinetics and peak current amplitudes that were not significantly different from WT hTRPA1 as well as slightly reduced AITC sensitivity (Fig. 4c, timepoint i and Supplementary Fig. 4). Thus, we compared AITC-evoked WT and Δ1109–1119 hTRPA1 activities by TEVC in the presence of extracellular Ca2+ to directly discern how loss of Ca2+/CaM binding affects channel activity. Application of extracellular Ca2+ triggered rapid potentiation of WT and Δ1109–1119 hTRPA1 channels with comparable peak current amplitudes, suggesting that loss of Ca2+/CaM binding to the TRPA1 DCTCaMBE does not impact potentiation (Fig. 4a–c, timepoint ii). While WT hTRPA1 channels subsequently exhibited nearly complete desensitization within 25 s of extracellular Ca2+ addition (Fig. 4a, timepoint iii), Δ1109–1119 channels remained open with 20–30% its initial activity 5 min after extracellular Ca2+ application (Fig. 4b, c, timepoint iv). This residual activity could be inhibited with the TRPA1 antagonist A-967079, demonstrating that the ongoing activity was due to hTRPA1 channels (Fig. 4b, timepoint v).

Representative time-traces at −80 and +80 mV holding potentials from oocytes expressing WT (a, black) or Δ1109–1119 hTRPA1 (b, red). Current evoked with 250 µM AITC in the absence (orange i) and presence (blue ii) of 1.8 mM extracellular Ca2+. Channels were blocked with 10 µM A-967079 (grey v). Dashed line denotes 0 µA current. Protocol of condition application indicated above. Boxed below are the corresponding current-voltage relationships from timepoints indicated by black o (baseline), orange i (AITC without Ca2+), blue ii (AITC with Ca2+), purple iii (25 s after Ca2+ addition), green iv (5 min after Ca2+ addition), and grey v (A-967079 inhibited). c Quantification of peak current amplitudes at +80 mV (above) and −80 mV (below) before (orange i, left), directly after (blue ii, middle), and 5 min after (green iv, right) Ca2+ addition. d Calculated time constants of desensitization at −80 mV (left) and +80 mV (right) from fitting data as in (a, b) to a single-exponential function. e Quantification of charge translocation (µA•s, µC, e.g., area under the curve) at −80 mV (left) and +80 mV (right) from oocytes used as in (a, b). c–e Colors as indicated in (a, b). n = 10 oocytes per condition. Data represent mean ± SEM. Two-tailed Student’s t-test. P-values indicated in figure panels.
To quantitatively characterize the effect of Ca2+/CaM binding on proper TRPA1 Ca2+ regulation, we calculated rate constants for potentiation and desensitization from baseline-corrected traces at −80 and +80 mV holding potentials. Such calculations reveal that WT hTRPA1 potentiated at a rate of 2.7 s (−80 mV) and 2.2 s (+80 mV) and desensitized at a rate of 11.9 s (−80 mV) and 12.5 s (+80 mV) following Ca2+ addition, consistent with literature precedence (Fig. 4d and Supplementary Fig. 5a, black bars)36,38. In contrast, Δ1109–1119 hTRPA1 exhibited significantly delayed desensitization kinetics with 10-fold slowing to desensitization with inward currents (i.e., −80 mV) and 16-fold slowing to desensitization kinetics with outward currents (i.e.,+80 mV) compared to WT hTRPA1 channels (Fig. 4d, red bars). This fold-slowing to Ca2+-dependent desensitization kinetics are on the order of that previously reported for a TRPA1 mutant exhibiting 100-fold reduced Ca2+ permeability and may, in part, reflect channel rundown independent of true Ca2+ desensitization36. Potentiation kinetics were more modestly slowed for Δ1109–1119 hTRPA1 compared to WT hTRPA1 (~3-fold at −80 mV and ~1.8-fold at +80 mV), though this can be largely accounted for by the greater effects to desensitization which would inherently provide more time for full potentiation (Supplementary Fig. 5a red bars). Thus, these results suggest that Ca2+/CaM binding at the TRPA1 distal cytoplasmic C-terminus specifically affects rapid desensitization.
Such drastic delays to channel desensitization allows for sustained ion conduction through active mutant channels over a prolonged period, which could have significant physiological effects, including delivery of additional channels to the plasma membrane. Δ1109–1119 hTRPA1 channels exhibited 8-fold and 5-fold increases in translocated charge at −80 mV and +80 mV, respectively (Fig. 4e, red bars). These collective differences in Δ1109–1119 hTRPA1 channel function are not due to changes in total protein expression (Supplementary Fig. 1d). Together, these results suggest that the hTRPA1 distal C-terminus contains a critical DCTCaMBE that is required for Ca2+/CaM binding and its negative regulation of channel activity through proper promotion of channel desensitization.
TRPA1 and CaM co-localize in cells
The pronounced Ca2+/CaM binding we observed at basal Ca2+ concentration (Fig. 1c, d, 100 nM Ca2+) raises the intriguing possibility that WT hTRPA1 and Ca2+/CaM form a stable complex at rest and that this interaction is lost in the Δ1109–1119 hTRPA1 mutant. Consistently, deconvoluted immunofluorescence (IF) images of Neuro2A cells co-expressing V5-tagged WT CaM and 3xFLAG-tagged WT hTRPA1 exhibited pronounced co-localization of WT hTRPA1 and WT CaM in resting cells (Fig. 5a, left). In contrast, the 3xFLAG-tagged Δ1109–1119 hTRPA1 channels showed greatly reduced co-localization with WT CaM in Neuro2A cells (Fig. 5a, right). To further assess the degree of co-localization between CaM and WT hTRPA1 or the Δ1109–1119 hTRPA1 mutant, we quantified the V5-CaM and 3xFLAG-TRPA1 intensities at every pixel in individual cells and calculated Pearson’s correlation coefficients (R) (Supplementary Fig. 6)70. These results revealed a significant reduction in the CaM:TRPA1 co-localization with the Δ1109–1119 hTRPA1 mutant compared to WT channels (Fig. 5b), suggesting that the loss of Ca2+/CaM binding in pulldown experiments translates to a significant disruption of the relative spatial distribution of TRPA1 and CaM in a cellular environment due to reduced protein–protein interaction.

a Representative deconvoluted Airyscan images of Neuro2A cells co-expressing 3xFLAG-WT (left) or Δ1109–1119 hTRPA1 (right) with V5-WT CaM. Cells were stained with anti-FLAG (red) and anti-V5 (green) antibodies. Scale bar indicates 2 µm. Images are representative of 3 independent experiments. b Pearson’s correlation coefficients (R) were determined for WT hTRPA1:WT CaM or Δ1109–1119 hTRPA1:WT CaM using raw images (see Supplementary Fig. 6). Data represent mean ± SD. n = 41 (WT) or 40 (Δ1109–1119) cells from 3 independent experiments, two-tailed Student’s t-test. n > 10,000 pixels in 1 cell per condition. c Proximity biotinylation approach. TurboID (orange star) fused to the CaM C-lobe (green circle) is co-expressed with hTRPA1 variants (grey) in cells. Addition of biotin (pink star) to the media will facilitate generation of a local reactive biotin cloud (purple) that will biotinylate TurboID-CaM C-lobe and TRPA1, pending an interaction. d Immunoblotting analysis of biotinylated 3xFLAG-WT, Δ1109–1119, or W1103A hTRPA1 co-expressed with empty vector (-), 3xFLAG-TurboID-CaM C-lobe, or 3xFLAG-TurboID in HEK293T cells. FLAG immunoprecipitated eluates were probed for biotinylation with Streptavidin-HRP. FLAG-tagged proteins were probed using an anti-FLAG antibody. Tubulin was the loading control. Blots are representative of four independent experiments. e Quantification of the percent change to TRPA1 biotinylation with TurboID-CaM C-lobe versus TurboID from experiments as in (d). Pulldown Streptavidin-HRP (e.g., biotinylation) was normalized to FLAG and Tubulin from inputs for each sample. Biotinylation by TurboID was subtracted from biotinylation by TurboID-CaM C-lobe. Data are presented as the percent change from biotinylation with TurboID. Data represent mean ± SEM. n = 4 independent experiments, one-way ANOVA with Bonferroni’s post hoc analysis. b, e P-values indicated in figure panels.
We next employed a proximity biotinylation assay to determine whether a direct protein-protein interaction between WT hTRPA1 and CaM occurs in intact cells at basal Ca2+ levels and whether this interaction is lost with the Δ1109–1119 hTRPA1 mutant71. TurboID-fused CaM C-lobe or free TurboID tag were co-expressed with WT hTRPA1 or the Δ1109–1119 hTRPA1 mutant and biotinylated proteins were detected in FLAG immunoprecipitation eluates (Fig. 5c). These experiments revealed that both TRPA1 constructs were biotinylated by the free TurboID tag, consistent with TRPA1 and cytoplasmic TurboID proteins occupying similar subcellular spaces (Fig. 5d). In contrast, WT hTRPA1 exhibited an approximately 40% increase in biotinylation when co-expressed with the TurboID-fused CaM C-lobe construct suggesting a direct interaction in cells (Fig. 5d, e). Interestingly, the Δ1109–1119 hTRPA1 mutant exhibited reduced biotinylation with the TurboID-fused CaM C-lobe, which may be due to sequestration of TurboID-tagged CaM C-lobe by endogenous CaM effector proteins that would limit the stochastic interaction of TRPA1 with cytoplasmic TurboID (Fig. 5d, e). Collectively, these results support the model that WT TRPA1 and Ca2+/CaM associate at basal Ca2+ concentrations in cells.
TRPA1 distal C-terminus has high affinity for Ca2+/CaM C-lobe
We next sought to quantify the affinity of the hTRPA1-Ca2+/CaM interaction. While quantitative binding assays with membrane proteins are particularly challenging, we developed hTRPA1 distal C-terminus peptides for such analyses. Maltose binding protein (MBP)-tagged versions of the full hTRPA1 unresolved C-terminus (hTRPA11079–1119) and smaller fragments (hTRPA11089–1119 and hTRPA11099–1119) were expressed in HEK293T cells and tested for their ability to bind Ca2+/CaM. Each bound CaM in a Ca2+-dependent manner with no binding observed with free MBP tag, confirming that the C-terminus contains a DCTCaMBE (Supplementary Fig. 7a). The hTRPA11089–1119 peptide was selected for binding studies as it afforded the highest-yield bacterial purifications (Supplementary Fig. 7b). Importantly, the purified hTRPA11089–1119 peptide competed with WT hTRPA1 for Ca2+/CaM binding confirming that the isolated element can adopt a conformation amenable to Ca2+/CaM binding after bacterial purification and liberation from MBP (Supplementary Fig. 7c, d).
To initially characterize the hTRPA1 DCTCaMBE:Ca2+/CaM interaction, we performed size exclusion chromatography (SEC)-based binding assays with the hTRPA11089–1119 peptide and a suite of purified CaM constructs (Supplementary Fig. 7e–h). These assays were performed by mixing equimolar amounts (100 µM) of peptide and CaM construct in the presence of 2 mM free Ca2+ to promote uniform complex formation and to simplify data interpretation. While hTRPA11089–1119 and WT CaM eluted as spatially separate peaks when analyzed independently (Supplementary Fig. 8a, blue and yellow traces), mixed hTRPA11089–1119 and WT CaM eluted as a single peak slightly larger than free WT CaM with higher 280 nm absorbance, indicating all available hTRPA11089–1119 peptide was bound in a stable complex with CaM (Fig. 6a, green trace and inset 1 and Supplementary Fig. 8a, green trace and insets 1 and 2). This complex was disrupted by chelating Ca2+ with 5 mM EGTA, confirming the Ca2+-dependence of the interaction (Fig. 5a, black trace). Moreover, the Ca2+-insensitive CaM1234 mutant failed to bind the TRPA1 peptide consistent with no interaction between apo CaM and the TRPA1 distal C-terminus (Supplementary Fig. 8b, red trace). Fractions from the SEC runs revealed co-elution of the hTRPA11089–1119 peptide with WT CaM in the presence, but not the absence of Ca2+ (Fig. 6a, inset 2). To simulate a low Ca2+ environment72, we performed similar experiments with CaM12, which revealed that it also forms a complex with all available hTRPA11089–1119 peptide only in the presence of Ca2+ (Fig. 6b and Supplementary Fig. 8c, green versus black trace). These results were similarly supported by fractions from the SEC runs (Fig. 6b, inset 2). Our Ca2+ imaging experiments suggested that the CaM N-lobe is not required for TRPA1 regulation by Ca2+/CaM. To ask whether the N-lobe is required for CaM binding to the hTRPA11089–1119 peptide, we performed SEC assays with the CaM C-lobe, which revealed that the CaM C-lobe can engage the hTRPA11089–1119 peptide in isolation (Fig. 6c, green trace). These results suggest that neither the N-lobe nor Ca2+ binding in the N-lobe is required for the hTRPA1 DCTCaMBE-Ca2+:CaM interaction and that the CaM C-lobe is likely responsible for mediating the Ca2+-dependent binding to the hTRPA1 distal C-terminus.

Superdex 75 chromatograms of WT CaM (a) or CaM12 (b) with hTRPA11089–1119 in the presence of 2 mM Ca2+ (green) or 5 mM EGTA (black). Inset 1 are zoom ins on the CaM and CaM:peptide peaks (peaks labeled 1 and 2). Inset 2 are Coomassie stains of fractions labeled 1 (green trace), 2 (black trace), and 3 (black trace). Arrows indicate CaM constructs and hTRPA11089–1119 peptide. c Superdex 75 chromatograms of CaM C-lobe alone (black) or with hTRPA11089–1119 in the presence of 2 mM Ca2+ (green). Chromatograms in a–c were generated from 100 µM protein for each construct. Data are representative of three independent replicates. d, e The TRPA1 distal C-terminus has a higher affinity for CaM12 than WT CaM. Representative isothermal titration calorimetry plots of CaM12 (d) or WT CaM (e) titrated by hTRPA11089–1119 peptide at 2 mM Ca2+ and fitted using the one-site binding model. Values for the number of binding sites (N) and the binding constant Kd are shown.
To quantitatively measure the hTRPA11089-1119 peptide:Ca2+/CaM interaction we used isothermal titration calorimetry (ITC). As with the SEC assays above, these assays were performed with 2 mM Ca2+ to reduce heterogeneity and to simplify data interpretation. Low and high Ca2+ levels were simulated with CaM12 and WT CaM, respectively as reported previously73,74. Three independent runs of the TRPA11089-1119 peptide with CaM12 and WT CaM revealed affinities centering on 100 nM and 613 nM, respectively (Fig. 6d, e, Supplementary Fig. 8d, e). The ITC results were not due to titration artifacts from the peptide (Supplementary Fig. 8f) and these binding affinities are within range of those reported for CaM-regulated ion channels73,75,76,77,78. The higher affinity of CaM12 than WT CaM for the hTRPA11089–1119 peptide suggests that CaM undergoes a conformational change upon full Ca2+-loading that reduces the binding affinity, which has been observed with some CaM complexes; however, we do not think that this change in affinity is enough to dissociate Ca2+/CaM from TRPA1 upon Ca2+ influx (see “Discussion”)79,80,81. These affinities also provide insight into why we saw a reduced TRPA1:Ca2+/CaM interaction at higher Ca2+ concentrations in our CaM-agarose assays (Fig. 1c, d). Notably, we only observed a single binding event with both CaM12 and WT CaM indicating that the CaM N-lobe does not also bind the effector to mediate a bridged interaction between adjacent subunits as shown previously for TRPV673. Such lobe specificity has been observed for Ca2+/CaM binding sites in other ion channels50,77,78,82. Together with our IF imaging and proximity biotinylation data (Fig. 5), these results indicate that hTRPA1 is a physiologically relevant binding partner for CaM, that the Ca2+/CaM interaction is predominantly mediated by the C-lobe, and that hTRPA1 and Ca2+/CaM may pre-associate in cells prior to channel activation.
TRPA1 distal C-terminus contains highly conserved DCTCaMBE
To initially determine how hTRPA1 binds CaM, we modeled the TRPA1-Ca2+/CaM interaction by using the TRPV1-Ca2+/CaM crystal structure as the template83. First, the hTRPA11089–1119 sequence was aligned with the TRPV1 distal C-terminal (CT) CaM binding segment peptide (rat TRPV1784–798), which revealed a best alignment with hTRPA11100–1114 (Supplementary Fig. 9a). Second, this minimal alignment was used to generate the homology model with the Modeller Comparative tool in ChimeraX (Fig. 7a)84. This model predicts that the CaM C-lobe binds to an hTRPA1 C-terminal alpha helix at a motif spanning amino acids 1102–1111 with W1103 forming a hydrophobic anchor akin to W787 in the TRPV1 CT (Fig. 7b). Interestingly, while the TRPV1 CT also engages the CaM N-lobe through a second hydrophobic anchor (L796), this site is an alanine in hTRPA1 (A1112) suggesting that the hTRPA1 distal C-terminus will not similarly engage the CaM N-lobe consistent with the binding results above (Figs. 7b, 1c, 1d, and 6).

a Homology model of TRPA1 distal C-terminus (residues 1100–1114, yellow) bound to Ca2+/CaM. WT CaM atomic model with N-lobe (dark green, residues 1–79) and C-lobe (light green, residues 80–149) denoted. Homology model built with the Modeller Comparative tool in ChimeraX with the TRPV1 distal C-terminal CaM binding segment (CT, blue)-Ca2+/CaM X-ray crystal structure as the template (PDB: 3SUI). Sequence alignment between TRPV1 and TRPA1 used for model building is in Supplementary Fig. 9a. This model yielded a z-DOPE score of 0.1256. Ca2+ ions bound to the four CaM EF hands are modeled in pink. b Ribbon diagram of homology model from (a). Key TRPV1 and TRPA1 residues involved in mediating these interactions are depicted as balls and sticks. c Immunoblotting analysis of the indicated 3xFLAG-hTRPA1 constructs after CaM-agarose pulldown in the absence or presence of Ca2+ from lysates of HEK293T cells transfected with 3xFLAG-WT, W1103A, L1107A, V1110A, W1103A/V1106A/L1107A/V1110A (Quad), or R1102A/K1111A (RK/AA) hTRPA1. Samples were probed as in Fig. 1c. d Quantification of CaM-agarose pulldowns represented in (c). Pulldown was normalized to the WT hTRPA1 with Ca2+ average. Data represent mean ± SEM. P values indicated in figure panel. n = 3 independent experiments, one-way ANOVA with Tukey’s post hoc analysis. e Sequence alignment of nine TRPA1 orthologues aligned to residues 1089–1112 of hTRPA1. Alignment generated with Sequence Logo. Box denotes TRPA1 DCTCaMBE. Hydrophobic (orange) and salt bridge (blue) residues proposed to form the DCTCaMBE are denoted with stars. f Immunoblotting analysis of the indicated MBP-tagged TRPA1 species orthologues after CaM-agarose pulldown in the absence or presence of Ca2+ from lysates of HEK293T cells transfected with MBP-WT mouse, zebrafish TRPA1 paralog a (zA1a), or C. elegans TRPA1 or the Δ1109–1119 equivalents of mouse (Δ1111–1125) or zebrafish (Δ1101–1115) TRPA1. Blot is representative of four independent experiments. Samples were probed using an anti-MBP primary antibody. Tubulin from whole cell lysates (10%, inputs) was the loading control.
Like the TRPV1 CT peptide, the residues forming the putative hTRPA1 DCTCaMBE contain a hydrophobic ridge comprised of residues W1103, V1106, L1107, and V1110 which bind the hydrophobic cleft of the CaM C-lobe (Fig. 7b). Additionally, this hydrophobic ridge is flanked on the N-terminal side by R1102 and on the C-terminal side by K1111, which adopt similar positions as the TRPV1 CT R785 and R797, respectively (Fig. 7b). This putative DCTCaMBE is consistent with loss of CaM binding in the Δ1109–1119 hTRPA1 truncation, which lacks the V1110 and K1111 residues (Fig. 2f).
To discern which of these residues comprise the DCTCaMBE, we built a suite of hTRPA1 alanine mutations and analyzed them for CaM binding. Individual mutation of W1103 or V1110 to alanine completely ablated CaM binding at basal Ca2+ concentration with a partial reduction in binding by a L1107A mutant (Fig. 7c, d). Mutating all four hydrophobic residues (Quad) or the flanking R1102 and K1111 residues (RK/AA) to alanine similarly ablated CaM binding at basal Ca2+ concentration (Fig. 7c, d). These data support that R1102, W1103, V1106, L1107, V1110, and K1111 form the hTRPA1 DCTCaMBE, consistent with the modeling prediction (Fig. 7e, box).
Prior work demonstrated that CaM regulates the mouse TRPA1 channel41. The hTRPA1 DCTCaMBE identified in this study is highly conserved among vertebrate TRPA1, including the mouse orthologue, but it is absent from C. elegans TRPA1 (Fig. 7e and Supplementary Fig. 9b). To ask whether the DCTCaMBE governs CaM binding in other TRPA1 species orthologues, we assayed the CaM binding ability of WT and partial DCTCaMBE truncations of mouse, zebrafish, and C. elegans TRPA1. WT mouse and zebrafish TRPA1 constructs bound CaM in a Ca2+-dependent manner (Fig. 7f). In contrast, WT C. elegans TRPA1 and the partial DCTCaMBE truncations of mouse and zebrafish TRPA1 failed to bind CaM (Fig. 7f). These results are consistent with the DCTCaMBE serving as a critical and highly conserved Ca2+/CaM binding element in many, but not all TRPA1 channels.
NMR spectroscopy supports DCTCaMBE-Ca2+/CaM C-lobe binding mode
We next empirically investigated the CaM lobe specificity of the Ca2+/CaM:TRPA1 C-terminal peptide interaction by collecting 15N-1H HSQC NMR spectra of 15N-labeled CaM12 or WT CaM alone and with unlabeled hTRPA11089–1119 peptide at 1:1 ratios (Fig. 8a, b). We chose to perform these experiments with a 1:1 CaM:hTRPA11089–1119 ratio since our SEC shift assays and ITC measurements demonstrated a single and saturated binding event at this ratio (Fig. 6a, b, d, e). Chemical shift perturbations (CSPs), which yield residue specific information regarding the binding interaction, were observed predominantly in the C-lobe for both CaM12 and WT CaM in the presence of the hTRPA11089–1119 peptide, consistent with a C-lobe driven interaction (Fig. 8c, red and black, respectively). Moreover, CaM12 exhibited more CSPs in the C-lobe than WT CaM, consistent with a more engaged binding event (Fig. 8c, red versus black). To determine whether there is a conformational change between the CaM12:TRPA1 peptide and the WT CaM:TRPA1 peptide complexes that might explain the differences in binding affinities observed above, we subtracted the CSPs for CaM12:TRPA11089–1119 (Fig. 8a) from those for CaMWT:TRPA11089–1119 (Fig. 8b), which yields differential binding effects between the two complexes (Supplementary Fig. 9c). This analysis was used previously to reveal CaM N-lobe engagement of a TRPV6 peptide with WT CaM73. Here, it revealed a loss of peaks for many C-lobe residues as well as some increased peaks at residues within the linker (residues 73–83), supporting a differential engagement of the TRPA1 peptide by the C-lobe, and only the C-lobe, in CaM12 versus WT CaM. This is likely due to a conformational change that may account for the weaker binding affinity in the fully Ca2+-loaded state.

a Overlay of the 15N-1H HSQC spectra of 15N-labled CaM12 (black) with the 15N-labeled CaM12:TRPA11089–1119 complex (red) at a 1:1 molar ratio. Insets depict expanded view of the CSPs boxed in blue. b Overlay of the 15N-1H HSQC spectra of 15N-labled CaMWT (black) with the 15N-labeled CaMWT:TRPA11089–1119 complex (purple) at a 1:1 molar ratio. Insets depict expanded view of the CSPs boxed in blue. a, b Higher resolution spectra are provided in Supplementary Fig. 16 and 17. c CaM12 (red) and CaMWT (black) chemical shift perturbations (CSPs) (δbound − δfree) as a function of residue number for the 15N-labeled CaM12:TRPA11089–1119 or 15N-labeled CaMWT:TRPA11089–1119 complex as in (a, b), respectively. Dark and light green shadings denote CaM N- and C-lobe residues, respectively. The mean value plus one standard deviation is the horizontal blue line. d Ribbon diagram of the homology model from Fig. 7b showing only the TRPA1 distal C-terminus (yellow, residues 1100–1114) bound to the Ca2+/CaM C-lobe (green, residues 80–149). CaM residues exhibiting CSPs from the 15N-labeled CaMWT:TRPA11089–1119 complex (black), the 15N-labeled CaM12:TRPA11089–1119 complex (teal), and both complexes (green) that face the TRPA1 C-terminus are depicted as balls and sticks. TRPA1 DCTCaMBE residues (yellow) are depicted as balls and sticks. Dashed lines denote putative H-bond interactions. e CaMWT (black) or CaM12 (red) CSPs for the C-lobe residues modeled in (d) from the 15N-labeled CaM12:TRPA11089–1119 or CaMWT:TRPA11089–1119 complexes from (a, b), respectively.
To identify residues within our NMR spectra that may directly face the TRPA1 C-terminal peptide, we mapped the residues with CSPs from the CaM12:TRPA11089-1119 and CaMWT:TRPA11089–1119 spectra to our homology model (Fig. 8d). This analysis identified five hydrophobic residues lining the CaM C-lobe hydrophobic pocket that showed chemical shifts in both the CaM12 and WT CaM spectra (Fig. 8d, green), an additional five hydrophobic residues and one acidic residue uniquely observed in the CaM12 spectra (Fig. 8d, teal), and one acidic residue uniquely observed in the WT CaM spectra (Fig. 8d, navy). Analysis of CSPs for these peptide-facing residues reveals more and larger shifts from the CaM12:TRPA11089–1119 complex than the CaMWT:TRPA11089–1119 complex further supporting a more engaged binding event (Fig. 8e).
Our model predicts that W1103 binds a hydrophobic pocket lined by residues identified in both the CaM12 and the WT CaM NMR data sets (A129, F142, and M145) as well as residues uniquely identified in the CaM12 data set (I101, L106, M125, and V137) (Fig. 8d). V1106 and L1107 are similarly located near residues identified in both NMR data sets (V92, F142, M145, and M146) while V1110 is near a residue identified in both (V92) and a residue uniquely identified in the CaM12 data set (A89) (Fig. 8d). Of these hydrophobic CaM C-lobe residues, A89, A129, I101, L106, F142, M145, and M146 also exhibited CSPs in the TRPV5-Ca2+/CaM12 complex and they mediated hydrophobic interactions with four TRPV5 residues to stabilize the complex74. Moreover, L106, M124, and M145 mediated hydrophobic interactions with the TRPV1 hydrophobic anchor W787 providing further support that the TRPA1 DCTCaMBE is predicted to engage the CaM C-lobe hydrophobic binding pocket83. The WT CaM NMR data set uniquely identified D81, which may form a salt bridge with K1111 in the hTRPA1 DCTCaMBE while the CaM12 data set identified E128 that may interact with R1102 (Fig. 8d, e). Though D81 was not identified in the CaM12 NMR data set, mutation of K1111 and R1102 in the hTRPA1 DCTCaMBE ablates Ca2+/CaM binding at basal Ca2+ concentration indicating that interactions with these residues are also important when cells are at rest (Fig. 7c, d). Collectively, these results suggest that Ca2+/CaM binds the TRPA1 distal C-terminus through its Ca2+-loaded C-lobe only and that further Ca2+-loading of the N-lobe induces a conformational change that weakens the binding event. Future structural work is needed to capture the TRPA1 DCTCaMBE and Ca2+/CaM complex at distinct Ca2+ concentrations, however, our NMR data suggest that this interaction is predominantly driven by hydrophobic interactions at basal Ca2+ concentrations and by a combination of fewer hydrophobic interactions and salt bridge interactions at higher Ca2+ concentrations.
Mutating DCTCaMBE hydrophobic anchor breaks TRPA1 desensitization
Mutation of the putative hTRPA1 DCTCaMBE hydrophobic anchor W1103 to alanine ablated Ca2+/CaM binding to the channel at basal Ca2+ concentration (W1103A, Fig. 7b–d). Consistently, the W1103A hTRPA1 mutant also showed a loss in biotinylation by TurboID-CaM C-lobe demonstrating it does not interact with Ca2+/CaM in cells (Fig. 5d, e, blue). Thus, we next characterized W1103A hTRPA1 activity and Ca2+ regulation by TEVC. Control experiments revealed that like the Δ1109–1119 hTRPA1 mutant, W1103A hTRPA1 exhibited similar gating kinetics and peak current amplitudes to WT hTRPA1 with a slightly reduced AITC sensitivity (Fig. 9a, b, timepoint i and Supplementary Fig. 4, blue data). Further, W1103A hTRPA1 channels exhibited rapid potentiation upon extracellular Ca2+ application and comparable peak current amplitudes to WT hTRPA1 (Fig. 9a, b, timepoint ii). Unlike WT hTRPA1, W1103A channels remained open with 10–20% their initial activity 5 min after extracellular Ca2+ application (Fig. 9a, b, timepoint iv). As with the Δ1109–1119 hTRPA1 mutant, this residual activity could be inhibited with the TRPA1 antagonist A-967079, demonstrating that the ongoing activity was due to hTRPA1 channels (Fig. 9a, timepoint v). It is possible that Ca2+ brought in during prolonged W1103A hTRPA1 channel activation triggers delivery of new channels to the plasma membrane or other Ca2+-dependent signaling pathways that contribute to the extended activation profile. Thus, we pre-incubated oocytes expressing W1103A hTRPA1 with 500 µM EGTA-AM for 20 min before TEVC recordings to buffer global Ca2+ concentrations while still allowing for local Ca2+ regulation50,85,86. These recordings reveal a delayed desensitization profile compared to WT hTRPA1 channels and a faster desensitization profile than W1103A hTRPA1 channels without intracellular Ca2+ buffering, demonstrating that prolonged Ca2+ entry has additional physiological effects (Fig. 9c).

a Overlay of representative time-traces at −80 and +80 mV holding potentials from oocytes expressing WT (transparent black) or W1103A hTRPA1 (blue). Current evoked with 250 µM AITC in the absence (orange i) and presence (blue ii) of 1.8 mM extracellular Ca2+. Channels were blocked with 10 µM A-967079 (grey v). Dashed line denotes 0 µA current. Protocol of condition application indicated above. Boxed below are the corresponding current-voltage relationships from timepoints indicated by black o (baseline), orange i (AITC without Ca2+), blue ii (AITC with Ca2+), purple iii (25 s after Ca2+ addition), green iv (5 min after Ca2+ addition), and grey v (A-967079 inhibited). b Quantification of peak current amplitudes at +80 mV (above) and −80 mV (below) before (orange i, left), directly after (blue ii, middle), and 5 min after (green iv, right) Ca2+ addition. WT hTRPA1 quantification is repeated from Fig. 4c. Colors as in (a). c Representative time-traces at −80 and +80 mV holding potentials from oocytes expressing W1103A hTRPA1 and pre-treated with 0.5 mM EGTA-AM (yellow) 20 min prior to recordings to chelate global intracellular Ca2+. Dashed line denotes 0 µA current. Protocol of condition application indicated above and timepoints marked as in (a). d Calculated time constants of desensitization at −80 mV (left) and +80 mV (right) from fitting data as in (a, c) to a single-exponential function. WT hTRPA1 quantification is repeated from Fig. 4d. Colors as in (a, c). e Quantification of charge translocation (µA•s, µC, e.g., area under the curve) at −80 mV (left) and +80 mV (right) from oocytes used as in (a, c). WT hTRPA1 quantification is repeated from Fig. 4e. Colors as in (b). b, d, e n = 10 (WT hTRPA1), 12 (W1103A hTRPA1) or 8 (W1103A hTRPA1 with 0.5 mM EGTA) oocytes per condition. Data represent mean ± SEM. P-values indicated in figure panels. b, e Two-tailed Student's t-test. d One-way ANOVA with Bonferroni’s post hoc analysis.
We next calculated rate constants for desensitization from baseline-corrected traces at −80 and +80 mV holding potentials, which revealed that W1103A hTRPA1 channels exhibited significantly delayed desensitization kinetics with 15-fold and 20-fold slowing to desensitization with inward (i.e., −80 mV) and outward (i.e., +80 mV) currents, respectively compared to WT channels (Fig. 9d, blue bars). These delays are even larger than those observed with the Δ1109–1119 hTRPA1 mutant (Fig. 4d). Even when intracellular Ca2+ was chelated with 500 µM EGTA, W1103A hTRPA1 exhibited a 10.5-fold and 10.7-fold slowing to desensitization at −80 and +80 mV, respectively compared to WT hTRPA1 channels (Fig. 9d, yellow bars). This sustained W1103A hTRPA1 channel activity resulted in significant 20- and 10-fold increases in translocated charge at −80 and +80 mV, respectively compared to WT channels and likely contributes to the delivery of additional channels to the plasma membrane as suggested above (Fig. 9e). These collective differences in mutant channel function were not due to changes in total protein expression (Supplementary Fig. 1d).
Loss of Ca2+/CaM binding has minor impact on potentiation
Like the Δ1109–1119 hTRPA1 mutant, W1103A hTRPA1 potentiation kinetics were more modestly slowed compared to WT hTRPA1 with 1.8 mM extracellular Ca2+ (~3-fold at −80 mV and ~1.8-fold at +80 mV) (Supplementary Fig. 5a, blue bars). W1103A hTRPA1 with buffered intracellular Ca2+ did not exhibit a significant change to potentiation kinetics compared to WT hTRPA1 channels (Supplementary Fig. 5a, yellow bars) consistent with a more modest effect on desensitization. These results suggest that disruption of the hTRPA1 DCTCaMBE does not affect potentiation, however, loss of Ca2+/CaM binding may affect channel potentiation in a way that is masked by the rapid WT hTRPA1 desensitization kinetics. The potentiation phase of TRPA1 Ca2+ regulation can be isolated by perfusing extracellular solutions with buffers containing lower Ca2+ concentrations (e.g., below 1 mM)36,41. Therefore, we recorded currents from oocytes expressing WT or W1103A hTRPA1 in the absence and presence of 100 or 500 µM extracellular Ca2+ (Supplementary Fig. 5b, c). WT hTRPA1 channels exhibited delayed desensitization rates with 500 µM extracellular Ca2+ and no desensitization with 100 µM extracellular Ca2+ (Supplementary Fig. 5b). We then quantified potentiation by determining the fold-change to current amplitudes in the presence of extracellular Ca2+ compared to those before Ca2+ addition, as reported previously36,41. At 100 µM extracellular Ca2+, where WT hTRPA1 exhibited no desensitization during the 5-min recordings, W1103A hTRPA1 channels exhibited a non-significant reduction to potentiation with inward currents than WT channels suggesting that mutation of the TRPA1 DCTCaMBE could have a slight negative effect to potentiation at low Ca2+ levels (Supplementary Fig. 5d). There was no difference between WT and W1103A hTRPA1 channels at 100 µM Ca2+ with outward currents (Supplementary Fig. 5e). At 500 µM extracellular Ca2+ where WT hTRPA1 exhibited a slower desensitization rate, WT and W1103A hTRPA1 channels exhibited nearly identical degrees of potentiation with both inward and outward currents (Supplementary Fig. 5d, e). Finally, at physiologically relevant extracellular Ca2+ levels (1.8 mM), W1103A hTRPA1 channels exhibited significant 22.8- and 1.6-fold higher degrees of potentiation than WT channels at the inward and outward currents, respectively (Supplementary Fig. 5d, e). We propose the latter is due to the greater effects to desensitization, which would inherently provide more time for full potentiation (Fig. 9d). Collectively, these results suggest that Ca2+/CaM binding at the TRPA1 DCTCaMBE specifically affects rapid desensitization with only minor effects on potentiation. This also provides compelling evidence that potentiation and desensitization are independent regulatory events, as previously proposed36.
Sequestering endogenous CaM delays TRPA1 desensitization
To independently characterize the effect of CaM binding on hTRPA1 Ca2+ regulation, we used the MBP-hTRPA11089–1119 peptide (called the MBP-peptide) as an experimental tool to sequester endogenous CaM since this peptide competed with WT hTRPA1 in CaM binding assays (Supplementary Fig. 7c, d). Importantly, a Δ1109–1119 version of the hTRPA1 peptide (MBP-hTRPA11089–1108) as well as a free MBP tag (control) failed to bind CaM confirming the specificity of the DCTCaMBE sequence to the Ca2+/CaM interaction (Supplementary Fig. 10a, b). Co-expression of MBP-WT hTRPA1 with the MBP-peptide in HEK293T cells yielded significantly enhanced basal and AITC-evoked activity by Ca2+ imaging compared to channels co-expressed with free MBP or the MBP-Δ1109–1119 peptide, presumably due to endogenous CaM sequestration and release from CaM regulation (Supplementary Fig. 10c–e). In contrast, the MBP-peptide had no effect on Δ1109–1119 hTRPA1 activity consistent with its insensitivity to Ca2+/CaM regulation (Supplementary Fig. 10f–h). We next asked how the MBP-peptide affects channel function by injecting oocytes expressing WT hTRPA1 with purified free MBP tag or MBP-peptide at a final concentration of 100 µM one hour prior to TEVC recordings. WT hTRPA1 profiles from oocytes injected with free MBP were indistinguishable from those with WT hTRPA1 alone (compare Fig. 4a and Supplementary Fig. 10i) and the channels exhibited similar desensitization rates (11.9 and 12.5 s at −80 and +80 mV for WT hTRPA1, respectively, versus 9.1 and 10.34 s at −80 and +80 mV for WT hTRPA1 with free MBP, respectively). In contrast, WT hTRPA1 channels from oocytes injected with the MBP-peptide exhibited 16- and 18-fold elongations to desensitization kinetics at −80 and +80 mV, respectively, compared to channels from oocytes injected with free MBP (Supplementary Fig. 10j–l). Thus, the MBP-peptide had a comparable effect on WT hTRPA1 activity as the Δ1109–1119 and W1103A mutants. Collectively, these results demonstrate that Ca2+/CaM is a critical regulator of hTRPA1 wherein Ca2+/CaM binding to the DCTCaMBE is required for proper hTRPA1 desensitization in intact cells.
High extracellular Ca2+ partially rescues desensitization
The free CaM C-lobe has a severely limited ability to respond to Ca2+ flux compared to free WT CaM, which raises questions about how it could confer desensitization through binding at the hTRPA1 DCTCaMBE. One possibility is that CaM does not directly transmit a Ca2+ signal to TRPA1, but instead its binding primes an intrinsic Ca2+ binding site in TRPA1 that triggers channel desensitization. Indeed, TRPA1 can be desensitized by direct perfusion of Ca2+ to its cytoplasmic domains in pulled patches, suggesting TRPA1 desensitization involves a direct Ca2+ binding event36,40,87. If true, desensitization of Ca2+/CaM binding deficient TRPA1 mutant channels should be at least partially rescued by increasing the extracellular Ca2+ concentration available to permeate through open channels. Consistent with literature precedence, we observed that WT hTRPA1 channels desensitize faster with 10 mM extracellular Ca2+ than with 1.8 mM extracellular Ca2+ (Fig. 10a, b)36,38. W1103A hTRPA1 channels were even more sensitive to extracellular Ca2+ concentration exhibiting an approximately 3-fold faster desensitization rate with 10 mM extracellular Ca2+ than with 1.8 mM extracellular Ca2+ (Fig. 10c, d). Despite these enhanced rates, W1103A hTRPA1 channels were still approximately 4-fold slower to desensitize than WT hTRPA1 with 1.8 mM extracellular Ca2+. Collectively, these results are consistent with loss of Ca2+/CaM binding affecting an intrinsic TRPA1 Ca2+ binding site that can only be partially rescued by the higher intracellular Ca2+ concentration permeated by W1103A hTRPA1 channels with 10 mM extracellular Ca2+. Future work will identify this intrinsic TRPA1 Ca2+ binding site and its contribution to channel desensitization.

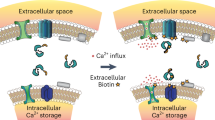
Representative time-traces at −80 and +80 mV holding potentials from oocytes expressing WT (a) or W1103A hTRPA1 (c). Current evoked with 250 µM AITC in the absence and presence of 1.8 mM (black/dark blue) or 10 mM (grey/light blue) extracellular Ca2+. Channels were blocked with 10 µM A-967079. Dashed line denotes 0 µA current. Protocol of condition application indicated above. Extracellular solutions contained 125 µM niflumic acid (NFA). b, d Calculated time constants of desensitization from fitting data as in (a, c) at 1.8 or 10 mM extracellular calcium to a single-exponential function. Data represent mean ± SEM. P values listed in figure panels. n = 9 (WT), 10 (W1103A, 1.8 mM Ca2+) or 11 (W1103A, 10 mM Ca2+) oocytes per condition, two-tailed Student’s t-test. e Model illustrating Ca2+ desensitization of TRPA1 (red inhibition arrow) in a WT channel (left) versus a Ca2+/CaM binding deficient channel (right). Channel activation facilitates permeation of sodium (Na+) and Ca2+ ions into the cytoplasm. WT CaM (dumbbell) or the CaM C-lobe (green circle) binding to the TRPA1 DCTCaMBE (yellow) in the structurally unresolved distal cytoplasmic C-terminus primes hTRPA1 for rapid desensitization following Ca2+ permeation (left, blue color on the Ca2+ concentration heat map). When the TRPA1:Ca2+/CaM interaction is disrupted, channels remain active, allowing intracellular Ca2+ concentrations to rise (right, orange color on the Ca2+ concentration heat map) before incomplete desensitization is observed.
Discussion
TRPA1 is a Ca2+-permeable ion channel implicated in chronic pain and inflammation whose activity is controlled by rapid desensitization following a cytosolic Ca2+ increase13,21,23,24,25,26,27,28,29,30,31,32,36,39,40,41. Despite the importance of desensitization to restricting proper TRPA1 functionality, this regulatory mechanism, and whether it involves the universal Ca2+ sensor CaM, has remained controversial36,39,40,41,87. Here, we find that Ca2+/CaM binds human TRPA1 best at basal Ca2+ concentration, co-localizes with TRPA1 at rest, and suppresses channel activity when co-expressed in heterologous cells, suggesting Ca2+/CaM is a bona fide TRPA1 regulator. Ca2+/CaM mediates this regulation through a previously unidentified, highly conserved, high-affinity binding site in the TRPA1 distal cytoplasmic C-terminus that we have named the distal C-terminal CaM binding element (DCTCaMBE). The TRPA1 DCTCaMBE resides in the final 18 amino acids of a 41-amino acid C-terminus that has yet to be resolved by structural studies (the TRPA1 structurally unresolved C-terminus, Fig. 1a)39,88,89. Quantitative binding assays, homology modeling, and NMR spectroscopy reveal that the TRPA1 DCTCaMBE exclusively engages the high Ca2+-affinity CaM C-lobe in a Ca2+-dependent manner, supporting a pre-association of TRPA1 and Ca2+/CaM in cells. Mutation of putative TRPA1 DCTCaMBE residues in the full-length channel or sequestration of CaM with a TRPA1 C-terminal peptide ablates Ca2+/CaM binding at basal Ca2+ concentration and yields hyperactive channels that exhibit a 10- to 20-fold slowing of Ca2+-mediated desensitization, which can only be partially rescued by an increased extracellular Ca2+ concentration (Fig. 10e, right). This functional perturbation is more drastic than that caused by all known TRPA1 channelopathic mutations and suggests that disruption of the TRPA1:Ca2+/CaM interaction would have profound neurological effects, which could provide a useful tool for studying TRPA1 hyperactivity in genetically tractable animal models10,11,12. Indeed, some TRPV4, Nav1.1, and Nav1.2 channelopathic mutations are thought to mimic loss of CaM binding90,91. Our work identifies the TRPA1 DCTCaMBE as the main Ca2+/CaM binding element and reveals that Ca2+/CaM or Ca2+/CaM C-lobe binding to this site is critically required for rapid TRPA1 desensitization upon Ca2+ influx (Fig. 10e, left).
CaM binds and regulates the activity of hundreds of effector proteins including many ion channels through several types of binding motifs, requiring a remarkable degree of plasticity for a protein that is less than 17 kDa48,72,77,78,82,83,91,92,93,94,95,96,97,98,99. The canonical binding mode involves both lobes of Ca2+-bound CaM wrapping around a single effector protein alpha helix through a series of hydrophobic interactions with the CaM lobes’ hydrophobic clefts though the N- and C-lobes can make a bridged interaction by engaging separate effectors or by engaging distinct binding motifs44,48,72,77,78,82,83,91,94,95,96,97,98,99,100. Our binding studies, homology modeling, and NMR data support exclusive engagement of the TRPA1 DCTCaMBE by the Ca2+/CaM C-lobe. The predicted TRPA1 DCTCaMBE is a 10-residue binding segment within a putative alpha helix with four hydrophobic residues at positions 2, 5, 6 and 9 and basic residues at positions 1 and 10 (Fig. 7b, e). It is predicted to bind the Ca2+/CaM C-lobe as an alpha helix with W1103 forming the main hydrophobic anchor akin to W787 in the TRPV1 C-terminal CaM binding segment (Fig. 7b)83. Following naming conventions, the TRPA1 DCTCaMBE is a 2-5-6-9 motif; it provides another example of CaM binding plasticity and it supports the growing body of literature that non-canonical CaM binding motifs are more common than previously expected92.
Our IF imaging and proximity biotinylation data suggest that Ca2+/CaM forms a stable interaction with TRPA1 at rest through an interaction between the TRPA1 DCTCaMBE and the CaM C-lobe (Fig. 5). Our CaM-agarose binding assays and ITC experiments reveal that this TRPA1-Ca2+/CaM interaction weakens six-fold as Ca2+ levels rise. However, we do not think that Ca2+/CaM will dissociate from TRPA1 upon Ca2+ influx. A crude estimate predicts that the local protein concentration at the DCTCaMBE-CaM interaction is ~2 mM, which is three-to-four orders of magnitude higher than the 600 nM TRPA1-Ca2+/CaM interaction at high Ca2+ (Supplementary Fig. 11). Thus, any bound Ca2+/CaM will remain associated with TRPA1 throughout the regulatory cycle thereby promoting rapid channel desensitization (Fig. 10e, left). This is in striking contrast to TRPA1 DCTCaMBE mutants that are unable to engage CaM at this site (Fig. 10e, right). Instead, our NMR data suggest that the TRPA1-Ca2+/CaM binding mode may change upon Ca2+ influx. This altered binding mode may contribute to desensitization. Indeed, Ca2+-mediated conformational changes in CaM and alterations to CaM-effector protein binding modes are fundamental to CaM-mediated regulation of proteins including in other channels45,46. Future work is needed to confirm the stability of the TRPA1-Ca2+/CaM interaction in cells throughout the regulatory cycle, to reveal the TRPA1-Ca2+/CaM binding mode at different Ca2+ concentrations, and to determine whether and how these change(s) contribute to desensitization.
Together, these results raise the intriguing possibility that CaM is a TRPA1 auxiliary subunit. Auxiliary subunits including CaM have been studied extensively and were shown to regulate ion channel folding, stability, trafficking, localization, stimulus sensitivity, and gating kinetics60,61,62,63,64,101,102. Unlike KCNQ channels, we did not find that Ca2+/CaM affects TRPA1 assembly or trafficking (Figs. 1g, h, 3c, d)103,104,105,106. Instead, our work reveals that Ca2+/CaM is an indispensable auxiliary subunit required for rapid channel desensitization that effectively tightens the TRPA1 functional window and limits total ion influx (Figs. 4d, 9d and 10e). In this way, Ca2+/CaM operates akin to the auxiliary subunits of voltage-gated ion channels47,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,107. Several lines of evidence support CaM serving as an endogenous TRPA1 regulator in native tissues. First, a prior study showed that introduction of a TRPA1 CaM binding peptide containing the CaMBD through the recording pipette disrupted proper TRPA1 desensitization in patch clamp recordings from mouse dorsal root ganglion neurons41. Our TRPA1 C-terminal peptide encoding the DCTCaMBE binds CaM at high affinity and could compete with WT TRPA1 to bind CaM in HEK cells and Xenopus oocytes yielding hyperactive channels with disrupted desensitization (Supplementary Fig. 10a–e, j, k); we anticipate that this peptide would exhibit a similarly robust effect in neurons. Second, spatial transcriptomics profiling of human dorsal root ganglia reveals that CaM is highly expressed in TRPA1+ neurons from the CALM1 and CALM3 genes, further supporting a role for CaM as an endogenous channel regulator in humans108. Future in vitro work on TRPA1 should ensure that CaM is expressed at sufficient levels to initiate rapid desensitization and to avoid artifactual results from hyperactive channels. This is particularly key for heterologous systems where endogenous CaM pools can be easily overwhelmed by overexpressed TRPA1 and must be supplemented by exogenous CaM.
We identified the TRPA1 DCTCaMBE in the distal C-terminus, which has never been resolved structurally, and thus it is unknown where the DCTCaMBE resides in relation to domains that govern channel gating39,88,89. In this way, TRPA1 joins the ranks of the many other ion channels with C-terminal CaM regulatory domains47,49,50,51,52,53,54,55,56,57,58,59,83,107,109,110,111. Unlike these other channels, however, the TRPA1 C-terminus is likely located quite far from the pore. Indeed, the 41 amino acid structurally unresolved distal cytoplasmic C-terminus extends from a centrally located coiled coil that terminates ~75 Å below the pore88. This raises mechanistic questions about how Ca2+/CaM binding to the TRPA1 DCTCaMBE could promote channel desensitization (Fig. 1a, b). One intriguing model is a coordinated response between the TRPA1 DCTCaMBE and the previously identified CaM binding domain (CaMBD)41, which resides near the ion conduction pathway, through sequential binding of the CaM C- and N-lobes, respectively as intracellular Ca2+ rises to trigger desensitization. Such mechanisms involving disparate binding elements within the same channel for the CaM N- and C-lobes have been proposed for CaM- and Ca2+-mediated SK channel activation, TRPV5 and TRPV6 inhibition, as well as regulation of voltage-gated ion channels42,43,45,46,48,50,78. However, we and others have shown that the CaM N-lobe is dispensable for TRPA1 regulation (Fig. 1e, f)41.
The lack of dependence of TRPA1 on the CaM N-lobe for rapid desensitization is strikingly distinct. Indeed, most CaM-regulated ion channels rely on conformational changes in CaM upon Ca2+ binding to the N- and C-lobes that either directly blocks the pore or induces a conformational change in the channel to regulate gating42,43,44,45,46,47,49,50,51,52,53,54,55,56,57,58,59. Thus, a second possible model is that the Ca2+/CaM C-lobe engages the TRPA1 DCTCaMBE with a Ca2+-loaded EF hand 4 and, upon Ca2+ influx, Ca2+ binding to EF hand 3 triggers a conformational change in the CaM C-lobe that contributes to rapid desensitization. EF hands 3 and 4 were previously shown to both be required for TRPA1 regulation and future mechanistic work will further explore this possible model41. A third and intriguing model is that CaM may not be functioning as a direct Ca2+ sensor, per se in rapid TRPA1 desensitization. Our data show that while ablation of Ca2+/CaM binding to the TRPA1 DCTCaMBE drastically slows the desensitization rate, it does not completely ablate desensitization; permeating Ca2+ ions are, albeit inefficiently, able to desensitize TRPA1 and this slowed desensitization can be partially rescued by increasing the extracellular Ca2+ concentration (Figs. 4, 9 and 10e, right, smaller red inhibition arrow). Collectively, we believe our results support a model wherein Ca2+/CaM binding at the DCTCaMBE primes the affinity and/or accessibility of an intrinsic TRPA1 Ca2+ binding site so that it can readily respond to Ca2+ flux through the channel pore and trigger rapid desensitization (Fig. 10e, left, larger red inhibition arrow). This model can explain why Ca2+ ions perfused directly onto the TRPA1 cytoplasmic domains in excised patches that were previously exposed to EGTA can still trigger desensitization36,40,87. Future structural and functional work are needed to identify this Ca2+ binding site, to determine how Ca2+ binding triggers rapid TRPA1 desensitization, and to uncover how Ca2+/CaM binding affects this process. Finally, while the CaM N-lobe is not required for rapid TRPA1 desensitization, it may serve to enhance Ca2+/CaM binding to TRPA1 through the CaMBD or other unidentified site that may contribute to other aspects of channel regulation, or it may remain available to interact with other local effectors and initiate Ca2+- and CaM-dependent signaling112.
Collectively, we identified a highly conserved and critical Ca2+/CaM regulatory element in the human TRPA1 distal unstructured C-terminus that is required for proper channel function. Localization of the TRPA1 DCTCaMBE to the distal C-terminus is serendipitous since it allowed us to completely ablate Ca2+/CaM binding through genetic modification of TRPA1 without affecting intrinsic channel function (e.g., WT and mutant hTRPA1 channels exhibited similar activities in Ca2+ free solution, Figs. 4a–c, 9a, b and Supplementary Fig. 4). Thus, we were able to directly reveal this critical role for Ca2+/CaM in TRPA1 regulation without relying on perturbation of CaM, which can have off-target effects. Moreover, our work demonstrates how short segments of structurally unresolved amino acids can drastically affect channel function in unexpected ways. Ion channels are highly enriched for intrinsically disordered segments at both their N- and C-termini with some of them, including TRPA1 as described here, shown to be indispensable for proper function113,114. For others, such as the related capsaicin receptor, TRPV1, a structurally unresolved element in the distal C-terminus governs phosphatidylinositol lipid and possibly Ca2+ regulation115,116,117. Perturbations therein promote TRPV1 sensitization and a splice variant that removes this regulatory element may underlie infrared detection capability in vampire bats115,117,118. Given their wide distribution amongst the ion channel proteome including the large TRP channel family, other intrinsically disordered segments are bound to have novel channel regulatory modes119. Such segments provide a ripe evolutionary source for new functionality as they are more permissive to mutations than structured regions, which is particularly useful for transmembrane proteins whose complex regulation has developed mainly through extra-membrane domains69,118,120,121,122. While the cryo-EM resolution revolution has been a boon for the ion channel and membrane protein fields, our work illustrates the need to study structurally unresolved and intrinsically disordered regions with renewed interest and vigor.
Methods
Plasmid construction
DNA sequences for human TRPA1, human TRPA1448–1078, and human Calmodulin were PCR amplified and subcloned into a p3xFLAG-eYFP-CMV-7.1 vector (Addgene) at the NotI/BamHI sites using In-Fusion EcoDry cloning (Takara) according to manufacturer protocols. Human TRPA1, mouse TRPA1, zebrafish TRPA1, and C. elegans TRPA1 were subcloned into 8xHis-MBP pFastBac1 modified with a CMV promoter (obtained from David Julius’ lab) at the BamHI/NotI sites following the same protocol. 8xHis-MBP pFastBac1 modified with a CMV promoter human TRPA11089–1119 was created by introducing a BamHI site at the −1 position of the codon encoding TRPA11089 and digesting the plasmid with BamHI restriction enzyme followed by gel purification. The linearized vector segment was then transformed into XL-10 Gold cells resulting in a repaired vector. A similar strategy was followed for the creation of 8xHis-MBP pFastBac1 with a CMV promoter human TRPA11079–1119, and human TRPA11099–1119. 8xHis-MBP-human TRPA11089–1108 in the CMV vector was built by site-directed mutagenesis from the 8xHis-MBP-human TRPA11089–1119 construct (Agilent). For bacterial protein purification, DNA sequences for 8xHis-MBP hTRPA11089–1119 and Flag-6xHis-WT CaM were PCR amplified and subcloned into a pET28a vector (Addgene) at the NcoI/NotI sites using In-Fusion EcoDry cloning. All mutants including hTRPA1Δ1109–1119, hTRPA1Δ1079–1119, hTRPA1Δ1089–1119, hTRPA1Δ1099–1119, and hTRPA1Δ1–61, TRPA1W1103A, TRPA1L1107A, TRPA1V1110A, TRPA1Quad, TRPA1RK/AA, CaM12, CaM34, and CaM1234 and CaM C-lobe were created using QuikChange Lightning site-directed mutagenesis (Agilent). 3xFLAG-TurboID-CaM C-lobe was generated by producing a MegaPrimer for TurboID that would replace the CaM N-lobe following a site-directed mutagenesis reaction with the 3xFLAG-WT hCaM template. 3xFLAG-TurboID was generated from 3xFLAG-TurboID-CaM C-lobe by site-directed mutagenesis to replace the CaM C-lobe sequence with a TAG stop codon. V5-tagged CaM was created by site-directed mutagenesis to replace the 3xFLAG-tag in the construct above. For expression in Xenopus laevis oocytes, 3xFLAG-hTRPA1 variant genes were subcloned into the combined mammalian/oocyte expression vector pMO (obtained from David Julius’ lab) prior to generating cRNAs.
All DNA primers were ordered from ThermoFisher and all constructs were sequence-verified using the Yale School of Medicine Keck DNA Sequencing Facility.
Mammalian cell culture and protein expression
Human embryonic kidney cells (HEK293T, ATCC CRL-3216) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% calf serum and 1x Penicillin-Streptomycin (Invitrogen) at 37 °C and 5% CO2. Neuro2A cells (ATCC CCL-131) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum and 1x Penicillin-Streptomycin (Invitrogen) at 37 °C and 5% CO2. Cells were grown to ~85–95% confluence before splitting for experiments or propagation. HEK293T cells cultured to ~95% confluence were seeded at 1:10 or 1:20 dilutions into 6- or 12-well plates (Corning), respectively. After 1–5 h recovery, cells were transiently transfected with 1 μg plasmid using jetPRIME (Polyplus) according to manufacturer protocols. For surface biotinylation experiments, cells were co-transfected with 0.5 µg 3xFLAG-tagged hTRPA1 variants and 0.5 µg 3xFLAG-CaM variants.
Cell lysis and pulldown experiments
16–24 h post-transfection, HEK293T cells were washed with PBS (calcium and magnesium free) and lysed in 75–150 μL of TRPA1 lysis buffer (40 mM Tris pH 8.0, 150 mM NaCl, 5 mM DDM, 500 μM EGTA, EDTA-free complete protease cocktail inhibitor tablet) at 4 °C while gently nutating. Cell debris were pelleted from the resulting lysates by centrifugation at 21,300 × g for 10 min at 4 °C. Total protein concentration in lysates were quantified using a BCA assay (Pierce). Equal concentrations of protein lysate (100 μg) from each experimental condition were added to resins as specified below. 10% of loaded protein amount was reserved as a whole-cell lysate loading control. Buffers were supplemented with free Ca2+ as indicated in each figure. Concentrations of CaCl2 required for each free Ca2+ concentration were calculated using Ca-EGTA calculator v1.3 using constants from Theo Schoenmakers’ Chelator on the MaxChelator website123.
CaM-agarose pulldown
100 µg lysates were incubated with 10 μL of lysis buffer-equilibrated Calmodulin-agarose (Sigma-Aldrich) for 1 h at 4 °C with gentle nutation. Resin beads were washed 3–5 times with lysis buffer prior to elution; resin was pelleted, and bound proteins were eluted in TRPA1 lysis buffer supplemented with 5 mM EGTA for 30 min at 4 °C with gentle nutation.
Biotinylation assays
Surface biotinylation assays were conducted as previously reported10. Briefly, HEK293T cells were seeded in a 6-well plate pre-coated with Poly-L-Lysine (Sigma) and transfected with expression plasmids. 16–24 h post-transfection, cells were washed with PBS, placed on ice, and incubated for 20 min with chilled 0.05 mg/mL Sulfo-NHS-LC-Biotin (ThermoFisher) in Ringer’s solution. Cells were then washed with chilled wash buffer (PBS + 100 mM Glycine) and the reaction was quenched on ice for 30 min with 100 mM Glycine and 0.5% BSA in PBS. Cells were then washed three times and lysed in TRPA1 lysis buffer containing 100 mM Glycine directly on the 6-well plate. Lysates were collected, protein concentration was determined by BCA assay, biotinylated proteins were isolated by Neutravidin pulldown and analyzed by immunoblot as described below. During quantification, any FLAG signal from CaM in the eluates were subtracted from all other conditions to account for probe internalization.
For proximity biotinylation assays, HEK293T cells co-expressing 3xFLAG-TurboID or 3xFLAG-TurboID-CaM C-lobe and 3x-FLAG-tagged WT, Δ1109–1119, or W1103A hTRPA1 were treated with 50 μM biotin supplemented into culture media ~48 h post-transfection. Cells were lysed 15 min later with TRPA1 lysis buffer and affinity purified via the 3xFLAG tags as described below. Biotinylated proteins were detected with HRP-conjugated Streptavidin (GE, 1:50,000).
Neutravidin pulldown
Cell lysates that were generated following surface labeling experiments were incubated with 15 μL of lysis buffer-equilibrated Neutravidin resin (Pierce) for 2 h at 4 °C with gentle nutation. The resin was then washed with lysis buffer three times, followed by a harsher wash with 1× PBS + 100 mM DTT. Resin was then washed once each with lysis buffer and 1× PBS. Biotinylated protein was eluted from the resin with a multi-step protocol to prevent TRPA1 aggregation while maximizing protein elution from the resin. First, resin was incubated with 10 μL of biotin elution buffer (TRPA1 lysis buffer, 100 mM Glycine, 10 mM Biotin, 1% SDS) on ice for 10 min, followed by addition of 1 μL ß-mercaptoethanol (BME; Sigma) to each sample and incubation on ice for 5 min, and finally by addition of 4 μL 4× Laemmli buffer + 10% BME with incubation at 65 °C for 10 min. The resin was centrifuged, supernatant was removed and combined with additional 4 μL Laemmli buffer + 10% BME for SDS-PAGE analysis.
FLAG immunoprecipitation
Lysates were incubated with 10 µL EZview Red Anti-FLAG M2 affinity resin (Sigma) for 1 h at 4 °C with gentle rotation. Resin beads were washed four times with TRPA1 lysis buffer prior to elution with 125 µg/ml 3xFLAG peptide (Sigma).
SDS-PAGE and immunoblot
Samples were combined with Laemmli Sample buffer + 10% BME and separated on pre-cast 4–20% SDS-PAGE gels (Bio-Rad). Gels were transferred onto PVDF membranes (Bio-Rad) by semi-dry transfer at 15 V for 40 min. Blots were blocked in 3% BSA prior to antibody probing. The following primary antibodies were used in PBST buffer (Boston Bioproducts): anti-MBP (mouse, 1:30,000, New England Biolabs), anti-FLAG (mouse, 1:30,000, Sigma), anti-tubulin (mouse, 1:5000 in 3% BSA, Sigma). HRP-conjugated IgG secondary anti-mouse antibody was used as needed (rabbit, 1:25,000, Invitrogen). Membranes were developed using Clarity Western ECL substrate (Bio-Rad) and imaged using a Chemidoc Imaging System (Bio-Rad). Densitometric quantifications were performed with ImageJ software. All quantified band intensities for eluted samples were divided by their tubulin-normalized input band intensities.
Immunofluorescence imaging and co-localization analysis
Neuro2A cells (ATCC CCL-131) transiently transfected with jetPRIME (Polyplus) according to manufacturer protocols were transferred to poly-L-lysine-coated cover slips and incubated for 16–20 h prior to immunostaining. Cells were fixed on coverslips with 4% paraformaldehyde for 20 min, then permeabilized with 0.1% Triton in PBS for 10 min. Cells were then blocked in 4% BSA in PBS followed by incubation in primary anti-V5 monoclonal antibody (mouse, 1:500, Invitrogen) in 4% BSA in PBS at room temperature for 45 min. Cells were then washed with PBS and incubated with Alexa-Fluor488-conjugated secondary anti-mouse antibody in 4% BSA in PBS at room temperature for 45 min (goat, 1:700, Invitrogen), followed by PBS washes. Finally, cells were incubated in Cy3-conjugated anti-FLAG monoclonal antibody (mouse, 1:500, Sigma) in 4% BSA in PBS at room temperature for 45 min. Images were acquired on a Zeiss Axio Observer Z1 inverted microscope equipped with an Airyscan detection unit, using a Plan-Apochromat 63x/1.40 NA oil objective. Image deconvolution was performed with ZEN 2.1 software. Pixel intensity-based co-localization analysis was performed using the colocalization module in ZEN, from which Pearson’s correlation coefficients were calculated for each analyzed cell. Co-localization scatterplot crosshairs were set based on mono-stained samples for green and red channels.
Ratiometric calcium imaging
16–24 h post-transfection, HEK293T cells were plated into isolated silicone wells (Sigma) on poly-L-lysine (Sigma)-coated cover glass (ThermoFisher). Remaining cells were lysed for anti-FLAG immunoblotting to ensure equivalent expression. 1 h later, cells were loaded with 10 μg/mL Fura 2-AM (ION Biosciences) in physiological Ringer’s solution (in mM: 120 NaCl, 5 KCl, 2 CaCl2, 25 NaHCO3, 1 MgCl2, 5.5 HEPES, 1 D-glucose, pH 7.4; Boston BioProducts) with 0.025% Pluronic F-127 (Sigma) and incubated for 1 h at room temperature, then rinsed twice with Ringer’s solution. Ratiometric calcium imaging was performed using a Zeiss Axio Observer 7 inverted microscope with a Hamamatsu Flash sCMOS camera at 20x objective. Dual images (340 and 380 nm excitation, 510 nm emission) were collected and pseudocolour ratiometric images were monitored during the experiment (MetaFluor software). After stimulation with agonist, cells were observed for 45–100 s. AITC was purchased from Sigma and was freshly prepared as a stock at 4× the desired concentration in 1% DMSO and Ringer’s solution. 5 μL of 4× agonist was added to wells containing 15 μL Ringer’s solution to give the final 1× desired concentration. For all experiments, a minimum of 60–90 cells were selected per condition per replicate for ratiometric fluorescence quantification in MetaFluor with 3–5 replicates per experiment. Background signal was quantified from unresponsive cells and subtracted from quantified cells for normalization.
Bacterial protein expression and purification
Bacterial expression vectors were transformed into BL21(DE3) cells and plated on Kanamycin (Kan) LB agar plates. Individual colonies were selected to inoculate LB Kan overnight starter cultures from which 10 mL was used to inoculate 1 L of Terrific Broth (TB) Kan. Once the inoculated TB Kan reached an OD of 0.8–1.0, protein expression was induced using 1 mM IPTG (GoldBio). After 4 h post-induction, bacteria were harvested at 2400 × g for 20 min, the supernatant was discarded, and cell pellets were snap frozen with liquid nitrogen and stored at −80 °C. Bacteria were cultured at 37 °C and 220 RPM for each step.
Bacterial pellets were thawed at 4 °C and resuspended in bacterial lysis buffer (50 mM Tris pH 8.0, 500 mM NaCl, 2 mM CaCl2, 5 mM β-ME, 1 mM PMSF, 10% glycerol, and 5 mg bovine DNase I). Bacteria were lysed on ice using a FisherBrand Model 120 Sonic Dismembrator (Fisher Scientific). The sonicator was set to 50% amplitude and cycled between 1 min on and 1 min off for a total of 20 min. Bacterial debris were pelleted using an Eppendorf centrifuge 5810 R that had been cooled to 4 °C at 2041 × g for 30 min. All constructs contained either a 6xHis- or 8xHis-tag and were purified by gravity-flow immobilized metal affinity chromatography (IMAC) using lysis buffer-equilibrated HisPur Ni-NTA resin (ThermoFisher). To remove non-specifically bound proteins, the resin was washed with low-molar imidazole wash buffer (50 mM Tris pH 8.0, 150 mM NaCl, 5 mM β-ME, 0.1 mM PMSF, and 20 mM imidazole). Bound protein was eluted in 2 mL fractions with high-molar imidazole elution buffer (50 mM Tris pH 8,0, 150 mM NaCl, 2 mM CaCl2, 5 mM β-ME, 0.1 mM PMSF, and 300 mM imidazole) and an A280 measurement was taken for each fraction using a NanoDrop One (ThermoFisher). Protein-containing fractions were pooled, exchanged into storage buffer (50 mM Tris pH 8.0 150 mM NaCl, 2 mM CaCl2, and 0.1 mM PMSF) or oocyte injection buffer (50 mM Tris pH 8.0, 150 mM KCl, and 0.1 mM PMSF) by dialysis overnight or diafiltration and concentrated using Amicon Ultra centrifugal filter with a 10,000 Da molecular weight cutoff (Millipore). Diafiltration is a technique where macromolecules are concentrated using a spin filter and subsequently diluted with the new buffer. This technique is repeated several times and allows for rapid buffer exchanges relative to dialysis methods. CaM and CaM12 were further purified through size exclusion chromatography using a Superdex 75 Increase 10/300 GL (Cytiva). MBP-tagged TRPA11089–1119 was incubated with TEV protease overnight at 4 °C to cleave the MBP tag. After TEV protease digestion, TRPA11089–1119 was isolated from free MBP using an Amicon Ultra centrifugal filter with a 30,000 Da molecular weight cutoff. SDS-Page and Coomassie staining was used for post-hoc analysis of each step of the purifications.
Size-exclusion chromatography binding assay
Each condition was prepared using 100 μM of each purified protein in a total volume of 500 μL using storage buffer as described above and mixed by gentle nutation at 4 °C for 5 min. Samples were then loaded onto a Superdex 75 Increase 10/300 GL column pre-equilibrated with either storage buffer or storage buffer supplemented with 5 mM EGTA and run at a flow rate of 0.5 mL/min. The A280 signal was monitored by the ÄKTA pure chromatography system (Cytiva), and fractions corresponding to observed peaks were collected, concentrated with the appropriate Amicon Ultra cut-off spin filter, and subjected to Coomassie stain analysis.
Isothermal titration calorimetry
All ITC experiments were carried out using a MicroCal VP-ITC (Malvern) at the Keck Biophysical Resource Center at Yale. The cell was loaded with 15 μM CaM or CaM12 and the titrant syringe with 810 μM hTRPA11089–1119. All proteins were buffer matched with storage buffer as described above. 35 injections of 3 μL were performed with 240 s between each injection and a stir speed of 290 RPM. Data were analyzed with Origin7 software and fit using the one-site model.
Molecular modeling of the hTRPA1 DCTCaMBE-Ca2+/CaM complex
Sequence for hTRPA11089–1119 was aligned to the rat TRPV1 C-terminal (CT) CaM binding segment (rTRPV1784–798) in Clustal Omega, which revealed a best alignment with hTRPA11100–1114 (Supplementary Fig. 9a). This minimal sequence alignment was then used to build a homology model using the TRPV1 CT-Ca2+/CaM crystal structure (PDB: 3SUI) as the template with Modeller Comparative in ChimeraX84. After obtaining models from Modeller, we used the model with the lowest z-DOPE score for all further analysis.
NMR spectroscopy
NMR spectra were recorded at 25 °C on a Varian Inova 700 MHz spectrometer equipped with a triple resonance cryoprobe. 15N-labeled samples were expressed and purified as described above except that cells were grown in M9 medium with the addition of 15N-labeled ammonium chloride. The purified samples were exchanged into the storage buffer supplemented with 10% (v/v) D2O and 2 mM CaCl2. Concentration of the samples ranged from 200 to 700 µM. Data were collected in 3- or 5-mm tubes for samples with concentration lower than 300 or higher than 400 µM, respectively. Binding between the 15N-labeled CaM variants and unlabeled hTRPA11089–1119 peptide was monitored by two-dimensional 15N − 1H HSQC experiments. The number of scans and time domain points of the 15N − 1H HSQC experiments on samples with concentration greater than 300 µM were set to 8 and 64, respectively. The sweep widths at the f2 and f1 dimensions were set to 17 and 40 ppm, respectively. The acquisition times were 85 and 180 ms at the f2 and f1 dimensions, respectively. The carrier was set to 4.6 ppm for the 1H and 120 ppm for 15N. The relaxation delay was 1.0 s, and the total experiment time was ~2 h. For samples lower than 300 µM, the number of scans and time domain points were set to 64 and 256, respectively. Here, the total experiment time was ~10 h.
The chemical assignments for 15N-WT CaM and 15N-CaM12 were obtained from the BMRB database: entries 6541 and 26685, respectively124,125. These reference sectra were used to assign peaks in our 15N-WT CaM and 15N-CaM12 samples. Chemical shift perturbations (CSPs) were analyzed using the nearest neighbor approach, which accurately maps CSPs due to protein-protein interactions126,127. CSPs were quantitatively analyzed using the formula:
where H and N represents 1H and 15N nuclei, respectively, \(\alpha\) is the scaling factor of 15N ppm (set to 0.14), and
\(\delta\) is the change in chemical shift of the respective nuclei.
CSPs of the amide protons and nitrogen were monitored. The spectra were processed in NMRFx analyst128. CSPs greater than one significant number above the average CSP were deemed significant.
Oocyte electrophysiology
Experiments were conducted as previously reported10. pMO vectors carrying 3×FLAG-tagged hTRPA1 constructs were linearized with PmeI, cRNAs were generated by in vitro transcription with the mMessage mMachine T7 transcription kit (Thermo) according to the manufacturer’s protocol and were purified with a RNeasy kit (Qiagen). cRNA transcripts were microinjected into surgically extracted Xenopus laevis oocytes (purchased from Ecocyte) with a Nanoject III (Harvard Apparatus). Oocytes were injected with 0.25 ng (WT hTRPA1) or 0.1 ng (DCTCaMBE variants) of cRNA per cell, and whole-cell currents were measured 24 h post-injection using two-electrode voltage clamp (TEVC). Currents were measured using an OC-725D amplifier (Warner Instruments) delivering a ramp protocol from −100 mV to +100 mV applied every second. Microelectrodes were pulled from borosilicate glass capillary tubes and filled with 3 M KCl. Microelectrode resistances of 0.7–1.2 MΩ were used for all experiments. Bath solution contained (in mM) 93.5 NaCl, 2 KCl, 2 MgCl2, 0.1 BaCl2, and 5 HEPES (pH 7.5). For experiments in the presence of calcium, BaCl2 was replaced with 1.8 mM CaCl2. For experiments comparing 1.8- and 10-mM extracellular calcium, BaCl2 was replaced with 1.8 or 10 mM CaCl2 and supplemented with 125 µM niflumic acid (NFA) to inhibit endogenous calcium-activated chloride channels. For experiments comparing 100 µM and 500 µM extracellular calcium, the solutions contained (in mM) 93.5 NaCl, 2 KCl, 2 MgCl2, and 5 HEPES (pH 7.5) with 0.1 EGTA and 0.6 CaCl2 or 1 EGTA and 1.1 CaCl2, respectively. For global calcium chelation experiments oocytes were incubated with 0.5 mM EGTA-AM for 20 min in calcium-free buffer before recordings were taken. Opening and closing times for hTRPA1 constructs were obtained by applying 100 µM AITC in calcium-free buffer to the oocytes for 10 s followed by AITC washout with calcium-free buffer. For experiments in Supplementary Fig. 10i–l, the purified free MBP tag or MBP-hTRPA11089–1119 peptide in oocyte injection buffer were microinjected at least 1 h prior to recording with a final concentration of 100 μM assuming an oocyte volume of 1 μL. Data were subsequently analyzed using pClamp11 software (Molecular Devices). Oocytes were individually collected after recordings, lysed in 20 µL TRPA1 lysis buffer, and subjected to anti-FLAG immunoblot analysis to confirm construct expression.
Sequence alignment
The sequence alignment in Fig. 7e was built by aligning the mouse, rat, chicken, fruit bat, ball python snake, boa snake, rat snake, and Drosophila melanogaster TRPA1 C-terminal sequences to residues 1089–1112 of human TRPA1 in Sequence Logo.
Statistical analysis
All data quantification was performed in Microsoft Excel. Quantified data presentation and statistical analyses were performed in GraphPad Prism. Criterion for statistical significance for all tests was p < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available as an accompanying Source data file. Uncropped blots and Coomassie gels are provided in Supplementary Figs. 12–15. The 6V9W and 3SUI PDB files were used in this study. Models were built with ChimeraX version 1.10. Source data are provided with this paper.
References
Clapham, D. E. Calcium signaling. Cell 131, 1047–1058 (2007).
Averbeck, B. Encyclopedia of Pain 1320-1322 (Springer, 2007).
Chiu, I. M., von Hehn, C. A. & Woolf, C. J. Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat. Neurosci. 15, 1063–1067 (2012).
Ji, R. R., Nackley, A., Huh, Y., Terrando, N. & Maixner, W. Neuroinflammation and central sensitization in chronic and widespread pain. Anesthesiology 129, 343–366 (2018).
Matsuda, M., Huh, Y. & Ji, R. R. Roles of inflammation, neurogenic inflammation, and neuroinflammation in pain. J. Anesth. 33, 131–139 (2019).
Richardson, J. D. & Vasko, M. R. Cellular mechanisms of neurogenic inflammation. J. Pharm. Exp. Ther. 302, 839–845 (2002).
Gao, R. et al. Potent analgesic effects of a store-operated calcium channel inhibitor. Pain 154, 2034–2044 (2013).
Hagenston, A. M. & Simonetti, M. Neuronal calcium signaling in chronic pain. Cell Tissue Res. 357, 407–426 (2014).
Yousuf, M. S. et al. Endoplasmic reticulum stress in the dorsal root ganglia regulates large-conductance potassium channels and contributes to pain in a model of multiple sclerosis. FASEB J. 34, 12577–12598 (2020).
Bali, A. et al. Molecular mechanism of hyperactivation conferred by a truncation of TRPA1. Nat. Commun. 14, 2867 (2023).
Kremeyer, B. et al. A gain-of-function mutation in TRPA1 causes familial episodic pain syndrome. Neuron 66, 671–680 (2010).
Nirenberg, M. J., Chaouni, R., Biller, T. M., Gilbert, R. M. & Paisan-Ruiz, C. A novel TRPA1 variant is associated with carbamazepine-responsive cramp-fasciculation syndrome. Clin. Genet. 93, 164–168 (2018).
Bautista, D. M., Pellegrino, M. & Tsunozaki, M. TRPA1: a gatekeeper for inflammation. Annu. Rev. Physiol. 75, 181–200 (2013).
Chen, J. & Hackos, D. H. TRPA1 as a drug target-promise and challenges. Naunyn Schmiedeb. Arch. Pharm. 388, 451–463 (2015).
Grace, M. S., Baxter, M., Dubuis, E., Birrell, M. A. & Belvisi, M. G. Transient receptor potential (TRP) channels in the airway: role in airway disease. Br. J. Pharm. 171, 2593–2607 (2014).
Julius, D. TRP channels and pain. Annu. Rev. Cell Dev. Biol. 29, 355–384 (2013).
Talavera, K. et al. Mammalian transient receptor potential TRPA1 Channels: from structure to disease. Physiol. Rev. 100, 725–803 (2020).
Zhang, X. Targeting TRP ion channels for itch relief. Naunyn Schmiedeb. Arch. Pharm. 388, 389–399 (2015).
Bandell, M. et al. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 41, 849–857 (2004).
Hinman, A., Chuang, H. H., Bautista, D. M. & Julius, D. TRP channel activation by reversible covalent modification. Proc. Natl. Acad. Sci. USA 103, 19564–19568 (2006).
Jordt, S. E. et al. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 427, 260–265 (2004).
Macpherson, L. J. et al. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 445, 541–545 (2007).
Andre, E. et al. Cigarette smoke-induced neurogenic inflammation is mediated by alpha,beta-unsaturated aldehydes and the TRPA1 receptor in rodents. J. Clin. Investig. 118, 2574–2582 (2008).
Balestrini, A. et al. A TRPA1 inhibitor suppresses neurogenic inflammation and airway contraction for asthma treatment. J. Exp. Med. 218, e20201637 (2021).
Gouin, O. et al. TRPV1 and TRPA1 in cutaneous neurogenic and chronic inflammation: pro-inflammatory response induced by their activation and their sensitization. Protein Cell 8, 644–661 (2017).
Kunkler, P. E., Ballard, C. J., Oxford, G. S. & Hurley, J. H. TRPA1 receptors mediate environmental irritant-induced meningeal vasodilatation. Pain 152, 38–44 (2011).
Meseguer, V. et al. TRPA1 channels mediate acute neurogenic inflammation and pain produced by bacterial endotoxins. Nat. Commun. 5, 3125 (2014).
Trevisani, M. et al. 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc. Natl. Acad. Sci. USA 104, 13519–13524 (2007).
Fernandes, E. S., Fernandes, M. A. & Keeble, J. E. The functions of TRPA1 and TRPV1: moving away from sensory nerves. Br. J. Pharm. 166, 510–521 (2012).
Julius, D. & Basbaum, A. I. Molecular mechanisms of nociception. Nature 413, 203–210 (2001).
Nassini, R., Materazzi, S., Benemei, S. & Geppetti, P. The TRPA1 channel in inflammatory and neuropathic pain and migraine. Rev. Physiol. Biochem. Pharm. 167, 1–43 (2014).
Asgar, J. et al. The role of TRPA1 in muscle pain and mechanical hypersensitivity under inflammatory conditions in rats. Neuroscience 310, 206–215 (2015).
Holzer, P. TRP channels in the digestive system. Curr. Pharm. Biotechnol. 12, 24–34 (2011).
Wang, Z. et al. The TRPA1 channel in the cardiovascular system: promising features and challenges. Front. Pharm. 10, 1253 (2019).
Nagata, K., Duggan, A., Kumar, G. & Garcia-Anoveros, J. Nociceptor and hair cell transducer properties of TRPA1, a channel for pain and hearing. J. Neurosci. 25, 4052–4061 (2005).
Wang, Y. Y., Chang, R. B., Waters, H. N., McKemy, D. D. & Liman, E. R. The nociceptor ion channel TRPA1 is potentiated and inactivated by permeating calcium ions. J. Biol. Chem. 283, 32691–32703 (2008).
Conaway, C. C., Krzeminski, J., Amin, S. & Chung, F. L. Decomposition rates of isothiocyanate conjugates determine their activity as inhibitors of cytochrome p450 enzymes. Chem. Res. Toxicol. 14, 1170–1176 (2001).
Sura, L. et al. C-terminal acidic cluster is involved in Ca2+-induced regulation of human transient receptor potential ankyrin 1 channel. J. Biol. Chem. 287, 18067–18077 (2012).
Zhao, J., Lin King, J. V., Paulsen, C. E., Cheng, Y. & Julius, D. Irritant-evoked activation and calcium modulation of the TRPA1 receptor. Nature 585, 141–145 (2020).
Zurborg, S., Yurgionas, B., Jira, J. A., Caspani, O. & Heppenstall, P. A. Direct activation of the ion channel TRPA1 by Ca2+. Nat. Neurosci. 10, 277–279 (2007).
Hasan, R., Leeson-Payne, A. T., Jaggar, J. H. & Zhang, X. Calmodulin is responsible for Ca(2+)-dependent regulation of TRPA1 Channels. Sci. Rep. 7, 45098 (2017).
Dang, S. et al. Structural insight into TRPV5 channel function and modulation. Proc. Natl. Acad. Sci. USA 116, 8869–8878 (2019).
Hughes, T. E. T. et al. Structural insights on TRPV5 gating by endogenous modulators. Nat. Commun. 9, 4198 (2018).
Kovalevskaya, N. V. et al. Structural analysis of calmodulin binding to ion channels demonstrates the role of its plasticity in regulation. Pflug. Arch. 465, 1507–1519 (2013).
Lee, C. H. & MacKinnon, R. Activation mechanism of a human SK-calmodulin channel complex elucidated by cryo-EM structures. Science 360, 508–513 (2018).
Singh, A. K., McGoldrick, L. L., Twomey, E. C. & Sobolevsky, A. I. Mechanism of calmodulin inactivation of the calcium-selective TRP channel TRPV6. Sci. Adv. 4, eaau6088 (2018).
Ben-Johny, M. et al. Conservation of Ca2+/calmodulin regulation across Na and Ca2+ channels. Cell 157, 1657–1670 (2014).
Chang, A. et al. A calmodulin C-Lobe Ca(2+)-dependent switch governs Kv7 channel function. Neuron 97, 836–852 e836 (2018).
DeMaria, C. D., Soong, T. W., Alseikhan, B. A., Alvania, R. S. & Yue, D. T. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature 411, 484–489 (2001).
Dick, I. E. et al. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature 451, 830–834 (2008).
Keen, J. E. et al. Domains responsible for constitutive and Ca(2+)-dependent interactions between calmodulin and small conductance Ca(2+)-activated potassium channels. J. Neurosci. 19, 8830–8838 (1999).
Lee, A. et al. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature 399, 155–159 (1999).
Liang, H. et al. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron 39, 951–960 (2003).
Peterson, B. Z., DeMaria, C. D., Adelman, J. P. & Yue, D. T. Calmodulin is the Ca2+ sensor for Ca2+ -dependent inactivation of L-type calcium channels. Neuron 22, 549–558 (1999).
Singh, A. et al. C-terminal modulator controls Ca2+-dependent gating of Ca(v)1.4 L-type Ca2+ channels. Nat. Neurosci. 9, 1108–1116 (2006).
Tadross, M. R., Ben Johny, M. & Yue, D. T. Molecular endpoints of Ca2+/calmodulin- and voltage-dependent inactivation of Ca(v)1.3 channels. J. Gen. Physiol. 135, 197–215 (2010).
Wahl-Schott, C. et al. Switching off calcium-dependent inactivation in L-type calcium channels by an autoinhibitory domain. Proc. Natl. Acad. Sci. USA 103, 15657–15662 (2006).
Xia, X. M. et al. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature 395, 503–507 (1998).
Zuhlke, R. D. & Reuter, H. Ca2+-sensitive inactivation of L-type Ca2+ channels depends on multiple cytoplasmic amino acid sequences of the alpha1C subunit. Proc. Natl. Acad. Sci. USA 95, 3287–3294 (1998).
Campiglio, M. & Flucher, B. E. The role of auxiliary subunits for the functional diversity of voltage-gated calcium channels. J. Cell Physiol. 230, 2019–2031 (2015).
Deutsch, C. Potassium channel ontogeny. Annu Rev. Physiol. 64, 19–46 (2002).
Dolphin, A. C. Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J. Physiol. 594, 5369–5390 (2016).
Isom, L. L., De Jongh, K. S. & Catterall, W. A. Auxiliary subunits of voltage-gated ion channels. Neuron 12, 1183–1194 (1994).
Yu, F. H. & Catterall, W. A. Overview of the voltage-gated sodium channel family. Genome Biol. 4, 207 (2003).
Wu, X. & Bers, D. M. Free and bound intracellular calmodulin measurements in cardiac myocytes. Cell Calcium 41, 353–364 (2007).
Liu, C. et al. A non-covalent ligand reveals biased agonism of the TRPA1 ion channel. Neuron 109, 273–284 e274 (2021).
Meents, J. E., Ciotu, C. I. & Fischer, M. J. M. TRPA1: a molecular view. J. Neurophysiol. 121, 427–443 (2019).
Gaudet, R. A primer on ankyrin repeat function in TRP channels and beyond. Mol. Biosyst. 4, 372–379 (2008).
Cordero-Morales, J. F., Gracheva, E. O. & Julius, D. Cytoplasmic ankyrin repeats of transient receptor potential A1 (TRPA1) dictate sensitivity to thermal and chemical stimuli. Proc. Natl. Acad. Sci. USA 108, E1184–E1191 (2011).
Kilpatrick, B. S., Yates, E., Grimm, C., Schapira, A. H. & Patel, S. Endo-lysosomal TRP mucolipin-1 channels trigger global ER Ca2+ release and Ca2+ influx. J. Cell Sci. 129, 3859–3867 (2016).
Branon, T. C. et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 36, 880–887 (2018).
Chin, D. & Means, A. R. Calmodulin: a prototypical calcium sensor. Trends Cell Biol. 10, 322–328 (2000).
Bate, N. et al. A novel mechanism for calmodulin-dependent inactivation of transient receptor potential vanilloid 6. Biochemistry 57, 2611–2622 (2018).
Bokhovchuk, F. M. et al. The structural basis of calcium-dependent inactivation of the transient receptor potential vanilloid 5 channel. Biochemistry 57, 2623–2635 (2018).
Chen, W. et al. Calmodulin binds to Drosophila TRP with an unexpected mode. Structure 29, 330–344 e334 (2021).
Lau, K., Chan, M. M. & Van Petegem, F. Lobe-specific calmodulin binding to different ryanodine receptor isoforms. Biochemistry 53, 932–946 (2014).
Marques-Carvalho, M. J. et al. Molecular insights into the mechanism of calmodulin inhibition of the EAG1 potassium channel. Structure 24, 1742–1754 (2016).
Sarhan, M. F., Van Petegem, F. & Ahern, C. A. A double tyrosine motif in the cardiac sodium channel domain III-IV linker couples calcium-dependent calmodulin binding to inactivation gating. J. Biol. Chem. 284, 33265–33274 (2009).
Andreasen, T. J., Luetje, C. W., Heideman, W. & Storm, D. R. Purification of a novel calmodulin binding protein from bovine cerebral cortex membranes. Biochemistry 22, 4615–4618 (1983).
Baudier, J., Deloulme, J. C., Van Dorsselaer, A., Black, D. & Matthes, H. W. Purification and characterization of a brain-specific protein kinase C substrate, neurogranin (p17). Identification of a consensus amino acid sequence between neurogranin and neuromodulin (GAP43) that corresponds to the protein kinase C phosphorylation site and the calmodulin-binding domain. J. Biol. Chem. 266, 229–237 (1991).
Xia, Z. & Storm, D. R. The role of calmodulin as a signal integrator for synaptic plasticity. Nat. Rev. Neurosci. 6, 267–276 (2005).
Gardill, B. R., Rivera-Acevedo, R. E., Tung, C. C. & Van Petegem, F. Crystal structures of Ca(2+)-calmodulin bound to Na(V) C-terminal regions suggest role for EF-hand domain in binding and inactivation. Proc. Natl. Acad. Sci. USA 116, 10763–10772 (2019).
Lau, S. Y., Procko, E. & Gaudet, R. Distinct properties of Ca2+-calmodulin binding to N- and C-terminal regulatory regions of the TRPV1 channel. J. Gen. Physiol. 140, 541–555 (2012).
Sali, A. & Blundell, T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993).
Neher, E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron 20, 389–399 (1998).
Song, L. S., Sham, J. S., Stern, M. D., Lakatta, E. G. & Cheng, H. Direct measurement of SR release flux by tracking ‘Ca2+ spikes’ in rat cardiac myocytes. J. Physiol. 512, 677–691 (1998).
Doerner, J. F., Gisselmann, G., Hatt, H. & Wetzel, C. H. Transient receptor potential channel A1 is directly gated by calcium ions. J. Biol. Chem. 282, 13180–13189 (2007).
Paulsen, C. E., Armache, J. P., Gao, Y., Cheng, Y. & Julius, D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 520, 511–517 (2015).
Suo, Y. et al. Structural insights into electrophile irritant sensing by the human TRPA1 channel. Neuron 105, 882–894 e885 (2020).
Loukin, S. H., Teng, J. & Kung, C. A channelopathy mechanism revealed by direct calmodulin activation of TrpV4. Proc. Natl. Acad. Sci. USA 112, 9400–9405 (2015).
Van Petegem, F., Lobo, P. A. & Ahern, C. A. Seeing the forest through the trees: towards a unified view on physiological calcium regulation of voltage-gated sodium channels. Biophys. J. 103, 2243–2251 (2012).
Tidow, H. & Nissen, P. Structural diversity of calmodulin binding to its target sites. FEBS J. 280, 5551–5565 (2013).
Yap, K. L. et al. Calmodulin target database. J. Struct. Funct. Genom. 1, 8–14 (2000).
Fallon, J. L. et al. Crystal structure of dimeric cardiac L-type calcium channel regulatory domains bridged by Ca2+* calmodulins. Proc. Natl. Acad. Sci. USA 106, 5135–5140 (2009).
Kim, E. Y. et al. Structures of CaV2 Ca2+/CaM-IQ domain complexes reveal binding modes that underlie calcium-dependent inactivation and facilitation. Structure 16, 1455–1467 (2008).
Liu, Y. et al. Crystal structure of calmodulin binding domain of orai1 in complex with Ca2+ calmodulin displays a unique binding mode. J. Biol. Chem. 287, 43030–43041 (2012).
Yu, Q., Anderson, D. E., Kaur, R., Fisher, A. J. & Ames, J. B. The crystal structure of calmodulin bound to the cardiac ryanodine receptor (RyR2) at residues Phe4246-Val4271 reveals a fifth calcium binding site. Biochemistry 60, 1088–1096 (2021).
Zhang, M. et al. Structural basis for calmodulin as a dynamic calcium sensor. Structure 20, 911–923 (2012).
Halling, D. B. et al. Determinants in CaV1 channels that regulate the Ca2+ sensitivity of bound calmodulin. J. Biol. Chem. 284, 20041–20051 (2009).
Meador, W. E., Means, A. R. & Quiocho, F. A. Target enzyme recognition by calmodulin: 2.4 A structure of a calmodulin-peptide complex. Science 257, 1251–1255 (1992).
Saimi, Y. & Kung, C. Calmodulin as an ion channel subunit. Annu. Rev. Physiol. 64, 289–311 (2002).
Feldkamp, M. D., Yu, L. & Shea, M. A. Structural and energetic determinants of apo calmodulin binding to the IQ motif of the Na(V)1.2 voltage-dependent sodium channel. Structure 19, 733–747 (2011).
Etxeberria, A. et al. Calmodulin regulates the trafficking of KCNQ2 potassium channels. FASEB J. 22, 1135–1143 (2008).
Ghosh, S., Nunziato, D. A. & Pitt, G. S. KCNQ1 assembly and function is blocked by long-QT syndrome mutations that disrupt interaction with calmodulin. Circ. Res. 98, 1048–1054 (2006).
Liu, W. & Devaux, J. J. Calmodulin orchestrates the heteromeric assembly and the trafficking of KCNQ2/3 (Kv7.2/3) channels in neurons. Mol. Cell Neurosci. 58, 40–52 (2014).
Shamgar, L. et al. Calmodulin is essential for cardiac IKS channel gating and assembly: impaired function in long-QT mutations. Circ. Res. 98, 1055–1063 (2006).
Orlov, S. N. & Kravtsov, G. M. [Participation of calmodulin in the regulation of plasma membrane electric potential by intracellular calcium]. Biokhimiia 48, 1447–1455 (1983).
Tavares-Ferreira, D. et al. Spatial transcriptomics of dorsal root ganglia identifies molecular signatures of human nociceptors. Sci. Transl. Med. 14, eabj8186 (2022).
Schonherr, R., Lober, K. & Heinemann, S. H. Inhibition of human ether a go-go potassium channels by Ca(2+)/calmodulin. EMBO J. 19, 3263–3271 (2000).
Whicher, J. R. & MacKinnon, R. Structure of the voltage-gated K(+) channel Eag1 reveals an alternative voltage sensing mechanism. Science 353, 664–669 (2016).
Zhang, S., Ehlers, M. D., Bernhardt, J. P., Su, C. T. & Huganir, R. L. Calmodulin mediates calcium-dependent inactivation of N-methyl-D-aspartate receptors. Neuron 21, 443–453 (1998).
Li, J. et al. Research Progress on TRPA1 in Diseases. J. Membr. Biol. 256, 301–316 (2023).
Lobley, A., Swindells, M. B., Orengo, C. A. & Jones, D. T. Inferring function using patterns of native disorder in proteins. PLoS Comput. Biol. 3, e162 (2007).
Magidovich, E., Orr, I., Fass, D., Abdu, U. & Yifrach, O. Intrinsic disorder in the C-terminal domain of the Shaker voltage-activated K+ channel modulates its interaction with scaffold proteins. Proc. Natl. Acad. Sci. USA 104, 13022–13027 (2007).
Cao, E., Cordero-Morales, J. F., Liu, B., Qin, F. & Julius, D. TRPV1 channels are intrinsically heat sensitive and negatively regulated by phosphoinositide lipids. Neuron 77, 667–679 (2013).
Chuang, H. H. et al. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 411, 957–962 (2001).
Prescott, E. D. & Julius, D. A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science 300, 1284–1288 (2003).
Gracheva, E. O. et al. Ganglion-specific splicing of TRPV1 underlies infrared sensation in vampire bats. Nature 476, 88–91 (2011).
Goretzki, B., Guhl, C., Tebbe, F., Harder, J. M. & Hellmich, U. A. Unstructural biology of TRP Ion channels: the role of intrinsically disordered regions in channel function and regulation. J. Mol. Biol. 433, 166931 (2021).
Allard, C. A. H. et al. Structural basis of sensory receptor evolution in octopus. Nature 616, 373–377 (2023).
Bellono, N. W., Leitch, D. B. & Julius, D. Molecular basis of ancestral vertebrate electroreception. Nature 543, 391–396 (2017).
Kang, G. et al. Sensory specializations drive octopus and squid behaviour. Nature 616, 378–383 (2023).
Schoenmakers, T. J., Visser, G. J., Flik, G. & Theuvenet, A. P. CHELATOR: an improved method for computing metal ion concentrations in physiological solutions. Biotechniques 12, 870–874, 876–879 (1992).
Kainosho, M. et al. Optimal isotope labelling for NMR protein structure determinations. Nature 440, 52–57 (2006).
Piazza, M., Guillemette, J. G. & Dieckmann, T. Chemical shift assignments of calmodulin constructs with EF hand mutations. Biomol. NMR Assign. 10, 193–198 (2016).
Cala, O., Guilliere, F. & Krimm, I. NMR-based analysis of protein-ligand interactions. Anal. Bioanal. Chem. 406, 943–956 (2014).
Williamson, R. A., Carr, M. D., Frenkiel, T. A., Feeney, J. & Freedman, R. B. Mapping the binding site for matrix metalloproteinase on the N-terminal domain of the tissue inhibitor of metalloproteinases-2 by NMR chemical shift perturbation. Biochemistry 36, 13882–13889 (1997).
Norris, M., Fetler, B., Marchant, J. & Johnson, B. A. NMRFx processor: a cross-platform NMR data processing program. J. Biomol. NMR 65, 205–216 (2016).
Acknowledgements
We thank Wendy Gilbert, Franziska Bleichert, Lily Kabeche, Tony Koleske, Enrique de la Cruz, and Yong Xiong for constructive suggestions, Fred Sigworth, and members of the Paulsen lab for helpful discussions and for critical reading of the manuscript, and Grover Paulsen-Sharpe and Marilee Pawsen for moral support. We thank Ewa Folta-Stogniew and Eric Paulson for technical assistance with ITC experiments and NMR data collection, respectively. We thank the referees for their thoughtful reviews, which strengthens the impact of this manuscript. J.H.S. is supported by an NIH NINDS NRSA pre-doctoral fellowship (1F31NS122412) and a Cellular and Molecular Biology pre-doctoral training grant (T32GM007223). C.G. and G.Q. are supported by a Biophysics pre-doctoral training grant (T32GM149438) and Gruber Science Fellowships from Yale. K.M.T. is supported by an Anderson Postdoctoral Fellowship and a Debra Fine ’81 Fund for Equality in Science fellowship from Yale University. A.B. is supported by a Biophysics pre-doctoral training grant (T32GM008283). This research was supported by an International Association for the Study of Pain Early Career Research Grant, a Rita Allen Foundation and American Pain Society Pain Scholar Award, and by NIH grant R35GM142825 to C.E.P. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
J.H.S. and C.E.P. planned the project, designed experiments and cloned all constructs used in the study. J.H.S. and C.G. carried out calcium imaging and surface biotinylation assays. J.H.S., C.G., G.A.A., and C.E.P. carried out Calmodulin binding assays. J.H.S. carried out size exclusion chromatography assays and isothermal titration calorimetry. J.H.S. and G.Q. performed the two-electrode voltage clamp work. J.H.S. and K.M.T. performed protein purifications. A.B. carried out immunostainings. A.B., K.M.T., and C.E.P. carried out proximity biotinylation assays. K.M.T. collected and analyzed the 15N-1H HSQC NMR spectroscopy data. C.E.P. generated the homology model. J.H.S. and C.E.P. wrote the manuscript with input from C.G., K.M.T., G.Q., G.A.A., and A.B.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xuming Zhang and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sanders, J.H., Garcia, C., Taiwo, K.M. et al. Calmodulin binding is required for calcium mediated TRPA1 desensitization. Nat Commun 16, 8640 (2025). https://doi.org/10.1038/s41467-025-63767-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63767-7
