
Abstract
Dysfunction of muscle satellite cells is linked to diabetic myopathy. The mechanisms vitiating muscle satellite cell proliferative activity in diabetes are not well understood. Here, we show that AS160, a key cytosolic Rab-GTPase activating protein (RabGAP) in insulin signaling, is a moonlighting protein regulating muscle satellite cell proliferation as a transcriptional co-factor. Deletion of AS160, but not its GAP-inactive mutation, impairs muscle satellite cell proliferation and consequent muscle regeneration, and exacerbates age-related sarcopenia. Mechanistically, Thr642 phosphorylation of AS160 promotes its translocation into the nucleus where AS160 functions as a co-factor of Signal Transducer and Activator of Transcription 3 (STAT3). AS160 binds to STAT3 to enhance the transcription of myogenic cascades and consequent muscle satellite cell proliferation. Disruption of the AS160-STAT3 interaction, or inhibition of AS160-Thr642 phosphorylation, inhibits muscle satellite cell proliferation and impairs muscle regeneration. Together, our findings reveal a moonlighting function of AS160 as a transcriptional co-factor in the nucleus, and have therapeutic implications for muscle regeneration.
Similar content being viewed by others

Introduction
Skeletal muscle mass and function are progressively declined in type 2 diabetes (T2D), which is known as diabetic myopathy and contributes to the progression of diabetes-associated comorbidities, including obesity and hyperglycemia1. Moreover, regeneration of injured muscle is impaired in diabetes, which may relate to diabetic complications such as foot ulcers and chronic limb-threatening ischemia1,2. Maintenance of skeletal muscle homeostasis and regeneration of injured muscle rely on muscle satellite cells (MuSCs) that are resident muscle stem cells3. MuSCs are normally in a quiescent state under physiological conditions, but become activated in response to microenvironmental changes4. Upon activation, quiescent MuSCs expressing PAX7 undergo asymmetric cell division for the first round to produce one quiescent MuSC for replenishment of the MuSC pool, and the other for generation of myoblasts through multiple rounds of symmetric division5,6. Subsequently, a family of transcription factors, including MYOD, MYF5, MYOG and MYF6 (also known as MRF4), drives the expression of muscle-specific genes in myoblasts for their differentiation into myotubes7,8. Dysfunction or exhaustion of MuSCs not only impairs muscle regeneration for repair in diabetes but also contributes to diabetes-/age-induced muscle loss9,10,11. However, it is still not well understood how diabetes impinges on MuSC proliferative activity.
AS160 (also known as TBC1D4) is a Rab-GTPase activating protein (RabGAP) that possesses two phospho-tyrosine binding (PTB) domains at its N-terminus and a RabGAP domain at its C-terminus12. This RabGAP is a crucial regulator of GLUT4 trafficking in adipocytes and skeletal muscle through its downstream targets Rab10 and Rab8a, respectively12,13,14,15. AS160 is phosphorylated by PKB on multiple sites, including Thr642, upon stimulation with insulin, which may regulate access of AS160 to downstream target Rabs associated with GLUT4 vesicles rather than affecting its GAP activity to facilitate GLUT4 trafficking12,16,17. Substitution of Thr642 with a non-phosphorylatable alanine inhibits insulin-stimulated GLUT4 trafficking and subsequent glucose uptake in skeletal muscle18. A pre-mature stop mutation Arg684Ter on a long isoform of AS160 is frequently present in Inuit, which confers muscle insulin resistance and type 2 diabetes19. An AS160R684X rat model has been developed to harbor the corresponding human mutation in Inuit, in which the nonsense mutation suppresses muscle glucose utilization through decreasing GLUT4 protein and simultaneously augments fatty acid oxidation in skeletal muscle through increasing insulin-stimulated translocation of lipid translocase CD3620. So far, the accumulative evidence shows that AS160 is a crucial regulator of cell metabolism functioning in the cytoplasm.
In this study, we demonstrate that AS160 is a moonlighting protein and translocates into the nucleus, functioning as a transcription cofactor to engage with the myogenic transcriptional cascades in MuSCs. AS160 deficiency, or inhibition of AS160-Thr642 phosphorylation, decreases MuSC proliferative activity, thereby impairing muscle regeneration upon injury and causing diabetes-/age-related sarcopenia.
Results
Nuclear entry of AS160 regulates proliferation and differentiation of myoblasts
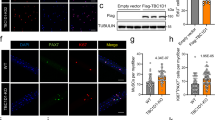
In an attempt to delineate potential functions of different domains of AS160, we expressed its N-terminal part AS160M1-I770 containing the two PTB domains and the C-terminal region AS160S771-end containing the RabGAP domain in HEK293 cells, respectively. In contrast to the cytosolic localization of the AS160S771-end, the AS160M1-I770 was mainly present in the nucleus (Fig. 1a). The nuclear localization of the AS160M1-I770 intrigued us to further investigate its mechanism and potential functions. Two bipartite nuclear localization signals (NLS) were predicted on AS160, NLS1 (KKVHENNQKTQPRRRHA) and NLS2 (RRRAHTFSHPPSSSRRK) (http://psort.hgc.jp) (Fig. 1b). The NLS1 and NLS2 were fused to a GST-GFP dual tag, respectively, and expressed in HEK293 cells. In contrast to the cytosolic localization of the GST-GFP and NLS1-GST-GFP, the NLS2-GST-GFP fusion protein was mainly found in the nucleus (Fig. 1c). Subcellular fractionation revealed the presence of a substantial amount of full-length Flag-AS160 in the nucleus (Supplementary Fig. 1a, b). Deletion of the NLS2, but not the NLS1, decreased the accumulation of Flag-AS160 in the nucleus, although a substantial amount of Flag-AS160ΔNLS2 was still present in the nucleus (Supplementary Fig. 1a, b). Moreover, a considerable amount of endogenous AS160 was detected in the nuclear fraction of wild-type (WT) soleus and EDL muscle but not AS160 knockout (KO) muscle (Fig. 1d). Similarly, endogenous AS160 was also found in the nuclear fraction of mouse C2C12 myoblasts and rat L6 myoblasts (Supplementary Fig. 1c, d). To further investigate the subcellular localization of AS160, we generated a 3xFlag-AS160 knockin mouse line in which a 3xFlag tag was added to the N-terminus of endogenous AS160 (Supplementary Fig. 1e, f). We isolated primary myoblasts from 3xFlag-AS160/+ mice and WT controls, and found that the 3xFlag tag did not affect the expression level of AS160 protein (Supplementary Fig. 1g). Notably, 3xFlag-AS160 could be detected in the nuclear fraction of primary myoblasts (Fig. 1e). Finally, endogenous AS160 could also be detected in the nucleus of a human HSkMC myoblast cell line (Supplementary Fig. 1h). These data show that AS160 possesses a functional NLS2 and a portion of this RabGAP is localized in the nucleus.

a Subcellular localization of GFP-GST, GFP-AS1601-770 and GFP-AS160771-1299 in HEK293 cells. Nuclei were stained with DAPI (blue). Bars indicate 10 µm in length. b Diagrammatic illustration of domain composition of AS160. NLS1 and NLS2 are predicted NLS sequences of AS160. c Subcellular localization of GFP-GST, NLS1-GFP-GST, and NLS2-GFP-GST in HEK293 cells. Nuclei were stained with DAPI (blue). Bars indicate 50 µm in length. d AS160 protein in the nuclear and cytosolic fractions of soleus and EDL muscle from the WT and AS160-KO mice. e Flag protein in the nuclear and cytosolic fractions of primary myoblasts isolated from 3-week-old WT and 3xFlag-AS160/+ mice. f, g Proliferative activities of shNC and AS160-KD L6 myoblasts assayed with EdU labeling. Nuclei were stained with DAPI (blue). f Representative images. g Quantitative results. n = 4 biological replicates. Bars indicate 50 µm in length. h, i Effects of nuclear AS160 on proliferation of L6 myoblasts. AS160-KD L6 myoblasts were infected with lentivirus to express mCherry (empty vector), NES-AS160-P2A-mCherry, or NLS-AS160-P2A-mCherry. Synonymous mutations were introduced in the AS160S220-C225 region (the corresponding sequence is TCAAGtCTtATtGAcGAtTGtATG, synonymous mutations were shown in lower case bold) to avoid targeting by the shRNA. shNC L6 myoblasts expressing mCherry (empty vector) was used as the control group. mCherry protein was stained with the anti-RFP antibody. h representative images. Bars indicate 50 µm in length. i quantitative results. n = 5(except for AS160-KD + NES-AS160 n = 4) biological replicates. Data were presented as mean values ± SEM. Numbers on the graphs represent p values. Statistical analyses were carried out via one-way ANOVA. Source data are provided as a Source Data file.
In order to explore the potential function of AS160 in the nucleus, we constructed two independent AS160 knockdown (KD) L6 cell lines using short hairpin RNA (shRNA) (Supplementary Fig. 2a, b), and performed deep RNA sequencing (RNA-Seq) (Supplementary Fig. 2f). A total of 11,025 genes were analyzed by RNA-seq, among which 121 genes were upregulated, and 142 genes were downregulated, in AS160-KD L6 cells (Supplementary Fig. 2f). The differentially expressed genes were enriched in the pathways of muscle development and growth factor response (Supplementary Fig. 2g), suggesting that AS160 might be involved in the proliferation and differentiation of myoblasts. To investigate such a possibility, we performed an EdU-based cell proliferation assay in AS160-KD and control cells, which revealed a lower proliferation rate, as evidenced by less EdU-positive signals, in AS160-KD L6 cells than in control cells (Fig. 1f, g). Similar results were obtained using a CFSE-based cell proliferation assay (Supplementary Fig. 2h). In agreement, proliferation regulators such as c-Myc and Cyclind1 were decreased in AS160-KD L6 cells at both protein and mRNA levels (Supplementary Fig. 2a, c, d, i). The effect of AS160 depletion on proliferation was also observed in mouse C2C12 cells as well as in human HSkMC cells (Supplementary Fig. 2m–u). Furthermore, the expression of differentiation markers such as Myog, Myf6, and Des were significantly lower in AS160-KD L6 cells, suggesting a defect in their differentiation (Supplementary Fig. 2j–l). The mammalian target of rapamycin (mTOR) is a critical regulator of proliferation and differentiation of myoblasts21, whose expression and phosphorylation remained unaltered in AS160-KD L6 cells, suggesting that mTOR probably does not account for the defects in these cells (Supplementary Fig. 2a, e).
The inhibitory effect of AS160 depletion on myoblast proliferation could be due to its deficiency either in the cytosol or in the nucleus. To distinguish these two possibilities, we re-expressed a cytosolic form, NES-AS160 and a nuclear form, NLS-AS160, in AS160-KD cells, respectively. Notably, the NLS-AS160, but not the NES-AS160, restored proliferation of AS160-KD L6 cells (Fig. 1h, i), suggesting that nuclear AS160 may play a critical role in regulating myoblast proliferation.
Deficiency of AS160 impairs muscle regeneration after injury
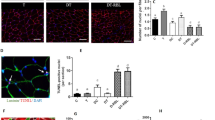
We next investigated whether AS160 might regulate the proliferation and differentiation of MuSCs that play crucial roles in muscle regeneration. Similar to L6 and C2C12 myoblasts (Supplementary Fig. 1c, d), a portion of endogenous AS160 was detected in the nucleus of primary myoblasts (Supplementary Fig. 3a). We isolated myofibers from WT and AS160-KO mice for in vitro culture to examine the proliferation of MuSCs attached to myofibers. After culture for 2 days, less total PAX7+ MuSCs and proliferative PAX7+/Ki67+ MuSCs were found on AS160-KO myofibers than on WT myofibers (Fig. 2a–c), suggesting impaired MuSC proliferation in the AS160-KO group. Moreover, PAX7+/MYOD1−, PAX7+/MYOD1+, and PAX7−/MYOD1+ cells were all decreased in the AS160-KO group when myofibers were cultured for 3 days (Fig. 2d, e). Since our AS160-KO is a global KO model, AS160 is deleted in both MuSCs and mature myofibers. To find out whether AS160 functions in MuSCs to regulate their proliferation, we isolated these cells from WT and AS160-KO mice for in vitro culture to proliferate as primary myoblasts. Notably, AS160-KO myoblasts contained less PAX7+ cells with lower PAX7 expression at both mRNA and protein levels than WT myoblasts (Fig. 2f, h, i and Supplementary Fig. 3b, c). We employed an EdU-based method to label proliferative PAX7+ myoblasts, and found less PAX7+/EdU+ cells in myoblasts from AS160-KO mice (Fig. 2g, h), suggesting that AS160 deficiency impairs myoblast proliferation. Moreover, myogenic regulators such as Pax7, Myod1, Myog, and Myf5, and cell proliferation regulators such as c-Myc, Cyclind1, and Cyclind2, were decreased in AS160-KO myoblasts (Fig. 2i and Supplementary Fig. 3d). A luciferase reporter assay showed that the promoter activities of Pax7 and Myod1 were significantly lower in AS160-KO myoblasts than in WT myoblasts (Fig. 2j, k), suggesting that lower transcript levels of Pax7 and Myod1 in AS160-KO myoblasts were most likely due to their transcriptional activities in the nucleus. We wondered whether the regulation of myoblast proliferation by AS160 depended on its entry into the nucleus. To this end, we overexpressed the NLS-tagged or NES-tagged AS160 in AS160-KO myoblasts for targeting AS160 in the nucleus and cytosol, respectively. Nuclear expression of AS160 in AS160-KO myoblasts markedly promoted cell proliferation, whereas cytosolic expression of AS160 had almost no such effect, as compared to non-transfected control cells (Fig. 2l, m). The GAP activity of AS160 is essential for metabolic regulation in skeletal muscle22. AS160 deficiency or a GAP-inactivating AS160R972K mutation causes degradation of GLUT4 but not GLUT1 in skeletal muscle22,23. In agreement with a previous report24, we found that GLUT1 was dominant in myoblasts while GLUT4 was mainly expressed in skeletal muscle (Supplementary Fig. 4a). Interestingly, protein expression of GLUT1 and GLUT4 displayed no changes in AS160-KO myoblasts with normal glucose uptake (Supplementary Fig. 4a–c), suggesting that the lack of AS160 GAP activity may not affect glucose utilization in myoblasts. More importantly, the GAP-inactivating AS160R972K mutation did not affect MuSC proliferation (Supplementary Fig. 4d–h). These data show that the nuclear localization of AS160 is essential, whereas its GAP activity is dispensable, for MuSC proliferation.

a–c Proliferation of MuSCs attached to the myofibers isolated from WT and AS160-KO mice (2-month-old) and in vitro cultured for 42 h. PAX7 (green), Ki67 (red), and DAPI (blue). a Representative images. Bars indicate 50 µm in length. b, c Quantitative results. n = 60 (WT) and 61 (AS160-KO). d, e Proliferation of MuSCs attached to the myofibers isolated from WT and AS160-KO mice (2-month-old) and in vitro cultured for 72 h. PAX7 (green), MYOD1 (red), and DAPI (blue). d Representative images. Bars indicate 50 µm in length. e Quantitative results. n = 37 (WT) and 45 (AS160-KO). f–h Proliferation of primary myoblasts isolated from WT and AS160-KO mice (3-week-old) and in vitro cultured for 4 days. PAX7 (red), EdU (green), and DAPI (blue). f, g Quantitative results. n = 4. h Representative images. Bars indicate 50 µm in length. i mRNA levels of Pax7, Myod1, Myog, and Myf5 in primary myoblasts isolated from WT and AS160-KO mice (3-week-old). n = 5. j, k Relative luciferase activities of Pax7 (j) and Myod1 (k) promoters in primary myoblasts isolated from the WT and AS160-KO mice. n = 3 (Blank) and 4 (Pax7-luci and Myod1-luci). l, m Proliferation of AS160-KO myoblasts overexpressing NES-AS160-P2A-mCherry and NLS-AS160-P2A-mCherry. EdU (green), anti-RFP (red) and DAPI (blue). l Representative images. m Quantitative results. n = 4. White arrows indicate EdU+RFP- cells. Red arrows indicate EdU+RFP+ cells. Bars indicate 50 µm in length. n–p Transplantation of primary myoblasts isolated from WT/tdTomato and AS160-KO/tdTomato mice. n Experimental flow chart. o Representative images of tdTomato (red) and DAPI (blue) in recipient TA muscle. Bars indicate 50 µm in length. p Quantitative results. n = 5. q–s Transplantation of AS160-KO primary myoblasts overexpressing NES-AS160-P2A-mCherry or NLS-AS160-P2A-mCherry. q Experimental flow chart. r Representative images of anti-RFP (red) and DAPI (blue) in recipient TA muscle. Bars indicate 50 µm in length. s Quantitative results. n = 4. Data were presented as mean values ± SEM. Numbers on the graphs represent p values. Statistical analyses were carried out via two-sided t-test for b, c, f, g, e, i, p, s, and two-way ANOVA for j, k, m. Source data are provided as a Source Data file.
We then employed a muscle injury model induced by cardiotoxin (CTX) to explore a possible role of AS160 in muscle regeneration. AS160 protein displayed dynamic changes during the course of muscle injury and repair. Its level in WT skeletal muscle was markedly decreased on the first day after CTX injection and recovered on the third day (Supplementary Fig. 3e, f). In agreement with the in vitro analyses, the in vivo injury model showed that the mRNA levels of key regulators of MuSCs, including Pax7, Myod1, Myog, Myf5, c-Myc, Cyclind1, and Cyclind2 were lower in AS160-KO muscle than in WT muscle on the third day after injury (Supplementary Fig. 3g, h). The protein levels of PAX7, MYOD1, and MYOG were also significantly decreased in injured AS160-KO muscle as compared to WT muscle (Supplementary Fig. 3i, j). The embryonic myosin (eMyHC) is expressed in immature myofibers during the repair of damaged muscle25, and the proportion of eMyHC+ myofibers culminated on day-3.5 after CTX injury and declined afterwards in WT mice (Supplementary Fig. 3k, l). The proportion of eMyHC positive myofibers in AS160-KO mice was lower on day-3.5 after injury, but higher on day-5, than in WT mice (Supplementary Fig. 3m–p), suggesting that AS160 deficiency might undermine muscle recovery after injury, probably in part due to impaired differentiation of MuSCs. On the seventh day after injury, AS160-KO mice had less PAX7+ MuSCs in their muscle than WT littermates (Supplementary Fig. 3q, r). Notably, the maximal cross-sectional area (CSA) of AS160-KO myofibers was significantly smaller than that of WT myofibers on the seventh day after injury (Supplementary Fig. 3s–u), suggesting an impairment of muscle regeneration in AS160-KO mice. In agreement with the observation that the GAP activity of AS160 is dispensable for MuSC proliferation (Supplementary Fig. 4d–h), the AS160R972K mice exhibited normal muscle regeneration after being injured with CTX (Supplementary Fig. 4i–k).
Together, these data show that whole-body AS160 deficiency inhibits the proliferation and differentiation of MuSCs and impairs muscle regeneration after injury.
Injury-induced muscle regeneration requires nuclear AS160 in MuSCs
The impaired muscle regeneration could be due to either alterations in MuSCs, changes in the microenvironment, or both, in whole-body AS160-KO mice. We first performed a transplantation experiment to examine the effect of AS160 deficiency in MuSCs on their proliferation in vivo. To this end, we generated WT and AS160-KO mice expressing a tdTomato transgene, and isolated MuSCs from these mice to proliferate as myoblasts in vitro. The same amounts of WT/tdTomato and AS160-KO/tdTomato myoblasts were transplanted into the tibialis anterior (TA) muscle of NCG immunodeficient recipient mice that were injured with CTX one day before transplantation, and the TA muscle was analyzed three weeks after transplantation (Fig. 2n). Notably, the fluorescence area of tdTomato+ myofibers were decreased in the recipients with AS160-KO/tdTomato myoblasts as compared to those in the recipients with WT/tdTomato MuSCs (Fig. 2o, p), showing that AS160 deficiency in myoblasts inhibits their proliferation in vivo. Furthermore, we confirmed that WT and AS160-KO myoblasts did not differ in a post-transplantation cell survival assay (Supplementary Fig. 4l–n).
Muscle regeneration involves multiple cell types such as myogenic cells, macrophages, endothelial cells and neuronal cells4. To further determine the importance of AS160 in myogenic cells during muscle regeneration after injury, we generated an AS160 myogenic-specific knockout (AS160-mKO) mouse model by mating AS160f/f mice with Mlc1f-Cre mice24. In line with a previous report that Mlc1f is expressed in myoblasts derived from various skeletal muscles26, we found that Mlc1f was expressed in freshly-isolated MuSCs (Supplementary Fig. 5a, b). Moreover, lineage tracing showed that the Mlc1f-Cre was expressed in PAX7+ MuSCs as well as in various skeletal muscles, including slow and fast muscles (Supplementary Fig. 5c–f). We also confirmed that AS160 was deleted in freshly-isolated MuSCs as well as in primary myoblasts from AS160-mKO mice (Supplementary Fig. 6a). Deletion of AS160 was also confirmed in both soleus (slow) and EDL (fast) muscles of AS160-mKO mice (Supplementary Fig. 6b, c). Again, primary myoblasts isolated from AS160-mKO mice exhibited a lower proliferative activity than AS160f/f control cells as evidenced by less PAX7+ and PAX7+/EdU+ cells (Supplementary Fig. 6d–f). Similarly, MuSCs attached to AS160-mKO myofibers also exhibited a lower proliferative activity (Supplementary Fig. 6g–i). In the CTX-induced muscle injury assay, the myofiber CSA was smaller in AS160-mKO mice than in AS160f/f control mice on the 7th day after injury (Supplementary Fig. 6j, k), showing that myogenic deficiency of AS160 impairs muscle regeneration in vivo. In the AS160-mKO mice, AS160 was deleted not only in MuSCs but also in mature myofibers. To further delineate the role of AS160 in MuSCs in muscle regeneration upon injury, we mated AS160f/f mice with inducible MuSC-specific Pax7CreERT2/+ mice27 to enable deletion of AS160 specifically in MuSCs but not in mature myofibers in the resultant AS160-imKO (AS160f/f/Pax7CreERT2/+) mice. MuSCs attached to AS160-imKO myofibers displayed a lower proliferative activity after in vitro culture for 2 or 3 days (Fig. 3a–d). Moreover, primary myoblasts from AS160-imKO mice exhibited a decreased proliferative activity with reduced mRNA and protein levels of PAX7 as compared to Pax7CreERT2/+ control cells (Fig. 3e, f and Supplementary Fig. 8a, b). AS160 deficiency had little effect on the symmetric and asymmetric divisions of MuSCs (Supplementary Fig. 8c, d). At 2 weeks after tamoxifen induction, AS160-imKO mice were subjected to CTX-induced muscle injury (Fig. 3g). Notably, AS160-imKO mice displayed a marked decrease in EdU+ myonuclei, a pronounced delay in eMyHC+ myofiber formation, and a significant reduction in myofiber CSA after injury (Fig. 3h–k and Supplementary Fig. 8e–l). Moreover, protein levels of PAX7, MYOD1, MYOG, and MYH3 were significantly decreased in skeletal muscle of AS160-imKO mice on day-3.5 after injury (Supplementary Fig. 8m, n). Together, these data show that AS160 deficiency in MuSCs impairs muscle regeneration.

a, b Proliferation of MuSCs attached to the myofibers isolated from Pax7CreERT2/+ and AS160-imKO mice (2-month-old) and in vitro cultured for 2 days. MuSCs were stained with PAX7 (green), MYOD1 (red), and DAPI (blue). a Representative images. Bars indicate 50 µm in length. b Quantitative results. n = 54 (Pax7CreERT2/+) and 55 (AS160-imKO). c, d Proliferation of MuSCs attached to the myofibers isolated from Pax7CreERT2/+ and AS160-imKO mice (2-month-old) and in vitro cultured for 3 days. MuSCs were stained with PAX7 (green), MYOD1 (red) and DAPI (blue). c Quantitative results. n = 48 (Pax7CreERT2/+) and 51 (AS160-imKO). d Representative images. Bars indicate 50 µm in length. e, f Proliferation of primary myoblasts isolated from Pax7CreERT2/+ and AS160-imKO mice (6-week-old) and in vitro cultured for 4 days. Myoblasts were stained with PAX7 (red), EdU (green) and DAPI (blue). e Representative images. Bars indicate 50 µm in length. f Quantitative results. n = 6. g Experimental scheme of Pax7CreERT2/+ and AS160-imKO mice. 6-week-old male Pax7CreERT2/+ and AS160-imKO mice were intraperitoneally injected Tamoxifen for 5 days. After recovery for 2 weeks, the two groups of mice were injured by CTX, and muscle was harvested on the ninth day. h–k Myofiber sizes in CTX-injured TA muscle of male Pax7CreERT2/+ and AS160-imKO mice (10-week-old) after CTX injury for 9 days. h Representative images of H&E-stained cross-sections of TA muscle. Bars indicate 50 µm in length. i–k Quantitative results. n = 4. Data were presented as mean values ± SEM. Numbers on the graphs represent p values. Statistical analyses were carried out via two-sided t-test. Source data are provided as a Source Data file.
We then investigated whether nuclear targeting of AS160 dictated myoblast proliferation in vivo. To this end, AS160-KO myoblasts expressing the nuclear NLS-AS160-P2A-mCherry or cytosolic NES-AS160-P2A-mCherry were transplanted into CTX-injured TA muscle of NCG immunodeficient recipient mice (Fig. 2q). The fluorescence area of mCherry+ myofibers were significantly larger in the recipients with AS160-KO/NLS-AS160-P2A-mCherry myoblasts than in the recipients with AS160-KO/NES-AS160-P2A-mCherry myoblasts (Fig. 2r, s), showing that nuclear AS160 in myoblasts promotes their proliferation in vivo.
Together, these data show that nuclear AS160 in MuSCs is crucial for muscle regeneration after injury.
AS160 is required for maintenance of the MuSC pool
Reduced proliferative activities of MuSCs may inhibit the replenishment of the MuSC pool, which would further dampen muscle regeneration with repeated injuries. Indeed, muscle regeneration in AS160-mKO mice became worse in a second injury than in the first injury as evidenced by smaller myofiber CSA, more eMyHC positive myofibers and less PAX7+ MuSCs on day-7 after injury (Supplementary Fig. 6l–r).
Chronically impaired replenishment of the MuSC pool would eventually jeopardize muscle health in aging and lead to age-related sarcopenia11. The MuSC pool size was comparable between AS160-mKO mice and their AS160f/f littermate controls at a young age (2-month-old), while it became smaller in aged AS160-mKO (21-month-old) than in their AS160f/f littermates (Supplementary Fig. 7a, b). Moreover, the proliferative activity of MuSCs attached to the myofibers was significantly decreased when cultured in vitro (Supplementary Fig. 7a, c, d). At a young age, the myofiber CSA showed no differences between the AS160-mKO mice and their littermates (Supplementary Fig. 7e, f). The exercise endurance and limb grip strength were also comparable between these two genotypes (Supplementary Fig. 7i, k). However, at the age of 21 months, the myofiber CSA in AS160-mKO mice was significantly smaller than that in their AS160f/f littermate controls (Supplementary Fig. 7e, g, h). In running and gripping tests, the AS160-mKO mice showed poorer muscle strength than their littermates (Supplementary Fig. 7j, l). These data suggest that AS160 deficiency in skeletal muscle might cause age-related sarcopenia. The transition from type II muscle fiber to type I muscle fiber is a key feature of sarcopenia28. In line with the development of sarcopenia in the aged AS160-mKO mice, we found that the proportion of Type IIX and Type IIA myofibers were lower, while Type I myofibers were higher, in the AS160-mKO mice than in their AS160f/f littermate controls (Supplementary Fig. 7m–r). Since AS160 was deleted in both MuSCs and myofibers of the AS160-mKO mice, it is currently unclear whether, if so, how AS160 deficiency in myofibers might contribute to the reduced MuSC pool size and the exacerbation of sarcopenia in aged mice.
AS160 interacts with STAT3 to increase STAT3 nuclear localization
The nuclear role of AS160 in regulating MuSC proliferation led us to hypothesize that AS160 might interact with certain transcription factor(s) to control myogenic gene transcription. We performed RNA sequencing to identify differentially expressed genes in AS160-KD L6 cells and predicted potential upstream transcription factors for these differentially expressed genes (Supplementary Fig. 2f). We also took a proteomic approach via mass spectrometry analysis to identify potential proteins that were co-immunoprecipitated with Flag-AS160 (Supplementary Fig. 9a–c). A conjoint analysis of those transcription factors and potential AS160 interactors revealed that signal transducer and activator of transcription 3 (STAT3) might be a candidate mediating the effect of AS160 on myogenic gene transcription (Fig. 4a). It has been reported that STAT3 plays a crucial role in regulating cell proliferation and differentiation during muscle recovery29,30,31. Purified recombinant Flag-AS160 and GST-STAT3 interacted with each other in the in vitro pulldown assay (Fig. 4b, c). When Flag-AS160 was immunoprecipitated from cell lysates, co-expressed GFP-STAT3 was found in the immunoprecipitates (Supplementary Fig. 9d). When Flag-STAT3 was immunoprecipitated from cell lysates, endogenous AS160 was detected in the immunoprecipitates (Supplementary Fig. 9e). Moreover, endogenous STAT3 was found in the AS160 immunoprecipitates of WT myoblasts but not AS160-KO myoblasts (Fig. 4d). We then used a split GFP system to visualize the interaction between AS160 and STAT3 in the nucleus. An NLS-STAT3-NLS-GFP1-10 designed for nuclear expression was co-expressed with GFP11X2-AS160 in HEK293 cells. GFP signals was reconstituted in the nucleus when NLS-STAT3-NLS-GFP1–10 was co-expressed with GFP11X2-AS160 while no GFP signals were observed for either protein alone, further showing that AS160 interacted with STAT3 in the nucleus (Fig. 4e). Together, these data demonstrate that STAT3 is an AS160-interacting protein.

a Overlap of the AS160-interacting proteins identified via mass spectrometry and the predicted upstream transcriptional factors (TFs) (based on ChEA3 database) for differentially expressed genes in AS160-KD myoblasts via RNA-Seq analysis. b, c In vitro interaction of purified Flag-AS160 and GST-STAT3 via Flag pulldown (b) or GST pulldown (c). d Co-immunoprecipitation of STAT3 with AS160 in primary myoblasts isolated from 3-week-old WT and AS160-KO mice. e Reconstitution of GFP signals in the nuclei of HEK293 cells expressing NLS-STAT3-GFP1-10 and GFP11X2-AS160. Nuclei were stained with DAPI (blue). Bars indicate 10 µm in length. f, g Nuclear STAT3 levels in AS160-KD L6 myoblasts (f) and AS160-KD C2C12 myoblasts (g). h Nuclear levels of AS160 and STAT3 in primary myoblasts isolated from Pax7CreERT2/+ and AS160-imKO mice (6-week-old). i–k Nuclear levels of AS160 and STAT3 in AS160-KO HEK293 cells overexpressing HA-STAT3 together with Flag-tagged AS160-WT or AS160R964k. i representative immunoblots. j, k quantitative results. n = 3 biological replicates. l, m Nuclear levels of STAT3 in AS160-KO HEK293 cells overexpressing HA-STAT3 together with Flag-tagged NLS-AS160 or NES-AS160. l Representative immunoblots. m Quantitative results. n = 6 biological replicates. n, o Nuclear levels of STAT3 in AS160-KO HEK293 cells overexpressing HA-STAT3 together with Flag-tagged AS160-WT, AS160ΔNLS1, or AS160ΔNLS2. n Representative immunoblots. o Quantitative results. n = 4 biological replicates. Data were presented as mean values ± SEM. Numbers on the graphs represent p values. Statistical analyses were carried out via one-way ANOVA for j, k, m, o. Source data are provided as a Source Data file.
We then examined whether AS160 might regulate the nuclear localization of STAT3. Knockdown of AS160 markedly decreased STAT3 level in the nucleus in L6, C2C12 as well as HSkMC myoblasts (Fig. 4f, g and Supplementary Fig. 9f). Similar changes of nuclear STAT3 were observed in myoblasts isolated from AS160-imKO mice (Fig. 4h). In contrast, overexpression of AS160 elevated nuclear STAT3 but reduced cytosolic STAT3 (Fig. 4i–k). Interestingly, overexpression of a GAP-inactive AS160R964K mutant exerted a similar effect on STAT3 subcellular localization (Fig. 4i–k), suggesting that the stimulative effect of AS160 on STAT3 nuclear localization did not require its GAP activity. We then wondered whether the subcellular localization of AS160 might regulate STAT3 nuclear level. Notably, overexpression of nuclear NLS-AS160 promoted STAT3 nuclear localization as compared to cytosolic NES-AS160 (Fig. 4l, m). Consistently, overexpression of an AS160ΔNLS2 mutant, but not an AS160ΔNLS1 mutant, markedly decreased nuclear accumulation of STAT3 as compared to WT AS160 (Fig. 4n, o).
AS160 enhances the binding of STAT3 to its cognate DNA for transcriptional activation
To gain insights whether this AS160–STAT3 complex might be involved in regulation of gene transcription, we performed ChIP-Seq analyses in L6 cells expressing HA-AS160 or HA-STAT3, and identified 3290 peaks for ChIP-Seq with HA-AS160 and 3998 peaks with HA-STAT3 (Supplementary Fig. 10a, b). Genome-wide analyses of these peaks revealed that 60% of the peaks with HA-AS160 and 47% of the peaks with HA-STAT3 were mapped to gene promoter regions, showing high enrichments of these two proteins on gene promoters (Supplementary Fig. 10a, b). Notably, 1955 peaks were common for HA-AS160 and HA-STAT3 (Fig. 5a and Supplementary Fig. 10c–e), and the canonical STAT3-binding motif was enriched in both groups (Fig. 5b). These data suggest a possible cooperation between these two proteins in the control of gene transcription. Moreover, GO analysis of genes derived from common peaks for HA-AS160 and HA-STAT3 showed an enrichment of cell cycle-related genes (Supplementary Fig. 10f), suggesting a potential role of this AS160–STAT3 complex in cell proliferation.

a, b ChIP-Seq analysis was performed using the HA antibody in L6 cells overexpressing HA-AS160 or HA-STAT3. a, heatmap analysis of AS160- and STAT3-binding sites within ±3 kb from the transcriptional start site (TSS) on the common downstream targets of AS160 and STAT3. b Motif analysis of AS160- and STAT3-binding sites. c, d EMSA analysis of the DNA binding capacity of AS160 and STAT3. The hot probe was based on the classical STAT3 motif (5’-GATCCTTCTGGGAATTCCTAGATC-3’) and labeled by ATP-cy5. The competitor was the unlabeled probe with the same sequence. c Quantitative results. n = 3 biological replicates. d Representative fluorescence image. e, f Interaction of endogenous AS160 with endogenous STAT3 via co-immunoprecipitation assay in injured skeletal muscle. e Representative immunoblots. f Quantitative results. n = 4 biological replicates. g, h EMSA analysis of the DNA binding capacity of AS160 and STAT3. The hot probe was based on the predicted binding sites in Pax7 promoter (g) and Myod1 promoter (h). i CUT-Tag QPCR analysis of Pax7 promoter region in primary myoblasts isolated from 3-week-old WT and AS160-KO mice with anti-STAT3. n = 3. j, k Relative luciferase activity of the Pax7 (j) and Myod1 (k) promoters in AS160-KO primary myoblasts overexpressing AS160 and STAT3. The empty vector 1 and empty vector 2 were the vector control for AS160 and STAT3, respectively. Quantitative results. n = 4 (except for the first control group n = 3) biological replicates. Data were presented as mean values ± SEM. Numbers on the graphs represent p values. Statistical analyses were carried out via one-way ANOVA for (c, f, j, k), and two-way ANOVA for (i). Source data are provided as a Source Data file.
We then employed an electrophoretic mobility shift assay (EMSA) to investigate how the AS160–STAT3 complex binds to DNA. As expected, a purified GST-STAT3 bound to the canonical binding motif of STAT3 in the EMSA. In contrast, a purified Flag-AS160 did not bind to the STAT3-binding motif, which was consistent with the lack of a DNA binding domain on AS160. Notably, the addition of purified Flag-AS160 significantly enhanced the binding of GST-STAT3 to its cognate DNA by over two folds (Fig. 5c, d).
The interaction between AS160 and STAT3 was markedly increased in skeletal muscle on the third day after CTX injury, and fell back to the basal level on the seventh day (Fig. 5e, f). This dynamic formation of the AS160–STAT3 complex after muscle injury suggested that it might contribute to the regulation of MuSC proliferation. The decreases of myogenic regulators Pax7 and Myod1 in AS160 deficient MuSCs prompted us to hypothesize that the AS160–STAT3 complex might regulate gene transcription of Pax7 and Myod1. Each promoter of Pax7 and Myod1 contains three potential STAT3-binding motifs (http:// https://jaspar.elixir.no/). Indeed, the purified STAT3 protein could bind to these motifs in the EMSA (Fig. 5g, h). In contrast, the purified AS160 protein alone exhibited no binding activities to these motifs in the EMSA (Fig. 5g, h). Notably, the addition of AS160 also increased the binding of STAT3 to all these motifs on the promoters of Pax7 and Myod1 (Fig. 5g, h). To find out whether AS160 regulates the binding of STAT3 to the promoter of Pax7 in vivo, we performed CUT-Tag qPCR in primary myoblasts isolated from Pax7CreERT2/+ and AS160-imKO mice. Notably, the binding of STAT3 to the three cis-elements in the Pax7 promoter was significantly lower in AS160 deficient myoblasts than in WT cells (Fig. 5i). We then examined whether AS160 and STAT3 might regulate transcription of Pax7 and Myod1 in a cooperative manner. The Pax7-luci and Myod1-luci reporters were expressed in AS160-KO myoblasts. Re-expression of AS160 in these AS160-KO myoblasts could significantly increase the activities of both Pax7-luci and Myod1-luci reporters as compared to the vector controls (Fig. 5j, k). Overexpression of STAT3 also increased the activities of both reporters in AS160-KO myoblasts, but to a lesser extent than re-expression of AS160 (Fig. 5j, k). Notably, co-expression of AS160 and STAT3 caused an additive effect on the activities of both Pax7-luci and Myod1-luci reporters in AS160-KO myoblasts (Fig. 5j, k). Together, these data show that nuclear AS160 may enhance transcription of STAT3-target genes, including Pax7 and Myod1, through forming a protein complex with STAT3.
Disruption of the AS160–STAT3 complex impairs muscle regeneration after injury
We next sought to find out the importance of the AS160–STAT3 complex formation in muscle regeneration in vivo. To this end, we first performed a biochemical dissection of segment requirements on both proteins for the formation of the AS160–STAT3 complex. It has been reported that STAT3 can form a homodimer as well as heterodimers with a number of proteins32,33. The formation of STAT3 homodimer depends on its SH2 domain and TAD region that contains two critical residues, namely Y705 and S727. Interestingly, the AS160-interacting domain(s) on STAT3 was also mapped to the SH2-TAD region (Supplementary Fig. 11a, b). Mapping of the STAT3-interacting region on AS160 showed that the PTB1 of AS160 most likely mediated the interaction with STAT3 (Supplementary Fig. 11c–f). Fine mapping of this region eventually led to the identification of six amino acids (aa) spanning from Cys207 to Val212 on AS160 that are essential for interacting with STAT3 (Fig. 6a and Supplementary Fig. 11g–m). Internal deletion of these 6 amino acids markedly decreased the interaction of AS160 with STAT3 (Fig. 6a). This AS160Δ(207-212) mutant exhibited normal nuclear localization, but caused a decreased nuclear accumulation of STAT3, as compared to the WT AS160 (Fig. 6b–d), suggesting that nuclear accumulation of STAT3 depended on its interaction with AS160. Notably, re-expression of the AS160Δ(207-212) mutant in the AS160-KO myoblasts exhibited a much weaker stimulative effect on the activities of both Pax7-luci and Myod1-luci reporters than the WT AS160 (Fig. 6e, f). The deletion of these six amino acids also significantly weakened the additive effect of AS160 and STAT3 on the activities of both Pax7-luci and Myod1-luci reporters in AS160-KO myoblasts (Fig. 6e, f).

a Mapping of STAT3-interacting region on AS160 via co-immunoprecipitation in HEK293 cells overexpressing Flag-STAT3 and GFP-tagged AS160 deletion mutants. b–d Nuclear levels of AS160 and STAT3 in AS160-KO HEK293 cells overexpressing HA-STAT3 together with Flag-tagged AS160-WT or AS160Δ207-212. b Representative immunoblots. c, d Quantitative results. n = 4 biological replicates. e, f Relative luciferase activity of the Pax7 (e) and Myod1 (f) promoters in AS160-KO MuSCs overexpressing STAT3 together with AS160-WT or AS160Δ207-212. The empty vector 1 and empty vector 2 were the vector control for AS160 and STAT3, respectively. n = 4 (except for the first control group n = 3) biological replicates. g, h Proliferation of AS160-KO myoblasts overexpressing AS160-P2A-mCherry or AS160Δ207-212-P2A-mCherry. g Representative immunostaining of EdU (green) and RFP (red) in the myoblasts. Bars indicate 50 µm in length. h Quantitative results. n = 4. i, j Transplantation of AS160-KO myoblasts overexpressing AS160-P2A-mCherry or AS160Δ207-212-P2A-mCherry. i Representative immunostaining of anti-RFP (red) and DAPI (blue) in recipient TA muscle. Bars indicate 50 µm in length. j Quantitative results. n = 4. k–m Proliferation of primary myoblasts isolated from WT and AS160Δ6aa mice (3-week-old) and in vitro cultured for 4 days. Myoblasts were stained with PAX7 (red), EdU (green) and DAPI (blue). k representative images. Bars indicate 50 µm in length. l, m Quantitative results. n = 4. n–p Proliferation of MuSCs attached to the myofibers isolated from WT and AS160Δ6aa mice (2-month-old) and in vitro cultured for 42 h. PAX7 (green), Ki67 (red), and DAPI (blue). n Representative images. Bars indicate 50 µm in length. o, p Quantitative results. n = 60 (WT) and 64 (AS160Δ6aa). q–s Myofiber sizes in CTX-injured TA muscle of male WT and AS160Δ6aa mice (2-month-old) after CTX injury for 7 days. q Representative images of H&E-stained cross-sections of TA muscle. Bars indicate 50 µm in length. r, s quantitative results. n = 4. Data were presented as mean values ± SEM. Numbers on the graphs represent p values. Statistical analyses were carried out via one-way ANOVA for (c–f, h), and two-sided t-test for (j, l, m, o, p, r, s). Source data are provided as a Source Data file.
Re-expression of WT AS160 in AS160-KO myoblasts significantly increased cell proliferation as evidenced by the EdU labeling experiment (Fig. 6g, h). This stimulative effect of AS160 on cell proliferation was dampened when the AS160Δ(207-212) mutant was re-expressed in AS160-KO myoblasts (Fig. 6g, h). We then transplanted AS160-KO myoblasts expressing the AS160-P2A-mCherry or AS160Δ(207-212)-P2A-mCherry into CTX-injured TA muscle of NCG immunodeficient recipient mice. The fluorescence area of mCherry+ myofibers were smaller in the recipients with AS160Δ(207-212)-P2A-mCherry myoblasts than in the recipients with AS160-KO/AS160-P2A-mCherry myoblasts (Fig. 6i, j). These data suggest that the AS160–STAT3 complex may promote myoblast proliferation in vitro as well as in vivo.
We next generated an AS160Δ6aa mouse model in which the region spanning from Cys207 to Val212 was deleted via CRISPR-Cas9-mediated knockin mutagenesis (Supplementary Fig. 12a–c). The AS160Δ6aa mice were born normally and displayed no overt phenotypic changes. Notably, when MuSCs were isolated and cultured in vitro, PAX7+ myoblasts as well as PAX7+/EdU+ myoblasts were significantly decreased in the AS160Δ6aa group as compared to those in the WT control group (Fig. 6k–m). Moreover, the numbers of both PAX7+ and PAX7+/Ki67+ MuSCs attached to the isolated myofibers were lower in the AS160Δ6aa group than in the WT control group after culture for 40 h (Fig. 6n–p). These data show an impaired proliferative activity of AS160Δ6aa MuSCs. We then subjected the AS160Δ6aa mice and their WT littermates to CTX injury. The protein levels of PAX7, MYOD1, and MYOG were significantly decreased in the AS160Δ6aa mice on the 3rd day after injury, indicating an impaired activation of AS160Δ6aa MuSCs (Supplementary Fig. 12d, e). The AS160Δ6aa mice had less EdU+ myonuclei and immature eMyHC+ myofibers on day-3.5 after injury (Supplementary Fig. 12f–i), but more eMyHC+ myofibers and still less EdU+ myonuclei on day-5 after injury (Supplementary Fig. 12j–m). Moreover, the AS160Δ6aa mice had smaller myofibers than the WT littermates on the seventh day after injury (Fig. 6q–s). Prior to CTX injury, the AS160Δ6aa mice exhibited normal endurance to a treadmill exercise (Supplementary Fig. 11n). CTX injury elicited a decline in exercise endurance within the first two days in both WT and AS160Δ6aa mice to similar extents. The exercise endurance was recovered in the WT mice on the fourth day after injury, but it remained impaired in the AS160Δ6aa mice (Supplementary Fig. 11n).
Together, these data show that the AS160–STAT3 complex plays a critical role in regulating MuSC proliferation during muscle regeneration.
Inhibition of AS160-Thr642 phosphorylation decreases MuSC proliferation and impairs muscle regeneration
Thr642 phosphorylation of AS160 is critical for insulin-induced muscle glucose uptake, and its inhibition causes insulin resistance and glucose intolerance18. Since Thr642 resides in the NLS2 of AS160, we examined whether its phosphorylation might affect the function of NLS2. Mutation of Thr642 to a non-phosphorylatable alanine or a phospho-mimetic aspartate did not alter nuclear targeting of NLS2 (Supplementary Fig. 13a), suggesting that Thr642 phosphorylation is likely not to directly affect the function of NLS2. However, Thr642 phosphorylation level and STAT3-binding of nuclear AS160 were higher than those of cytosolic AS160, although only a fraction of AS160 was localized in the nucleus of primary myoblasts (Fig. 7a and Supplementary Fig. 13b). Macrophages secrete insulin-like growth factor-1 (IGF-1) after muscle injury34,35. Notably, both protein expression and Thr642 phosphorylation of AS160 were markedly elevated in MuSCs freshly isolated from injured muscle than in MuSCs from uninjured muscle (Fig. 7b). Stimulation of C2C12 myoblasts with IGF-1 increased AS160-Thr642 phosphorylation, enhanced the interaction between these two proteins, and also caused nuclear accumulation of AS160 and STAT3 (Supplementary Fig. 13c–g). Insulin-stimulated Thr642 phosphorylation of AS160 is impaired in skeletal muscle of type 2 diabetic patients36. We first employed Db/Db mice as a diabetic model, and found that Thr642 phosphorylation of AS160 was decreased in myoblasts from Db/Db mice, which was associated with decreased myoblast proliferative activity and impaired muscle regeneration when injured (Fig. 7c and Supplementary Fig. 14a–e). We then utilized HFD and streptozotocin (STZ) to induce T2D for further examination of dynamic changes of AS160 expression and Thr642 phosphorylation in MuSCs in T2D (Supplementary Fig. 14f–n). HFD/STZ successfully induced diabetes in mice as shown by the elevated blood glucose level (Supplementary Fig. 14k), and caused diabetic sarcopenia as evidenced by decreased body weight, muscle weight, grip strength and myofiber CSA (Supplementary Fig. 14l–p). Treatment with HFD/STZ also impaired muscle regeneration upon CTX injury (Supplementary Fig. 14q). We then isolated MuSCs via FACS from HFD/STZ-treated 3xFlag-AS160/+ mice, and determined the expression and phosphorylation of AS160 in these cells. Notably, AS160 expression and Thr642 phosphorylation were both decreased in MuSCs isolated from diabetic mice (Fig. 7d). Immunofluorescence staining revealed significant decreases of nuclear AS160 and STAT3 in MuSCs from diabetic mice (Fig. 7e–g). These data suggest that the impaired AS160 phosphorylation is associated with impairment of MuSC proliferation and diabetic sarcopenia.

a AS160-Thr642 phosphorylation and STAT3-binding of 3xFlag-AS160 in both nuclear and cytosolic fractions of primary myoblasts isolated from 3xFlag-AS160/+ mice. b AS160 protein level and AS160-T642 phosphorylation level of MuSCs isolated from uninjured gastro and 3-day injured gastro via FACS. c AS160-Thr642 phosphorylation in primary myoblasts isolated from WT and Db/Db mice. d. Flag protein level and AS160-Thr642 phosphorylation in MuSCs isolated from NC (normal control) group and DM (diabetes mellitus) group of 3xFlag-AS160/+ mice via FACS. e–g Immunofluorescence staining of Flag and STAT3 in MuSCs isolated from NC group and DM group of 3xFlag-AS160/+ mice via FACS. e Representative images. f, g Quantitative results. n = 41 (NC) and 45 (DM). Bars indicate 5 µm in length. h–j Proliferation of primary myoblasts isolated from WT and AS160T642A mice (3-week-old) and in vitro cultured for 4 days. Myoblasts were stained with PAX7 (red), EdU (green), and DAPI (blue). h, i Quantitative results. n = 4. j Representative images. Bars indicate 50 µm in length. k–m Proliferation of MuSCs attached to the myofibers isolated from WT and AS160T642A mice (2-month-old) and in vitro cultured for 42 h. MuSCs were stained with PAX7 (green), Ki67 (red), and DAPI (blue). k Representative images. Bars indicate 50 µm in length. l, m Quantitative results. n = 61 (WT) and 58 (AS160T642A). n, o Transplantation of MuSCs isolated from WT/tdTomato and AS160T642A/tdTomato mice into TA muscle of male NCG mice (2-month-old) after injury for 1 day. n Quantitative results. n = 4. o Representative images of tdTomato (red) and DAPI (blue) in recipient TA muscle. Bars indicate 50 µm in length. p–r Myofiber sizes in CTX-injured TA muscle of male WT and AS160T642A mice (2-month-old) after CTX injury for 7 days. p Representative images of H&E-stained cross-sections of TA muscle. Bars indicate 50 µm in length. q, r, quantitative results. n = 6. Data were presented as mean values ± SEM. Numbers on the graphs represent p values. Statistical analyses were carried out via a two-sided t-test. Source data are provided as a Source Data file.
To further delineate possible roles of AS160-Thr642 phosphorylation in MuSC proliferation and muscle regeneration, we utilized a previously-reported insulin-resistant AS160T642A knockin mouse model in which Thr642 was substituted with a non-phosphorylatable alanine18. The AS160T642A mutation did not affect the AS160–STAT3 interaction in skeletal muscle (Supplementary Fig. 15a). Notably, AS160 protein was found less in the nucleus, while remained unchanged in the cytoplasm, in skeletal muscle of the AS160T642A mice as compared to that in the WT mice (Supplementary Fig. 15b, c). The in vitro proliferative activity, as evidenced by PAX7+ and PAX7+/EdU+ myoblast numbers, was significantly decreased in the AS160T642A myoblasts (Fig. 7h–j). Moreover, the isolated AS160T642A myofibers had less PAX7+ and PAX7+/Ki67+ MuSCs attached to them than the WT myofibers after in vitro culture for 40 h (Fig. 7k–m). To examine the effect of AS160T642A mutation on myoblast proliferation in vivo, we isolated MuSCs from WT and AS160T642A mice expressing the tdTomato transgene to culture in vitro, and transplanted the resultant myoblasts into CTX-injured TA muscle of NCG immunodeficient recipient mice. Fluorescence area of tdTomato+ myofibers were lower in the recipients with AS160T642A/tdTomato myoblasts than in the recipients with WT/tdTomato myoblasts (Fig. 7n, o). These data show that inhibition of AS160-Thr642 phosphorylation impairs the proliferation of MuSCs in vitro as well as in vivo.
We next investigated whether inhibition of AS160-Thr642 phosphorylation might affect muscle regeneration after injury. The myogenic regulators, including PAX7, MYOD1, and MYOG, were lower in the AS160T642A mice than in the WT mice on the third day after injury (Supplementary Fig. 15d, e). In accordance, the EdU+ myonuclei and eMyHC+ myofibers were significantly decreased in the AS160T642A mice as compared to those in the WT mice on day-3.5 after injury (Supplementary Fig. 15f–h). Moreover, the number of PAX7+ cells and EdU+ myonuclei were significantly decreased, while the proportion of eMyHC+ myofibers were markedly increased, in the AS160T642A mice as compared to those in the WT controls on day-5 after injury (Supplementary Fig. 15i–m). On day-7 after injury with CTX, the CSA of AS160T642A myofibers was significantly decreased as compared to that of the WT controls (Fig. 7p–r), which contrasted to the normal CSA of AS160T642A myofibers under uninjured conditions with saline injection (Supplementary Fig. 15n). The AS160T642A mice had normal exercise endurance before injury (Supplementary Fig. 15o). However, their exercise endurance became lower than the WT controls on the sixth day after CTX injury (Supplementary Fig. 15o), suggesting a poor recovery of injured muscle in the AS160T642A mice.
Together, these data show that inhibition of AS160-Thr642 phosphorylation plays a critical role in the impairment of muscle regeneration.
Discussion
In this study, we find that AS160, a well-known critical cytosolic regulator of insulin action, may act as a transcription cofactor in the nucleus, engaging with the myogenic transcriptional cascades to regulate MuSC proliferation for muscle regeneration (Supplementary Fig. 16). Deficiency of AS160 or inhibition of its Thr642 phosphorylation decreases MuSC proliferation and impairs regeneration of injured muscle.
The major finding of our study is that AS160 functions as a crucial transcription regulator of myogenic genes in the nucleus of MuSCs. Initially identified as a key component of insulin action, AS160 regulates membrane trafficking in the cytosol of various types of cells, including adipocytes, myocytes and renal epithelial cells12,13,37. These cytosolic roles of AS160 in regulating membrane trafficking depend on its RabGAP function. In contrast, the nuclear role of AS160 in the control of myogenic genes does not require its RabGAP function. Thus, AS160 functions as a moonlighting protein. AS160 contains a functional NLS that spans from Pro634 to Leu654. Though this NLS drives nuclear localization of the GFP-GST recombinant protein, it causes only a portion of the full-length AS160 protein to be imported into the nucleus. Given the nuclear localization of the AS160M1-I770 fragment, we suspect that there might be some inhibitory sequences in the GAP-containing C-terminal part of AS160. Such inhibitory effect on nuclear localization of AS160 might be relieved through conformational changes of AS160 upon its Thr642 phosphorylation, which might expose the NLS on AS160. In agreement with such a possibility, phosphorylation of Thr642 does not affect the nuclear localization of NLS-GFP-GST fusion protein that might contain no inhibitory sequence and constitutively expose the NLS. In contrast, mutation of Thr642 to a non-phosphorylatable alanine decreases nuclear localization of AS160, suggesting that phosphorylation of Thr642 might cause some conformational changes that lead to exposure of the NLS. Interestingly, Thr642 phosphorylation may also be critical for regulating access of AS160 to its target Rabs associated with GLUT4 vesicles to facilitate consequent trafficking of GLUT4 in response to insulin12,16,18. Thus, Thr642 phosphorylation might coordinately regulate both nuclear and cytosolic functions of AS160.
The involvement of AS160 in the regulation of gene transcription is intriguing. No DNA-binding domain and transcriptional activation domain are found on AS160, suggesting that it has to bind to transcription factors to regulate gene expression. In this study, we show that AS160 binds to STAT3 to regulate myogenic gene expression in MuSCs through a twofold mechanism in which AS160 increases the nuclear level of STAT3 and enhances the transcriptional activity of STAT3. Previous studies show that STAT3 may regulate MuSC differentiation through MYOD1 and MYOG30,31. A recent study provides evidence that STAT3 binds to the promoter region of the Pax7 gene, and that deletion of STAT3 in MuSCs decreases Pax7 expression and consequently inhibits MuSC proliferation after muscle injury29. AS160 increases the binding of STAT3 to its cognate DNA motif in vitro and enhances the transcriptional activity of STAT3 in cells. Presumably, AS160 binding might cause conformational changes of STAT3 to expose its DNA-binding domain. Both AS160 and STAT3 are expressed in many cell types, which raises the question of whether AS160 might regulate gene expression in other cell types through interaction with STAT3. Almost half of AS160-interacting genomic loci overlap with STAT3-binding sites in the ChIP-Seq experiment, suggesting that STAT3 is a major AS160-interacting transcription factor in myoblasts. The ChIP-Seq data also suggest that AS160 might bind to other transcription factors to regulate the rest of the AS160-interacting genomic loci. It would be essential in the future to delineate the dynamics of AS160-binding sites in the genome together with the corresponding transcription factors in different cells for elucidation of the potential roles of AS160 as a transcription cofactor.
TBC1D1 is a RabGAP closely related to AS160, and also regulates membrane trafficking to exert its metabolic functions in a manner similar to AS16038. TBC1D1 contains a predicted NLS spanning from Pro747 to His753. Insulin also induces phosphorylation of TBC1D1 on Thr596, a site paralogous to Thr642 on AS16039. Deficiency of TBC1D1 confers leanness to mice while a TBC1D1R125W mutation is linked to obesity predisposition40,41. Given the similarity between TBC1D1 and AS160, it would be intriguing to find out whether TBC1D1 might also be targeted to the nucleus to regulate MuSCs for muscle regeneration.
In summary, we show that AS160 is a critical transcriptional regulator of MuSC proliferation and that its deficiency or hypophosphorylation impairs muscle regeneration.
Methods
This study was carried out under the approval of the Ethics Committee of Nanjing University, complying with all relevant ethical regulations. All animal procedures in this study were approved by the Institutional Animal Care and Use Committee (IACUC) at Model Animal Research Center of Nanjing University.
Materials
Cardiotoxin (CTX) was bought from Latoxan (Valence, France). The EdU Cell Proliferation Kit was from Beyotime (Shanghai, China). Protein G-Sepharose was from Cytiva (Shanghai, China), GFP-Trap-Agarose was from ChromoTek (Germany), and anti-Flag affinity beads was from Smart-Lifesciences (Changzhou, China). All other chemicals were from Sigma-Aldrich (Shanghai, China) or Sangon Biotech (Shanghai, China). The commercial antibodies and affinity resins used in this study are listed in Supplementary Table 1.
Molecular biology
The cDNAs encoding human AS160 and STAT3 were cloned into pcDNA5-FRT/TO vectors with different tags for expression in mammalian cells. The numbering of key residues on AS160 was according to the sequence of the human long isoform (NP_055647.2). The split GFP system consists of a GFP1-10 (M1-K215 of EGFP (Accession No: MN968812) containing the following mutations: S31R, Y40I, Q81R, F100S, N106K, E112V, I129T, Y146F, M154T, V164A, K167T, I168V, I172V, S206T, and A207V) and a tandem GFP11 (GFP11X2, R216-T231 of EGFP containing the following mutations: L222H, F224Y, and T226T). For Firefly luciferase reporters, the −12 to −2511 bp region of mouse Pax7 (NM_011039.2) promoter and the 133 to −1867 bp region of mouse Myod1 (NM_010866.2) promoter, as shown in Supplementary Table 2, were cloned into a pGL3-basic vector to construct Pax7-luci and Myod1-luci, respectively. All plasmids were verified via sequencing at AZENTA Life Science (Suzhou, China).
Animals
All animal procedures carried out in this study were reviewed and approved by the Ethics Committee at the Model Animal Research Center of Nanjing University. Mice and rats were produced and maintained in a facility with a light/dark cycle of 12 h, ambient temperature of 23 ± 2 °C and humidity 40–70%.
The AS160-KO mice22 AS160R972K mice22, and AS160T642A mice18 were previously described. The AS160-mKO mice were generated by mating AS160f/f mice22 with Mlc1f-Cre mice42. The AS160-imKO mice were generated by mating AS160f/f mice with Pax7CreERT2/+ mice43. The AS160-imKO mice were continuously injected with tamoxifen (80 mg/kg) for 5 days to induce deletion of As160 in MuSCs, and then allowed to recover for 1–2 weeks. The 3xFlag-AS160 mice were generated on a C57Bl/6J background using the CRISPR/Cas9-based strategy by Gempharmatech (Jiangsu, China). The 3xFlag sequence was inserted into the N-terminus of AS160 via CRISPR/Cas9-assisted knockin substitution to generate 3xFlag-AS160 mice. AS160Δ6aa mice were generated on a C57Bl/6J background using the CRISPR/Cas9-based strategy by Gempharmatech (Jiangsu, China). The 207th to 212th amino acids (the corresponding sequence is CGRVIV) of As160 was deleted via CRISPR/Cas9-assisted knockin substitution to generate AS160Δ6aa mice. WT mice (primers for WTs: 5ʹ-GGGTTTCAGGTACGAGTCAAC-3ʹ and 5ʹ-GGTCACGATCACCCGCCCAC-3ʹ, 394 bp) and AS160Δ6aa mice (primers for AS160Δ6aa mice: 5ʹ-AGAAATTCGAAGTGCTTTACAC-3ʹ and 5ʹ-TGACTGTACCATCTGCCTTC-3ʹ, 207 bp) were genotyped using the two sets of specific primers. AS160-KO/Tdtomato mice and AS160T642A/Tdtomato mice were generated by mating Rosa26mTmG mice44 with AS160-KO mice and AS160T642A mice, respectively. Db/Db mice on the BKS background and BKS WT controls were bought from Gempharmatech (Jiangsu, China).
Muscle injury and regeneration
Muscle injury was performed through the injection of CTX (L8102, Latoxan) as previously described45. Briefly, mice (2 to 3-month-old) were anesthetized with isoflurane, and the TA muscle was subjected to CTX injury. CTX was dissolved in saline (50 µL of 10 µM per TA muscle) and injected in situ into the middle of the left TA muscle via a 29-G needle. The right TA muscle was injected with an equal volume of saline as a control. Mice were sacrificed within 7 days after injury, and TA muscles were harvested for subsequent analyses. For the 2nd injury, the same amount of CTX was injected into the TA muscles 21 days after the first injury. A similar protocol for muscle injury was performed in rats, in which a dose of CTX (150 µL of 10 µM per TA muscle) was used.
MuSC isolation and culture
MuSCs were isolated as previously described46. Briefly, limb muscles of 3–4 weeks old mice or rats were minced into small pieces with scissors, and digested in 0.2% collagenase II and 1 g/L dispase II at 37 °C for 45 min. The resultant cell suspension was neutralized with adequate suspension medium (2% FBS, 1% penicillin-streptomycin in DMEM). Tissue debris was removed with a 40-μm filter, and cells were collected by centrifugation at 350×g for 5 min at 4 °C. Cells were further washed twice with the suspension medium. For direct analyses of MuSCs, resultant cell suspensions were incubated with the flow cytometry antibodies for 30 min on ice, as previously described47, including anti-Cd11b (BD, FITC), anti-CD31 (BD, FITC), anti-CD45 (BD, FITC), anti-Sca1 (BD, FITC), and anti-Integrin-α7 (R&D, APC). MuSCs (Cd11b−CD31−CD45-Sca1−Integrin-α7+) were isolated via fluorescence-activated cell sorting (FACS) using a BD FACSAriaTM III flow sorter (BD Biosciences), and used for subsequent analyses. For the culture of primary myoblasts without FACS, fibroblasts in resultant cells were removed through preculture in non-coated dishes for about 1 h. Remaining MuSCs were then seeded in dishes coated with rat tail collagen I, and cultured with growth medium (20% FBS, 1% chicken embryo extract, 1% Glutamax, 5 ng/mL bFGF, 1% penicillin-streptomycin in Ham’s F-10 media) to obtain enough primary myoblasts.
Single myofiber isolation and culture
Single myofibers were isolated from mouse EDL muscle as previously described48. Briefly, the EDL muscle with intact tendon was isolated immediately after mice were sacrificed, and subjected to digestion with 0.2% collagenase I in a shaking water bath at 37 °C for 60 min. The digestion was then stopped through neutralization with 10% FBS in high-glucose DMEM. Individual myofibers were gently separated through pipetting under a Versatile Stereo microscope (SMZ-168, Motic, China). Isolated myofibers were washed three to four times in DMEM to remove tissue debris, and cultured in horse serum-coated dishes with myofiber growth medium (20% FBS, 1% chicken embryo extract, 1% penicillin-streptomycin in high-glucose DMEM). Individual myofibers were fixed either immediately after isolation for analysis of MuSC pool size or after culture for 2–3 days for analysis of MuSC proliferation.
Cell culture, differentiation, and transfection
Human embryonic kidney HEK293 cells and mouse C2C12 myoblasts were obtained from the Cell Resource Center, Chinese Academy of Medical Sciences and Peking Union Medical College (China), and rat L6 myoblasts were from Dr. Amira Klip (University of Toronto, Toronto, Canada). Human HSkMC were purchased from Shanghai Cell Bank of the Chinese Academy of Sciences. Cell lines were maintained in DMEM (Dulbecco’s Modified Eagle Medium) supplemented with 10% (v/v) fetal bovine serum and 1% penicillin-streptomycin. For differentiation, L6 myoblasts and C2C12 myoblasts were cultured in differentiation medium (2% horse serum, 1% penicillin-streptomycin in DMEM) for 7 days. Muscle cells were collected on the first, third, fifth, and seventh days after differentiation.
Cells were transfected with plasmids using a Lipofectamine-3000 (Thermo Fisher Scientific) mediated method. Lentiviral delivery of shRNAs was used to knock down target genes. The shRNA sequences targeting mouse AS160 are 5′-GCATTCAGGATGAGCCTTTCC-3′ (shAs160-1m) and 5′-GCATCAGGCAGTTATCCAAGG-3′(shAs160-2m). The shRNA sequences targeting rat AS160 are 5′-GCCTCATTGATGACTGCAAGG-3′ (shAs160-1r) and 5′-GCTCGAGAAGAATTTCAAAGA-3′(shAs160-2r). The shRNA sequences targeting human AS160 are 5′-GCCAACTAGCATGTTGCATCT-3′ (shAS160-1h) and 5′-GCATTTCAACTCTTCACTTCC-3′(shAS160-2h).
EdU-based proliferation assays
For the in vitro EdU-based proliferation assay, myoblasts and MuSCs were cultured on glass coverslips for a day, and then supplemented with fresh growth medium containing 10 µM EdU for the indicated time. EdU labeling was performed for 1 h for C2C12 myoblasts, 1.5 h for L6 myoblasts and human HSkMC, and 3.5 h for primary myoblasts. After EdU labeling, cells were washed with pre-cold PBS and fixed in 4% paraformaldehyde for 15 min. EdU-labeled nuclei were stained using an EdU Cell Proliferation Kit (Beyotime, C0071S).
For the in vivo EdU-based proliferation assay, on the first two days after injury, EdU (1 mg/mL in sterile PBS) was injected intraperitoneally into mice at 10 mg/kg body weight. TA muscles were prepared into freezing slices with a thickness of 10 µm and stained according to the provided instructions of EdU Cell Proliferation Kit.
CFSE cell proliferation assay
For the CFSE-based cell proliferation assay, 4 × 105/mL myoblasts were suspended in DMEM containing 5 µM CFSE for 10 min at room temperature with gentle shaking. The blank control group of cells were incubated without CFSE. After staining, cells were pelleted down via centrifugation at 400×g for 3 min, and then resuspended in DMEM containing 10% FBS. Cells were then seeded in 6-well dishes for 48 h. Afterwards, cells were mildly trypsinized, resuspended in DPBS, and then spun down via centrifugation at 150×g for 3 min. Cell pellets were resuspended in FACS buffer and analyzed using a flow cytometer (LSRFortessa™ cell analyzer, BD).
Glucose uptake
Glucose uptake into primary myoblasts was performed using a fluorescent D-glucose analog 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino]−2-deoxy-D-glucose (2-NBDG). Briefly, primary myoblasts were incubated in uptake buffer containing 2-NBDG (200 μM) for 1 h. Afterwards, cells were washed three times and lysed. Fluorescent signals of 2-NBDG in cell lysates were measured at 475 nm excitation and 550 nm emission using a microplate reader (Synergy H1, BioTek Instruments Inc). Protein concentrations in cell lysates were determined and used for normalization of glucose uptake.
Primary myoblast transplantation
Primary myoblast transplantation were performed as previously described47. Briefly, primary myoblasts were isolated from donor mice and cultured for 2 days before transplantation. TA muscles of recipient NCG mice were injured with CTX one day before transplantation to induce a suitable microenvironment. Primary myoblasts were collected and resuspended in suspension buffer (10% FBS in Ham’s F-10 media). After incubation on ice for 2 h, an equal amount of cells was injected in situ into the injured TA muscle of recipient NCG mice (2–3 × 104 of cells per leg). After transplantation for 3 weeks, TA muscles were harvested for histological analysis via a confocal microscope (LSM880, ZEISS).
Chromatin immunoprecipitation (ChIP)
The ChIP assay was performed as previously described49. Briefly, L6 myoblasts overexpressing HA-AS160 or HA-STAT3 were fixed in 1% formaldehyde for 15 min at room temperature. Fixation was stopped by adding glycine to a final concentration of 140 mM and incubated for 10 min at room temperature. Cells were washed with pre-cold PBS twice. 1 × 107 cells were resuspended in Buffer A (50 mM HEPES, pH 7.6, 1 mM EDTA, 140 mM NaCl, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100 and protease inhibitors cocktails), and lysed on ice for 10 min to release the nuclei that were collected via centrifugation at 1000×g for 10 min at 4 °C. The collected nuclei were washed with Buffer B (10 mM Tris-Cl, pH 7.8, 1 mM EDTA, 200 mM NaCl, 1 mM EGTA, and protease inhibitors cocktails) twice, and resuspended in Buffer C (50 mM Tris-Cl, pH 7.8, 10 mM EDTA, 1% SDS, and protease inhibitors cocktails). Chromatin DNA was fragmented into 150–300 bp via a Bioruptor. Subsequently, the fragmented Chromatin lysates were diluted 10-fold with Buffer D (20 mM Tris-Cl, pH 7.8, 2 mM EDTA, 150 mM NaCl, and 1% Triton X-100) and incubated with anti-HA (Cell Signaling Technology #3724, 1:50) for 16 h at 4 °C. Protein G magnetic beads were added to the Chromatin lysates for another 4-h incubation at 4 °C. The immunoprecipitates were washed with Buffer E (20 mM Tris-Cl, pH 8.0, 2 mM EDTA, 150 mM NaCl, 0.1% SDS, and 1% Triton X-100) twice, followed by another two washes with Buffer F (50 mM Tris-Cl, pH 8.0, 1 mM EDTA). The crosslinked DNA was released from HA-AS160 or HA-STAT3 in Buffer G (50 mM Tris-Cl, pH 8.0, 10 mM EDTA, and 1% SDS) at 65 °C overnight, and the resultant DNA was isolated via a QIAGEN PCR product purification kit (#28104).
ChIP-seq
The library was prepared using a KAPA Hyper Prep Kit (Roche, KK8503) following the manufacturer’s instructions with adapters and primer sequences from Roche. All ChIP libraries were pair-end sequenced via the HiSeq3000 systems at Novogene (Beijing, China). ChIP-Seq datasets were analysed using HOMER.
Luciferase reporter assay
The Firefly luci reporters together with the Renilla luciferase expressing vector were co-transfected into mouse primary myoblasts. The Renilla luciferase was used as an internal control for normalization. Primary myoblasts were harvested 36 h after transfection and subjected to dual reporter luciferase assay by using Nano-Glo® Dual-Luciferase® Reporter Assay System (Promega, E1910).
CUT-Tag qPCR
The Cut-Tag qPCR assay was carried out in primary myoblasts using the Hyperactive Universal CUT-Tag Assay Kit from Vazyme (TD904, TD904-C1, TD904-C2). Briefly, primary myoblasts (2 ×105) were used for nuclear extraction and cross-linking for each individual sample. The Cut-Tag qPCR experiments were conducted according to the manufacturer’s instructions, and the primers for qPCR were listed in Supplementary Table 3.
EMSA
EMSA was performed as previously described50. For the preparation of DNA probes, primers were annealed to form a double-stranded DNA whose 3’-end was labeled with Cy5-dATP (AAT Bioquest #17038) using the Klenow DNA Polymerase (NEB M0210). Unlabeled probes with the same sequence were used as competitors. The sequence of the classical STAT3 motif is 5’-GATCCTTCTGGGAATTCCTAGATC-3’. The primers used in the preparation of DNA probes were listed in Supplementary Table 4. Purified proteins and DNA probes were incubated at room temperature in protein-DNA binding buffer (20 mM Tris-Cl, pH 8.0, 150 mM KCl, 0.5 mM EDTA, 0.1% Triton X-100, 12.5 % glycerol (v/v), and 0.2 mM DTT) for 30 min. DNA-protein mixtures were separated via electrophoresis in 6% polyacrylamide gel on ice for 1 hour. Gels were visualized using Odyssey® CLx Infrared Imaging System (LI-COR Biosciences), and band intensities were quantified using ImageJ.
Subcellular fractionation
Subcellular fractionation was performed as previously described51. Briefly, fractionation of cells and skeletal muscle was carried out using NE-PERTM Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, Cat No. #78833) to obtain nuclear and cytosolic fractions that were analyzed via immunoblotting.
Grip strength assay
Limb grip strength of mice was measured using the computerized force transducer (Grip Strength Meter, Bioseb). Mice were trained three times and fully recovered before the assay. Limb grip strength was measured on each mouse for at least three times to obtain an average as the final measurement.
Treadmill exercise
Maximal exercise performance in mice was measured using an Exer5/9 mouse treadmill (Techman Soft, China) as previously described52. The treadmill was set at a +5° incline and 0.3 mA aversive electrical stimulation. After acclimating to treadmill running (5 min per day at 9 m/min for 5 days), mice were subjected to an endurance test, in which the belt speed of the treadmill was set at 9 m/min initially and increased by 1 m/min every 7 min. Running tests were stopped when mice did not re-engage on the treadmill after aversive stimulation for more than 15 s.
Histology, immunofluorescence staining, and imaging
Fresh isolated TA muscles were frozen in isopentane cooled with liquid nitrogen and stored in a −80 °C refrigerator immediately. The frozen muscles were sectioned into slices with a thickness of 10 µm using a Leica RM2016 microtome for H&E staining or immunofluorescence. For immunofluorescence, muscle slices were air-dried, fixed in 4% PFA for 15 min at room temperature, and then permeabilized with 0.1% Triton X-100 or pre-cold methyl alcohol. Antigen repair was performed with citric acid antigen repair buffer for PAX7 staining. Muscle slices were blocked with 4% bovine serum albumin containing 50% goat serum for 1 h, and incubated in primary antibodies overnight at 4 °C. After washing with 0.1% Tween-20 in PBS five times, muscle slices were incubated with secondary antibodies for 1 h at room temperature. Nuclei were stained with DAPI. Immunofluorescence staining of isolated myofibers were performed using the same protocol. H&E staining of muscle slices were photographed using an Olympus BX53F microscope (Japan), and pictures of immunofluorescence staining were taken using a Leica confocal microscope.
Immunoprecipitation
Protein immunoprecipitation was performed through incubating tissue or cell lysates with antibody-coupled protein G-Sepharose (Cytiva), GFP-Trap-Agarose (ChromoTek, Germany) or anti-Flag affinity beads (Smart-Lifesciences, Changzhou, China) for 16 h at 4 °C. The beads were collected by centrifugation for 2 min at 3000×g and washed with a low-salt buffer (25 mM Tris-Cl, pH = 7.4, and 150 mM NaCl) and a high-salt buffer (25 mM Tris-Cl, pH = 7.4, and 500 mM NaCl) to remove non-specific binding proteins. Subsequently, immunoprecipitated proteins were denatured in SDS sample buffer and eluted from the beads for immunoblotting analysis.
Immunoblotting
Protein lysates were electrophoretically separated on SDS-PAGE gels and immunoblotted onto nitrocellulose membranes. After being blocked with milk or BSA, membranes were sequentially incubated with primary antibodies and horseradish-peroxidase-conjugated secondary antibodies. Membranes were incubated with ECL® (enhanced chemiluminescence reagent; GE Healthcare), and chemiluminescent signals were recorded by a gel documentation system (Tanon, China). Intensities of chemiluminescent signals were quantified using ImageJ as a measure of protein expression levels.
Mass spectrometry
Proteins were separated via SDS-PAGE and subjected to in-gel trypsin digestion. After trypsin digestion, peptides were analysed via LC-MS/MS, in which a TripleTOF 5600+ System (AB SCIEX) was coupled with a NanoLC.2D (Eksigent Technologies) for MS data acquisition. Original MS/MS data were analysed using ProteinPilot Software (version 4.5, AB Sciex), and peptides/proteins were identified via searching against UniProt database.
Real-time quantitative PCR
Total RNA was extracted from mouse tissues or cells using the TRIzol® Reagent (Life Technologies). PrimeScript® RT reagent kit (DRR047A, TaKaRa) was used for reverse transcription of RNA into cDNA. Expression levels of target genes were measured via real-time Quantitative PCR using a Roche Lightcycler Real-Time PCR system. The primers for quantitative PCR were listed in Supplementary Table 5.
RNA-seq
RNA-sequencing was performed via the Illumina platform by Annoroad Gene Tech (Beijing) Co., Ltd. Statistical analysis was carried out using the cuffdiff tool from Cufflinks. Differentially expressed genes (DEGs) identified using the criteria |log2FC| >0.585 and q < 0.05 were subjected to gene function enrichment analysis by the Metascape database.
Statistics and reproducibility
Statistical analyses were carried out via two-sided t-test for two groups or via one-way ANOVA or two-way ANOVA for multiple groups using Prism v9.5.1 software (GraphPad, San Diego, CA, USA). Differences were considered statistically significant at p < 0.05. Error bars, P values, n numbers and statistical tests used are reported in the figures or figure legends. No statistical method was used to predetermine sample size. Samples were randomly allocated into experimental groups. The Investigators were not blinded to allocation during experiments and outcome assessment. Similar results were obtained from at least two experiments for Fig. 1a, c, d, e; 4b–h; 5g, h; 6a; 7a–d.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The proteomic data generated in this study have been deposited in the MassIVE Repository (University of California, San Diego) under accession code MSV000098225 [https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=954870423f3548c680638bac8c0cfb1e]. The RNA-Seq data generated in this study have been deposited in the NCBI database under the accession code PRJNA1273628. The ChIP-Seq data generated in this study have been deposited in the NCBI database under the accession code PRJNA1273619. All supplementary and unprocessed data generated in this study are provided in the Supplementary Information/Source Data file. Source data are provided with this paper.
References
Espino-Gonzalez, E. et al. Impaired skeletal muscle regeneration in diabetes: from cellular and molecular mechanisms to novel treatments. Cell Metab. 36, 1204–1236 (2024).
Teng, S. & Huang, P. The effect of type 2 diabetes mellitus and obesity on muscle progenitor cell function. Stem Cell Res. Ther. 10, 103 (2019).
Wang, Y. X. & Rudnicki, M. A. Satellite cells, the engines of muscle repair. Nat. Rev. Mol. Cell Biol. 13, 127–133 (2011).
Sousa-Victor, P., Garcia-Prat, L. & Munoz-Canoves, P. Control of satellite cell function in muscle regeneration and its disruption in ageing. Nat. Rev. Mol. Cell Biol. 23, 204–226 (2022).
Kuang, S., Kuroda, K., Le Grand, F. & Rudnicki, M. A. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell 129, 999–1010 (2007).
Evano, B., Khalilian, S., Le Carrou, G., Almouzni, G. & Tajbakhsh, S. Dynamics of asymmetric and symmetric divisions of muscle stem cells in vivo and on artificial niches. Cell Rep. 30, 3195–3206 e3197 (2020).
Yin, H., Price, F. & Rudnicki, M. A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 93, 23–67 (2013).
Hernandez-Hernandez, J. M., Garcia-Gonzalez, E. G., Brun, C. E. & Rudnicki, M. A. The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin. Cell Dev. Biol. 72, 10–18 (2017).
Chakkalakal, J. V., Jones, K. M., Basson, M. A. & Brack, A. S. The aged niche disrupts muscle stem cell quiescence. Nature 490, 355–360 (2012).
Sousa-Victor, P. et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506, 316–321 (2014).
Feige, P., Brun, C. E., Ritso, M. & Rudnicki, M. A. Orienting muscle stem cells for regeneration in homeostasis, aging, and disease. Cell Stem Cell 23, 653–664 (2018).
Sano, H. et al. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J. Biol. Chem. 278, 14599–14602 (2003).
Kramer, H. F. et al. AS160 regulates insulin- and contraction-stimulated glucose uptake in mouse skeletal muscle. J. Biol. Chem. 281, 31478–31485 (2006).
Sano, H. et al. Rab10, a target of the AS160 Rab GAP, is required for insulin-stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metab. 5, 293–303 (2007).
Sun, Y., Bilan, P. J., Liu, Z. & Klip, A. Rab8A and Rab13 are activated by insulin and regulate GLUT4 translocation in muscle cells. Proc. Natl Acad. Sci. USA 107, 19909–19914 (2010).
Eickelschulte, S. et al. AKT/AMPK-mediated phosphorylation of TBC1D4 disrupts the interaction with insulin-regulated aminopeptidase. J. Biol. Chem. 296, 100637 (2021).
Geraghty, K. M. et al. Regulation of multisite phosphorylation and 14-3-3 binding of AS160 in response to IGF-1, EGF, PMA and AICAR. Biochem. J. 407, 231–241 (2007).
Chen, S., Wasserman, D. H., MacKintosh, C. & Sakamoto, K. Mice with AS160/TBC1D4-Thr649Ala knockin mutation are glucose intolerant with reduced insulin sensitivity and altered GLUT4 trafficking. Cell Metab. 13, 68–79 (2011).
Moltke, I. et al. A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature 512, 190–193 (2014).
Yang, X. et al. Tissue-specific splicing and dietary interaction of a mutant As160 allele determine muscle metabolic fitness in rodents. Diabetes 70, 1826–1842 (2021).
Zhang, P. et al. mTOR is necessary for proper satellite cell activity and skeletal muscle regeneration. Biochem. Biophys. Res. Commun. 463, 102–108 (2015).
Xie, B. et al. The inactivation of RabGAP function of AS160 promotes lysosomal degradation of GLUT4 and causes postprandial fyperglycemia and hyperinsulinemia. Diabetes 65, 3327–3340 (2016).
Wang, H. Y. et al. AS160 deficiency causes whole-body insulin resistance via composite effects in multiple tissues. Biochem. J. 449, 479–489 (2013).
Guillet-Deniau, I., Leturque, A. & Girard, J. Expression and cellular localization of glucose transporters (GLUT1, GLUT3, GLUT4) during differentiation of myogenic cells isolated from rat foetuses. J. Cell Sci. 107, 487–496 (1994).
Sartore, S., Gorza, L. & Schiaffino, S. Fetal myosin heavy chains in regenerating muscle. Nature 298, 294–296 (1982).
Zhang, S. Z. et al. Regulation of cell proliferation by fast Myosin light chain 1 in myoblasts derived from extraocular muscle, diaphragm and gastrocnemius. Exp. Biol. Med. 233, 1374–1384 (2008).
Nishijo, K. et al. Biomarker system for studying muscle, stem cells, and cancer in vivo. FASEB J. 23, 2681–2690 (2009).
Hosoi, T. et al. Sarcopenia phenotype and impaired muscle function in male mice with fast-twitch muscle-specific knockout of the androgen receptor. Proc. Natl Acad. Sci. USA 120, e2218032120 (2023).
Zhu, H. et al. STAT3 regulates self-renewal of adult muscle satellite cells during injury-induced muscle regeneration. Cell Rep. 16, 2102–2115 (2016).
Price, F. D. et al. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat. Med. 20, 1174–1181 (2014).
Tierney, M. T. et al. STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat. Med. 20, 1182–1186 (2014).
Reich, N. C. & Liu, L. Tracking STAT nuclear traffic. Nat. Rev. Immunol. 6, 602–612 (2006).
Laudisi, F., Cherubini, F., Monteleone, G. & Stolfi, C. STAT3 interactors as potential therapeutic targets for cancer treatment. Int. J. Mol. Sci. https://doi.org/10.3390/ijms19061787 (2018).
Wosczyna, M. N. & Rando, T. A. A muscle stem cell support group: coordinated cellular responses in muscle regeneration. Dev. Cell 46, 135–143 (2018).
Baht, G. S. et al. Meteorin-like facilitates skeletal muscle repair through a Stat3/IGF-1 mechanism. Nat. Metab. 2, 278–289 (2020).
Karlsson, H. K. et al. Insulin-stimulated phosphorylation of the Akt substrate AS160 is impaired in skeletal muscle of type 2 diabetic subjects. Diabetes 54, 1692–1697 (2005).
Alves, D. S., Thulin, G., Loffing, J., Kashgarian, M. & Caplan, M. J. Akt substrate of 160 kD regulates Na+,K+-ATPase trafficking in response to energy depletion and renal ischemia. J. Am. Soc. Nephrol. 26, 2765–2776 (2015).
Chen, S., Synowsky, S., Tinti, M. & MacKintosh, C. The capture of phosphoproteins by 14-3-3 proteins mediates actions of insulin. Trends Endocrinol. Metab. 22, 429–436 (2011).
Chen, S. et al. Complementary regulation of TBC1D1 and AS160 by growth factors, insulin and AMPK activators. Biochem. J. 409, 449–459 (2008).
Chadt, A. et al. Tbc1d1 mutation in lean mouse strain confers leanness and protects from diet-induced obesity. Nat. Genet. 40, 1354–1359 (2008).
Stone, S. et al. TBC1D1 is a candidate for a severe obesity gene and evidence for a gene/gene interaction in obesity predisposition. Hum. Mol. Genet. 15, 2709–2720 (2006).
Bothe, G. W., Haspel, J. A., Smith, C. L., Wiener, H. H. & Burden, S. J. Selective expression of Cre recombinase in skeletal muscle fibers. Genesis 26, 165–166 (2000).
Lepper, C., Conway, S. J. & Fan, C. M. Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature 460, 627–631 (2009).
Muzumdar, M. D., Tasic, B., Miyamichi, K., Li, L. & Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 (2007).
Shang, M. et al. Macrophage-derived glutamine boosts satellite cells and muscle regeneration. Nature 587, 626–631 (2020).
Motohashi, N., Asakura, Y. & Asakura, A. Isolation, culture, and transplantation of muscle satellite cells. J. Vis. Exp. https://doi.org/10.3791/50846 (2014).
Feige, P. & Rudnicki, M. A. Isolation of satellite cells and transplantation into mice for lineage tracing in muscle. Nat. Protoc. 15, 1082–1097 (2020).
Wu, R. et al. MicroRNA-431 accelerates muscle regeneration and ameliorates muscular dystrophy by targeting Pax7 in mice. Nat. Commun. 6, 7713 (2015).
Wang, X. et al. DDB1 binds histone reader BRWD3 to activate the transcriptional cascade in adipogenesis and promote onset of obesity. Cell Rep. 35, 109281 (2021).
Wang, H. Y. & Williams, J. G. Identification of a target for CudA, the transcription factor which directs formation of the Dictyostelium tip organiser. Int. J. Dev. Biol. 54, 161–165 (2010).
Wei, W. et al. TRIM24 is an insulin-responsive regulator of P-bodies. Nat. Commun. 13, 3972 (2022).
Chen, Q. et al. A Tbc1d1 (Ser231Ala)-knockin mutation partially impairs AICAR- but not exercise-induced muscle glucose uptake in mice. Diabetologia 60, 336–345 (2017).
Acknowledgements
We thank members of the resource unit at Nanjing University for technical assistance. Thanks to the National Natural Science Foundation of China (Grant Nos. 32025019 to S.C. and 32401080 to X.Y.Y.), the Science and Technology Foundation of Jiangsu Province of China (Grant No. BK20243041 (Key Research Program) to S.C. and H.-Y.W.), the Ministry of Science and Technology of China (Grant Nos. 2021YFF0702100 and 2018YFA0801100 to S.C. and H.-Y.W.), and the Fundamental Research Funds for the Central Universities (021414380533 to S.C.), for financial support.
Author information
Authors and Affiliations
Contributions
X.Y.Y., Y.C., Y.Z., Q.Y., P.R., X.W., Q.C., W.F., L.Z., H.A., L.F. and X.H.Y. performed experiments, analyzed data, reviewed and edited the manuscript. D.Z. and T.-J.Z. reviewed and edited the manuscript. H.-Y.W. and S.C. designed experiments, analyzed data, and wrote the manuscript. All the authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Davy Vanhoutte and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, X., Cao, Y., Zhou, Y. et al. Nuclear entry of AS160 as a transcriptional regulator of satellite cells for muscle regeneration. Nat Commun 16, 9162 (2025). https://doi.org/10.1038/s41467-025-64220-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64220-5
This article is cited by
-
TBC1D1 functions as a negative regulator of satellite cells for muscle regeneration
Nature Communications (2025)
