
Abstract
A hallmark of obesity is a pathological expansion of white adipose tissue (WAT), accompanied by marked tissue dysfunction and fibrosis. Autophagy promotes adipocyte differentiation and lipid homeostasis, but its role in obese adipocytes and adipose tissue dysfunction remains incompletely understood. Using a mouse model, we demonstrate that autophagy is a key tissue-specific regulator of WAT remodelling in diet-induced obesity. Importantly, loss of adipocyte autophagy substantially exacerbates pericellular fibrosis in visceral WAT. Change in WAT architecture correlates with increased infiltration of macrophages with tissue-reparative, fibrotic features. We uncover that autophagy restrains purine nucleoside metabolism in obese adipocytes. This ultimately leads to a reduced release of the purine catabolites xanthine and hypoxanthine. Purines signal cell-extrinsically for fibrosis by driving macrophage polarisation towards a tissue reparative phenotype. Our findings in mice reveal a role for adipocyte autophagy in regulating tissue purine nucleoside metabolism, thereby limiting obesity-associated fibrosis and maintaining the functional integrity of visceral WAT. Purine signals may serve as a critical balance checkpoint and therapeutic target in fibrotic diseases.
Similar content being viewed by others


Introduction
Excess weight and obesity represent a major global health and socioeconomic burden1. Obesity pathogenesis is characterized by a marked increase in white adipose tissue (WAT) mass, predominantly in subcutaneous and visceral locations, with the latter being more detrimental in obesity pathophysiology2. Excess adiposity is considered a major risk factor for metabolic complications, including type II diabetes mellitus and fatty liver disease3. While traditionally viewed as a highly specialized tissue for energy storage and mobilization, adipose tissue is now recognized as a dynamic endocrine and paracrine organ4. During excess nutrient availability, WAT mass increases through cell growth (hypertrophy) and number (hyperplasia) of adipocytes, which shift their metabolism to meet the energetic demands of the organism5. The obesity-associated metabolic shift predominantly includes core lipid and glucose metabolism to support energy storage and mobilisation, and these processes are tightly linked to functional mitochondria6. However, adipocyte metabolism and obesity-related metabolic rewiring beyond these pathways remain poorly understood. Adipose tissue architectural changes are supported by a dynamic remodelling of the extracellular matrix (ECM). Rapid and chronic expansion leads to hypoxia, chronic low-grade inflammation, and fibrosis, rendering WAT inflexible and dysfunctional7. Besides obesity-induced changes in adipocytes, adipose tissue dysfunction is also characterized by an accumulation of adipose tissue macrophages (ATMs)8. ATMs create an inflammatory milieu by releasing inflammatory cytokines and support adipose tissue fibrogenesis through ECM remodelling and fibroblast stimulation9. While the exact sequence of these processes is still unclear10,11, their detrimental impact on adipose tissue is indisputable.
Autophagy is a fundamental process for the regulation of cellular metabolism and energy homeostasis12. Through a highly dynamic regulation of cellular recycling and degradation, autophagy controls metabolic adaptation, differentiation, homeostasis, and ultimately the overall function of cells and organs13. Autophagy is activated by various cellular and environmental stress signals, including nutrient and energy deprivation, and oxidative stress14. When initiated, it recycles organelles and macromolecules either as a quality control mechanism or to replenish energy and anabolic precursor pools. Through these processes, it can both rewire metabolic processes as well as supply nutrients, deeming it a master regulator of cellular metabolism15,16. Notably, autophagy can supply nutrients both in a cell-intrinsic as well as in a cell-extrinsic manner17.
Autophagy supports adipocyte differentiation and lipid homeostasis, as well as facilitating communication between adipose tissue and the liver18,19,20,21; however, its function in obese adipocytes and adipose tissue dysfunction remains unclear and controversial22,23. Here, we demonstrate that in obese conditions, adipocytes upregulate autophagy to support their metabolic and structural adaptation. Failure to meet their metabolic demands in the absence of autophagy leads to elevated purine nucleoside production and release. Xanthine and hypoxanthine-mediated adipocyte-macrophage crosstalk drives a tissue-reparative macrophage phenotype and ultimately leads to excessive pericellular adipose tissue fibrosis.
Results
Autophagy is dysregulated in obesity and shapes WAT remodelling by limiting pericellular fibrosis
Adipocytes undergo significant structural, metabolic, and functional remodelling during obesity. To gain deeper insight into how obesity alters human WAT adipocytes, we re-analysed a recently published human WAT single-nuclei RNAseq (sn-RNAseq) atlas24. Comparison of white adipocytes between lean and obese states revealed macroautophagy as one of the most notable, significantly dysregulated pathways, together with multiple pathways known to be impacted by weight gain, including insulin signalling, lipid metabolism, and tissue repair (Fig. 1A, B). Accordingly, we observed that in mice fed with high-fat diet (HFD) to induce obesity, autophagy initially increased alongside adiposity (Fig. 1C and Supplementary Fig. 1A). This observation correlated with previous reports of autophagy upregulation during obesity22,25,26,27,28,29,30. Prolonged HFD feeding, however, led to a significant downregulation of autophagic flux (Fig. 1C). To investigate its pathophysiological role, we generated a mouse model with an inducible, adipocyte-specific deletion of Atg7 (Atg7Ad) to circumvent defective adipogenesis. Specificity of Cre expression in adipose tissues and Atg7 expression in metabolic tissues were assessed prior to this study31. Atg7 encodes for an E1-like enzyme required for autophagosome formation, and its deletion leads to a profound loss of autophagic activity13. Through its ligase activity, Atg7 is essential for the conjugation of LC3 to autophagic membranes, which is a critical step in the autophagy pathway32. Following the induction of Atg7 deletion in mature adipocytes (Supplementary Fig. 1B), the obesity-associated increase in autophagy flux was abrogated in gonadal WAT (gWAT) adipose depot after the tamoxifen treatment, as assessed by western blot analysis at 16 weeks after HFD treatment (Supplementary Fig. 1C, D). Loss of autophagy in adipocytes had a profound impact on the tissue structure of obese Atg7Ad gWAT (Fig. 1D). Obese Atg7Ad mice showed exacerbated pericellular fibrosis compared to littermate controls (Fig. 1E, F). The onset of fibrosis correlated with an increase in obesity-induced gWAT autophagy in WT mice (Fig. 1C). Fibrosis in Atg7Ad gWAT was obesity-dependent, as control (NCD)-fed Atg7Ad mice did not develop fibrotic adipose tissue (Supplementary Fig. 1E, F). Pericellular fibrosis onset in gWAT developed during the initial body weight gain between six and nine weeks of HFD feeding (Fig. 1E, F). In line with the aggravated ECM accumulation, qPCR analysis revealed that several ECM components and enzymes, including Col3a1, Fn1, Mmp14 and Timp1, were strongly increased (Fig. 1G). In WT mice, we noted that fibrosis significantly increased between 16 and 30 weeks of HFD feeding and then persisted over chronic HFD exposure up to 60 weeks (Supplementary Fig. 1G), suggesting that a decline in autophagy at 60 weeks may contribute to the maintenance of fibrosis. Taken together, these data point towards a critical protective role of obesity-induced autophagy in the control of ECM remodelling and tissue fibrosis.

A UMAP projection of human white adipocytes from lean (BMI < 30; 12822 adipocytes) and obese (BMI > 40; 9191 adipocytes) subjects. Single-nucleus RNA-seq data has been obtained from a deposited dataset (GSE176171). B Enrichment GO analysis of differentially regulated pathways in human adipocytes isolated from obese compared to lean WAT. Data analysis was conducted using the Seurat package v5.0.1. The number of genes identified for each term is labelled. C WT mice were fed a normal chow diet (NCD) or high-fat diet (HFD) for 10, 30 or 60 weeks before autophagy flux in gonadal white adipose tissue (gWAT) was assessed as explained in Materials and Methods. Western blot analysis of autophagy flux was calculated as (LC3-II (Inh) – LC3-II (Veh)). n = 3 (NCD-16), 5 (HFD-60), 6 (HFD-16), 7 (HFD10, HFD30), 8 (NCD-10, HFD-10) and 9 (NCD-30, NCD-60) mice. arb. = arbitrary D Photograph of gWAT fat pads of WT and Atg7Ad mice fed with high-fat diet (HFD) for 16 weeks. E Picrosirius red staining (PSR), specifically staining collagen I and III, of gWAT depots harvested from HFD-fed WT and Atg7Ad mice after 6, 9 and 16 weeks of feeding. Representative images are shown. Scale bar, 200 µm. F Quantification of picrosirius red positive area as a percentage of total area from (E). n = 8 (WT-6), 10 (WT-9, Atg7Ad-6, Atg7Ad-9), 12 (WT-16) and13 (Atg7Ad-16) mice. G Relative mRNA levels of ECM-related genes in gWAT after 16 weeks of HFD measured by qRT-PCR. n = 4 mice. Data are presented as mean ± SEM (C) or mean ± SD (F, G). Dots represent individual biological replicates. Data are representative (D, G) or merged from 3 independent experiments (C, F). Statistical analysis by two-way ANOVA with Tukey multi comparisons (C) or Fisher (F) test or multiple unpaired t-test (G).
Multi-OMICS analysis reveals a key role for autophagy in adipocyte metabolic adaptation and nucleoside homeostasis during obesity
We next asked whether a striking shift in fibrotic processes in obese Atg7Ad gWAT was due to an autophagy-mediated cell-intrinsic process. To explore the role of autophagy in adipocyte cellular remodelling, we conducted a multi-OMICS analysis of obese WT and Atg7Ad adipocytes (Fig. 2). Proteomics analysis showed that classical autophagy receptors such as SQSTM1, which are typically degraded during autophagy, accumulated in autophagy-deficient adipocytes, confirming a lack of autophagy function (Supplementary Fig. 2A). Among the significantly enriched proteins (Fig. 2A), loss of adipocyte autophagy prominently altered metabolic processes, particularly nucleoside and lipid metabolism, as well as responses to oxidative stress (Fig. 2B, C and Supplementary Fig. 2A). Furthermore, the analysis highlighted impaired mitochondrial homeostasis in Atg7Ad adipocytes, evidenced by reduced expression of the electron transport chain subunit complexes I-V following autophagy ablation (Supplementary Fig. 2B). Impaired metabolism in Atg7Ad adipocytes was accompanied by a decrease in adipokine production and secretion, including leptin, adiponectin, and DPP4 (Supplementary Fig. 2C, D). To exclude that these findings could be solely explained by increased cell death of the autophagy-deficient adipocytes, we assessed adipocyte viability by Perilipin-1 staining, a widely used method in the field33. Clearly and as expected, adipocyte viability was markedly reduced due to HFD feeding, while autophagy depletion only increased the cell death minimally (Supplementary Fig. 2E, F). Perilipin-1 staining is based on morphological distinction of unilocular adipocytes and thus gives limited information about the type of cell death induced by loss of autophagy. To investigate whether it occurs via necrosis or programmed cell death (apoptosis), we also treated adipose tissue with the pan-caspase inhibitor Q-VD-OPh34, which reduced adipocyte caspase 3 cleavage (Supplementary Fig. 2G, H), suggesting that at least some cell death occurred via caspase 3-induced apoptosis.

A–C Hierarchical clustering of proteomics profiles of enriched proteins in adipocytes isolated from gWAT of WT and Atg7Ad mice fed with HFD for 16 weeks (1886 proteins identified with adjusted p-value ≤ 0.01). Colour-coding represents the log2 fold difference between WT and Atg7Ad mice (A). Enrichment GO analysis of differentially regulated pathways based on upregulated (B) or downregulated (C) proteins in adipocytes isolated from Atg7Ad compared to WT gWAT. The number of genes identified for each term is labelled. n = 4 mice. D Z-score heatmap of significantly (p < 0.05) abundant nucleotide and nucleoside metabolites in adipocytes. Metabolomics analysis was performed on adipocytes isolated from gWAT of WT and Atg7Ad mice following HFD feeding for 16 weeks. n = 3 mice. E Log2 fold change heatmap of significantly differentially abundant proteins between WT and Atg7Ad involved in pentose phosphate pathway (PPP) and purine nucleoside metabolism in adipocytes as measured by proteomics analysis (as in A). F Schematic summary of adipocyte proteome and metabolome changes upon loss of autophagy depicting simplified pentose phosphate and purine nucleoside metabolic pathways. Representative enzymes and metabolic products are colour-coded based on the fold change. G Pie chart representation of metabolic pathways identified in the enrichment GO analysis of differentially regulated pathways in human adipocyte snRNA-seq isolated from obese compared to lean WAT. H Differentially regulated GO biological pathways related to purine nucleoside metabolism in human adipocytes isolated from obese and lean subjects. The number of genes identified for each term is labelled. I, J Concentration of intracellular ATP (I) and xanthine and hypoxanthine (J) in gWAT adipocytes from WT and Atg7Ad mice fed with HFD for 16 weeks. n = 5 (I) and 11 (J) mice. Data are presented as mean ± SD. Dots represent individual biological replicates. Data are representative (I) or merged from 3 independent experiments (J). All heatmap values were scaled by row (protein/metabolite) using z-score. Statistical data analysis performed using the limma (v3.54.1) and clusterProfiler (v4.6.0) packages (A–C, E), one-way ANOVA (D), Seurat package v5.0.1 (H), unpaired t-test (I) or Mann–Whitney test (J).
In line with our proteomics data, we found that adipocyte autophagy loss in obese mice profoundly impacted their cellular metabolome (Fig. 2D and Supplementary Fig. 3A). We observed that loss of autophagy led to a global reduction in both essential and non-essential amino acids, alongside enzymes supporting the TCA cycle and fatty acid oxidation in obese adipocytes assessed by metabolomics and proteomics, respectively (Supplementary Fig. 3B–D). Furthermore, we found key components for RNA synthesis, including nucleotides UMP and AMP, adenosine, as well as ribose, significantly reduced (Fig. 2D).
Surprisingly, the loss of autophagy resulted in a pronounced upregulation of metabolites primarily associated with nucleoside metabolism (Fig. 2D). We found a strong accumulation of purine and pyrimidine nucleosides in autophagy-deficient adipocytes, including guanosine, cytidine, uridine, as well as xanthine, a downstream product of purine catabolism (Fig. 2D). The dysregulation in nucleoside metabolism was further emphasised by altered protein levels of several critical enzymes involved in the pentose phosphate pathway (PPP) and intracellular purine metabolism (Fig. 2E). A prominent increase in PPP and purine metabolism enzymes was observed alongside elevated glycolytic enzymes that support this metabolic axis (Supplementary Fig. 3D). Notably, these profound changes in adipocyte metabolism revealed that autophagy plays a critical role in the maintenance of functional nucleotide pools in adipocytes during obesity (Fig. 2F). Remarkably, when examining snRNA-seq data of human adipocytes, we found a similar pattern; out of 19 dysregulated metabolic pathways with obesity, more than one-fifth were related to purine metabolism (Fig. 2G, H). This parallel between mouse and human data underscores the central role of purine nucleoside metabolism in adipocytes and its regulation by autophagy during obesity.
To validate the role of autophagy in regulating purine metabolism, we measured both upstream and downstream intermediate metabolites with enzymatic assays, including ATP, hypoxanthine, xanthine, and uric acid. Obese Atg7Ad adipocytes showed a significant reduction in the energy-rich purine nucleotide ATP (Fig. 2I). In contrast, downstream intermediates of purine catabolism, xanthine and hypoxanthine, were significantly increased (Fig. 2J). However, there was no difference in the end product of purine catabolism, uric acid, or in the enzymatic activity of xanthine oxidase (Supplementary Fig. 3E, F), suggesting that purine catabolism was not upregulated to generate excess uric acid. Taken together, autophagy is indispensable to maintaining balanced purine nucleoside metabolism in obese adipocytes. Its impairment shifts the metabolism towards the increased catabolic activity of the nucleotide metabolic pathway, leading to the accumulation of downstream products, including nucleosides and purine bases.
Autophagy limits obesity-induced xanthine and hypoxanthine release from adipocytes
Based on the intracellular changes in nucleoside metabolism, we next investigated whether this has paracrine or endocrine effects on the tissue environment and systemically. Xanthine and hypoxanthine levels progressively accumulate in mouse serum over the time course of HFD feeding (Fig. 3A). Strikingly, serum xanthine and hypoxanthine were also found elevated in the absence of adipocyte autophagy (Fig. 3B), reflecting the intracellular metabolic rewiring in Atg7Ad adipocytes. Since we did not observe a shift of purine catabolism towards increased synthesis of its end product, uric acid, in Atg7Ad adipocytes, we postulated that xanthine might be, rather than converted to uric acid intracellularly, released into the extracellular milieu by adipocytes. To understand whether these nucleobases were adipocyte-derived and controlled by autophagy, we performed targeted metabolomics on the adipocyte secretome. Similar to their intracellular levels, we found a notable accumulation of cytidine, uridine, guanosine, and xanthine in the secretome derived from Atg7Ad compared to WT adipocytes (Fig. 3C). Xanthine and hypoxanthine levels more than doubled in the secretome of obese Atg7Ad adipocytes (Fig. 3D), and we observed a strong negative correlation between the activity of the autophagy pathway and their release from obese gWAT in WT and Atg7Ad mice (Fig. 3E). Purine nucleoside phosphorylase (PNP) plays a key role in purine catabolism, limiting the production of purine nucleobases35. Inhibition of PNP activity by the clinically approved drug forodesine resulted in a significantly lower xanthine and hypoxanthine release from both WT and Atg7Ad adipocytes, with the latter being reduced to almost WT levels upon PNP inhibition (Fig. 3F). To exclude that increased apoptosis of Atg7Ad adipocytes is the reason for elevated nucleoside production and/or release, we inhibited gWAT apoptosis by pan-caspase inhibitor Q-VD-OPh or induced gWAT apoptosis by staurosporine (STS) (Supplementary Fig. 2G, H and Supplementary Fig. 3G, H). Neither treatment had an impact on xanthine and hypoxanthine efflux from WT or Atg7Ad adipocytes (Fig. 3G, H). However, when gWAT explants were lysed with 0.1% Triton-X, this resulted in markedly elevated extracellular levels in Atg7Ad explants (Fig. 3I). These results suggest that the mild increase in apoptosis due to loss of autophagy does not contribute to increased extracellular nucleoside levels. Furthermore, autophagy-depleted adipocytes actively generate xanthine and hypoxanthine through PNP activity that can be released when cells are fully lysed. Taken together, these assays suggest that while autophagy is not essential for efflux, it limits the excessive release of xanthine and hypoxanthine from visceral adipocytes during obesity.

A, B Concentration of serum xanthine and hypoxanthine in WT mice were fed NCD or HFD for 10, 30 or 60 weeks (A) or WT and Atg7Ad mice fed with HFD for 16 weeks (B). n = 5 (HFD60, B-WT), 6 (B-Atg7Ad), 8 (HFD10, HFD30, NCD10) and 9 (NCD30, NCD60) mice. C Relative abundance of nucleosides released by gWAT adipocytes isolated from WT and Atg7Ad mice following HFD feeding for 16 weeks and measured in metabolomics analysis. n = 3 mice. D Concentration of xanthine and hypoxanthine released from gWAT adipocytes cultured over 24 h ex vivo. Adipocytes were isolated as in (C). n = 5 (WT) and 6 (Atg7Ad) mice. E Correlation analysis of the level of autophagy flux in gWAT and concentration of xanthine and hypoxanthine released from gWAT adipocytes as in (D). Western blot analysis of autophagy flux was calculated as (LC3-II (Inh) – LC3-II (Veh)). n = 9 mice. F Concentration of xanthine and hypoxanthine as in (D) treated with either PBS or 10 µM forodesine, a purine nucleoside phosphorylase (PNP) inhibitor. n = 6 mice. G Concentration of xanthine and hypoxanthine released from gWAT explants cultured overnight ex vivo and treated with either DMSO or 20 µM Q-VD-OPh, a pan-caspase inhibitor. n = 5 (Atg7Ad) and 6 (WT) mice. H Concentration of xanthine and hypoxanthine after treatment of gWAT explants with DMSO or 10 µM staurosporine (STS), an apoptosis inducer, for 24 h ex vivo. n = 11 mice. I Concentration of released xanthine and hypoxanthine from gWAT explants after cell lysis with 0.1 % Triton-X for 1 h before the end of incubation. n = 4 mice. Data are presented as mean ± SD. Dots represent individual biological replicates. Data are representative (D, I) or merged from 2 to 3 independent experiments (A, B, E–H). Statistical analysis by two-way ANOVA with Tukey multi comparisons (A), Mann–Whitney test (B), unpaired t-test (D), Pearson R correlation analysis (E), Fisher test (F) or Šídák multi comparisons test (I).
Adipocyte autophagy controls WAT remodelling by limiting immune cell expansion
Given the dysregulated intra- and extracellular purine nucleoside signalling in Atg7Ad mice and the pronounced fibrosis, we set out to investigate whether these factors impact broader tissue remodelling and have a body-wide impact. We found that WAT body distribution exhibited remarkable differences with a reduced gWAT but expanded inguinal WAT (iWAT) deposition in obese Atg7Ad mice (Supplementary Fig. 4A). This was independent of weight gain or energy consumption between WT and Atg7Ad mice on either control (NCD) or HFD, where no differences were observed (Supplementary Fig. 4B, C). Deposition of fat into the visceral/gonadal area has been recognized to be more detrimental for obesity pathology, and it has been suggested that iWAT expansion could potentially buffer the deleterious effects of visceral fat increase1,9. In line with this hypothesis, changes in fat deposition in obese Atg7Ad mice were associated with improved obesity-induced metabolic syndrome, as demonstrated by increased glucose tolerance and lessened ectopic fat deposition in the liver (Supplementary Fig. 4D–G). Yet, no reduction in serum triglycerides and HDL cholesterol was observed (Supplementary Fig. 4H, I). In addition, obese Atg7Ad mice displayed no significant changes in circulating levels of adiponectin or leptin, as measured by ELISA, compared to controls (Supplementary Fig. 4J, K). Furthermore, autophagy-deficient adipocytes displayed no notable differences in cell size in gWAT (Supplementary Fig. 4L). These data suggest that autophagy-mediated adipocyte metabolic and tissue structural remodelling impact fat distribution in the pathological visceral WAT, alleviating obesity-induced metabolic syndrome.
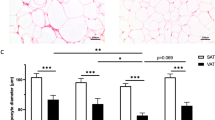
Excessive ECM deposition in most tissues is commonly associated with increased secretion of pro-fibrotic cytokines that act to modulate the activity of ECM remodelling cells, such as transforming growth factor β (TGFβ) and osteopontin (OPN)7,36,37. To assess the production and release of these cytokines, we cultured gWAT ex vivo for six hours and measured their secretion by ELISA. Both TGFβ and OPN secretion were increased in Atg7Ad obese mice (Fig. 4A, B). The increase in these two cytokines was associated with a significant elevation in nuclear density in Atg7Ad gWAT (Fig. 4C, D). This nearly threefold difference in nuclear density could not be attributed to adipocyte hyperplasia, as the number of unilocular adipocytes in the tissue remained constant (Fig. 4E). The fibroblast population (PDGFRα+), the main cell type involved in ECM dynamics, decreased in the Atg7Ad gWAT (Fig. 4F, G). This observation was further supported by the expression of Acta2, a gene largely restricted to myofibroblasts, which was not increased in gWAT of obese Atg7Ad mice (Fig. 1G). Flow cytometry analysis also demonstrated that the endothelial cell population (CD31+) remained steady (Fig. 4H). Notably, the immune cell (CD45+) population, which can also be implicated in ECM dynamics9, expanded significantly within the stromal vascular fraction of the Atg7Ad gWAT (Fig. 4I).

WT and Atg7Ad mice were fed HFD for 16 weeks before gWAT was isolated for analysis. A, B Secretion of TGFβ (A) and osteopontin (OPN) (B) measured by ELISA. n = 7 (A-Atg7Ad), 8 (A-WT), 13 (B-Atg7Ad) and 15 (B-WT) mice. C H&E staining of gWAT. Scale bar, 200 µm. D, E Quantification of nuclei and adipocyte number from (C). n = 5 (E-WT), 6 (D-WT) and 8 (Atg7Ad - D, E) mice. F Flow cytometry analysis of CD45+, CD31+, and PDGFRα+ populations in gWAT. n = 5 mice. G–I Absolute numbers of PDGFRα+ (G), CD31+ (H), and CD45+ (I) populations normalized to gram of WAT as in (F). n = 5 (H-WT), 6 (H-Atg7Ad, I-Atg7Ad), 7 (I-WT), 15 (G-Atg7Ad) and 16 (G-WT) mice. J Flow cytometry analysis of immune cell (CD45+) composition. NKT natural killer T cell. n = 3 (WT) and 7 (Atg7Ad) mice. K Flow cytometry analysis of F4/80+ CD64+ macrophage number in gWAT. n = 9 (WT) and 11 (Atg7Ad) mice. L Representative immunofluorescence staining of F4/80, CD45 and CD68 of gWAT sections from WT and Atg7Ad mice following HFD feeding for 16 weeks. M Flow cytometry analysis of F4/80+ CD64+ macrophage frequency labelled with CD45.1 (donor) and CD45.2 (host) congenic markers in gWAT after adoptive transfer. CD45.1 bone marrow cells were transferred in CD45.2 WT and Atg7Ad hosts, where conditional knockout was induced 25 days following the transfer and mice were fed HFD for an additional 12 weeks. n = 6 mice. Data are presented as mean ± SD. Dots represent individual biological replicates. Data are representative (C, F, J, L) or merged from 2 to 3 independent experiments (A, B, D, E, G–I, K, M). Statistical analysis by unpaired t-test (A, B, D, E, G–I, K) or two-way ANOVA with Šídák multi comparisons test (F, J, M).
Assessing the composition of the CD45+ compartment in gWAT by flow cytometry revealed that macrophages were the most prevalent population upon obesity, which further increased in abundance in Atg7Ad mice (Fig. 4J, K and Supplementary Fig. 5). The increase in macrophage numbers was confirmed by in-situ imaging (Fig. 4L), with macrophage numbers more than doubling (Fig. 4K). To test whether these cells were derived from tissue-resident macrophages or infiltrating monocytes, we transplanted congenic bone marrow into WT or Atg7Ad hosts. Reconstitution of Atg7Ad mice with congenic CD45.1 bone marrow revealed that the majority of tissue-infiltrating macrophages were monocyte-derived, and of those, there were twice as many in the obese adipose tissue of the Atg7Ad host compared to the WT host (Fig. 4M). Collectively, these observations highlight the critical role of adipocyte autophagy in modulating the inflammatory environment during obesity by controlling macrophage infiltration.
ATMs switch to a tissue-reparative phenotype in Atg7 Ad gWAT
To better understand the identity and function of accumulated macrophages in Atg7Ad gWAT, we isolated F4/80+ CD64+ macrophages from gWAT of WT and Atg7Ad mice by fluorescence-activated cell sorting (FACS) and performed transcriptomics. Surprisingly, gene enrichment analysis of significantly dysregulated genes (Fig. 5A) revealed that loss of adipocyte autophagy induces downregulation of pathways associated with inflammation and cytokine production (Fig. 5B) while upregulating proliferative and tissue-remodelling processes as well as purine/nucleotide metabolism in ATMs (Fig. 5C). To validate the results, we first cultured ATMs from gWAT of WT and Atg7Ad mice ex vivo and measured their secreted cytokines. We confirmed that macrophages from obese Atg7Ad mice notably decreased their cytokine production of IL-1β, IL-6, TNFα, and IL-10 (Fig. 5D–G). In contrast, ATMs from obese Atg7Ad mice increased transcription of key pro-fibrotic tissue remodelling genes Col3a1 and Mmp14 (Fig. 5H).

A–C Transcriptomics analysis of F4/80+ CD64+ macrophages isolated from gWAT of WT and Atg7Ad mice fed with HFD for 16 weeks. Hierarchical clustering of transcriptional profiles of the top 1000 differentially expressed genes (A). Colour coding represents the log2 fold difference between WT and Atg7Ad mice. Enrichment GO analysis of differentially regulated pathways based on downregulated (B) or upregulated (C) genes in macrophages isolated from Atg7Ad compared to WT gWAT. The number of genes identified for each term is labelled. n = 6 mice. D–G Secretion of IL-1β (D), IL-6 (E), TNFα (F), and IL-10 (G) by macrophages enriched from gWAT of WT and Atg7Ad mice fed HFD for 16 weeks. n = 3 (E), 5 (D, F, G-WT) and 6 (D, F, G-Atg7Ad) mice. H Relative mRNA levels of extracellular matrix (ECM)-related genes in sorted F4/80+ CD64+ macrophages isolated from gWAT of WT and Atg7Ad mice fed HFD for 16 weeks measured by qRT-PCR. Data presented as log2 fold difference. n = 6 (Atg7Ad) and 7 (WT) mice. Representative of 3 independent experiments. I–M Representative plots of lipid-associated macrophages (LAM) (I), identified as CD63+ CD9+, perivascular (PVM) and non-perivascular macrophages (NPVM) (J), identified as MHCIIlow Lyve1high and MHCIIhigh Lyve1low, respectively, assessed by flow cytometry. Quantification of LAM (K), PVM (L) and NPVM (M) frequency in the gates shown. n = 5 (L-Atg7Ad), 7 (K-WT, L-WT, M-Atg7Ad), 8 (M-WT) and 9 (K-Atg7Ad) mice. Data are presented as mean ± SD. Dots represent individual biological replicates. Data are representative (D–G) or merged from 2 to 3 independent experiments (H, K–M). Statistical analysis by the limma (v3.54.1) and clusterProfiler (v4.6.0) packages (A–C), unpaired t-test (D–G, K–M) or two-way ANOVA with Šídák multi comparisons test (H).
The growing recognition of ATM plasticity and heterogeneity has revealed a complexity that renders the traditional M1/M2 (pro- and anti-inflammatory) paradigm overly simplistic and outdated38,39. The recent classification obtained under both normal chow and HFD using single-cell RNA sequencing (scRNA-Seq) suggests three main macrophage subtypes, including perivascular-like macrophages (PVM), non-perivascular-like macrophages (NPVM), and lipid-associated macrophages (LAM)40,41,42,43,44. While NPVMs and LAMs mediate inflammatory processes, PVMs control tissue repair8. Analysis of these macrophage populations by flow cytometry (Fig. 5I, J) revealed no difference in LAM (marked as F4/80+ CD64+ CD9+ CD63+) abundance between obese Atg7Ad and WT gWAT (Fig. 5I, K). In contrast, we found tissue-reparative PVM (marked as F4/80+ CD64+ Lyve1high MHCIIlow) more than sevenfold increased and antigen-presenting NPVM (marked as F4/80+ CD64+ Lyve1low MHCIIhigh) threefold decreased among macrophages isolated from Atg7Ad gWAT (Fig. 5J, L, M). In contrast to gWAT, iWAT displayed no increase in F4/80+ CD64+ ATM or Lyve1high MHCIIlow PVM accumulation, suggesting that macrophage abundance and phenotypes notably differ between the depots in Atg7Ad mice (Supplementary Fig. 6A, B). A significant, but much lower release of xanthine and hypoxanthine was observed from Atg7Ad iWAT adipocytes compared to gWAT (Fig. 3D and Supplementary Fig. 6C). This further underscored the intrinsic differences between these depots. In summary, we uncovered that in gWAT of obese Atg7Ad mice, macrophages switch from a predominantly pro-inflammatory to a tissue-reparative pro-fibrotic phenotype, which is accompanied by a strong ECM transcriptional signature.
Metabolic dysregulation of Atg7 Ad adipocytes is signalled through xanthine and hypoxanthine to macrophages for a tissue-reparative phenotypic switch
Observing that autophagy significantly impacted adipocyte purine nucleoside metabolism, which might, in turn, influence the surrounding microenvironment, we aimed to determine whether purine nucleosides could induce a tissue-reparative phenotype in macrophages. In pursuit of this goal, we first tested whether the adipocyte secretome could switch macrophages in vivo by cultivating ATMs isolated from lean adipose tissue in the presence of the secretome derived from either obese WT or Atg7Ad adipocytes. Three days after the exposure, we observed a significant increase in the Lyve1high MHCIIlow tissue repair macrophage population as well as the upregulation of ECM-related genes Col3a1, Mmp14, and Timp1 in macrophages exposed to Atg7Ad adipocyte-derived secretome (Fig. 6A–C). These results mimicked our observations in vivo, suggesting that adipocyte-derived soluble signals are responsible for the macrophage phenotype.

A–C Macrophages were isolated from lean WT gWAT and cultivated in vitro in the presence of conditioned medium (CM) generated by 24 h ex vivo incubation of obese gWAT WT and Atg7Ad adipocytes. Representative plots of tissue repair macrophages assessed by flow cytometry (A). Quantification of flow cytometry analysis of MHCIIlow Lyve1high F4/80+ CD64+ macrophage number after 72 h of treatment with CM from WT or Atg7Ad adipocytes (B). Relative mRNA levels of ECM-related genes in macrophages after 72 h of treatment with CM from WT or Atg7Ad adipocytes or baseline full medium (C). RNA levels measured by qRT-PCR. n = 3 (C-medium only), 5 (C-WT), 7 (C-Atg7Ad) and 9 (B) mice. D Macrophages were isolated from lean WT gWAT as in (A–C) and cultivated in vitro for 72 h in baseline full medium supplemented with 50 ng/ml of M-CSF and 100 µM of either adenosine, guanosine, hypoxanthine or xanthine. n = 3 mice. E Relative mRNA levels of ECM-related genes in macrophages as in (A–C) with or without 100 µM supplementation of both xanthine and hypoxanthine (XHX). Data presented as log2 fold difference. n = 6 mice. F, G Transcriptomics analysis of macrophages isolated from lean WT gWAT and cultivated in vitro in the presence of 50 ng/ml M-CSF and treated with 100 µM xanthine or hypoxanthine for 72 h. F Enrichment GO analysis of upregulated pathways in macrophages treated with either xanthine or hypoxanthine compared to the control based on shared DEGs. The number of genes identified for each term is labelled. G Dot plot of scaled expression of key DEGs regulating GTPase-mediated signal transduction, MAPK signalling pathway and tissue remodelling. n = 3 (Ctrl, Hypox) and 4 (Xanth) mice. Data are presented as mean ± SD. Dots represent individual biological replicates. Data are merged from 2 to 3 independent experiments (B, C, E). Statistical analysis by unpaired t-test (B), multiple unpaired t-tests (C, E), two-way ANOVA with Dunnett’s multiple comparisons test (D), or the limma (v3.54.1) and clusterProfiler (v4.6.0) packages (F, G).
We next aimed to determine whether purine nucleosides could be responsible for these observations. To this end, lean ATMs were cultured in vitro for 72 h in 50 ng/ml of M-CSF supplemented with 100 µM of either adenosine, guanosine, hypoxanthine or xanthine (Fig. 6D). Xanthine, and to a lesser extent hypoxanthine, led to a significant upregulation of ECM-related genes, whereas adenosine and guanosine did not. To further test whether hypoxanthine and xanthine could indeed trigger a tissue-reparative switch, lean ATMs were treated in vitro with the secretome of obese WT adipocytes for 72 h, supplemented with a mixture of 100 µM xanthine and hypoxanthine each. We observed a marked increase in the pro-fibrotic signature genes Mmp14, Col3a1 and Timp1 (Fig. 6E).
To shed light on signalling pathways induced by xanthine and hypoxanthine in macrophages, we performed transcriptomic profiling of ATMs treated with xanthine or hypoxanthine in vitro. This revealed significant upregulation of MAPK and ERK signalling pathways, alongside enrichment in GTPase activity and Ras signalling transcripts (Fig. 6F, G). These pathways are commonly triggered by G-protein coupled receptors (GPCRs), including purinergic receptors45. ERK activation is associated with a reparative macrophage phenotype and fibrosis46,47,48. Notably, P2Y10, a purinergic GPCR and one of the most upregulated genes following xanthine/hypoxanthine treatment in our data, has been linked to RhoA activation and downstream ERK signalling49. These findings suggest that extracellular purines may reprogram macrophages via purinergic GPCR-mediated MAPK-ERK signalling.
Collectively, these results suggest that increased release of xanthine and hypoxanthine from adipocytes can promote a tissue-reparative switch in macrophages during obesity. While the release is autophagy-independent, autophagy controls the purine nucleoside metabolism in adipocytes. Dysregulation of this metabolic pathway leads to excessive nucleoside release, which in turn shifts the balance from tissue inflammation toward fibrosis.
Discussion
In this study, we have identified autophagy as a major brake on WAT fibrosis. Combining a genetic model and dietary intervention with proteomic, metabolomic, and functional analyses, we uncovered a critical role of autophagy in supporting adipocyte metabolic needs during excessive growth, limiting purine nucleoside catabolism. By studying the nucleoside metabolic changes upon loss of autophagy, we identified (hypo)xanthine-driven adipocyte-to-macrophage crosstalk. Finally, our work revealed a critical role of autophagy in limiting WAT ECM pathological remodelling through a (hypo)xanthine-induced macrophage tissue repair phenotype.
Understanding the changes in autophagy activity in adipose tissues during obesity in both humans and mice remained elusive, despite numerous reports22,23,25,26,27,28,29,30,50. In addition, the lack of clarity on the mechanism and function of autophagy in obese WAT highlighted the complex and poorly understood role of autophagy. While it has been reported that adipocyte autophagy supports adipose tissue-liver crosstalk, contradictory conclusions were drawn in the different studies20,21. Our data suggest that obesity dysregulates autophagy both in humans and mice and that autophagy primarily increases with obesity in mice, with an eventual drop after prolonged HFD feeding, perhaps explaining a few studies that showed decreased autophagy levels with obesity23,50. We find that the primary function of autophagy is the support of the high metabolic demands of adipocytes during fat mass expansion. Adipocyte metabolism underlying fat storage and turnover is well understood6, and is majorly determined by an increase in WAT mass. Nevertheless, our understanding of metabolic rewiring beyond glucose and lipid metabolism remains limited, with only scarce evidence for the role of other key metabolic processes in adipocytes51,52,53. We find that in humans, purine nucleoside metabolism represents one of the main dysregulated metabolic pathways in obese adipocytes. Our proteomics and metabolomics analyses revealed that autophagy critically supports nucleotide and amino acid pools in obese adipocytes. Similar autophagy-dependent changes have been previously observed in lung cancer cells under starvation and haematopoietic stem cells54,55,56. Similar to these cell types, mature adipocytes have a highly dynamic metabolic demand and can enter a pseudo-starvation state through adipokine signalling in obesity57. Furthermore, increased production of purine nucleoside catabolic intermediates, such as hypoxanthine and xanthine, has been previously suggested to relate to ATP depletion54,58,59,60, which we also observed. Therefore, we believe these observations spanning several different cell types share a common molecular mechanism and highlight the indispensable role of autophagy in the provision of bioenergetic and biosynthetic substrates, responding to stress, maintaining redox homeostasis and survival.
Failure of adipocyte autophagy induction resulted in gWAT fibrosis, which is the more fibrosis-prone WAT depot9. The role of autophagy in fibrosis is controversial and highly context-dependent61,62. While the relationship between autophagy and adipose tissue fibrosis has not been experimentally addressed to date, their potential link has been proposed recently63. Tissue fibrosis develops when either ECM deposition or turnover become dysfunctional and is difficult to reverse10. Fibrosis of adipose tissue has been traditionally seen as detrimental as it mechanically stiffens the tissue, thereby negatively impacting its critical plasticity feature in the response to nutrient status7. Nevertheless, fibrosis is an essential component of tissue repair that limits tissue damage and aims to restore functional tissue architecture, improve recovery, and enhance survival64. In a chronic setting, when damage is persistent or severe, however, fibrosis leads to disruption of tissue architecture, interferes with organ function, and can ultimately lead to organ failure65. While chronic fibrosis occurs in gWAT of HFD-fed wild-type mice progressively, this fibrotic development was accelerated in adipose tissue deficient for adipocyte autophagy. The early fibrotic onset observed in Atg7Ad gWAT, preceding elevation of autophagy flux in WT mice, suggests there is a demand for autophagy to maintain adipocyte cellular homeostasis early on during dietary challenge. This requirement for adipocyte autophagy is further exacerbated with prolonged HFD feeding, likely to reduce diet-induced cellular stress. We suggest that, initially, fibrosis acts to prevent acute and excessive tissue damage due to impaired adipocyte homeostasis and function upon autophagy depletion. Eventually, however, chronic accumulation of ECM likely leads to a broader adipose tissue dysfunction. Similar observations have recently been made in the pancreas66. Since WAT is not functionally compartmentalized, the detrimental effects of chronic fibrosis at the organismal level are difficult to discern. Indeed, increased deposition in the subcutaneous area positively correlates with more favourable disease outcomes compared to visceral deposition3. It has been proposed that the expansion of subcutaneous WAT could potentially help reduce the detrimental impact of visceral WAT expansion9. Concomitant with this, we observed the physical limitation of the pathological visceral WAT expansion by fibrosis, improving glucose homeostasis, and reducing ectopic fat deposition in the liver. The role of autophagy in adiposity remodelling has also been observed in humans, where genome-wide association studies strongly linked Atg7 to body fat distribution67. In addition, autophagy impairment, as seen in patients with biallelic MFN2 mutation (mitofusin 2, involved in mitochondrial fusion), was similarly found to impact depot-specific fat expansion68,69. Thus, a better understanding of autophagy as a determinant of WAT remodelling and fibrosis holds important therapeutic potential to improve obesity management and health outcomes of obese patients.
Excessive pericellular fibrosis positively correlated with a pronounced accumulation of tissue-reparative macrophages. Increased macrophage accumulation in adipocyte autophagy-deficient gWAT has been observed before, but never studied in detail20,21. In addition, caloric restriction studies in humans, known to induce autophagy, demonstrated reduced macrophage activation and, consequently, protection from adipose tissue fibrosis, but the link to autophagy was not made70. Macrophages are known as key regulators of tissue repair, regeneration, and fibrosis71, and this may be true for adipose tissues as well9,72. The evidence, however, remains scarce, with elastin and TLR4 signalling being proposed to play a role in macrophage-induced WAT fibrosis during obesity72,73. On the other hand, ATMs have also been proposed to prevent pathological changes of ECM and limit the development of gWAT fibrosis74. Nevertheless, it remains unclear which signals induce the macrophage pro- or anti-fibrotic phenotypic switch that could serve as important balance checkpoints and therapy targets in fibrotic diseases. Local metabolic signals contributing to immune cell fates are becoming an area of increasing interest75,76. We show here for the first time that products of nucleoside catabolism, xanthine and hypoxanthine, can act as determinants of adipose tissue macrophage fate, resulting in a tissue-reparative phenotype. Our data suggest that this effect may be mediated via purinergic GPCR signalling, particularly through P2Y10, leading to activation of the MAPK-ERK pathway and downstream pro-fibrotic transcriptional programs. Notably, we found xanthine and hypoxanthine increased with obesity progression in mouse serum, and similar observations have been made before in humans, identifying adipose tissue as one of the main contributing factors77,78,79,80. Adipocytes have been previously described to actively release nucleosides upon stress, including hypoxanthine, xanthine, inosine, guanosine, and uridine53,81,82,83,84. Our data show that the release of xanthine and hypoxanthine occurs independently of autophagy, but instead, autophagy-dependent metabolic rewiring results in their increased intracellular accumulation. We uncover for the first time that autophagy acts as a brake on the active accumulation of nucleosides and nucleobases, which are subsequently released, either actively or passively. Release of xanthine by T cells has been identified to relay cell-extrinsic effects under stress conditions85. These results, together with our observations, suggest a common molecular signalling mechanism of cellular stress to the microenvironment via purine nucleobase signals. Our data further indicate that autophagy serves as a key modulator of this extracellular purine signal, which helps the cell to adapt to altered metabolic challenges such as excessive storage of fat. It is plausible that by activating nucleotide degradation, autophagy-deficient adipocytes salvage NADPH or ribose through the PPP. This enables them to partially sustain their metabolism and oxidative stress, sourcing carbon for energy, antioxidant molecules, and anabolic precursor generation. In turn, nucleoside catabolites signal the altered adipocyte state to the macrophages, which by remodelling the ECM, shut down the tissue and limit systemic dysregulation. While autophagy-dependent metabolic signals have been previously reported to play a role in cancer and inflammatory bowel disease31,86,87, pro-fibrotic purine nucleoside catabolites have not been described before.
Yet, this study contains a few limitations warranting future investigations. First, we could not fully elucidate the exact molecular mechanism by which purine catabolites regulate macrophage function, as we were using in vivo material only and this physiologically relevant WAT microenvironment cannot be easily recapitulated in vitro. Second, the xanthine and hypoxanthine downstream signalling pathways are poorly understood and lack an experimental toolset at the moment; however, our findings provide a strong basis for future mechanistic work when this toolset becomes available. Finally, our findings were not experimentally validated in human samples due to a lack of access to clinical materials, but we mitigated this by in silico analysis of published human datasets, strengthening the translational relevance of our findings.
In conclusion, our work highlights the key role of autophagy acting as a brake in the control of adipocyte nucleoside metabolism and tissue integrity in diet-induced obesity. When dysfunctional, this leads to uncontrolled activation of metabolic rewiring that generates purine catabolites xanthine and hypoxanthine, signalling tissue repair. By depleting autophagy, we uncover a purine nucleobase-mediated pro-fibrotic signalling pathway, and further research is necessary to elucidate whether these signalling molecules control fibrosis of other tissues and organs, potentially deeming them druggable targets.
Methods
Lead contact
Information and requests for reagents and resources should be directed to the Lead Contact, Anna Katharina Simon (katja.simon@kennedy.ox.ac.uk).
Mouse models
Adipoq-CreERT2 mice88 were purchased from Charles River, UK (JAX stock number: 025124) and crossed to Atg7fl/fl mice89. Genetic deletion was induced at 6–8 weeks of age by oral gavage of 4 mg tamoxifen per mouse for five consecutive days. Tamoxifen was given to all groups of mice. Two days after receiving the last tamoxifen dose, mice were subjected to an altered diet regime with either a HFD with 60 kcal% fat (D12492i, Research Diets) or a complementary normal chow diet with 10 kcal% fat (D12450Ji, Research Diets) for the duration stated in the text. Wild-type C57BL/6J or B6.SJL.CD45.1 mice were bred in-house. Experimental cages were sex- and age-matched and balanced for genotypes. All data shown, except proteomics and metabolomics data are pooled from both sexes. Mice were maintained on a 12 h dark/light cycle and housed in groups of 3–5 with unlimited access to water and food under specific pathogen-free conditions. The temperature was kept between 20 and 24 °C, with a humidity level of 45–65%. All experiments were performed in accordance with approved procedures by the Local Review Committee and the Home Office under the project license (PPL30/3388 and P01275425).
Bone marrow chimera generation
Recipient WT and Atg7Ad mice were lethally irradiated with 11 Gray dose before intravenously injecting between 250 and 300,000 B6.SJL.CD45.1 donor cells (equal numbers in the same experiment to allow comparison between the two groups). Cell replenishment was followed bi-weekly. Five weeks after irradiation, mice were treated with tamoxifen for genetic deletion and fed HFD for a total of 12 weeks (as described above).
Tissue processing, macrophage isolation and primary cell culture
Adipose tissue digestion was performed as previously described31. In brief, depots were digested in DMEM containing 1% fatty acid-free BSA, 5% HEPES, 0.2 mg/ml Liberase TL (Roche), and 20 μg/mL DNaseI. Tissues were minced and incubated for 25–30 min at 37 °C at 180 rpm. Digested tissue was strained through a 300 μm mesh and the digestion was quenched by the addition of PBS with 0.5% BSA and 2 mM EDTA. Adipocyte and stromal vascular fraction were separated by 7 min centrifugation at 500 × g and collected for further analysis.
To generate a conditioned medium, adipocytes were collected with wide-bore tips and washed three times with PBS. The floating fraction was collected and 250 µl of packed adipocytes were seeded in 500 µl of RPMI containing 10% foetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S) and incubated for 24 h at 37 °C. Alternatively, the medium was supplemented with 10 µM forodesine. After incubation, the medium and cells were harvested, centrifuged at 300 × g for 5 min and purified medium was collected and snap-frozen for in vitro experiments.
For primary cell culture, the stromal vascular fraction was enriched for CD11b+ cells with CD11b MicroBeads (Miltenyi Biotec) according to manufacturers’ instructions after red blood cell lysis. 350,000 cells were seeded in 24-well plates in RPMI containing 10% FBS and 1% P/S and incubated overnight to allow macrophages to attach. The following day, macrophages were enriched by washing the wells with room temperature PBS and treated with experimental conditions. For conditioned medium treatment, RPMI containing 10% FBS and 1% P/S was mixed with the conditioned medium in a 2:1 ratio and applied to the cells for 72 h. For nucleoside treatment, macrophages were cultured in RPMI containing 10% FBS and 1% P/S and 50 ng/ml M-CSF and treated with 100 µM of either adenosine, guanosine, hypoxanthine or xanthine over 72 h.
Glucose tolerance test
Mice were subjected to 12 h fast before measuring fasted glucose levels. To monitor response to bolus glucose, 1.5 × g of glucose per kg of body mass was injected intraperitoneally. Blood glucose levels were measured via tail clip at 15, 30, 60, 90, 120, and 180 min after injection with a glucose meter (Freestyle Lite, Abbott).
Histology and immunohistochemistry
Adipose tissues and liver were fixed in 10% neutral buffered formalin for 24 and 48 h, respectively. All tissues were transferred to 70% ethanol and sent to the Kennedy Institute of Rheumatology Histology service for paraffin embedding, sectioning (5 µm), and staining. Haematoxylin and eosin (H&E) and picrosirius red staining were performed according to standard protocols. Images were acquired with a Zeiss Axioscan 7 scanning microscope. Image analysis and quantification were performed using QuPath, ImageJ, and an in-house developed script (available at https://github.com/Oxford-Zeiss-Centre-of-Excellence/pyHisto). In brief, the blind colour deconvolution method was used based on a stain vector estimation90, followed by Otsu thresholding and determination of collage-to-area ratio.
For immunofluorescence staining, WAT was fixed in 4% paraformaldehyde for 24 h, embedded and sectioned as above. Tissues were subsequently deparaffinized and heat-retrieved at 100 °C for 20 min in Citrate antigen retrieval solution (Vector Laboratories, pH 6.0), and allowed to cool naturally in Tris antigen retrieval solution (10 mM, pH 8.8–9.0). Slides were washed in PBS and blocked overnight in 10% donkey serum and 3% BSA. The next day, slides were incubated with DAPI for 15 min to record a background scan. After background imaging, slides were incubated for 20 min with Fc block reagent (1:200 in 3% BSA), followed by primary antibody incubation overnight at 4 °C. Slides were washed in PBST, and incubated with secondary antibodies diluted in 3% BSA for one hour at room temperature. Following washes in PBST, slides were mounted, and images were acquired with a GE Cell DIVE multiplex imager. See the Supplementary Table 1 for a list of primary and secondary antibodies.
Western blot
Autophagy activity was assessed by measuring autophagy flux in WAT. Adipose tissue was incubated in full DMEM supplemented with 100 nM Bafilomycin A1 and 20 mM NH4Cl in DMSO for 4 h to inhibit lysosomal activity. DMSO was used as a “vehicle” control. To assess LC3-turnover, autophagic flux was calculated as: (LC3-II (Inh) – LC3-II (Veh)). To determine the contribution of apoptosis, WAT explants were incubated in full RPMI supplemented with either DMSO (vehicle control) or 20 µM Q-VD-OPh overnight or 10 µM staurosporine for 24 h. Protein extraction was performed as published91. Briefly, 500 μL of lysis buffer containing protease inhibitors and PhosphoStop was added per 100 mg of tissue. Cells were lysed using Qiagen TissueLyser II and lipid contamination was removed through serial centrifugation. Protein content was determined using a BCA Protein Assay kit and 15 μg of protein was separated on a 4–12% Bis-Tris SDS PAGE gel. After wet-transferred onto a PVDF membrane, membranes were blocked in 5 % milk in TBST, and incubated with primary antibodies overnight. Proteins were visualized on a membrane using IRDye 800 or IRDye 680 (LI-COR Biosciences) secondary antibodies at the dilution 1:10,000 (LI-COR). Quantification was performed with ImageStudio software (LI-COR). See the Supplementary Table 1 for a list of primary and secondary antibodies.
Enzyme-linked immunosorbent assay (ELISA)
Adipose tissue secretome was generated by incubating 200 mg of adipose tissue explants (each explant ~ 10 mg) in 1 ml of RPMI containing 10% FBS and 1% P/S for 6 h at 37 °C. Cytokine levels in supernatant were measured by commercially available ELISA kits. See the Supplementary Table 1 for a list of ELISA kits. All cytokine levels were normalized to input tissue weight.
Serum chemistry
After eight hours of fasting, serum was collected by a cardiac puncture, collected in Microtainer tubes, and centrifuged for 90 s at 15,000 × g. Triglycerides and high-density lipoprotein were measured using a Beckman Coulter AU680 clinical chemistry analyser.
Gene expression analysis (qRT-PCR)
Tissues were homogenized in TRI reagent (Sigma) with ceramic beads (Bertin Instruments) using a Precellys 24 homogenizer (Bertin Instruments). RNA was extracted using RNeasy Mini or Micro Kit (Qiagen). RNA yield and quality were assessed using a NanoDrop and cDNA was synthesized using a High-Capacity RNA-to-cDNA™ kit (ThermoFisher). Gene expression was measured using TaqMan Fast Advanced Master Mix on a ViiA7 real-time PCR system. Values were normalized to Ppia reference gene using the comparative Ct method. Primers are listed in the Supplementary Table 1.
Flow cytometry and cell sorting (FACS)
Cells for flow cytometry staining were isolated as described above. For surface staining, cells were incubated with fluorochrome-conjugated antibodies, LIVE/DEAD Fixable Stains and Fc receptor block antibody for 20 min at 4 °C. This was followed by a 10 min fixation with 4% PFA at room temperature. Samples were acquired on the Fortessa X-20 flow cytometer (BD Biosciences). Data were analysed with FlowJo v10.8 according to the gating strategy (Supplementary Fig. 5). See the Supplementary Table 1 for a list of flow cytometry antibodies.
Transcriptomics (bulk RNA sequencing)
Macrophages were isolated by FACS or cultured in vitro and RNA was extracted as described above. To generate PolyA libraries, cDNA was end-repaired, A-tailed and adapter-ligated. Libraries were then size-selected, multiplexed, quality-controlled, and sequenced using a NovaSeq6000. Quality control of raw reads was performed with a pipeline readqc.py (https://github.com/cgat-developers/cgat-flow). The resulting reads were aligned to the GRCm38/Mm10 reference genome using the pseudoalignment methods Salmon92 and Kallisto93. DEseq2 (v1.38.3) was used for differential gene expression analysis94. The workflow included the estimation of size factors, dispersion estimation, and fitting of a negative binomial generalized linear model. Prior to differential expression analysis, batch effects attributed to sex were corrected by incorporating them within the experimental design of deseq2 analysis, using the limma package (v3.54.1) for visualisation purposes95. Genes were considered significantly differentially expressed based on an adjusted p-value threshold of <0.05, after correcting for multiple testing using the Benjamini–Hochberg procedure. To explore the biological implications of the differentially expressed genes, gene set enrichment analysis was performed using the clusterProfiler package (v4.6.0).
Single-nucleus RNA-seq analysis
The adipocyte dataset was downloaded from the GEO database (GSE176171), originally published by Emont et al.24. The dataset was categorized into lean or obese groups based on BMI, following the methodology outlined in the source paper24. While we adopted the original study’s BMI cut-offs and confounding effects, we recognise that this study has limitations in sample size, sex, age, and ethnicity distribution. To facilitate visualization, UMAP (Uniform Manifold Approximation and Projection) was recalculated, and further data analysis was conducted using the Seurat package v5.0.196 for single-nuclei RNA-seq analysis. Functional enrichment analysis was performed using the ClusterProfiler package (v4.6.0)97.
Proteomics
Adipocytes were isolated as a floating fraction upon digestion and lysed and digested in SDC buffer. Specifically, a pellet of 100 µl packed adipocytes was lysed in SDC-buffer containing 2% (w/v) sodium deoxycholate (SDC; Sigma-Aldrich), 20 mM dithiothreitol (Sigma-Aldrich), 80 mM chloroacetamide (Sigma-Aldrich), and 200 mM Tris-HCl (pH 8). After being heated at 95 °C for 10 min, the lysates were digested enzymatically using endopeptidase LysC (Wako) and sequence grade trypsin (Promega) at a protein:enzyme ratio of 50:1. The digestion process occurred overnight at 37 °C. For reversed-phase liquid chromatography coupled to mass spectrometry (LC-MS) analysis, each sample replicate was injected with 1 µg of peptide amount into an EASY-nLC 1200 system (Thermo Fisher Scientific) for separation, using a 110-min gradient. Mass spectrometric measurements were carried out using an Exploris 480 (Thermo Fisher Scientific) instrument in data-independent acquisition (DIA) mode, which utilised an isolation scheme with asymmetric isolation window sizes. The raw files were analysed using DIA-NN version 1.8.198 in library-free mode, with a false discovery rate cutoff of 0.01 and relaxed protein inference criteria, while employing the match-between runs option. The spectra were compared to a Uniprot mouse database (2022-03), which included isoforms. The protein intensities were normalised using MaxLFQ and filtered to ensure that each protein had at least 50% valid values across all experiments, with an additional filter to retain at least 3 valid values in at least one experimental group. The limma package95 was used to calculate two-sample moderated t-statistics for significance calling. The nominal P-values were adjusted using the Benjamini–Hochberg method.
Mass spectrometry
Adipocytes and conditioned medium were obtained as described above. Metabolite extractions from frozen cell pellets were performed through a two-phase extraction with 80% MeOH and chloroform. In brief, when just thawed, cells were resuspended in 500 µl ice-cold 80% MeOH, vortexed and sonicated in an ice bath for 6 min. Following 1 h of incubation on dry ice, tubes were centrifuged at 16,000 × g for 10 min at 4 °C, and supernatants were mixed with water and chloroform in 1:1:1 ratio. Each sample was vortexed for 1 min and centrifuged at top speed for 15 min at 4 °C. Finally, 600 μl of the top aqueous layer was transferred to a glass vial and evaporated using an EZ-2Elite evaporator (Genevac). Samples were stored at −80 °C before analysis. The BCA assay was performed on the airdried pellets, resuspended in 200 µl of 0.2 M NaOH and heated at 95 °C for 20 min. Metabolite extractions from frozen conditioned medium precleared from cells and cell debris were performed after removing cells and cell debris by mixing 20 µl of clarified medium with 500 µl ice-cold 80% MeOH/20% H2O. After vortex and 30 min incubation at −80 °C, samples were centrifuged at 16,000 × g for 10 min at 4 °C, transferred to a glass vial and evaporated using an EZ-2Elite evaporator (Genevac). Dried extracts were stored at −80 °C before analysis. Dried metabolites were resuspended in 100 μL 50% ACN:water and 5 μL was loaded onto a Luna NH2 3 μm 100 A (150 × 2.0 mm) column (Phenomenex) using a Vanquish Flex UPLC (Thermo Scientific). The chromatographic separation was performed with mobile phases A (5 mM NH4AcO pH 9.9) and B (ACN) at a flow rate of 200 μl/min. A linear gradient from 15% A to 95% A over 18 min was followed by 7 min isocratic flow at 95% A and re-equilibration to 15% A. Metabolites were detected with a Thermo Scientific Q Exactive mass spectrometer run with polarity switching in full scan mode using a range of 70–975 m/z and 70.000 resolution. Maven (v 8.1.27.11) was used to quantify the targeted polar metabolites by AreaTop, using expected retention time and accurate mass measurements (< 5 ppm) for identification. Data analysis, including principal component analysis and heat map generation, was performed using in-house R scripts. In brief, metabolite intensities (area under the curve) were normalized to protein content and analysed with one-way ANOVA (ANOVA column). Metabolites with a p-value < 0.05 were termed as significant. Heatmaps are z-score normalized, assuming a normal distribution. Bar plots display relative amounts for each metabolite, calculated by averaging amounts for each condition (condition with the lowest average value set to 1). Raw metabolomics data is provided as Supplementary Data 1.
Metabolic assays
To measure metabolite levels in cells, cells were resuspended in an ice-cold homogenization medium (0.32 M sucrose, 1 mM EDTA, and 10 mM Tris–HCl, pH 7.4) and homogenized with 72 strokes using a tight pestle. After brief sonication at 40% Amp, homogenates were centrifuged at 2000 × g for 5 min at 4 °C to remove the lipid layer and protein concentration was determined using the BCA assay to normalize between conditions. 24 µg of protein was used per assay condition. Levels of xanthine and hypoxanthine (Abcam) in cell homogenates were measured using commercially available kits. Intracellular ATP was measured in living cells using the ATP bioluminescence assay kit CLS II (Roche) according to the manufacturer’s instructions. Commercial kits were also used to measure xanthine and hypoxanthine (Abcam) directly in serum and conditioned medium. For more information, see the Supplementary Table 1.
Quantification and statistical analysis
Experiments were conducted as 3 independent repeats or as indicated. Mice were randomly grouped in experimental groups and data were pooled from both sexes. Data were analysed and visualized using GraphPad Prism 9. The normal distribution of data was tested before applying parametric or nonparametric testing. For comparison between two independent groups, the unpaired Student’s t-test or the Mann–Whitney test were applied. Comparisons across multiple groups were performed using one-way or two-way ANOVA with Dunnett, Šídák or Tukey multiple testing correction. Data were considered statistically significant when p-value < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA sequencing data generated in this study have been deposited in the GEO database under accession codes GSE263837 and GSE306424. The proteomics data generated in this study have been deposited in the PRIDE database under accession code PXD052894. The metabolomics data generated in this study are provided in Supplementary Data 1. The single-nuclei RNA sequencing data used in this study are available in the GEO database under accession code GSE176171. Source data are provided with this paper.
Change history
05 February 2026
In the version of this article initially published, Ghada Alsaleh and Anna Katharina Simon were listed as equally contributing authors, rather than jointly supervising authors, as is now amended in the HTML and PDF versions of the article.
References
González-Muniesa, P. et al. Obesity. Nat. Rev. Dis. Primers 3, 17034 (2017).
Rosen, E. D. & Spiegelman, B. M. What we talk about when we talk about fat. Cell 156, 20–44 (2014).
Sakers, A., De Siqueira, M. K., Seale, P. & Villanueva, C. J. Adipose-tissue plasticity in health and disease. Cell 185, 419–446 (2022).
Ghaben, A. L. & Scherer, P. E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 20, 242–258 (2019).
Chouchani, E. T. & Kajimura, S. Metabolic adaptation and maladaptation in adipose tissue. Nat. Metab. 1, 189–200 (2019).
Morigny, P., Boucher, J., Arner, P. & Langin, D. Lipid and glucose metabolism in white adipocytes: pathways, dysfunction and therapeutics. Nat. Rev. Endocrinol. 17, 276–295 (2021).
Gliniak, C. M., Pedersen, L. & Scherer, P. E. Adipose tissue fibrosis: the unwanted houseguest invited by obesity. J. Endocrinol. 259, e230180 (2023).
Matz, A. J., Qu, L., Karlinsey, K., Vella, A. T. & Zhou, B. Capturing the multifaceted function of adipose tissue macrophages. Front. Immunol. 14, 1148188 (2023).
Marcelin, G., Gautier, E. L. & Clement, K. Adipose tissue fibrosis in obesity: etiology and challenges. Annu. Rev. Physiol. 84, 135–155 (2022).
Reggio, S., Pellegrinelli, V., Clément, K. & Tordjman, J. Fibrosis as a cause or a consequence of white adipose tissue inflammation in obesity. Curr. Obes. Rep. 2, 1–9 (2013).
Chait, A. & den Hartigh, L. J. Adipose tissue distribution, inflammation and its metabolic consequences, including diabetes and cardiovascular disease. Front. Cardiovasc. Med. 7, 22 (2020).
Kaur, J. & Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 16, 461–472 (2015).
Dikic, I. & Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 19, 349–364 (2018).
Klionsky, D. J. et al. Autophagy in major human diseases. EMBO J. 40, e108863 (2021).
Rabinowitz, J. D. & White, E. Autophagy and metabolism. Science 330, 1344–1348 (2010).
Deretic, V. & Kroemer, G. Autophagy in metabolism and quality control: opposing, complementary or interlinked functions?. Autophagy 18, 283–292 (2022).
Piletic, K., Alsaleh, G. & Simon, A. K. Autophagy orchestrates the crosstalk between cells and organs. EMBO Rep. 24, e57289 (2023).
Singh, R. et al. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Investig. 119, 3329–3339 (2009).
Zhang, Y. et al. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc. Natl. Acad. Sci. USA 106, 19860–19865 (2009).
Cai, J. et al. Autophagy ablation in adipocytes induces insulin resistance and reveals roles for lipid peroxide and Nrf2 signaling in adipose-liver crosstalk. Cell Rep. 25, 1708–1717.e1705 (2018).
Sakane, S. et al. White adipose tissue autophagy and adipose-liver crosstalk exacerbate nonalcoholic fatty liver disease in mice. Cell. Mol. Gastroenterol. Hepatol. 12, 1683–1699 (2021).
Clemente-Postigo, M., Tinahones, A., Bekay, R. E., Malagón, M. M. & Tinahones, F. J. The Role of autophagy in white adipose tissue function: implications for metabolic health. Metabolites 10, 179 (2020).
Soussi, H., Clément, K. & Dugail, I. Adipose tissue autophagy status in obesity: expression and flux—two faces of the picture. Autophagy 12, 588–589 (2016).
Emont, M. P. et al. A single-cell atlas of human and mouse white adipose tissue. Nature 603, 926–933 (2022).
Jansen, H. J. et al. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology 153, 5866–5874 (2012).
Kosacka, J. et al. Autophagy in adipose tissue of patients with obesity and type 2 diabetes. Mol. Cell. Endocrinol. 409, 21–32 (2015).
Kovsan, J. et al. Altered autophagy in human adipose tissues in obesity. J. Clin. Endocrinol. Metab. 96, E268–E277 (2011).
Mizunoe, Y. et al. Involvement of lysosomal dysfunction in autophagosome accumulation and early pathologies in adipose tissue of obese mice. Autophagy 13, 642–642 (2017).
Nuñez, C. E. et al. Defective regulation of adipose tissue autophagy in obesity. Int. J. Obes. 37, 1473–1480 (2013).
Öst, A. et al. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol. Med. 16, 235–246 (2010).
Richter, F. C. et al. Adipocyte autophagy limits gut inflammation by controlling oxylipin and IL-10. EMBO J. 42, e112202 (2023).
Levine, B. & Klionsky, D. J. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 6, 463–477 (2004).
Cinti, S. et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 46, 2347–2355 (2005).
Caserta, T. M., Smith, A. N., Gultice, A. D., Reedy, M. A. & Brown, T. L. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis 8, 345–352 (2003).
Korycka, A., Blonski, J. Z. & Robak, T. Forodesine (BCX-1777, Immucillin H)-a new purine nucleoside analogue: mechanism of action and potential clinical application. Mini. Rev. Med. Chem. 7, 976–983 (2007).
Meng, X. M., Nikolic-Paterson, D. J. & Lan, H. Y. TGF-beta: the master regulator of fibrosis. Nat. Rev. Nephrol. 12, 325–338 (2016).
Icer, M. A. & Gezmen-Karadag, M. The multiple functions and mechanisms of osteopontin. Clin. Biochem. 59, 17–24 (2018).
Maniyadath, B., Zhang, Q., Gupta, R. K. & Mandrup, S. Adipose tissue at single-cell resolution. Cell Metab. 35, 386–413 (2023).
Nance, S. A., Muir, L. & Lumeng, C. Adipose tissue macrophages: regulators of adipose tissue immunometabolism during obesity. Mol. Metab. 66, 101642 (2022).
Hill, D. A. et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc. Natl. Acad. Sci. USA 115, E5096–E5105 (2018).
Jaitin, D. A. et al. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell 178, 686–698.e614 (2019).
Chakarov, S. et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 363, eaau0964 (2019).
Xu, X. et al. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 18, 816–830 (2013).
Sarvari, A. K. et al. Plasticity of epididymal adipose tissue in response to diet-induced obesity at single-nucleus resolution. Cell Metab. 33, 437–453.e435 (2021).
Sugden, P. H. & Clerk, A. Regulation of the ERK subgroup of MAP kinase cascades through G protein-coupled receptors. Cell Signal 9, 337–351 (1997).
Shirakawa, K. et al. MerTK expression and ERK activation are essential for the functional maturation of osteopontin-producing reparative macrophages after myocardial infarction. J. Am. Heart Assoc. 9, e017071 (2020).
Sun, Y. et al. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept Signal Transduct. Res. 35, 600–604 (2015).
Wen, X., Jiao, L. & Tan, H. MAPK/ERK pathway as a central regulator in vertebrate organ regeneration. Int. J. Mol. Sci. 23, 1464 (2022).
Gurusamy, M. et al. G-protein-coupled receptor P2Y10 facilitates chemokine-induced CD4 T cell migration through autocrine/paracrine mediators. Nat. Commun. 12, 6798 (2021).
Soussi, H. et al. DAPK2 downregulation associates with attenuated adipocyte autophagic clearance in human obesity. Diabetes 64, 3452–3463 (2015).
Nagao, H. et al. Increased dynamics of tricarboxylic acid cycle and glutamate synthesis in obese adipose tissue: IN VIVO METABOLIC TURNOVER ANALYSIS. J. Biol. Chem. 292, 4469–4483 (2017).
Park, Y. J., Choe, S. S., Sohn, J. H. & Kim, J. B. The role of glucose-6-phosphate dehydrogenase in adipose tissue inflammation in obesity. Adipocyte 6, 147–153 (2017).
Kather, H. Pathways of purine metabolism in human adipocytes. Further evidence against a role of adenosine as an endogenous regulator of human fat cell function. J. Biol. Chem. 265, 96–102 (1990).
Guo, J. Y. et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 30, 1704–1717 (2016).
Borsa, M. et al. Autophagy preserves hematopoietic stem cells by restraining MTORC1-mediated cellular anabolism. Autophagy 20, 45–57 (2024).
Zhang, N. et al. Increased amino acid uptake supports autophagy-deficient cell survival upon glutamine deprivation. Cell Rep. 23, 3006–3020 (2018).
James, D. E., Stöckli, J. & Birnbaum, M. J. The aetiology and molecular landscape of insulin resistance. Nat. Rev. Mol. Cell Biol. 22, 751–771 (2021).
Harkness, R. A. Hypoxanthine, xanthine and uridine in body fluids, indicators of ATP depletion. J. Chromatogr. 429, 255–278 (1988).
Harkness, R. A., Geirsson, R. T. & McFadyen, I. R. Concentrations of hypoxanthine, xanthine, uridine and urate in amniotic fluid at caesarean section and the association of raised levels with prenatal risk factors and fetal distress. Br. J. Obstet. Gynaecol. 90, 815–820 (1983).
Harkness, R. A. & Lund, R. J. Cerebrospinal fluid concentrations of hypoxanthine, xanthine, uridine and inosine: high concentrations of the ATP metabolite, hypoxanthine, after hypoxia. J. Clin. Pathol. 36, 1–8 (1983).
Li, Y., Liu, R., Wu, J. & Li, X. Self-eating: friend or foe? The emerging role of autophagy in fibrotic diseases. Theranostics 10, 7993–8017 (2020).
Sun, M., Tan, L. & Hu, M. The role of autophagy in hepatic fibrosis. Am. J. Transl. Res. 13, 5747–5757 (2021).
Oh, J. et al. High levels of intracellular endotrophin in adipocytes mediate COPII vesicle supplies to autophagosome to impair autophagic flux and contribute to systemic insulin resistance in obesity. Metabolism 145, 155629 (2023).
Henderson, N. C., Rieder, F. & Wynn, T. A. Fibrosis: from mechanisms to medicines. Nature 587, 555–566 (2020).
Medzhitov, R. The spectrum of inflammatory responses. Science 374, 1070–1075 (2021).
Baer, J. M. et al. Fibrosis induced by resident macrophages has divergent roles in pancreas inflammatory injury and PDAC. Nat. Immunol. 24, 1443–1457 (2023).
Dornbos, P. et al. Evaluating human genetic support for hypothesized metabolic disease genes. Cell Metab. 34, 661–666 (2022).
Rocha, N. et al. Human biallelic MFN2 mutations induce mitochondrial dysfunction, upper body adipose hyperplasia, and suppression of leptin expression. Elife 6, e23813 (2017).
Sebastian, D. et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 35, 1677–1693 (2016).
Yu, L. et al. IgG is an aging factor that drives adipose tissue fibrosis and metabolic decline. Cell Metab. 36, 793–807.e795 (2024).
Lech, M. & Anders, H. J. Macrophages and fibrosis: how resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim. Biophys. Acta 1832, 989–997 (2013).
Vila, I. K. et al. Immune cell Toll-like receptor 4 mediates the development of obesity- and endotoxemia-associated adipose tissue fibrosis. Cell Rep. 7, 1116–1129 (2014).
Martinez-Santibanez, G. et al. Obesity-induced remodeling of the adipose tissue elastin network is independent of the metalloelastase MMP-12. Adipocyte 4, 264–272 (2015).
Chen, Q. et al. Resident macrophages restrain pathological adipose tissue remodeling and protect vascular integrity in obese mice. EMBO Rep. 22, e52835 (2021).
Bacigalupa, Z. A., Landis, M. D. & Rathmell, J. C. Nutrient inputs and social metabolic control of T cell fate. Cell Metab. 36, 10–20 (2024).
Richter, F. C., Obba, S. & Simon, A. K. Local exchange of metabolites shapes immunity. Immunology 155, 309–319 (2018).
Nagao, H., et al. Hypoxanthine secretion from human adipose tissue and its increase in hypoxia. Obesity 26, 1168–1178 (2018).
Furuhashi, M. et al. Differential regulation of hypoxanthine and xanthine by obesity in a general population. J. Diabetes Investig. 11, 878–887 (2020).
Ho, J. E. et al. Metabolomic profiles of body mass index in the Framingham Heart Study reveal distinct cardiometabolic phenotypes. PLoS ONE 11, e0148361 (2016).
Xie, G. et al. The metabolite profiles of the obese population are gender-dependent. J. Proteome Res. 13, 4062–4073 (2014).
Deng, Y. et al. Adipocyte Xbp1s overexpression drives uridine production and reduces obesity. Mol. Metab. 11, 1–17 (2018).
Pfeifer, A., Mikhael, M. & Niemann, B. Inosine: novel activator of brown adipose tissue and energy homeostasis. Trends Cell Biol. 34, 72–82 (2024).
Fromme, T. et al. Degradation of brown adipocyte purine nucleotides regulates uncoupling protein 1 activity. Mol. Metab. 8, 77–85 (2018).
Kather, H. Purine accumulation in human fat cell suspensions. Evidence that human adipocytes release inosine and hypoxanthine rather than adenosine. J. Biol. Chem. 263, 8803–8809 (1988).
Fan, K. Q. et al. Stress-induced metabolic disorder in peripheral CD4(+) T cells leads to anxiety-like behavior. Cell 179, 864–879.e819 (2019).
Sousa, C. M. et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483 (2016).
Poillet-Perez, L. et al. Autophagy maintains tumour growth through circulating arginine. Nature 563, 569–573 (2018).
Sassmann, A., Offermanns, S. & Wettschureck, N. Tamoxifen-inducible Cre-mediated recombination in adipocytes. Genesis 48, 618–625 (2010).
Komatsu, M. et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434 (2005).
Ruifrok, A. C. & Johnston, D. A. Quantification of histochemical staining by color deconvolution. Anal. Quant. Cytol. Histol. 23, 291–299 (2001).
An, Y. A. & Scherer, P. E. Mouse adipose tissue protein extraction. Bio Protoc. 10, e3631 (2020).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015).
Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020).
Acknowledgements
We thank Patricia Cotta Moreira, Luke Barker, Emily Wyeth, Daniel Andrew, and Mino Medghalchi from the Biomedical Services for their responsible care and assistance with animal well-being. Histology was performed with the help of the Kennedy Institute Histology Facility, with special thanks to Dr Ida Parisi. Dr Johanna ten Hoeve–Scott at UCLA Metabolomics Center, Dr Ulrike Brüning and Dr Jennifer Kirwan at BIH Charité Berlin for their help with metabolomics sample analysis. Dr Moustafa Attar for his help with the experimental design of the transcriptomics experiment. Jonathan Webber for his help with the flow cytometry experimental design. Cell DIVE Facility team at the Kennedy Institute of Rheumatology for their help with immunofluorescent staining. This work was supported by grants from the Wellcome Trust to A.K.S. (Investigator award 220784/Z/20/Z) and M.B. (Sir Henry Wellcome Fellowship 220452/Z/20/Z), the Kennedy Trust for Rheumatology Research (KTTR) Studentship to K.P. (KEN192001) and J.K.L.K. (awarded to Marco Fritzsche), The Kenneth Rainin Foundation, and Helmholtz Association REK-0157 to A.K.S. (20220003/20230038), Clarendon Fund Scholarship to K.P., Medical Research Council Doctoral Training Partnership Grant (BRT00030) to K.P., Ramage Scholarship to K.P., PhD studentship award 203803/Z16/Z to F.C.R., Versus Arthritis grant 22617 to G.A., EPA Cephalosporin Fund to J.K.L.K., EMBO Postdoctoral Fellowship (ALTF115-2019) to A.V.L.V. Li-cor Odyssey imager was funded by ERC AdG 670930. Flow cytometry and microscopy facilities were supported by KTTR. Graphical summaries were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
Conceptualization: K.P., F.P., G.A. and A.K.S. Methodology, investigation, analysis, visualization, and validation: K.P., A.H.K., F.C.R., M.B., A.V.L.-V., O.P., S.G., J.K.L.K., L.L., K.Z., M.K., H.K.H., L.K. and G.A. Essential reagents and support: O.P., L.K., P.M. and S.S. Writing of original draft: K.P., G.A. and A.K.S. Funding acquisition, supervision, and project administration: F.P., G.A. and A.K.S. Editing of the manuscript: all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Swapan Das and the other anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Piletic, K., Kayvanjoo, A.H., Richter, F.C. et al. Autophagy acts as a brake on obesity-related fibrosis by controlling purine nucleoside signalling. Nat Commun 16, 9220 (2025). https://doi.org/10.1038/s41467-025-64266-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64266-5
