
Abstract
The insertion of an oxygen atom into carbon-carbon (C-C) σ-bonds of readily available ketones to form esters represents a fundamental transformation known as Baeyer-Villiger (BV) oxidation. While this classical reaction serves as a cornerstone in organic synthesis, its scope remains limited to single oxygen-atom insertion into ketone substrates. We herein report a versatile catalytic protocol that enables the insertion of alkynyl phenol analogues into unstrained C-C σ-bonds of diverse carbonyl compounds, including ketones, esters, and amides. This method provides modular access to an array of structurally varied products ranging from linear esters to medium- and macrocyclic lactones. This methodology displays broad substrate scope, excellent functional group tolerance, direct applicability to bioactive molecule modification with effective transfer of axial chirality. An in-depth computational study provides insights into the reaction mechanism.
Similar content being viewed by others
Introduction
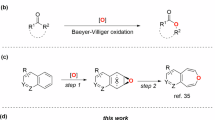
The creation of structurally diverse molecular libraries that explore expansive chemical space is fundamental to advances in chemical biology and drug discovery1,2,3,4. Among classical transformations for skeletal editing, the Baeyer–Villiger (BV) oxidation stands as a textbook example of C–C σ-bond cleavage, providing straightforward access to esters from linear ketones and lactones from cyclic ketones5,6. Despite its operational simplicity and intermolecular nature, conventional BV oxidation suffers from inherent limitations in product diversity, as the structural complexity remains constrained by the parent ketone framework (Fig. 1A). Given the privileged status of ester motifs in bioactive natural products and pharmaceuticals7,8, the development of efficient methods capable of generating structurally diverse ester libraries from readily available feedstocks represents an important synthetic objective.

A Baeyer–Villiger oxidation. B Concept on diversity-oriented bridging C–C bond activation. C Diversity-oriented ester synthesis through alkyne-bridging C–C bond activation.
Building upon our success in C–H bond activation9,10,11,12,13,14,15,16, we envisioned an alkyne-bridging C–C bond activation strategy for ester synthesis using readily available feedstock chemicals. This approach capitalizes on easily accessible reactants derived from abundant starting materials through simple transformations. Guided by this designed principle, we anticipated access to diverse, value-added products with structural complexity (Fig. 1B). Our previous work established a modular route to dibenzo-fused 7- to 8-membered lactones via palladium-catalyzed reactions of ortho-halobenzaldehydes with salicylaldehyde-derived carbene precursors10. Extending this strategy, we hypothesized that condensation of 1 with abundant ketones 2 would provide modular access to α,β-unsaturated carbonyl intermediates 5. Subsequent palladium-catalyzed reaction with unsymmetrical internal alkynes 6, which were readily prepared via Sonogashira coupling of 3 and 4, could enable the envisioned transformation. Key challenges in this design include: (i) Achieving regioselective intermolecular insertion of unsymmetrical alkyne 6 to form intermediate I, (ii) controlling 6-endo-trig cyclization (I→II) over the typically favored 5-exo-trig pathway, and iii selective cleavage of the unstrained C(acyl)-C σ-bond despite its high dissociation energy17,18,19,20,21,22. The ortho-phenol moiety in 6 was designed to serve multiple functions: directing regioselective alkyne insertion, facilitating subsequent C–C bond cleavage, and participating in ester bond formation. We anticipated that careful ligand selection could promote six-membered ring formation, while aromaticity restoration, driving β-carbon elimination23,24,25,26,27,28, would enable selective C–C cleavage29,30,31. Unlike conventional approaches such as BV oxidation or diazo-based ketone homologation32, multi-atom insertions into unstrained carbonyls remain challenging33,34,35,36,37,38,39, typically requiring reactive intermediates like arynes40 or strained alkenes41,42 as coupling partners. While Dong’s elegant work demonstrated two-carbon insertion into 1-indanones using ethylene/alkynes43,44, broader substrate scope with more unstrained carbonyl compounds remains unexplored. In 2015, Wu and co-workers developed an innovative method for synthesizing 8-membered lactones via palladium-catalyzed C–C bond cleavage of strained cyclobutanone derivatives37. Being aware of the thermodynamic driving force to form an aromatic ring, we expect that a variety of readily available unstrained carbonyl compounds45,46,47,48,49,50, including linear or cyclic ketones, esters, and amides51,52 might be fragmented through our conceived strategy. The feedstock abundance of all starting materials makes this alkyne-bridging C–C activation particularly attractive for rapid diversity generation (Fig. 1C). Notably, BV oxidation cannot cleave C–C bonds in esters and amides. Moreover, while BV reactions insert single oxygen atoms, our approach introduces modular building blocks at the original C(acyl)–C bond. We believe this bridging C–C bond activation principle may inspire new methodological developments.
Results
Reaction development
We began our investigation with the use of α, β-unsaturated ketone 5a and 2-alkynylphenol 6a as the initial substrates. After systematic survey of the reaction parameters (for details, see Supplementary Information Tables S1–S5), ester 7a was obtained in 97% GC yield using Pd(TFA)2 (5 mol%) as the precatalyst, Ad2Pn-Bu (10 mol%) as the ligand and Na2CO3 (2.0 equiv.) as the base in DCE (0.1 M) at 120 °C for 10 h (Table 1, entry 1). A number of control experiments were subsequently carried out. Firstly, the reactions cannot proceed without the addition of Pd salt or Ad2n-BuP. Other Pd(II) precatalysts or phosphine-based ligands were less efficient, and the yields of 7a decreased significantly (Table 1, entries 4–7). Lower the reaction temperature to 100 °C led to a lower yield of 7a. If the reaction was carried out in the open air, a more complex reaction mixture was obtained, and 7a was produced in 47% GC yield (Table 1, entry 9).
With optimized conditions established, we evaluated the generality and limitations of this palladium-catalyzed alkyne-bridging C–C activation using linear ketones (Fig. 2A). The reaction demonstrated remarkable tolerance toward diverse 2-alkynylphenols, accommodating substrates with varying electronic and steric properties, affording the desired products 7b–7j in moderate to high yields (41–98%). The efficiency seems to be not affected by slight variation of the electron property of the substituents. While the introducing of electron-withdrawing groups, such as p-CF3, p-CO2Me, and p-CN, the products (7g, 7h, and 7i) were obtained in diminished yields. Notably, sterically demanding substrates (1-naphthyl, o-tolyl) proved compatible, delivering 7k and 7l in 85% and 98% yields, respectively. Similarly, substituents ranging from 4-F to 4-Me and 5-Cl on the phenol ring were tolerated as well, giving the corresponding products 7m–7o in 50–90% yields upon isolation. Particularly encouraging was the compatibility of aliphatic alkynes bearing functional groups like chloro, labile THP-ether, silyl ether, cyclopropyl groups, the reactions furnishing the desired products 7p–7t in high yields. Encouraged by the above results, we thought such an alkyne-bridging C–C bond activation strategy might not be limited to ketones, and the carbon-acyl bonds in esters or amides might be cleavable under current conditions. As illustrated in Fig. 2B, esters 5b and 5e were indeed viable substrates to react with 2-alkynylphenol 6a, and the product 8a with a free hydroxyl group on the phenyl ring was obtained in high yields after hydrolysis under basic conditions. It is of note to mention that a one-pot procedure for the bridging C–C bond activation of ketone 5a with 6a with subsequent hydrolysis, could also give 8a in high efficiency. Amide 5c and Weinreb amide 5d could react with 6a, albeit in lower overall yields. Given the higher efficiency when α,β-unsaturated ketones and esters were employed as the reactants, we decided to test the reactivity of other analogs. As depicted in Fig. 2C, the reaction accommodated diverse aryl substituents (7v–7z), with electronic perturbations showing minimal impact on efficiency. Similarly, 5 deriving from ketones with different substituents on the phenyl ring (R1) could react with internal alkyne 6a smoothly, giving the corresponding benzoyl esters in good yields (7aa–7ae). Ketone or ester bearing naphthyl and thiophenyl rings were viable reactants. α,β-Unsaturated ester bearing Isoxepac (an anti-inflammatory agent) motif was a good substrate, giving the corresponding pharmaceutically relevant product 8ah in 68% yield. Our protocol was also applicable for ketones bearing aliphatic groups (R1 = Me, nBu or Bn, R2 = Me), and the corresponding products (7ai–7ak, 7am) were obtained up to 90% isolated yields. The embedding of a benzyloxy group in the reactant is noteworthy, as a product of binol derivatives 7al could be prepared facilely.

A Scope of alkynylphenols. B Reaction with different α,β-unsaturated carbonyl compounds. C Scope of α,β-unsaturated ketones or esters. *Upon the completion of the reaction, the mixture was treated with NaOH in MeOH. †α,β-unsaturated methyl carboxylate was used as the initial material. ‡Reaction conducted with Pd(OAc)2 (5 mol%), Ad2Pn-Bu (10 mol%), K2HPO4 (2.0 equiv.), 1,4-dioxane (0.05 M), at 130 °C, 7 h, under an atmosphere of Argon.
While our previously reported carbene-bridging C–H activation provided efficient access to seven- and eight-membered lactones10, this approach proved ineffective for larger ring systems. Having achieved selective C–C bond cleavage in linear ketones, we envisioned that our alkyne-bridging C–C bond activation strategy could be adapted for functionalizing unstrained cyclic ketones, potentially enabling modular synthesis of medium-to-macrocyclic lactones through simple variation of the ketone ring size. In line with this assumption, reactants deriving from cyclopentanone and cyclohexanone were prepared and subjected to test the reactivity. To our delight, the corresponding 9-membered and 10-membered lactones 9a and 9b could be obtained in 56% and 64% yields upon isolation under a set of slightly different conditions by using Pd(OAc)2 as the palladium source and K2HPO4 as base (Fig. 3). This success prompted a thorough evaluation of substrate scope, which revealed remarkable generality.

Scope of the cyclic ketones.
As demonstrated in Fig. 3, indanone and tetralone derivatives could react to give the corresponding 9- and 10-membered lactones 9c and 9d in good isolated yields. Acetal moiety, and potentially catalyst poisoning sites containing S and N atoms are tolerated in the reactions (9e–9g). Compared with the reactions of linear carbonyl compounds, α, β-unsaturated ester containing a δ-lactone moiety reacts well with 2-alkynylphenol. Besides the corresponding 10-membered lactones 9h, an intriguing decarboxylated 9i bearing an 8-membered O-heterocycle was obtained in 40% isolated yield. Even a bridged bicyclic ketone participated effectively, affording 9j and demonstrating the method’s tolerance for complex architectures. Most significantly, systematic variation of cyclic ketone size, ranging from cycloheptanone to cyclopentadecanone, enabled straightforward access to diverse medium- and macrocyclic lactones 9k–9p.
Recognizing that this alkyne-bridging C–C activation constructs multi-substituted biaryl scaffolds, we investigated the potential for controlling axial chirality using commercially available chiral ketones (Fig. 4). We selected (D)-camphor as our initial substrate due to its cost-effectiveness, rigid backbone, and well-defined stereochemistry. Remarkably, the corresponding chiral α,β-unsaturated ketone reacted smoothly with various alkynylphenols under standard conditions, affording products 10a–10f as single diastereomers in moderate to high yields (40–70%). Single-crystal X-ray analysis of 10a unambiguously confirmed both the structure and absolute configuration. The methodology proved general for bioactive chiral ketones. Epiandrosterone and estrone derivatives reacted efficiently with 6a under standard conditions. The relatively remote chiral centers in the reactants seem to well induce the generation of axial chirality. Both 10g and 10h were obtained as single diastereoisomers in high yields after ring opening with MeOH under basic conditions. Pleasingly, we could obtain the crystal structure of 10g. Methylated derivative 10h also provided suitable crystals for X-ray analysis.

Axial chirality transfer by using chiral ketones as starting materials.
Computational studies
Density functional theory (DFT) calculations were performed to understand the mechanism of this C–C bond activation of linear carbonyl substrates, particularly regarding the role of 2-alkynylphenols in promoting unstrained C–C cleavage. The DFT calculations were performed at the B3LYP-D3/SDD-6-311++ G(d,p)/SMD(DCE)//B3LYP-D3/SDD-6-311G(d,p) level of theory53,54,55 using α, β-unsaturated ketone 5a and 2-alkynylphenol 6a as model substrates. The reaction commences with the oxidative addition of ArBr with Pd(0) species via TS1, requiring a barrier of 22.7 kcal/mol (Fig. 5A). Under the basic condition (Na2CO3), the 2-alkynylphenol substrate 4a is assumed to be deprotonated to afford its sodium phenolate, which is thermodynamically favored by 1.8 kcal/mol (see details in Supplementary Information Fig. S1). The formed Pd(II) intermediate (INT1) derived from ArBr oxidative addition undergoes ligand exchange with the sodium phenolate to generate the phenoxy Pd(II) species (INT2) with alkyne coordination. Subsequently, the O-directed alkyne migratory insertion proceeds via TS2 (ΔG‡ = 22.9 kcal/mol with respect to INT1). Based on the alkenyl Pd(II) intermediate (INT3), the straightforward C(alkenyl)-C(acyl) oxidative addition can be excluded due to the extremely high barrier (TS3, ΔG‡ = 47.4 kcal/mol). In contrast, the intramolecular alkene migratory insertion into the Pd-C(alkenyl) bond is more favorable. This process includes two competing pathways, i.e., 6-endo-trig cyclization versus 5-exo-trig cyclization. The computational results show that the 5-exo-trig cyclization (TS4b) is superior to the 6-endo-trig cyclization (TS4a). The higher barrier of TS4a is mostly due to the greater conformational deformations. Although the unproductive 5-exo-trig cyclization (TS4b) has a lower barrier, it is a reversible process because the ensuing β-H elimination (TS5) requires a much higher barrier. The desired 6-endo-trig cyclization (TS4a) leads to a seven-membered palladacycle (INT4a), which may proceed via β-C elimination to cleave the targeted C–C bond. However, the computed barrier of β-C elimination (TS6b) cannot compete with TS5 along the 5-exo-trig pathway. Alternatively, the nucleophilic attack at the carbonyl carbon by the phenolate oxygen shows a much lower barrier (TS6a, ΔG‡ = 9.7 kcal/mol), generating a polyfused ring system (INT5). More importantly, this nucleophilic attack results in two tertiary carbon centers in INT5, leading to the preactivation of the targeted C(sp3)-C(sp3) bond. This is supported by the elongated C–C bond length (1.61 Å) in INT5 compared to that (1.56 Å) in INT4a (Fig. 5B). The subsequent retro-oxidative cyclization via TS7 with a barrier of 20.0 kcal/mol delivers the corresponding product 7a. In addition, although the C–C bond in INT5 is relatively preactivated, the lower barrier of TS7 (retro-oxidative cyclization) compared to TS6b (β-C elimination) can also be attributed to the distinct steric environments around the Pd center. Due to the bulky phosphine ligand (Ad2Pn-Bu), TS6b bearing a four-coordinated Pd(II) suffers from greater steric hindrance between ligand and substrate (Fig. 5B, see NCI plots in Supplementary Information). In contrast, the three-coordinated Pd(II) center in TS7 is less steric demanding.

A DFT-computed pathways for the reaction of linear carbonyls with 2-alkynylphenols. B Optimized structures for key intermediates and transition states. Key bond distances are given in Å.
Discussion
In summary, we have developed a versatile platform for modular ester synthesis via alkyne-bridging C–C bond activation of unstrained carbonyl compounds. This method enables efficient construction of biaryl-containing esters from readily available starting materials while offering good functional group tolerance and structural diversity. Compared to our previous carbene-bridging C–H activation approach, which was limited to seven- and eight-membered lactones, the current strategy provides access to a broad range of medium-to-macrocyclic lactones through simple variation of cyclic ketone precursors. Notably, the axial chirality in products can be precisely controlled through chiral elements in the reactants. We envision that this general platform will inspire new strategies for activating other challenging inert bond systems.
Methods
General procedure for the synthesis of 7
To an oven-dried Schlenk tube was added Pd (TFA)2 (5 mol%, 3.3 mg), Ad2Pn-Bu (10 mol%, 7.2 mg), Na2CO3 (2.0 equiv., 42.4 mg), starting materials 5 (0.2 mmol) and 6 (0.3 mmol). The tube was degassed and filled with dry argon, repeated three times. DCE (2.0 mL) was added via syringe. This mixture was stirred at 120 °C for 10 h. When the reaction was complete, the reaction mixture was cooled to RT and filtered through a pad of Celite. The filtrate was evaporated under reduced pressure to give the crude mixture, which was purified by flash column chromatography on silica gel to give the desired product 7.
General procedure for the synthesis of 9
To an oven-dried Schlenk tube was added Pd (OAc)2 (5 mol%, 2.3 mg), Ad2Pn-Bu (10 mol%, 7.2 mg), K2HPO4 (2.0 equiv., 69.6 mg), starting materials 5 (0.2 mmol) and 6 (0.3 mmol). The tube was degassed and filled with dry argon, repeated three times. 1,4-Dioxane (4.0 mL) was added via syringe. This mixture was stirred at 130 °C for 7 h. When the reaction was complete, the reaction mixture was cooled to RT and filtered through a pad of Celite. The filtrate was evaporated under reduced pressure to give the crude mixture, which was purified by flash column chromatography on silica gel to give the desired product 9.
Data availability
The authors declare that the data supporting the findings of this study, including experimental procedures and compound characterization, are available within the paper and its Supplementary Information files, or from the corresponding author upon request. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Center (CCDC), under deposition numbers 2418470 (9i), 2418477 (10a), 2418491 (10b), 2418471 (10g), and 2418476 (10h). These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif. Source data are provided with this paper.
References
Schreiber, S. L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 287, 1964–1969 (2000).
Galloway, W. R., Isidro-Llobet, A. & Spring, D. R. Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun. 1, 80 (2010).
Reymond, J.-L., van Deursen, R., Blum, L. C. & Ruddigkeit, L. Chemical space as a source for new drugs. MedChemComm 1, 30–38 (2010).
Yi, S., Varun, B. V., Choi, Y. & Park, S. B. A brief overview of two major strategies in diversity-oriented synthesis: build/couple/pair and ring-distortion. Front. Chem. 6, 507 (2018).
Baeyer, A. & Villiger, V. Einwirkung des Caro’schen Reagens auf Ketone. Chem. Ber. 32, 3625–3633 (1899).
Zhou, L., Lin, L., Liu, X. & Feng, X. Baeyer–Villiger (BV) oxidation/rearrangement in organic synthesis in molecular rearrangements in organic synthesis. in Molecular Rearrangements in Organic Synthesis (ed. Rojas, C. M.) 35–58 (John Wiley & Sons, 2016).
Tsakos, M., Schaffert, E. S., Clement, L. L., Villadsen, N. L. & Poulsen, T. B. Ester coupling reactions—an enduring challenge in the chemical synthesis of bioactive natural products. Nat. Prod. Rep. 32, 605–632 (2015).
Sartori, S. K., Diaz, M. A. N. & Diaz-Muñoz, G. Lactones: classification, synthesis, biological activities, and industrial applications. Tetrahedron 84, 132001 (2021).
Yu, Y., Lu, Q., Chen, G., Li, C. S. & Huang, X. Palladium-catalyzed intermolecular acylation of aryl diazoesters with ortho-bromobenzaldehydes. Angew. Chem. Int. Ed. 57, 319–323 (2018).
Yu, Y. et al. Easy access to medium-sized lactones through metal carbene migratory insertion enabled 1,4-palladium shift. Nat. Commun. 11, 461 (2020).
Yu, Y. et al. A modular approach to dibenzo-fused ε-lactams: palladium-catalyzed rridging-C-H activation. Angew. Chem. Int. Ed. 59, 18261–18266 (2020).
Zhang, F., Xin, L., Yu, Y., Liao, S. & Huang, X. Recent advances in palladium-catalyzed bridging C–H activation by using alkenes, alkynes or diazo compounds as bridging reagents. Synthesis 53, 238–254 (2021).
Xin, L. et al. Construction of protoberberine alkaloid core through palladium carbene bridging C–H bond functionalization and pyridine dearomatization. ACS Catal. 11, 1570–1577 (2021).
Zhang, F. et al. Divergent isoindolinone synthesis through palladium-catalyzed isocyanide bridging C–H activation. Cell Rep. Phys. Sci. 3, 100776 (2022).
Ding, M. et al. Alkyne insertion enabled vinyl to acyl 1,5-palladium migration: rapid access to substituted 5-membered-dihydrobenzofurans and indolines. Angew. Chem. Int. Ed. 62, e202300703 (2023).
Mi, Y. et al. Three-component Diels-Alder reaction through palladium carbene migratory insertion enabled dearomative C(sp3)-H bond activation. Nat. Commun. 15, 10844 (2024).
Xia, Y., Lu, G., Liu, P. & Dong, G. Catalytic activation of carbon-carbon bonds in cyclopentanones. Nature 539, 546–550 (2016).
Song, F., Gou, T., Wang, B.-Q. & Shi, Z.-J. Catalytic activations of unstrained C-C bond involving organometallic intermediates. Chem. Soc. Rev. 47, 7078–7115 (2018).
Deng, L. & Dong, G. Carbon‒carbon bond activation of ketones. Trends Chem. 2, 183–198 (2020).
Wang, B., Perea, M. A. & Sarpong, R. Transition metal-mediated C−C single bond cleavage: making the cut in total synthesis. Angew. Chem. Int. Ed. 59, 18898–18919 (2020).
Xia, Y. & Dong, G. Temporary or removable directing groups enable activation of unstrained C-C bonds. Nat. Rev. Chem. 4, 600–614 (2020).
Feng, Q., Wang, Q. & Zhu, J. Oxidative rearrangement of 1,1-disubstituted alkenes to ketones. Science 379, 1363–1368 (2023).
De Voss, J. J. & Cryle, M. J. Carbon-carbon bond cleavage by P450 Systems. Met. Ions Life Sci. 3, 397–435 (2007).
Xu, Y. et al. Deacylative transformations of ketones via aromatization-promoted C-C bond activation. Nature 567, 373–378 (2019).
Li, L., Fang, L., Wu, W. & Zhu, J. Visible-light-mediated intermolecular radical conjugate addition for the construction of vicinal quaternary carbon centers. Org. Lett. 22, 5401–5406 (2020).
Bhunia, A. & Studer, A. Recent advances in radical chemistry proceeding through pro-aromatic radicals. Chem 7, 2060–2100 (2021).
Zhou, X., Xu, Y. & Dong, G. Olefination via Cu-mediated dehydroacylation of unstrained ketones. J. Am. Chem. Soc. 143, 20042–20048 (2021).
Zhou, X., Xu, Y. & Dong, G. Deacylation-aided C-H alkylative annulation through C-C cleavage of unstrained ketones. Nat. Catal. 4, 703–710 (2021).
O’Reilly, M. E., Dutta, S. & Veige, A. S. β-Alkyl elimination: fundamental principles and some applications. Chem. Rev. 116, 8105–8145 (2016).
Nogi, K. & Yorimitsu, H. Carbon–carbon bond cleavage at allylic positions: retro-allylation and deallylation. Chem. Rev. 121, 345–364 (2020).
Karimzadeh-Younjali, M. & Wendt, O. F. α- and β-eliminations in transition metal complexes: strategies to cleave unstrained C−C and C−F bonds. Helv. Chim. Acta 104, e2100114 (2021).
Candeias, N. R., Paterna, R. & Gois, P. M. Homologation reaction of ketones with diazo compounds. Chem. Rev. 116, 2937–2981 (2016).
Souillart, L. & Cramer, N. Catalytic C-C bond activations via oxidative addition to transition metals. Chem. Rev. 115, 9410–9464 (2015).
Fumagalli, G., Stanton, S. & Bower, J. F. Recent methodologies that exploit C-C single-bond cleavage of strained ring systems by transition metal complexes. Chem. Rev. 117, 9404–9432 (2017).
Xue, Y. & Dong, G. Deconstructive synthesis of bridged and fused rings via transition-metal-catalyzed “cut-and-sew” reactions of benzocyclobutenones and cyclobutanones. Acc. Chem. Res. 55, 2341–2354 (2022).
Murakami, M. & Ishida, N. Potential of metal-catalyzed C-C single bond cleavage for organic synthesis. J. Am. Chem. Soc. 138, 13759–13769 (2016).
Pan, X., Luo, Y., Xia, H. G. & Wu, J. A palladium-catalyzed tandem reaction of 2-(2-bromobenzylidene)cyclobutanone with 2-alkynylphenol. Chem. Commun. 51, 16483–16485 (2015).
Zhao, W. T., Gao, F. & Zhao, D. Intermolecular sigma-Bond cross-exchange reaction between cyclopropenones and (benzo)silacyclobutanes: straightforward access towards sila(benzo)cycloheptenones. Angew. Chem. Int. Ed. 57, 6329–6332 (2018).
Li, R. et al. A ring expansion strategy towards diverse azaheterocycles. Nat. Chem. 13, 1006–1016 (2021).
Tambar, U. K. & Stoltz, B. M. The direct acyl-alkylation of arynes. J. Am. Chem. Soc. 127, 5340–5341 (2005).
Murphy, S. K., Park, J. W., Cruz, F. A. & Dong, V. M. Rh-catalyzed C-C bond cleavage by transfer hydroformylation. Science 347, 56–60 (2015).
Ito, Y., Nakatani, S., Shiraki, R., Kodama, T. & Tobisu, M. Nickel-catalyzed addition of C-C bonds of amides to strained alkenes: the 1,2-carboaminocarbonylation reaction. J. Am. Chem. Soc. 144, 662–666 (2022).
Xia, Y., Ochi, S. & Dong, G. Two-carbon ring expansion of 1-indanones via insertion of ethylene into carbon-carbon bonds. J. Am. Chem. Soc. 141, 13038–13042 (2019).
Zhang, R., Xia, Y. & Dong, G. Intermolecular [5+2] annulation between 1-indanones and internal alkynes by rhodium-catalyzed C-C activation. Angew. Chem. Int. Ed. 60, 20476–20482 (2021).
Li, H. et al. Transformations of aryl ketones via ligand-promoted C-C bond activation. Angew. Chem. Int. Ed. 59, 14388–14393 (2020).
Xu, H. et al. Ligand-promoted alkynylation of aryl ketones: a practical tool for structural diversity in drugs and natural products. ACS Catal. 11, 1758–1764 (2021).
Li, H. et al. Enones as alkenyl reagents via ligand-promoted C–C bond activation. ACS Catal. 12, 82–88 (2022).
Wang, Z. Y. et al. Palladium-catalyzed deuteration of arylketone oxime ethers. Angew. Chem. Int. Ed. 63, e202319773 (2024).
Tao, K.-L. et al. Multisite modifications of arenes using ketones as removable handles enabled by Pd and norbornene cooperative catalysis. Nat. Synth. 4, 209–218 (2025).
Kuninobu, Y. et al. Rhenium- and manganese-catalyzed carbon–carbon bond formation using 1,3-dicarbonyl compounds and alkynes. Pure Appl. Chem. 82, 1491–1501 (2010).
Zhang, R., Yu, T. & Dong, G. Rhodium catalyzed tunable amide homologation through a hook-and-slide strategy. Science 382, 951–957 (2023).
Huang, Z.-C., Ruan, Z.-L., Xu, H. & Dai, H.-X. Ring expansion of 3-hydroxyoxindoles to 4-quinolones via palladium-catalyzed C–C(acyl) bond cleavage. Chem. Commun. 61, 109–112 (2025).
Miehlich, B., Savin, A., Stoll, H. & Preuss, H. Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 157, 200–206 (1989).
Yang, W., Parr, R. G. & Lee, C. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 37, 785–789 (1988).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (Grant Nos. 22371069 and 22173103), Science and Technology Planning Project of Hunan Province (2018TP1017), Science and Technology Innovation Program of Hunan Province (2021RC4059), the Key Project of Developmental Biology and Breeding (2022XKQ0205) from Hunan Normal University and State Key Laboratory of Elemento-Organic Chemistry, Nankai University (Grant No. 202303).
Author information
Authors and Affiliations
Contributions
X.H. conceived and directed the project; X.H. designed the experiments; M.D., M.N., Q.G., S.Y., and M.X. performed the experiments; L.H. and G.L. performed the theoretical studies; Y.Y. performed the single-crystal X-ray analysis; all authors analyzed the experimental results and prepared the manuscript. M.D., M.N., L.H., and Q.G. contributed equally to this work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Pui Ying Choy and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ding, M., Niu, M., Hu, L. et al. Modular esterification of unstrained carbonyls through palladium-catalyzed alkyne bridging C-C bond activation. Nat Commun 16, 9260 (2025). https://doi.org/10.1038/s41467-025-64279-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64279-0
