
Abstract
ERDRP-0519 is a potent nonnucleoside inhibitor active against measles virus (MeV) and other Morbilliviruses. Here we report cryo-EM structures of the compound bound to MeV polymerase complexes at 2.73 Å and 2.48 Å resolution, revealing a unique binding pocket in the RdRp palm subdomain that overlaps the catalytic GDN motif. These findings clarify the basis of resistance mutations, including W671, and provide a foundation for designing next-generation Paramyxovirus antivirals.
Similar content being viewed by others

Introduction
Paramyxoviruses, a family of negative-stranded RNA viruses, include significant human pathogens such as measles virus (MeV), mumps virus (MuV), human parainfluenza viruses (HPIVs), and Nipah virus (NiV)1. MeV is particularly concerning due to its high infectivity and potential to cause severe complications, including pneumonia and encephalitis, especially in infants and young children. Measles was declared eliminated in the U.S. in 2000 following widespread MMR vaccination; however, declining vaccination rates have led to resurgences. In 2025 alone, 14 outbreaks and over 1000 cases were reported, often linked to unvaccinated communities and international travel2. Currently, no FDA-approved antiviral treatment exists for MeV, and ribavirin, the only antiviral option for Paramyxovirus, offers limited efficacy and is rarely used for measles3,4. This highlights the urgent need for more effective antiviral therapies.
Two small-molecule antivirals, AS-485 and ERDRP-05196, were developed to target different viral components of MeV: the fusion (F) protein and the L-P polymerase complex, respectively. AS-48 emerged from structure-based design, while ERDRP-0519 was identified through high-throughput screening, with both compounds undergoing multiple rounds of optimization to improve toxicity, solubility and potency5,6,7,8 (Fig. 1a). The F protein, in conjunction with hemagglutinin, facilitates viral entry into host cells, whereas the L-P complex is essential for viral replication. Although AS-48 inhibits the F protein, resistance has emerged, including a resistant MeV isolate from Sub-Saharan Africa and rapid escape mutants observed in vitro9. Structural studies have elucidated the mechanism of resistance, leading to reduced prioritization of AS-48 development10. Conversely, polymerase inhibitors like ERDRP-0519 show greater promise due to the high conservation of the L-P complex across MeV strains. ERDRP-0519, a chemically optimized polymerase inhibitor, has demonstrated strong activity against multiple MeV isolates, with evidence of broader efficacy against other Morbilliviruses, positioning it as a leading therapeutic candidate11. However, despite its potential, structural information on ERDRP-0519’s binding to the L-P complex remains elusive, constraining further optimization efforts (Fig. 1b).

a Development of small-molecule inhibitors targeting MeV. b Chemical structure of the MeV RdRp lead compound ERDRP-0519 and its neutralization potency compared to other prodrugs. c Structures of the MeV polymerase complex with ERDRP-0519. The MeV Lcore-P (left) and Lfull-P-C (right) complexes are shown in cartoons with different subunit colors and ERDRP-0519 as magenta spheres. d The RdRp catalytic core is shown in cartoons: NTD (gray), fingers (blue), palm (lime), thumb (burlywood), and active site (red). ERDRP-0519 is represented as magenta sticks with transparent gray density. Zoom-in: Superimposition of the palm subdomain of the MeV Lfull-P-C-ERDRP-0519 complex (lime) and apo-MeV Lfull-P-C (gray). The active site loop (GDN) shifts upon compound binding (black arrow).
In this work, we determine cryo-EM structures of the MeV polymerase complexes bound to ERDRP-0519 at 2.73 Å and 2.48 Å resolution, revealing a unique binding pocket overlapping the RdRp catalytic GDN motif. Furthermore, cell-based assays demonstrate that a critical hydrogen bond with W671 plays a key role in mediating drug resistance.
Results and discussion
Structural basis of ERDRP-0519 bound to MeV polymerase complexes
Previous resistance profiling identified the L protein as the target of ERDRP-0519, with resistance mutations clustering around the conserved GDN motif in the RNA-dependent RNA polymerase (RdRp) and adjacent PRNTase domains12. A truncated MeV L variant (L1708), containing only those two domains, yielded a dissociation constant of 78-140 nM for ERDRP-0519, indicating strong binding affinity13. Docking analysis predicted that ERDRP-0519 binds between the RdRp and PRNTase domains, near PRNTase motifs A and D13. To test this, we assembled the MeV ternary polymerase complex14 with ERDRP-0519 and analyzed it by cryo-EM. The maps revealed two complexes: Lcore-P and Lfull-P-C, with clear ERDRP-0519 density on the L protein (Fig. 1c, Supplementary Fig. 1, and Supplementary Table 1). Contrary to predictions, ERDRP-0519 was observed binding within the RdRp domain rather than at the predicted PRNTase site (Fig. 1d). Additionally, C protein presence did not alter the binding mode, suggesting stable drug association across RNA synthesis states.
Molecular interactions and mechanism of ERDRP-0519 inhibition
Structural analysis revealed that ERDRP-0519 binds to the palm subdomain of the RdRp, targeting the catalytic GDN motif and a nearby hydrophobic channel (Figs. 1d, 2a). This binding mode differs from FDA-approved nucleoside RdRp inhibitors, such as Sofosbuvir for hepatitis C virus (HCV)15 and Ribavirin for foot-and-mouth disease virus (FMDV)16, which simply occupy the central polymerase cavity (Supplementary Fig. 2). Our data show the binding pocket is framed by escape mutations around the GDN motif, consistent with the resistance observed in the L-T751I mutant, which exhibited a 20-fold increase in EC90 values11. Mutations in the PRNTase domain also affect drug binding13, likely due to their proximity to the central polymerase cavity. The morpholine ring of ERDRP-0519 extends into this cavity, suggesting resistance may arise from structural changes that indirectly influence binding.

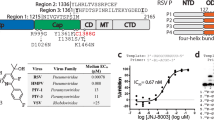
a Interactions between the palm subdomain and ERDRP-0519, with the hydrogen bond to W671 shown as a dashed line. b List of residues interacting with ERDRP-0519’s functional groups. c Superimposition of MeV Lfull-P-C-ERDRP-0519 and apo-MeV Lfull-P-C (PDB: 9DUT), showing D773 displaced outward, forming a hydrophobic interaction with the compound. d Superimposition of MeV Lfull-P-C-ERDRP-0519 and NiV L-P-RNA (PDB: 9GJU), with 3’ leader RNA shown as yellow sticks and the clash between ERDRP-0519 and RNA marked. The clip surface of MeV L protein (gray) displays the ERDRP-0519 binding pocket and the potential 3’ leader RNA channel.
Our structures also explain the observation that modifications to the central ring or pyrazole moiety of ERDRP-0519 reduce its antiviral potency8 (Fig. 2a, b). The central ring forms strong hydrophobic interactions with residue Y667, so any changes at this position are likely to cause steric clashes, disrupting binding. Additionally, the trifluoromethylated pyrazole group fits precisely into a hydrophobic pocket, engaging six surrounding residues and forming more interactions than any other part of the molecule (Fig. 2b). Altering or removing this moiety disrupts these interactions, markedly reducing binding and potency. In contrast, modifications to the piperidine ring are better tolerated, likely because extending it allows the compound to project into the central polymerase cavity, which has enough space to accommodate additional groups. Incorporating a morpholine ring into this region did not create new interactions with the L protein but instead occupied the solvent-accessible channel, partially explaining ERDRP-0519’s superior pharmacokinetic profile compared to AS-136A.
Notably, our structural data reveals two key mechanisms by which ERDRP-0519 exerts antiviral activity. First, it inhibits the catalytic center by interacting with residue D773 in the GDN motif (Fig. 2c). The piperidine group displaces D773, creating space for hydrophobic interaction that directly impairs active site function. It is noted that ERDRP-0519 does not affect the viral transcription gradient13, suggesting that residue D773 may be positioned differently when the polymerase is in an elongation conformation and/or that ERDRP-0519 cannot bind an elongating polymerase. Second, although the morpholine ring does not directly contact the L protein, structural comparison with the NiV L-P-RNA complex17 shows steric clashes, suggesting the morpholine ring obstructs RNA binding (Fig. 2d). Thus, ERDRP-0519 likely impairs RNA synthesis through direct engagement of the catalytic center and by inducing allosteric effects.
Conserved binding and antiviral efficacy of ERDRP-0519
Interestingly, ERDRP-0519 demonstrates broad-spectrum activity against various MeV strains6,11. Mapping its contact residues onto a sequence alignment of MeV isolates (Supplementary Fig. 3) reveals high conservation, explaining its consistent antiviral efficacy. These residues are also conserved in Canine distemper virus (CDV), a related Morbillivirus, supporting ERDRP-0519’s effectiveness against both MeV and CDV11. Extending its antiviral activity to other Paramyxoviridae or even Mononegavirales, will require further chemical optimization. Sequence comparisons (Fig. 3a) highlight key differences in binding site residues across viral genera, indicating that structural modifications will be necessary for broader activity.

a RdRp sequence alignment across Mononegavirales. MeV contact residues in red; conserved and non-conserved sites marked red and blue. Sequence sources: MeV (GenBank: K01711.1); CDV (KJ147057.1); HPIV3 (ARA15380.1); HPIV1 (ARB07783.1); NiV (AAY43917.1); MuV (AWI67642.1); HPIV5 (YP_138518.1); RSV (YP_009518860.1); HMPV (Q6WB93.1); VSV (Q98776.1); RABV (AAT48626.1); EBOV (NP_066251.1). b Dose-dependent inhibition of MeV polymerase by ERDRP-0519 (0, 1, 10, 100, 1000 nM) using the MeV NanoLuc minigenome assay. c Effect of ERDRP-0519 (100 nM) on MeV L mutants assessed by the MeV NanoLuc minigenome system. D773A serves as a negative control. Bars (b, c) show mean ± SD. Black circles indicate individual values (n = 3). Significance by two-tailed unpaired t-test (*P ≤ 0.05, **P ≤ 0.01; ***P ≤ 0.001; ns, not significant). Source data are provided as a Source Data file.
In addition, our structural analysis showed that the linker region of ERDRP-0519 is critical for binding. A strong hydrogen bond (2.8 Å) forms between the compound’s carboxyl group in the linker and residue W671 of the L protein (Fig. 2a). To test the importance of W671, we first assessed ERDRP-0519’s effect on wild-type MeV polymerase using a cell-based minigenome assay, observing dose-dependent inhibition (Fig. 3b). We then created a W671F mutant polymerase (Supplementary Table 2), disrupting the hydrogen bond since phenylalanine cannot form this interaction. Additional nearby mutations altering hydrophobicity or causing steric hindrance were introduced to evaluate their impact on binding. As shown in Fig. 3c, ERDRP-0519 failed to inhibit the W671F mutant, confirming W671’s essential role in binding and antiviral activity. ERDRP-0519 also does not bind to RSV L protein13, which has phenylalanine at position 671 (F671), supporting the importance of tryptophan at this site. These findings suggest modifying ERDRP-0519’s linker region could expand its antiviral spectrum, especially against viruses lacking W671. Additionally, mutations in the binding pocket impaired polymerase activity, likely due to disruptions near the GDN catalytic site.
Overall, our structural study provides in-depth insights into the binding site and inhibitory mechanism of ERDRP-0519. High-resolution structures of the MeV polymerase complexed with ERDRP-0519 reveal a unique binding pocket within the catalytic GDN motif of the RdRp palm subdomain, distinct from previously predicted sites and known RdRp inhibitors. We demonstrate how key interactions, including a hydrogen bond with W671, underpin compound efficacy and align with resistance mutations observed in functional assays. Our findings also support ERDRP-0519’s pan-Morbillivirus activity and offer a framework for future drug modifications to extend its antiviral spectrum beyond Morbillivirus. This study lays a solid foundation for designing next-generation polymerase inhibitors and advancing ERDRP-0519 as a promising therapeutic for measles and related viral diseases.
While our manuscript was under review, a similar study reporting the MeV polymerase structure bound to ERDRP-0519 was published18. They also determine the NiV polymerase-ERDRP-0519 complex, highlighting the compound’s broader antiviral potential across Mononegavirales.
Methods
Protein expression and purification
The non-codon-optimized L gene (GenBank: AB052820.1), with an N-terminal twin Strep-tag, and the P gene (GenBank: AB046113.1), with a C-terminal 6×His tag, from the Measles virus strain Edmonston, were subcloned into the pFastBac Dual vector and used for the Bac-to-Bac baculovirus expression system, as previously described14,19. Briefly, the amplified baculovirus expressing the complexes was used to infect Trichoplusia ni (Tni) cells at a multiplicity of infection (MOI) of 1 at 27 °C for 72 hours. Cells were harvested and resuspended in lysis buffer (25 mM HEPES, pH 7.8, 500 mM NaCl, 5% glycerol, 4 mM MgCl₂, 1 mM tris(2-carboxyethyl)phosphine (TCEP), 0.01% Tween 20, 10 mM imidazole), supplemented with cOmplete™ EDTA-free protease inhibitor cocktail (Roche). Following sonication and centrifugation, the lysate was loaded onto a HisTrap column pre-equilibrated with buffer A (25 mM HEPES, pH 7.8, 500 mM NaCl, 5% glycerol, 1 mM TCEP, 4 mM MgCl₂). The bound protein was eluted with buffer B (buffer A supplemented with 500 mM imidazole). Eluted fractions were pooled and incubated with Strep-Tactin XT resin (IBA, 2-5030-010) for 30 minutes, followed by four washes with wash buffer (25 mM HEPES, pH 7.8, 500 mM NaCl, 5% glycerol, 1 mM TCEP, 4 mM MgCl₂). The protein was then eluted with elution buffer (wash buffer supplemented with 50 mM biotin). Further purification was carried out by size-exclusion chromatography (Superose 6 Increase 10/300 GL, GE Healthcare) using a buffer containing 25 mM HEPES, pH 7.8, 500 mM NaCl, 5% glycerol, 1 mM TCEP, and 4 mM MgCl₂. Fractions near the peak maximum were analyzed by SDS–PAGE, and those containing the MeV polymerase complex were pooled and concentrated to 1 mg/mL. The final sample was flash-frozen and stored at −80 °C.
Cryo-EM sample preparation and data acquisition
The purified MeV RNA polymerase complex was diluted 1:1 with the buffer (50 mM HEPES pH 7.8, 50 mM NaCl, 1 mM TCEP) containing 50 µM ERDRP-0519 (AOBIOUS INC, Cat No. AOB2063) and incubated on ice for one hour. 4 µl mixture was applied to freshly glow-discharged Quantifoil R1.2/1.3 300-mesh copper grids (EM Sciences) and blotted for 4 seconds at 4 °C under 100% chamber humidity and plunge-frozen in liquid ethane using a Vitrobot Mark IV (FEI). Images were collected using the K3 Summit detector (Gatan) in super-resolution mode with binning 2 and CDS, along with a Gatan BioContinuum GIF energy filter (slit width of 20 eV) at the Hormel Institute, University of Minnesota20. The data collection was performed using the EPU software (Thermo Fisher Scientific) with a pixel size of 0.664 Å (nominal magnification of 130,000×) and a nominal defocus value between −1.0 to −2.0 μm. Each image consists of 40 dose-framed fractions and was recorded with a total dose of 50 e−/Å2. Cryo-EM data collection statistics are summarized in Supplementary Table 1.
Image processing
Cryo-EM data were processed using cryoSPARC v4.5.121, and the detailed procedure is outlined in Supplementary Fig. 1. Briefly, a total of 8,046 dose-fractionated movies were subjected to Patch motion correction using MotionCor222 and Patch CTF estimation using CTFFIND-4.1.1323, with data downsampled by 3/4 (0.885333 Å/pixel after downsampling). Images with the defocus values outside of −0.5 to −3.2 μm or CTF fit resolutions worse than 7 Å were excluded from further steps. A total of 4,333,399 particles were then picked using both the Blob picker and then Template picker in cryoSPARC v4.5.1 and subjected to the Remove Duplicate Particles Tool. Junk particles were removed through one round of 2D classification. A total of 1,325,963 particles from the good 2D classes were used for Ab-initio reconstruction of four maps. The heterogeneous refinement produced 524,675 and 358,252 particles from the good classes, which were then subjected to further non-uniform and CTF refinements to generate final maps of 2.48 Å and 2.73 Å resolution, respectively. Map resolution was determined by gold-standard Fourier shell correlation (FSC) at 0.143 between the two half-maps. Local resolution variation was estimated from the two half-maps in cryoSPARC v4.5.1.
Model building and refinement
The MeV polymerase machinery model was docked using our previously determined MeV L-P and L-P-C models (PDB codes: 9DUS and 9DUT), and then manually rebuilt in COOT-0.8.924. The cryo-EM densities were sufficient to define ERDRP-0519. Real-space refinements in Phenix-1.1625 were performed to obtain the final models. In the real-space refinement, minimization global, local grid search, and adp were performed with the secondary structure, rotamer, and Ramachandran restraints applied throughout the entire refinement. The stereochemistry of all structural models was evaluated using MolProbity26 in Phenix. Model and map statistics are summarized in Supplementary Table 1. Figures were generated using UCSF Chimera X v1.727.
Minigenome assay
The MeV minigenome encoding NanoLuc was constructed with the NanoLuc gene flanked by a T7 promoter and the 5′- and 3′-terminal untranslated regions of the MeV genome, as described previously14. 293T cells were seeded in 24-well plates and transfected with the minigenome system plasmids, including 100 ng pCAGGS-minigenome, 40 ng pCAGGS-T7, 40 ng pCAGGS-N, 40 ng pCAGGS-P, 450 ng pCAGGS-L or L mutants, and 1 ng pGL4.54-luc (firefly luciferase control), using Lipofectamine 3000 (Thermo Fisher Scientific, L3000001). The compound ERDRP-0519 was diluted in DMSO and added to the culture medium 12 hours post-transfection at the indicated concentrations. A consistent concentration of DMSO was maintained across all transfection reactions. L mutations were introduced via site-directed mutagenesis, with primers listed in Supplementary Table 2. At 48 hours post-transfection, cells were lysed with Passive Lysis Buffer (Promega, E1941) for 15 minutes at room temperature. The lysates were centrifuged at 13,000 × g for 10 minutes. For Luciferase assays, 40 µL of lysate supernatant was measured using the Nano-Glo Dual-Luciferase Reporter Assay (Promega, N1610) according to the manufacturer’s instructions. Luciferase activity was calculated as the NanoLuc/Firefly luciferase ratio. For Western blot analysis, the lysate supernatant was denatured in 2× sample loading buffer, boiled at 100 °C for 5 minutes, separated by SDS-PAGE, and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, IPVH00010). Membranes were blocked with 5% skim milk in Tris-buffered saline containing 0.1% Tween 20 (TBST) for 1 h, followed by incubation with primary antibodies at 4 °C overnight: rabbit anti-HA (1:2000, Cell Signaling Technology, C29F4), mouse anti-Flag (1:2000, Sigma, F1804), mouse anti-His (1:3000, Invitrogen, His.H8, MA1-21315), and mouse anti-Strep (1:1000, IBA, 2-1507-001). Detection of the primary antibodies was carried out using HRP-conjugated secondary antibodies (Cell Signaling Technology), including goat anti-rabbit (7074S) and rabbit anti-mouse (58802S), each at 1:5000. Blot images were captured using a ChemiDoc MP imager (Bio-Rad).
Statistical analyses
All data were analyzed and generated using GraphPad Prism 5 software. Statistical analysis was performed using student’s t-test for comparisons between two groups and one-way analysis of variance (ANOVA) for comparisons among multiple groups. Graphs display the means of the experiments, with error bars representing one standard deviation. Significance is indicated as follows: ns, not significant; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Sequence alignment analysis
Sequence alignments were performed using online tools, Clustal Omega, https://www.ebi.ac.uk/Tools/msa/clustalo/28 and ESPript 3.0 https://espript.ibcp.fr/ESPript/ESPript/index.php29.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The atomic coordinate of MeV Lcore-P-ERDRP-0519 and Lfull-P-C-ERDRP-0519 complexes have been deposited in the PDB with accession numbers 9OCF and 9OCE, respectively. The corresponding cryo-EM density maps have been deposited in the Electron Microscopy Data Bank with accession numbers EMD-70313 and EMD-70312, respectively. The NiV L-P-RNA structure used in this study is available in the PDB under accession code 9GJU. The HCV NS5B-Sofosbuvir structure used in this study is available in the PDB under accession code 4WTG. The FMDV RdRp-Ribavirin structure used in this study is available in the PDB under accession code 2E9R. Source data are provided with this paper.
References
Lamb, R. & Kolakofsky, D. The paramyxoviruses. Fields virology, 3rd ed. Lippincott-Raven Publishers, Philadelphia, Pa, 577-604 (1996).
Kuppalli, K. & Omer, S. B. Measles: the urgent need for global immunisation and preparedness. Lancet 405, 1565–1567 (2025).
Plemper, R. K. Measles resurgence and drug development. Curr. Opin. Virol. 41, 8–17 (2020).
Anderson, L. J., Parker, R. A. & Strilms, R. L. Association between respiratory syncytial virus outbreaks and lower respiratory tract deaths of infants and young children. J. Infect. Dis. 161, 640–646 (1990).
Sun, A. et al. Nonpeptide inhibitors of measles virus entry. J. medicinal Chem. 49, 5080–5092 (2006).
Ndungu, J. M. et al. Non-nucleoside inhibitors of the measles virus RNA-dependent RNA polymerase: Synthesis, structure–activity relationships, and pharmacokinetics. J. medicinal Chem. 55, 4220–4230 (2012).
Plemper, R. K. et al. A target site for template-based design of measles virus entry inhibitors. Proc. Natl Acad. Sci. 101, 5628–5633 (2004).
Sun, A., Chandrakumar, N., Yoon, J.-J., Plemper, R. K. & Snyder, J. P. Non-nucleoside inhibitors of the measles virus RNA-dependent RNA polymerase complex activity: Synthesis and in vitro evaluation. Bioorg. medicinal Chem. Lett. 17, 5199–5203 (2007).
Doyle, J. et al. Two domains that control prefusion stability and transport competence of the measles virus fusion protein. J. Virol. 80, 1524–1536 (2006).
Hashiguchi, T. et al. Structures of the prefusion form of measles virus fusion protein in complex with inhibitors. Proc. Natl Acad. Sci. 115, 2496–2501 (2018).
Krumm, S. A. et al. An orally available, small-molecule polymerase inhibitor shows efficacy against a lethal morbillivirus infection in a large animal model. Sci. Transl. Med. 6, 232ra252–232ra252 (2014).
Chattopadhyay, A., Raha, T. & Shaila, M. Effect of single amino acid mutations in the conserved GDNQ motif of L protein of Rinderpest virus on RNA synthesis in vitro and in vivo. Virus Res. 99, 139–145 (2004).
Cox, R. M., Sourimant, J., Govindarajan, M., Natchus, M. G. & Plemper, R. K. Therapeutic targeting of measles virus polymerase with ERDRP-0519 suppresses all RNA synthesis activity. PLoS Pathog. 17, e1009371 (2021).
Wang, D., Yang, G. & Liu, B. Structure of the measles virus ternary polymerase complex. Nat. Commun. 16, 3819 (2025).
Appleby, T. C. et al. Viral replication. Structural basis for RNA replication by the hepatitis C virus polymerase. Science 347, 771–775 (2015).
Ferrer-Orta, C. et al. Sequential structures provide insights into the fidelity of RNA replication. Proc. Natl Acad. Sci. USA 104, 9463–9468 (2007).
Sala, F. A., Ditter, K., Dybkov, O., Urlaub, H. & Hillen, H. S. Structural basis of Nipah virus RNA synthesis. Nat. Commun. 16, 2261 (2025).
Wang, Y. et al. Structures of the measles virus polymerase complex with non-nucleoside inhibitors and mechanism of inhibition. Cell, https://doi.org/10.1016/j.cell.2025.06.017 (2025).
Yang, G., Wang, D. & Liu, B. Structure of the Nipah virus polymerase phosphoprotein complex. Nat. Commun. 15, 8673 (2024).
Bhandari, J. et al. Efficient strategies and troubleshooting for single particle cryoEM data collection using EPU. BMC Methods 2, 3 (2025).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Rubinstein, J. L. & Brubaker, M. A. Alignment of cryo-EM movies of individual particles by optimization of image translations. J. Struct. Biol. 192, 188–195 (2015).
Rohou, A. & Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. Biol. Crystallogr 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. Biol. Crystallogr 66, 213–221 (2010).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D. Biol. Crystallogr 66, 12–21 (2010).
Goddard, T. D. et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Sievers, F. & Higgins, D. G. Clustal omega. Curr Protoc Bioinformatics 48, 3.13.11-16. https://doi.org/10.1002/0471250953.bi0313s48. (2014)
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42, W320–W324 (2014).
Acknowledgements
We thank the staff at the cryo-EM facility and instrument core facility in the Hormel Institute, University of Minnesota. This work was supported by the funding granted to B.L. from the Hormel Institute, University of Minnesota.
Author information
Authors and Affiliations
Contributions
B.L. and D.W. conceived the project and designed the experiments. D.W. prepared the proteins and performed functional studies. B.L., D.W., and G.Y. conducted cryo-EM data collection, cryo-EM image processing, atomic model building and refinement. F.B., D.W., and B.L. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jeremy Keown and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, D., Bu, F., Yang, G. et al. Structural basis of measles virus polymerase inhibition by nonnucleoside inhibitor ERDRP-0519. Nat Commun 16, 9061 (2025). https://doi.org/10.1038/s41467-025-64128-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64128-0
