
Abstract
BioPROTACs are heterobifunctional proteins designed for targeted protein degradation (TPD). They are useful not only for probing protein functions but also offer a therapeutic avenue for modulating disease-related proteins. To extend the use of TPD beyond just protein attenuation, we introduce a synthetic framework for logic-gated, switchable TPD to achieve conditional control of protein content. By exploiting both the cleavage and ligation functionalities of Sortase A (SrtA), we present a new strategy utilizing SrtA as the control input to direct bioPROTAC activity for switchable TPD. Furthermore, by layering the SrtA input with protease gating, conditional degradation phenotypes can be readily adapted with minimal modifications to the design. This Logic-gated AdPROM deploying SrtA-mediated Element Recombination (LASER) platform allows us to expand the possible protein degradation outcomes in mammalian cells using Boolean logic operations depending on the input combinations. The flexibility to modulate the level of multiple native intracellular proteins can potentially lead to applications from therapy to diagnostics and biotechnology.
Similar content being viewed by others
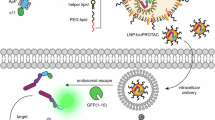


Introduction
Cell physiology is programmed through the dynamic interplay of intracellular protein expression and activity, directing virtually all cellular functions1,2. Rewiring these cellular networks requires the design of synthetic circuits to confer new cellular outputs3. While most efforts have focused on transcriptional regulation because of the relative ease to rewire gene expression to recognize different input signals4,5,6, protein-level circuits, in which post-translational modifications can specifically modify protein activity, localization, and/or stability, can often achieve much faster signaling processing7,8.
Protein degradation is an effective native mechanism used in modulating intracellular information, and plays an essential role in maintaining cellular homeostasis9. Repurposing native protein degradation in a synthetic context is gaining attention as a new strategy to manipulate cellular behavior for a wide range of applications including disease detection and therapy10. Targeted protein degradation (TPD) is a particularly attractive strategy for modulating protein contents by hijacking the native ubiquitin-proteasome system (UPS) to achieve a range of synthetic outcomes in mammalian systems11,12,13,14,15,16. Typically, a bifunctional small molecule proteolysis-targeting chimera (PROTAC) or a protein fusion equivalent (bioPROTAC) shepherds the colocalization of the native ubiquitination pathway with a specific protein of interest (POI) for degradation14,15,16,17,18. While this strategy is well documented, the current state of the art is static in nature and only limited to applications where protein attenuation is desired. The inability to toggle TPD precludes the feasibility to modulate the level of multiple protein targets in a conditional manner.
Recently, we have demonstrated the integration of functional protease linker sequences into bioPROTACs, such as the Affinity directed Protein Missile (AdPROM) system19, to allow tunable modulation of intracellular protein levels. AdPROM is a bifunctional bioPROTAC composed of an endogenous CUL2 recognition motif (Von Hippel−Lindau (VHL) protein) fused to a designer binding protein (DBP) that recognizes the target protein for degradation19,20. Deploying functional linkers also expands the input responsiveness to include small molecule or optogenetic control via Tobacco Etch Virus protease (TEVp)-mediated signal transduction. While this approach enables an effective way to turn off TPD, it remains limited by the lack of ON-and-OFF tuning or multi-target functionality21.
To address these challenges, we report here a robust and modular approach to toggle the ON-and-OFF behavior of TPD via Sortase A (SrtA)-mediated transpeptidation22,23. SrtA is an enzyme originating from Gram-positive bacteria that participate in the covalent attachment of proteins to the bacterial cell wall24,25. The canonical Staphylococcus aureus SrtA recognizes an LPXTG motif and harbors a catalytic cysteine residue to cleave the recognition motif between the threonine and glycine residues. This proteolytic cleavage reaction yields a thioacyl intermediate, which is resolved via the creation of a new peptide bond with an N-terminal triglycine nucleophile26,27,28. SrtA has been used in protein engineering applications to elicit conjugation of disparate protein components, primarily in a bacterial or in vitro context28,29.
While prior studies have overwhelmingly focused on the ligation property of SrtA, we exploit here the often neglected but powerful protease-like cleavage function to enable the reversible switching of TPD phenotypes in a SrtA-dependent fashion. The dual functionality of SrtA also offers the flexibility to interchange the degradation target by swapping the binding domain on AdPROM via sequential cleavage and ligation. By masking the required N-terminus triglycine motif for ligation that is activated only by removing the ENLYFQ | G blocking sequence by TEVp, we further integrate protease-masked SrtA responsiveness into Boolean logic operations.
Here, we present a new platform for toggling TPD based on the Logic-gated AdPROM deploying SrtA-mediated Element Recombination (LASER) bioPROTAC system (Fig. 1). First, we generated SrtA-responsive AdPROMSrtA designs that could be either activated or inactivated for TPD. By exploiting the dual cleavage and ligation functionalities of SrtA, we established a switchable AdPROMSrtA design, allowing intracellular selection between two different protein targets for TPD. Finally, we integrated the signal processing functionalities of both TEVp and SrtA into an AND-gated design to perform tunable targeting of multiple intracellular proteins by TPD.

Programmable targeted protein degradation outcomes with integrated target selection and safety modes can be achieved using different input combinations. LASER is comprised of the Von Hippel Lindel (VHL) E3 ligase recruitment protein fused to a designer binding protein (DBP1) that recruits a target protein (T1) for degradation. These proteins are genetically fused together using a Sortase A (SrtA) recognition motif linker. Concurrently, a DPB2 capable of binding a different target protein (T2) is expressed with an N-terminal tobacco etch virus protease (TEVp) responsive motif as a protective cap and a triglycine sequence for Sortase ligation. The default LASER state (1) redirects the native ubiquitin proteasome system to degrade T1 with no impact on T2. Expression of TEVp (2) removes the protective cap from DPB2 but no switching occurs without SrtA. However, LASER can be deactivated via addition of SrtA alone (3), resulting in rescue of T1. When both inputs are introduced (4), the LASER targeting is switched – T1 is rescued and the orthogonal T2 is now degraded.
Results
Demonstrate effective SrtA-mediated bioconjugation in human cells via a splitFAST fluorescent reporter system
While intracellular ligation in HEK293T cells has been reported using the calcium-dependent Streptococcus pyogenes SrtA, a large excess amount of G5-eGFP is needed to drive a significant level of ligation with Rac1-LPETG-Myc24. The recent creation of Ca2+-independent Staphylococcus aureus SrtA mutants (notably the 7 M and 7+ hepta-mutant variants)30 with up 140-fold increase in activity has allowed improved intracellular ligation of LPETG-tagged proteins in C. elegans31. Using this improved SrtA7+ variant, we first sought to demonstrate the efficient cleavage and ligation activities in HeLa cells. SrtA7+ activity was evaluated using the splitFAST fluorescent reporter system, which has been shown to provide real-time and reversible visualization of protein-protein interactions in live cells (Fig. 2a)32,33.

a The FAST protein reversibly binds HMBR to produce a fluorescent signal upon excitation at 481 nm and emission at 540 nm. b Schematic description of reporter protein sequences. FASTSrtA is expressed as a full-length protein with the LPETGGG SrtA recognition motif inserted after nFAST. An FKBP domain and a (G4S)2 linker were inserted before cFAST to match the design of the splitFAST pair as it is not possible to express the 10 amino acid cFAST without a fusion partner. To create splitFASTSrtA, the nFAST and FKBP-cFAST fragments are fused to the LPETGGGPV and triglycine motifs, respectively, for separate expression. An HA (hemagglutinin)-tag was inserted into both the full-length FASTSrtA and splitFASTSrtA sequences for probing protein expression. c SrtA-mediated ligation of splitFASTSrtA fragments results in a tethered splitFAST protein and fluorescence activation. d Insertion of a SrtA recognition motif into the same full-length FAST protein enables SrtA cleavage to separate the nFAST and cFAST fragment and induce inactivation of fluorescent signal. e HeLa cells expressing either the full length FASTSrtA or the splitFASTSrtA fragments were used to confirm intracellular SrtA-mediated transpeptidation activity. In both cases, co-expression of TEVp was used as a negative control. Ligation of the splitFASTSrtA fragments proceeds only under the condition where SrtA7+ is present, resulting in reconstitution of FAST protein fluorescence as confirmed by flow cytometry. Similarly, successful cleavage of the full-length FASTSrtA protein by SrtA7+ is accompanied by a decrease in FAST fluorescence detected using a plate-reader based fluorescence measurements as described in the supplementary methods. Data are biological triplicates (three separately transfected wells) of the relative fluorescence units (RFU) as measured on a per-well basis using a plate-reader set to 480/540 nm excitation/emission wavelengths. Values are normalized to the RFU for inactive expression in HeLa cells. For comparison of split and full-length expressions, background fluorescence is subtracted prior to normalization. Error bars represent standard deviation (SD) of the mean. Statistical significance was evaluated using the ordinary one-way ANOVA test; P < 0.0001 is denoted as ****. Source data are provided as a Source Data file.
We first investigated whether the splitFAST pair could be activated by SrtA-mediated ligation. To facilitate this, an LPETGGG motif was appended to the C-terminus of the larger nFAST fragment, and a triglycine motif was appended to the N-terminus of the smaller cFAST fragment (Fig. 2b, c). An FK506 binding protein (FKBP) domain was added in front of cFAST (10 amino acids) to enable stable expression of the fusion protein in HeLa cells32. As expected, only background fluorescence was observed when the splitFASTSrtA pair was expressed in the absence of SrtA7+. In contrast, co-expression with SrtA7+ resulted in successful ligation of the splitFASTSrtA pair, leading to HMBR binding and reconstituted fluorescence 24 h post transfection (Fig. 2e). We next sought to confirm the cleavage activity of SrtA7+ using a full-length FASTSrtA design that would respond to SrtA in the opposite manner. We constructed a full-length splitFAST fusion protein with the same LPETG3 motif inserted at the split site between nFAST and FKBP-cFAST (Fig. 2b, d). As expected, HeLa cells transfected with FASTSrtA and the non-interacting TEVp exhibited strong fluorescence at a similar level with the ligated splitFASTSrtA pair (Fig. 2e). However, co-expression and cleavage by SrtA7+ separated the two domains and reduced the fluorescent signal by more than 2-fold (Fig. 2e). Collectively, these results demonstrated that SrtA7+ could efficiently direct both ligation and cleavage reactions intracellularly.
Sortase A as a bioPROTAC control switch for targeted protein degradation
We next sought to exploit the use of SrtA7+ to achieve “ON and OFF” TPD in HeLa cells. To achieve OFF control, we inserted an LPETGG motif into the linker region of the GFP-targeting AdPROMSrtA in lieu of our previously used TEVp cleavage site (Fig. 3a). Since substrates containing two glycines has been shown to have significantly lower ligation efficiency, the number of glycines in the SrtA cleavage motif is reduced to two (LPETGG) to minimize re-ligation34. This design achieved a similar level of SH2-GFP knockdown as with the original AdPROM (Fig. 3b). Only co-expression with SrtA7+ resulted in the proper cleavage and deactivation, as demonstrated by a recovery in GPF signal, while strong attenuation of the GFP fluorescence was still detected with co-expression of the non-interacting TEVp (Fig. 3b). Western blotting was used to interrogate the cleavage reaction and disappearance of the full-length AdPROMSrtA band was observed only when SrtA7+ was co-expressed, consistent with the SH2-GFP degradation outcome (Supplementary Fig. 1).

a To achieve OFF control, a SrtA cleavable AdPROMSrtA is created via insertion of a LPETGG motif between the E3 ligase recruitment VHL domain and the GFP-targeting DBP (aGFP) in the original GFP-targeting AdPROM. Cleavage by SrtA separates the two domains and deactivates SH2-GFP degradation. b HeLa cells were transiently transfected with plasmid DNA expressing an SH2-GFP fluorescent reporter. TPD was induced through co-expression of either AdPROM, the TEVp-responsive AdPROMTEV, or the SrtA-responsive AdPROMSrtA. All three AdPROM variants demonstrate similar levels of SH2-GFP attenuation in the absence of both TEVp and SrtA. When the appropriate input is given, TPD inactivation is demonstrated by the three- to four-fold recovery in fluorescent signal. c For ON control, the VHL-LPETGG fusion and the GGG-ɑGFP fusion are independently expressed in the split AdPROMSrtA scheme. Here, the two components only assemble in the presence of a SrtA input to degrade SH2-GFP. d HeLa cells were transiently transfected with plasmid DNA expressing an SH2-GFP fluorescent reporter. TPD was induced via co-transfection of SrtA and with different ratios of the split AdPROMSrtA fusion proteins. Successful ligation of split AdPROMSrtA fragments is observed only at a GGG- ɑGFP /VHL-LPETGG transfection ratio higher than five as reflected by a similar level of SH2-GFP degradation to that of the full-length AdPROMSrtA. e To verify that the compatibility of this approach is not limited to a single DBP, a split AdPROMSrtAɑSH2 system was constructed using an SH2-targeting DBP (ɑSH2) domain. Here, full-length AdPROMSrtAɑSH2 could target SH2-GFP for degradation but would not interact with YFP. f Both split AdPROMSrtA variants can target the SH2-GFP reporter after SrtA ligation. However, co-expression of ligated AdPROMSrtAɑSH2 with the orthogonal YFP target results in no signal ablation. The specificity of the response confirms both the generalizability of this approach, and that the observed changes in fluorescence are degradation based. All data are presented as the average of biological triplicates for the geometric mean fluorescence intensity (MFI), normalized against the strongest fluorescence condition as a reference point. Error bars represent standard deviation (SD) of the mean. Statistical significance was evaluated using the ordinary one-way ANOVA test; P < 0.05 is denoted as *, P < 0.0001 is denoted as ****, no statistical significance is denoted as (ns). Source data are provided as a Source Data file.
To reverse the outcome into an ON control, we generated a split AdPROMSrtA pair comprised of the VHL domain with a C-terminus LPETGG motif, and the GFP-targeting nanobody GBP1 (aGFP) with an N-terminus triglycine (Fig. 3c). Unlike the cleavage reaction, SrtA-mediated ligation is typically pushed toward >85% completion by adding an excess amount (~5-fold) of one of the ligation partners35. For split AdPROMSrtA ligation, the level of ligated AdPROMSrtA can directly impact the extent of SH2-GFP degradation. To optimize ligation, experiments were performed by transfecting HeLa cells with different ratios of plasmid DNAs expressing either VHL-LPETGG or GGG- ɑGFP. Consistent with other reports, detectable SH2-GFP degradation was observed only when the GGG- ɑGFP to VHL-LPETGG transfection ratio is greater than 5:1 and SrtA7+ was co-expressed, indicative of efficient splitAdPROMSrtA ligation. Ratios lower than 5:1 resulted in no significant degradation (Fig. 3d).
We next confirmed that the ligation approach is compatible with the usage of different DBPs by replacing the GFP-targeting ɑGFP nanobody with an SH2-targeting NSa1 monobody (aSH2)36,37,38 (Fig. 3e). Ligation of VHL-LPETGG with GGG-aSH2 to generate the full-length SH2-targeting AdPROMSrtAɑSH2 resulted in a level of SH2-GFP degradation (Fig. 3f) comparable to that of ligation with GGG- ɑGFP. In contrast, only ligation of VHL-LPETGG with GGG- ɑGFP but not GGG- ɑSH2 could target the YFP reporter for degradation. This result confirms that TPD is a direct result of specific target binding and suggests the possibility of switching the degradation target simply by replacing the DBP.
Sortase A-mediated intracellular retargeting for conditional targeted protein degradation
The most exciting capability of SrtA is to elicit ON and OFF control simultaneously to mix and match different binding domains for TPD. This can be executed by first expressing a full-length AdPROMSrtA containing ɑGFP to target YFP for degradation. Inclusion of a LPETGG motif into the linker region enables not only SrtA-mediated cleavage of ɑGFP but also the simultaneous ligation with a compatible GGG- ɑSH2. In this manner, we could toggle protein degradation between two different targets via SrtA7+ expression as degradation of YFP is halted and degradation of SH2-miRFP670 is induced (Fig. 4a).

a Schematic representation of SrtA-mediated target selection for TPD. A full length AdPROMSrtA targeting YFP is redirected to target SH2-miRFP670 via sequential SrtA-mediated cleavage to remove the GFP-targeting ɑGFP and ligation with the SH2-targeting ɑSH2 to generate AdPROMSrtAɑSH2. b HeLa cells were transfected with plasmids expressing the full length GFP-targeting AdPROMSrtA and GGG-aSH2. Co-expression of the non-interacting TEVp has no impact on AdPROMSrtA as YFP remains degraded and SH2-miRFP670 is unaffected. In contrast, co-expression with SrtA7+ resulted in cleavage of GFP-targeting AdPROMSrtA and formation of AdPROMSrtAɑSH2 – toggling degradation away from YFP and toward SH2-miRFP670. Data are biological triplicates (three separately transfected wells) of the geometric mean fluorescence intensity (MFI). Normalization is done using the strongest fluorescence condition as a reference point. Error bars represent standard deviation (SD) of the mean. Statistical significance was evaluated using the ordinary one-way ANOVA test; P = 0.0022 is denoted as **, P < 0.0001 is denoted as ****. Source data are provided as a Source Data file.
To demonstrate this capability, we designed a system to toggle degradation between YFP and a near-infrared SH2-miRFP670 fusion protein. In the absence of SrtA7+, expression of the full length GFP-targeting AdPROMSrtA depleted YFP to a low level without any impact on SH2-miRFP670 even when GGG- ɑSH2 was co-expressed (Fig. 4b). However, expression of SrtA7+ effectively removed the GFP-targeting domain by cleavage and replaced it with the alternative Nsa1 monobody by ligation to drive formation of an SH2-targeting AdPROMSrtAɑSH2. Toggling between two target-binding domains rescued YFP from degradation and redirected the SH2-targeting AdPROMSrtAɑSH2 to promote degradation of the near-infrared SH2-miRFP670 fusion protein (Fig. 4b).
Expanding Sortase A ligation with logic functions
While SrtA7+ is efficient in offering simple ON and OFF control, it lacks the ability to perform more complex logic functions such as an AND gate operation. To overcome this limitation, we introduced a TEVp-cleavable masking motif in front of the triglycine recognition sequence to block ligation mediated by SrtA7+. Only cleavage of the TEVp recognition motif ENLYFQ | GGG39 liberates the masked triglycine tag for SrtA ligation (Fig. 5a), effectively creating an AND-gate design requiring both TEVp and SrtA7+ to generate a functional outcome.

a Sortase A ligation requires a free N-terminus triglycine to complete the nucleophilic attack for product formation. Insertion of a TEV protease cleavable peptide, ENLYFQ/G, upfront masks the triglycine motif. A fluorescent protein Electra2 is added in front of the TEVp cleavage site to provide better steric blocking of SrtA-mediated ligation. Cleavage of the masking domain by TEVp exposes the triglycine motif, enabling an AND-gated design requiring both TEVp and SrtA7+ for ligation. b HeLa cells were transiently transfected with plasmid DNA expressing SH2-miRFP670 fluorescent reporter and components of AND-gated split AdPROMSrtA ɑSH2. Both conditions where only a single input is present show no significant decrease in SH2-miRFP670 signal. However, when both TEVp and SrtA7+ are expressed, a significant reduction in SH2-miRFP670 signal, on par with that achieved using the full-length AdPROMSrtAɑSH2 alone, was detected only when Electra2-ENLYFQ | GGGG-aSH2 was delivered at a 6:1 transfection ratio relative to VHL-LPETGG to ensure efficient AND-gated ligation. Data are biological replicates of n = 4 (four separately transfected wells) of the geometric mean fluorescence intensity (MFI) normalized to the MFI for a SH2-miRFP670 expression control in HeLa cells. Error bars represent standard deviation (SD) of the mean. Ordinary one-way ANOVA was performed with P < 0.05 considered statistically significant throughout the experiments. P = 0.0030 was denoted as **, and P < 0.001 was denoted as ****. Source data are provided as a Source Data file.
To first test the effectiveness of the AND-gate architecture, we adapted an assay based on a cyclically permutated firefly luciferase (cycLucSbMV) that could be activated by the Soybean Mosaic Virus protease (SbMVp) to interrogate split protease ligation (Supplementary Fig. 2a)7. We constructed a SrtA-responsive split SbMVp pair and demonstrated this split pair had almost no background reconstitution as no activation of cycLucSbMV was detected (Supplementary Fig. 2b). In comparison, full activation was achieved when a full-length SbMVp was co-expressed. Co-expression of SrtA7+, on the other hand, resulted in successful ligation of the split SbMVp pair and cycLucSbMV activation (Supplementary Fig. 2b).
We next designed a masked split SbMVp pair to achieve cycLucSbMV activation using a two-input, AND-gate design. A TEVp-cleavage peptide motif was appended in front the triglycine tag of the cSbMVp fragment. For this masked split SbMVp pair, expression of either SrtA7+ or TEVp alone had no impact on cycLucSbMV activation (Supplementary Fig. 2b). However, co-expression of both SrtA7+ and TEVp resulted in ligation of the split SbMVpT-SrtA and cycLucSbMV activation. Successful ligation was further confirmed by western blotting (Supplementary Fig. 2b).
Logic-gated targeted protein degradation via protease-masked Sortase A ligation
We next implemented the AND-gate design to elicit improved control over TPD in a similar manner. A TEVp-cleavable motif was added in front of the SH2-targeting GGG-ɑSH2 (Fig. 5a). A fluorescent protein Electra2 was also added in front of the TEVp cleavage site (Electra2-ENLYFQ | GGGG-aSH2) to provide better steric blocking of SrtA ligation. Expressing the two split protein fragments (VHL-LPETGG and Electra2-ENLYFQ | GGGG-aSH2) even in the presence of TEVp had no ablative effect on levels of the near-infrared reporter (SH2-miRFP670). However, where both SrtA7+ and TEVp are co-expressed, full-length AdPROMSrtAɑSH2 assembly proceeded, and SH2-miRFP670 degradation was activated (Fig. 5b). Similar to the direct ligation results, full SH2-miRFP670 degradation was observed only when an Electra2-ENLYFQ | GGGG-aGFP to VHL-LPETGG transfection ratio higher than 5:1 was employed. Both TEVp and SrtA are required for cleavage and ligation, as co-expression with a similar Soybean Mosaic Virus protease (SbMVp) and SrtA could not activate TPD. This result confirms that SH2-miRFP670 degradation is solely due to formation of the new full-length AdPROMSrtAɑSH2, which requires specific cleavage by TEVp and ligation by SrtA. While the current design allows selective activation of targeted protein degradation based on an AND-gate architecture, an additional layer of control can be easily added by using split TEVp pairs that are responsive to either small molecules or light21.
While SrtA7+ can be used to toggle degradation between two different protein targets, this approach was constrained by the coupled deactivation and activation of the two degradation phenotypes. The compatibility of the AND-gate design with the toggling behavior presents an opportunity to decouple the deactivation and activation steps and generate LASER: Logic-gated AdPROM deploying SrtA mediated Element Recombination (Fig. 6a). By utilizing an AND-gated SH2-targeting monobody Nsa1 in conjunction with the SrtA-responsive GFP-targeting AdPROMSrtA, we could deactivate GFP-targeting separately from initiating SH2-targeting. This LASER strategy enables us to expand the possible outcomes from merely degrading either one of the two targets to now include a third potential outcome wherein neither target is degraded.

a Execution of the LASER system. Integrating AND-gated nucleophile ligation with a full-length AdPROMSrtA enables decoupling of intracellular targeting. b HeLa cells were transfected with plasmid DNAs expressing different LASER components, in conjunction with YFP and SH2-miRFP670 reporter proteins. Expression of TEVp alone enabled YFP-targeting to proceed as expected. Expression of both TEVp and SrtA7+ toggled targeting to SH2-miRFP670. However, logic-gated assembly elicited a third possible state: expression of only SrtA7+ turned OFF YFP degradation, while allowing SH2-miRFP670 to remain unperturbed – switching to a SAFE mode. Data are biological quintuplets (four separately transfected wells) of the geometric mean fluorescence intensity (MFI) normalized to the MFI for a high YFP and SH2-miRFP670 expression control in HeLa cells. Error bars represent standard deviation (SD) of the mean. Ordinary one-way ANOVA was performed with P < 0.05 considered statistically significant throughout the experiments. P = 0.0003 was denoted as *** and P < 0.0001 was denoted as ****. c Western blot analysis of the LASER experiment. TEVp cleavage and SrtA ligation were interrogated by probing for the HA-tagged ɑSH2. Full-length Electra2-ENLYFQ/GGG-HA- ɑSH2 intensity diminished and a 15 kDa cleavage band was detected upon TEVp co-expression (T), but no impact on the full-length protein was observed when only SrtA was co-expressed (S). A 38 kDa band resulting from SrtA mediated ligation of a TEVp cleavage product (GGGG-aSH2) to an independent SrtA-dependent ligation partner (VHL-SrtAs) was detected when both TEVp and SrtA were co-expressed (T + S). Western blotting with a VHL antibody elucidates the disappearance of a 39 kDa band upon cleavage of VHL-ɑGFP by SrtA(S), and restoration of a similar sized band when ligation of VHL-aSH2 occurs (S + T). Changes in YFP and SH2-miRFP670 levels were probed using the GFP and SH2 antibodies, respectively. A GAPDH control blot indicates consistent protein loading. Representative blot of n = 3 biological replicates with similar results. Source data are provided as a Source Data file.
To implement LASER, Electra2-ENLYFQ | GGGG-ɑSH2 was expressed in conjunction with the full length AdPROMSrtA to elicit a switchable targeting system with an additional OFF-state (Fig. 6b). In the presence of TEVp alone, the GFP-targeting AdPROMSrtA remained intact and only YFP was targeted for degradation. In the presence of SrtA7+ alone, the GFP-targeting AdPROMSrtA was cleaved and both YFP and SH2-miRFP670 signals remained high (Fig. 6b). However, SrtA-mediated cleavage and ligation occurred in the presence of both TEVp and SrtA7+, resulting in complete toggling between YFP and SH2-miRFP670 degradation 24 h post transfection (Fig. 6b). The ability to produce three distinct TPD states within a single cell growth cycle is the most unique of feature of LASER, making it useful to elicit a wider range of cellular outcomes compared to other reported TPD strategies using bioPROTACs.
Using a HA tag inserted in front of ɑSH2, successful cleavage and ligation of Electra2-ENLYFQ | GGGG- ɑSH2 for the LASER experiments were further verified by western blotting (Fig. 6c). A cleavage band was detected upon TEVp co-expression, but no impact was observed when only SrtA was co-expressed. A new ligated product corresponding to the concurrent cleavage of Electra2-ENLYFQ | GGGG- ɑSH2 and ligation of GGGG- ɑSH2 was detected when TEVp and SrtA were co-expressed. Similarly, western blotting was used to demonstrate the cleavage of VHL-ɑGFP and changes in YFP and SH2-miRFP670 levels. This result is consistent with the observed changes in fluorescent signals, indicating only the corresponding full-length AdPROM variant degrades its specific target.
To better assess the kinetics of LASER, we carried out a time course experiment by probing YFP and miRFP670 signals 4, 8, 12, 16, and 24 h post transfection (Supplementary Fig. 3). Reliable expression of both fluorescent proteins (>15% cells) was not detected until 12 h post transfection. Switching between different TPD states was detected at 16 h post transfection, indicating SrtA-mediated cleavage and ligation occurred within 4 h after protein expression. While the level of SH2-miRFP670 continued to increase from 16 to 24 h for the control, SH2-miRFP670 levels declined slightly for cells co-expressing both TEVp and SrtA, highlighting the efficiency of the LASER system. This is also reflected in the continuous rescue of YFP at 24 h.
Discussion
Proteins play a diverse role in controlling cell physiology, responsible for virtually all cellular functions. Their precise function within a complex cellular environment is typically investigated through alterations in the protein’s levels followed by analysis of the associated phenotypes40. Overexpression of proteins, for instance, is easy to implemented but may lead to gain-of-function artifacts41. Protein inactivation, on the other hand, is more reliable in providing insights on protein functions. Gene-level inactivation via RNA interference or genome editing offers high specificity and holds promise for treating genetic disorders42,43,44, but is not ideal due to lack of conditional control. Furthermore, knockout strategies are irreversible and gene silencing is slow in reducing stable proteins already present within cells45. Targeted protein degradation has recently emerged as an alternative strategy to modulate protein levels as it offers the possibility of exquisite control over the kinetics of protein knockdown46. BioPROTACs have been garnering increased attention as a viable alternatives to small molecule PROTACs for TPD due to the highly modular nature of targeting17. However, the current strategies are static in nature and a key challenge moving forward is to create a new synthetic biology framework that can elicit conditional protein degradation to permit targeting of essential proteins that is not presently possible.
Sortase A provides a unique solution to elicit conditional control of TPD as it possesses the ability to catalyze both protein cleavage and ligation reactions. Here, we exploit the inherent dual functionality of SrtA as a new synthetic biology tool to toggle the assembly and disassembly of the bioPROTAC AdPROM in human cells to achieve conditional TPD on two different protein targets. A broad range of TPD outcomes can be achieved by tuning both the affinity of the protein-binding domains and SrtA expression. The flexibility to modulate the level of many native intracellular proteins of interest can potentially lead to applications from therapy to diagnostics and biotechnology.
By exploiting the need for a free N-terminus triglycine motif for ligation, we further layer a protease-gated mechanism to activate SrtA ligation by removing the ENLYFQ | GGG blocking sequence by TEVp. The resulting LASER platform allows us to expand the possible degradation outcomes using Boolean logic operations depending on the input combinations. The ability of LASER to perform intracellularly switchable targeting with AND gate logic operations open up the possibility to interrogate novel protein targets previously untenable due to their essential nature. Furthermore, exploring intracellular protein interactions and dependencies could allow the dynamic fine-tuning of protein cascades.
Another advantageous feature of SrtA is its amenability to logic-gated outputs. Integrating signal transduction would present even greater opportunities for expanded input responsiveness in various cellular reprogramming applications. We expect that a split TEVp would enable small molecule and light responsive LASER outcomes7,47. Such an approach would enable an expanded logic-gated communication system to direct intracellular reprogramming.
Methods
Chemicals and reagents
Reagents and solvents used in this study were obtained from commercial sources and used without further purification.
Plasmid construction
A list of all plasmids used in this study is provided in Supplementary Table 1. A list of amino acid sequences for each construct is provided in Supplementary Table 2 and all full-length gene sequences are available in the Supplementary Data 1. For HeLa studies, all expression vectors were constructed using pcDNA3.1(+) as the backbone. This was purchased from Invitrogen via ThermoFisher Scientific.
The desired DNA (purchased from TWIST or PCR’d from existing lab plasmid stocks) and desired backbone were mated using digestion with standard restriction enzymes (New England BioLabs) and ligation via T4 DNA ligase (M0202S, New England BioLabs). All PCRs were performed using Q5 High-Fidelity Hot-Start DNA Polymerase (New England BioLabs) according to manufacture protocols. Sequence verification was done by Sanger sequencing (Azenta).
Cell culture
HeLa cells were cultivated as specified by ATCC. HeLa cells were maintained in complete growth media consisting of MEM (Corning 10-101CV), supplemented with 10% FBS (fetal bovine serum), and 1% penicillin—streptomycin. Cells were passaged at 90% confluency, typically every fourth or fifth day. For subculturing, aspirate and discard the culture media. Wash the cell layer with PBS (phosphate-buffered saline) (1X pH 7.4) and aspirate. One milliliter of trypsin—ethylenediaminetetraacetic acid (EDTA) (Corning Trypsin—EDTA 1X, 0.25% Trypsin/2.21 mM EDTA) was added to cover the cell layer and incubated for 5 min at 37 °C. Upon detachment of the cells, 5 mL of complete growth media was added to inactivate the trypsin. Appropriate aliquots of the cell suspension were seeded into new culture vessels prepared with 12 mL of fresh complete growth media, typically T-75 flasks (CellTreat Scientific Products, Pepperell MA). Seeding densities and culture volumes are as described by ThermoFisher Scientific Cell Culture Protocols.
Transient transfection in HeLa cell lines
HeLa cells were seeded in 12-well plates (CellTreat Scientific Products, Pepperell MA) at 1 × 105 cells/mL in 1 mL of complete growth media. For dosage studies and condition screening, HeLa cells were seeded in 96-well plates (Santa Cruz Biotechnology, Santa Cruz CA) at 1 × 105 cells/mL in 0.1 mL of complete growth media. After 20 h, cells were transfected between 80 and 90% confluency. Transfection was carried out with Lipofectamine 3000 (ThermoFisher) according to the manufacturer recommended protocols. Transfection ratios can be found in Supplementary Table 3. Cells were aspirated and fresh growth media added between 8 and 18 h post transfection. Cells were generally analyzed 24 h after transfection, unless otherwise noted.
Luminescence assays
For luminescence assays, HeLa cells were generally transfected in 24-well plates (CellTreat). 24 h post transfection, HeLa cells were washed with PBS before being trypsinized and 100 mL was transferred to 96 W plates for imaging. Subsequently, an equal volume of freshly reconstituted ONE-Glo assay reagent (Promega) added to each well. Reconstituted ONE-Glo Luciferase assay reagent was prepared according to manufacturer recommendations at room temperature. Quantification was performed using a Synergy H4 (Biotek) plate reader at a standard sensitivity setting.
Flow cytometry
For flow cytometry, HeLa cells were generally transfected in 12-well plates (CellTreat Scientific Products). 24 h post transfection, HeLa cells were washed with 0.5 mL/well PBS (phosphate-buffered saline) (1×, pH 7.4), trypsinized with 200 μL of trypsin–EDTA (Corning Trypsin–EDTA 1×, 0.25% trypsin/0.53 mM EDTA) for 5 min, with incubation at 37 °C. Upon cell detachment, 0.8 mL of complete growth media was added to inactivate trypsin. Cells were centrifuged for 5 min at 4 °C at 800 g. The media was removed, and cells were resolubilized in cold PBS, before transfer to a flow cytometry tube. The flow cytometer used for all experiments is the ACEA NovoCyte Flow Cytometer (Agilent, Santa Clara, CA). The cells were gated for live cells and then gated to exclude any doublets. Gated events (>15,000) were collected for each sample. YFP and GFP fusion protein fluorescence was detected using the configuration for fluorescein isothiocyanate (FITC) (Ex: 488 nm/detection: 530 nm). The mean fluorescence intensity values are shown as the results. miRFP670 and miRFP670 fusion protein fluorescence was detected using the 640 nm laser and 660 nm filter. The gating strategy is provided in Supplementary Fig. 4.
Western blot analysis
HeLa cells were plated at 1 × 105 cells/cm2 and transfected as described above before lysis with NP40 lysis buffer (Thermo Fisher Scientific). Lysates were separated on hand-cast 12% Tris-acrylamide gels and transferred to PVDF membranes. Transferred proteins were blocked with TBST + 5% milk for 8 h at 22 °C. α-VHL (Invitrogen #MA5-13940), α-SH2 (Invitrogen #MA5-49146), α-GFP (BioLegend #902602), α-FLAG (Abcam #ab49763), α-HA (Invitrogen #32-6700), and α-GAPDH (Invitrogen #MA1-16757) antibodies were diluted, 1:1000, 1:1000, 1:5000, 1:3000, 1:1000, and 1:1000 respectively, in TBST and incubated for 8 h at 4 °C. Secondary antibody goat α-mouse IgG with HRP conjugation (Bio-Rad #170-6516) was diluted at 1:3000 and used as needed.
Statistical analysis
All experiments were performed in biological triplicates, except for one as noted performed with n = 4. Results were also expressed as means ± standard deviations (SD). The Ordinary One-way ANOVA was performed with P < 0.05 considered statistically significant throughout the experiments. P < 0.05 was denoted as *, P < 0.01 was denoted as **, P < 0.001 was denoted as ***, P < 0.0001 was denoted as ****, and no statistically significant difference as (NS).
Data availability
Sequences of all constructs studied are included in the Supplementary Data 1. All data supporting the findings of this study are available within the article and its Supplementary Information. Additional data are available from the corresponding author upon request. Source data are provided with this paper.
References
Towbin, B. D. et al. Optimality and sub-optimality in a bacterial growth law. Nat. Commun. 8, 14123 (2017).
Keren, L. et al. Massively parallel interrogation of the effects of gene expression levels on fitness. Cell 166, 1282–1294.e1218 (2016).
Weinberg, B. H. et al. Large-scale design of robust genetic circuits with multiple inputs and outputs for mammalian cells. Nat. Biotechnol. 35, 453–462 (2017).
Kitada, T., DiAndreth, B., Teague, B. & Weiss, R. Programming gene and engineered-cell therapies with synthetic biology. Science 359, eaad1067 (2018).
Khalil, A. S. et al. A synthetic biology framework for programming eukaryotic transcription functions. Cell 150, 647–658 (2012).
Haellman, V. & Fussenegger, M. Synthetic biology—engineering cell-based biomedical devices. Curr. Opin. Biomed. Eng. 4, 50–56 (2017).
Fink, T. et al. Design of fast proteolysis-based signaling and logic circuits in mammalian cells. Nat. Chem. Biol. 15, 115–122 (2019).
Gao, X. J., Chong, L. S., Kim, M. S. & Elowitz, M. B. Programmable protein circuits in living cells. Science 361, 1252–1258 (2018).
Zhao, L., Zhao, J., Zhong, K., Tong, A. & Jia, D. Targeted protein degradation: mechanisms, strategies and application. Signal Transduct. Target. Ther. 7, 113 (2022).
Chen, R. P., Gaynor, A. S. & Chen, W. Synthetic biology approaches for targeted protein degradation. Biotechnol. Adv. 37, 107446 (2019).
Deshaies, R. J. Protein degradation: prime time for PROTACs. Nat. Chem. Biol. 11, 634–635 (2015).
Sakamoto, K. M. et al. Protacs: chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl Acad. Sci. USA 98, 8554–8559 (2001).
Xiao, M., Zhao, J., Wang, Q., Liu, J. & Ma, L. Recent advances of degradation technologies based on PROTAC mechanism. Biomolecules 12, 1257 (2022).
Sakamoto, K. M. et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 98, 8554–8559 (2001).
Hines, J., Gough, J. D., Corson, T. W. & Crews, C. M. Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proc. Natl Acad. Sci. USA 110, 8942–8947 (2013).
Schneekloth, A. R., Pucheault, M., Tae, H. S. & Crews, C. M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med Chem. Lett. 18, 5904–5908 (2008).
Lim, S. et al. bioPROTACs as versatile modulators of intracellular therapeutic targets including proliferating cell nuclear antigen (PCNA). Proc. Natl Acad. Sci. 117, 5791–5800 (2020).
Fletcher, A. et al. A TRIM21-based bioPROTAC highlights the therapeutic benefit of HuR degradation. Nat. Commun. 14, 7093 (2023).
Fulcher, L. J. et al. An affinity-directed protein missile system for targeted proteolysis. Open Biol. 6, 160255 (2016).
Fulcher, L. J., Hutchinson, L. D., Macartney, T. J., Turnbull, C. & Sapkota, G. P. Targeting endogenous proteins for degradation through the affinity-directed protein missile system. Open Biol. 7, 170066 (2017).
Yang, H. & Chen, W. Protease-responsive toolkit for conditional targeted protein degradation. ACS Synth. Biol. 13, 2073–2080 (2024).
Clancy, K. W., Melvin, J. A. & McCafferty, D. G. Sortase transpeptidases: insights into mechanism, substrate specificity, and inhibition. Biopolymers 94, 385–396 (2010).
Mazmanian, S. K., Liu, G., Ton-That, H. & Schneewind, O. Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science 285, 760–763 (1999).
Strijbis, K., Spooner, E. & Ploegh, H. L. Protein ligation in living cells using sortase. Traffic 13, 780–789 (2012).
Marraffini, L. A., Dedent, A. C. & Schneewind, O. Sortases and the art of anchoring proteins to the envelopes of gram-positive bacteria. Microbiol. Mol. Biol. Rev. 70, 192–221 (2006).
Ton-That, H., Liu, G., Mazmanian, S. K., Faull, K. M. & Schneewind, O. Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif. Proc. Natl. Acad. Sci. USA 96, 12424–12429 (1999).
Ton-That, H. & Schneewind, O. Anchor structure of staphylococcal surface proteins. IV. Inhibitors of the cell wall sorting reaction. J. Biol. Chem. 274, 24316–24320 (1999).
Popp, M. W. & Ploegh, H. L. Making and breaking peptide bonds: protein engineering using sortase. Angew. Chem. Int Ed. Engl. 50, 5024–5032 (2011).
Wang, H. H., Altun, B., Nwe, K. & Tsourkas, A. Proximity-based sortase-mediated ligation. Angew. Chem. Int. Ed. Engl. 56, 5349–5352 (2017).
Jeong, H. J., Abhiraman, G. C., Story, C. M., Ingram, J. R. & Dougan, S. K. Generation of Ca2+-independent sortase A mutants with enhanced activity for protein and cell surface labeling. PLoS One 12, e0189068 (2017).
Wu, Q., Ploegh, H. L. & Truttmann, M. C. Hepta-mutant Staphylococcus aureus sortase A (SrtA(7m)) as a tool for in vivo protein labeling in Caenorhabditis elegans. ACS Chem. Biol. 12, 664–673 (2017).
Tebo, A. G. & Gautier, A. A split fluorescent reporter with rapid and reversible complementation. Nat. Commun. 10, 2822 (2019).
Tebo, A. G. et al. Orthogonal fluorescent chemogenetic reporters for multicolor imaging. Nat. Chem. Biol. 17, 30–38 (2021).
Huang, X. et al. Kinetic mechanism of Staphylococcus aureus sortase SrtA. Biochemistry 42, 11307–11315 (2003).
Levary, D. A., Parthasarathy, R., Boder, E. T. & Ackerman, M. E. Protein-protein fusion catalyzed by sortase A. PloS One 6, e18342 (2011).
Sha, F. et al. Dissection of the BCR-ABL signaling network using highly specific monobody inhibitors to the SHP2 SH2 domains. Proc. Natl. Acad. Sci. USA 110, 14924–14929 (2013).
Hunt, V. M. & Chen, W. Deciphering the design rules of toehold-gated sgRNA for conditional activation of gene expression and protein degradation in mammalian cells. ACS Synth. Biol. 11, 397–405 (2022).
Hunt, V. M. & Chen, W. A microRNA-gated thgRNA platform for multiplexed activation of gene expression in mammalian cells. Chem. Commun. 58, 6215–6218 (2022).
Parks, T. D., Leuther, K. K., Howard, E. D., Johnston, S. A. & Dougherty, W. G. Release of proteins and peptides from fusion proteins using a recombinant plant virus proteinase. Anal. Biochem. 216, 413–417 (1994).
Kampmann, M. CRISPRi and CRISPRa screens in mammalian cells for precision biology and medicine. ACS Chem. Biol. 13, 406–416 (2018).
Prelich, G. Gene overexpression: uses, mechanisms, and interpretation. Genetics 190, 841–854 (2012).
Gaj, T., Gersbach, C. A. & Barbas, C. F. 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31, 397–405 (2013).
Bumcrot, D., Manoharan, M., Koteliansky, V. & Sah, D. W. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat. Chem. Biol. 2, 711–719 (2006).
Deleavey, G. F. & Damha, M. J. Designing chemically modified oligonucleotides for targeted gene silencing. Chem. Biol. 19, 937–954 (2012).
Olson, E. J. & Tabor, J. J. Post-translational tools expand the scope of synthetic biology. Curr. Opin. Chem. Biol. 16, 300–306 (2012).
Ludwicki, M. B. et al. Broad-spectrum proteome editing with an engineered bacterial ubiquitin ligase mimic. ACS Cent. Sci. 5, 852–866 (2019).
Wehr, M. C. et al. Monitoring regulated protein-protein interactions using split TEV. Nat. Methods 3, 985–993 (2006).
Acknowledgements
This work was supported by grants from NSF (CBET1803008, MCB1817675 and MCB2013991). Research reported in this publication was also supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number T32GM133395.
Author information
Authors and Affiliations
Contributions
H.Y., P.M. and W.C. conceived the project, designed experiments, analyzed the data, and wrote the paper. H.Y. and P.M. performed the experiments. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, H.K., Muthukumar, P.K. & Chen, W. Synthetic protein degradation circuits using programmable cleavage and ligation by Sortase A. Nat Commun 16, 8682 (2025). https://doi.org/10.1038/s41467-025-63819-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63819-y
