
Abstract
The oxygen evolution reaction, as the anodic reaction of many electrochemical devices, plays a crucial role in energy conversion. However, the insufficient stability of non-iridium-based materials during the oxygen evolution reaction has severely limited the large-scale application of such devices. Here, using a home-made operando differential electrochemical mass spectrometry system, we show a temperature dependent mechanism evolution effect of RhRu3Ox in the oxygen evolution process, which highlights the role of temperature in triggering mechanism evolution. This effect enriches the strategies for pathway manipulation. Since different kinetic pathways can influence catalyst stability, this finding suggests that temperature-dependent pathway regulation may serve as an approach to optimize stability. To evaluate the potential of RhRu3Ox for practical applications, we assemble it into a proton exchange membrane electrolyzer and demonstrate its stability at room temperature for over 1000 hours at a current density of 200 mA cm−2. Density functional theory studies suggest that the existence of a kinetic barrier related to lattice oxygen activation might be the reason for the observed temperature dependent behavior of RhRu3Ox at elevated temperatures.
Similar content being viewed by others

Introduction
To get rid of the dependence on fossil fuels, the directly use of renewable energy to drive substance conversion and realize an efficient and clean energy utilization system is the ultimate vision to achieve sustainable development1,2,3,4,5,6,7,8,9,10. As a clean, recyclable energy, hydrogen (H2) fuels can be generated through water electrolysis with zero-carbon emission. Proton exchange membrane water electrolysis (PEM-WE) is a technique that can convert intermittent power into H2, as its high current density and hydrogen purity are unmatched by conventional alkaline water electrolysis (AWE) technology6,11. Nevertheless, its large-scale application is strongly constrained by the anodic oxygen evolution reaction (OER), as iridium (Ir)-based materials, with scarce reserves12 (0.02 − 0.10 ngIr/g) and high price, are currently the only catalysts considered OER-suitable13,14,15. So far, ruthenium (Ru)-based materials have been recognized as potential Ir-alternatives in PEM-WE devices with even much active OER activity11. Many efforts have been made to improve the stability of Ru-based catalysts, especially in three-electrode systems. However, as revealed by experimental analysis and the Pourbaix diagram1,16, under the potential of OER, Ru species will inevitably undergo peroxidation to form dissolvable RuO4 with poor stability, making them unstable when applied in PEM-WE devices for practical H2 evaluation. This Ru dissolution process is related to its OER lattice oxygen mechanism (LOM). On the contrary, the OER adsorbate-evolving mechanism (AEM) can benefit a more stable metal status as the lattice oxygen won’t evolve in the reaction11,17. Thus, the mechanism evolution might be an efficient route to boost the acidic stability of Ru-based electrocatalysts, which has been seldomly reported so far.
Here, we report the synthesis of a binary metal oxide (BMO), RhRu3Ox and its temperature-dependent mechanism evolution (TDME) effect in the OER process. In a three-electrode system, RhRu3Ox exhibited an overpotential of only 184 mV at a current density of 10 mA cm−², with stability exceeding 200 h, outperforming pristine RuO2, which sustained less than 50 h. When integrated as an anode catalyst into a proton exchange membrane water electrolyzer (PEM-WE) with Pt/C as the cathode, RhRu3Ox maintained industrially relevant current densities18 of 200 mA cm⁻² for over 1000 h at room temperature. Techno-economic analysis further confirmed its feasibility for practical applications19. However, under elevated temperatures typical of industrial conditions, Ru-based materials used as anodes led to operational instability in the PEM-WE system. To investigate the underlying mechanisms, we developed a temperature-controlled electrochemical reactor coupled with a mass spectrometer11,20,21,22,23. Using this tandem experimental setup, operando isotope labeling experiments revealed a TDME effect. That is, at room temperature, RhRu3Ox catalyzes the OER through the relatively stable AEM mechanism, while at higher temperatures, the LOM mechanism emerges, leading to a decrease in catalyst stability. Further suggested by density functional theory (DFT) calculations, we propose a plausible explanation for the high-temperature behavior of Ru-based anode catalysts and their potential applications in PEM-WE systems.
Results
Preparation and characterization of RhRu3Ox
Herein, a series of binary metal oxides, MRu3Ox, were synthesized by a three-step method as previously reported6. Firstly, the precursor salts were wet-impregnated on the structural promoter-carbon black, and then the precursor salts were reduced to form an alloy loaded on the carbon black at 900 °C in an H2/Ar atmosphere (Fig. 1a). The presence of the structural promoter can prevent the agglomeration of the catalyst at high temperatures, thus exposing more specific surface area. Secondly, in order to eliminate the effect of non-OER current caused by carbon oxidation at the OER potential, MRu3/C was annealed at 450 °C in air to remove carbon species, and the alloy is oxidized to the corresponding binary metal oxide in this process. Thirdly, the obtained binary metal oxides were acid-leached in 1 M HCl to remove the unstable components and obtain the MRu3Ox. Eventually, RhRu3Ox was optimized from a range of platinum group element binary metal oxides.

a Schematic diagram of MRu3Ox synthesis process. b XRD patterns of RhRu3Ox, Hom-RuO2 and Com-RuO2 (from up to bottom). c, d HAADF-STEM images of RhRu3Ox. The inserted histogram in (c) is the particle size distribution statistics of the corresponding RhRu3Ox NPs. e The fast Fourier transform pattern corresponds to the (d). f HAADF image and corresponding EDS elemental mapping images of RhRu3Ox. g, h Operando X-ray absorption near edge structure (XANES) (g) and extended X-ray absorption fine structure (EXAFS) (h) spectra of RhRu3Ox at the Ru K-edge. The black rectangle inserted in (g) shows the magnified operando spectrum. i Ru oxidation states as a function of the Q1 edge positions. The Q1 edge position is defined as the energy at normalized absorption of 0.5. j Energy shift of the Q1 edge positions and Ru oxidation states shift in RhRu3Ox and Hom-RuO2 at different potentials compared with the corresponding sample at 1.24 VRHE.
The X-ray diffraction (XRD) patterns (Fig. 1b) of RhRu3Ox and Hom-RuO2 match well with pure RuO2 (PDF#43-1027) and RhO2 (PDF#21-1315). Transmission electron microscope (TEM) and aberration corrected scanning TEM (AC-STEM) showed that our samples have a smaller size compared to Com-RuO2, and the average particle size of RhRu3Ox was 4.4 nm (Fig. 1c, d and Supplementary Fig. 1-4). From the corresponding Fast Fourier Transform (FFT) image of Fig. 1c, we can clearly see the exposed facets of RhRu3Ox with Miller indices of {110} and {\(\bar{1}\)10}, together with [001] orientation (Fig. 1e). Energy dispersive spectrometer (EDS) mapping and line-scan analysis demonstrate that Ru and Rh are evenly distributed (Fig. 1f and Supplementary Fig. 5). The specific surface area of RhRu3Ox was determined by the Brunauer-Emmett-Teller (BET) method to be 197.1 m2 g−1 (Supplementary Fig. 6).
The electronic structure of the catalysts was studied by X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) (Supplementary Fig. 7–10). Compared to Hom-RuO2 and Com-RuO2, the binding energies of Ru4+ 3d3/2 and 3d5/2 in RhRu3Ox show a slight negative shift (Supplementary Fig. 9), which indicates that the Ru oxidation state on the surface of RhRu3Ox is the lowest. Peak A in the XAS spectrum (Supplementary Fig. 10) at the O K-edge represents the electronic transitions from O 1s to unoccupied O 2p-Ru 4d_t2g hybridized orbitals24,25. RhRu3Ox has the lowest peak A intensity, indicating that the 4d orbital of its metal atom is occupied more by electrons, which is consistent with the conclusion obtained in XPS.
In order to better understand the potential dependent oxidation of Ru during OER, we performed the operando XAS studies. The Ru K-edge XANES spectrum26 (Fig. 1g and Supplementary Fig. 11a) corresponds to the transition from 1s to 5p. The EXAFS spectrum of RhRu3Ox and Hom-RuO2 shows a dominant peak at around 1.5 Å (Fig. 1h and Supplementary Fig. 11b), which was assigned to the Ru-O coordination. Figure 1i shows the relationship between the Ru valence state of the catalyst and Q1 absorption edge position at different applied potentials. With the increase of applied potential, the Q1 edge position of RhRu3Ox and Hom-RuO2 both move towards higher energy (Fig. 1j), but the slope of RhRu3Ox is larger, indicating that it is easier to store oxidation charges under positive bias, and more oxidation charges generally lead to higher OER performance13,14.
Oxygen evolution performance of RhRu3Ox
The OER performance of RhRu3Ox and control samples (Hom-RuO2, Com-RuO2) were carried out in 0.1 M HClO4 using a standard RDE protocol27. It can be seen from the linear sweep voltammetry (LSV) and Tafel slope curves (Fig. 2a, b, and Supplementary Figs. 12, 13) that the overpotential of RhRu3Ox delivering 10 mA cm−2 is 184 mV and the Tafel slope is 54.7 mV/decade (dec). Lower than Hom-RuO2 (208 mV, 58.1 mV/dec) and Com-RuO2 (253 mV, 66.4 mV/dec) and other platinum group elements BMO (Supplementary Fig. 14), which is in line with the operando XAFS studies discussed above. Electrochemical impedance spectroscopy (EIS) shows that RhRu3Ox has the smallest semicircle radius compared with Hom-RuO2 and Com-RuO2 (Fig. 2c), which indicates that it has the lowest charge transfer resistance and the best OER kinetics. In order to fully understand the differences between RhRu3Ox and comparison samples, we also tested the electrochemical capacitance double layer (Cdl), which is proportional to the electrochemically active surface area (ECSA). The Cdl of RhRu3Ox is 18.77 mF, which is higher than the 12.03 mF and 3.76 mF of Hom-RuO2 and Com-RuO2 (Fig. 2d and Supplementary Fig. 15 and Supplementary Table 1). This indicates that forming BMO with Rh can increase the number of active sites of Hom-RuO2.

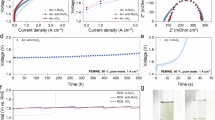
a–c LSV curves (a), Tafel slope (b) and EIS Nyquist plots (c) of RhRu3Ox, Hom-RuO2 and Com-RuO2. The solution resistance was determined from the intersection of the EIS curve with the real axis when the imaginary part (Z″) equals zero, yielding a value of 25.0 ± 0.4 Ω. d Cdl plots calculated from CV curves. e Tafel plots of RhRu3Ox at different temperatures. f Arrhenius plot of OER exchange current density of RhRu3Ox and Hom-RuO2. g Stability tests of RhRu3Ox, Hom-RuO2 and Com-RuO2. All the error bars mentioned above represent the standard deviations of 3 independent experimental data.
To assess the kinetic optimization introduced by Rh incorporation, we further investigated the Tafel slopes (Fig. 2e and Supplementary Fig. 16) of RhRu3Ox and Hom-RuO2 at elevated temperatures (30, 40, 50, 60 and 70 °C). The Arrhenius plots calculated from the temperature-dependent Tafel slope show that the apparent activation energy (Ea) of RhRu3Ox is 10.9 ± 0.5 kJ mol−1 (Fig. 2f), only about a quarter of that of Hom-RuO2 (46.7 ± 3.7 kJ mol−1), indicating enhanced reaction kinetics.
In addition, we also tested the stability of the catalysts using chronopotentiometry, as shown in the Fig. 2g, RhRu3Ox showed stability of over 200 h at 10 mA cm−2 in 0.1 M HClO4, which is much higher than Hom-RuO2 and Com-RuO2. In addition, the S number of RhRu3Ox at 200 h is 2.01 × 105 (Supplementary Fig. 17 and Supplementary Note 1), even higher than that of rutile IrO2 (~1 × 105, Sigma-Aldrich)15. Through the characterization of RhRu3Ox after stability test, we found that neither the morphology nor the electronic structure had obvious changes (Supplementary Figs. 18–20).
H2 production evaluation of RhRu3Ox under practical electrolyzer conditions
Given the comparable performance of RhRu3Ox in RDE, we decided to evaluate its activity under practical electrolyzer conditions employing a membrane electrode assembly (MEA), which better represents real device architecture. We assembled the PEM-WE devices with RhRu3Ox as anode catalyst and Pt/C as cathode catalyst, with platinized Ti fiber felt as anode porous transport layer (PTL) and carbon paper as cathode PTL (Fig. 3a, b and Supplementary Fig. 21). As shown by the current–voltage characteristic (I–V) curves (Fig. 3c), MEA using RhRu3Ox as the anode catalyst layer requires only 1.76, 1.90, 1.99, and 2.06 V to achieve a current density of 500, 1000, 1500, and 2000 mA cm−2 at room temperature, respectively. While compared to Com-RuO2 (2.15 V) and Hom-RuO2 (2.36 V) to achieve 1 A cm−2 at room temperature RhRu3Ox shows apparently stack voltage drop.

a, b photograph (a) and Schematic diagram (b) of the PEM electrolyzer. c I–V curves of the PEM electrolyzer using RhRu3Ox, Hom-RuO2 or Com-RuO2 as anodic catalyst and Pt/C as cathodic catalyst obtained in 18 MΩ DIW at room temperature with Nafion 115 membrane. d Chronopotentiometry curve of the PEM electrolyzer using RhRu3Ox as anodic catalyst and Pt/C as the cathode catalyst operated at 200 mA cm−2 at room temperature with Nafion 115 membrane. e, f Cross-sectional SEM and corresponding EDX mapping of Pt/C || Nafion 115 || RhRu3Ox without stability test (e) and after 1000 h stability test (f). g TEA for H2 production by PEM technology at 200 mA cm−2, where the black horizontal solid line represents the voltage required by RhRu3Ox at 200 mA cm−2, the black diagonal dashed line represents DOE’s 2026 H2 production cost target, and the region with a diagonal pattern represents the fossil fuel cost range. h The subdivision cost of H2 production by PEM technology at a given electricity price of 0.049 $ kWh−1 and a current density of 200 mA cm−2.
The current density-high frequency resistance (J-HFR) curve (Supplementary Fig. 22) shows the shape of first decreasing, then increasing and then decreasing, and it also shows the trend of gradually shifting to the right with Com-RuO2, Hom-RuO2, RhRu3Ox. This indicates that the mass transfer resistance of the catalyst layer composed of RhRu3Ox is also the lowest among the three (Supplementary Note 2).
What is more, in practical electrolyzer production, stability may play a more important role. Therefore, we performed the chronopotentiometry test at 200 mA cm−2 with RhRu3Ox as the anode catalyst and DIW as the electrolyte at room temperature (Fig. 3d). During its 1000 h operation time we do not see the obvious stack voltage increase, indicating that RhRu3Ox is one of the few catalysts with stability more than three orders of magnitude (103 h) in PEM-WE devices, which also proves that RhRu3Ox ranks among the most advanced Ru based OER catalysts available today (Supplementary Fig. 23 and Supplementary Table 2)1,2,3,4,5,6,7,8,9,10. From the cross-section of the Pt/C || Nafion 115 || RhRu3Ox (Fig. 3e, f), there is no significant change even after 1000 h of operation. In contrast, Hom-RuO2 showed obvious deterioration within 150 h under the same conditions (Supplementary Fig. 24).
Techno-economic analysis
To confirm the economic potential of producing H2 with RhRu3Ox as a PEM-WE anode catalyst, based on a recently reported model19 and our integrated test system, we simulated techno-economic analysis (TEA) for H2 production on an industrial scale (Supplementary Table 3 and Supplementary Note 3). The parameters used in TEA are derived from literature reports18,19,28,29 and the industrial scale pilot data we described above. H2 production costs are assessed using the International Renewable Energy Agency’s global weighted average levelized cost of electricity (LCOE) for solar photovoltaic (Supplementary Note 3), which is 0.049 $ kWh−1, at an industry-relevant current density (0.2 A cm−2). The LCOE for solar photovoltaic and onshore wind power has already met the US Department of Energy (DOE)‘s 2026 H2 production target30 of 2 $ \({{{\rm{kg}}}}_{{H}_{2}}\) −1 (Fig. 3g), and the LCOE for offshore wind power is also approaching the target.
In order to know whether our process can be profitable after scaling up production, we have listed the subdivision cost of the entire process in Fig. 3h, in which the electricity cost accounts for the highest proportion (62.1%). With the development of renewable energy generation technology, we believe that the cost of H2 will be further reduced. In addition, according to the assessment of the International Energy Agency (Supplementary Note 3), the global average levelized cost of hydrogen production from coal is between 1.9 to 2.5 $ \({{{\rm{kg}}}}_{{H}_{2}}\) −1, and the cost of H2 production with RhRu3Ox as the PEM-WE anode catalyst is 1.7 $ \({{{\rm{kg}}}}_{{H}_{2}}\) −1, which is lower than the cost of hydrogen production from coal, so the process is profitable.
Investigation of stability difference
After verifying the stability and profitability of RhRu3Ox at 200 mA cm−2, we wondered if there were other ways to increase the current density and thus further widen the profitability range. We have two options: one is to increase the stack voltage, and the other is to increase the operation temperature. However, as indicated by Pourbaix diagram, excessive stack voltage may cause the dissolve of Ru species16, so it seems not a proper option. We thus focus on changing the stack operation temperature to increase the current density. When we raise the temperature, the current density does increase at the same voltage (Supplementary Fig. 25). Nevertheless, the stack voltage showed an apparently increase when we performed the chronopotentiometry test (Supplementary Fig. 26). These phenomena have aroused our great curiosity why does the temperature increase also lead to the performance of RhRu3Ox to be unstable.
As reported previously20, if lattice oxygen participates in surface reconfiguration processes such as bonding and bond breaking in the OER process, that is, following the lattice oxygen oxidation mechanism (LOM), the stability of the catalyst will be notably affected. The relatively stable catalysts generally follow the adsorbate evolution mechanism (AEM) in which lattice oxygen does not participate in the reaction11. However, these two different mechanisms are often used to explain the stability differences between different catalysts21. This inspires us to ask whether the same catalyst will follow different reaction mechanisms at different temperatures, resulting in changes in catalyst stability when the temperature changes.
In order to uncover the mystery of the stability deterioration of RhRu3Ox at high temperatures, we decided to conduct the isotope 18O labeling experiment. However, soon we found that we were stuck in another swamp, the reactor connected to the mass spectrometer could not meet our requirements for collecting signals under variable temperature. There are two reasons: First, the traditional configuration cannot accurately control the temperature; Second, both the mainstream Wolter-Heitbaum configuration22 and the rarely used Bruckenstein configuration23 require a porous, hydrophobic polymer membrane to separate the reaction chamber from the vacuum chamber, but in our tests, we found that the airtightness of the polymer membrane hardly meets the vacuum requirements of the mass spectrometer at high temperatures23.
Therefore, we designed a electrochemical reactor configuration coupling with mass spectrometer (Fig. 4a, b), we name it operando temperature-controlled differential electrochemical mass spectrometry (TC-DEMS). With the help of this tandem experimental device (Supplementary Fig. 27), we conducted our isotope 18O labeling experiments. RhRu3Ox was labeled with 18O in H218O/0.1 M HClO4 (Supplementary Fig. 28), followed by thorough rinsing with H216O to remove all the possible 18O species absorbed by the van der Waals force.

a, b photograph (a) and Schematic illustration (b) of the operando TC-DEMS set up. c, d Triangular potential applied at room temperature (c) and 60 °C (d) and corresponding MS signal. The upper panel shows the potential (black, left axis) and current density (red, right axis) in relation to time. The lower panel shows the MS signals for 32O2 (m/z = 32, orange) and 34O2 (m/z = 34, blue). e, f The integrated peak area of m/z = 32 (e) and m/z = 34 (f) at different cycles and different temperatures. g Ratio of peak area of 34O2: 32O2. h Recommended protocol for Operando TC-DEMS experiments in OER.
We then applied a triangular wave potential to the reactor containing H216O/0.1 M HClO4 (Fig. 4c, d) to dynamically scan the redox region of the catalyst while the mass spectrometer collected the mass signal of 34O2 (16O + 18O) and 32O2 (16O + 16O). This waveform was chosen as it enables continuous access to both metal oxidation and charge transfer from lattice oxygen ligands to the metal centers, thereby providing real-time insights into potential-dependent mechanistic evolutions. Compared with static methods like chronopotentiometry or at open circuit potential, triangular scanning offers broader mechanistic coverage across a wide potential window.
At room temperature, the 34O2 signal closely followed the shape and change trend of 32O2 (Fig. 4c and Supplementary Fig. 29a). We integral the peak area of 34O2 and 32O2 (Fig. 4e, f), and find the ratio of 34O2:32O2 is about 0.4% (Supplementary Table 4), corresponding to the natural abundance of 18O of 0.2%. Moreover, the ratio of 34O2:32O2 is independent of the number of cycles (Fig. 4g blue line).
However, when we follow the same procedure except to elevated the temperature to 60 oC (Fig. 4d), we get a notably different information than at room temperature, the signal shape of 34O2 is no longer the same as that of 32O2 (Fig. 4f and Supplementary Fig. 29b), and the peak area gradually decreases with the number of cycles (Fig. 4g gray line). At the outset, the ratio of the integral peak area of 34O2:32O2 is higher than 0.4%, but gradually decreases to around 0.4% as the cycle happens31. This means that 18O was already present in the lattice of RhRu3Ox during the labeling process and was gradually replaced by 16O during the application of triangular wave potentials.
It is worth noting that the gradual decline in the 34O2:32O2 ratio over time may also be influenced by thermal isotope exchange or structural reorganization. For example, ICP confirms that Ru dissolution slightly increases under elevated temperatures (Supplementary Fig. 30), which may contribute to the concurrent loss of lattice incorporated 18O. However, XPS reveals no obvious changes in the electronic structure of RhRu3Ox, suggesting that reorganization remains relatively moderate (Supplementary Fig. 31). Although these factors may affect the rate or extent of 34O2 release, they do not undermine the mechanistic implication. The observed 34O2:32O2 signal, notably exceeding the natural abundance baseline after H216O rinsing, can be unequivocally attributed to the incorporation of 18O into the catalyst lattice. Furthermore, to exclude the possibility that the elevated 34O2:32O2 signal at 60 °C originated from thermal isotope fractionation, we conducted a control experiment using H216O/0.1 M HClO4 electrolyte under identical electrochemical conditions. As shown in Supplementary Fig. 32, the 34O2:32O2 ratio remained consistently at ~0.4% throughout the measurement, which matches the natural isotopic background. This control experiment establishes a temperature-specific baseline at 60 °C, confirming that no isotope fractionation occurs at 60 °C. Accordingly, the elevated 34O2:32O signal observed in the labeling experiment (Fig. 4g) should be attributed to the participation of lattice oxygen.
Together, these findings provide compelling evidence that the OER mechanism of RhRu3Ox at high temperatures changes from a complete AEM to lattice-involved mechanism (Supplementary Fig. 33 and Supplementary Note 4). This awakened us that a single catalyst may exhibit distinct mechanistic pathways depending on the operating temperature—a phenomenon herein referred to as the TDME effect. Specifically, the TDME effect can be considered as a gradual mechanistic evolution, in which lattice oxygen becomes increasingly involved in the reaction as temperature rises, while the AEM pathway may still remain operative. This phenomenon reflects a progressive shift in the relative contributions of different OER pathways at different temperatures. Notably, this TDME behavior provides a reasonable mechanistic explanation for the aforementioned instability of RhRu3Ox under high-temperature PEM-WE conditions. Considering the great potential of the TDME effect in researching and developing water oxidation catalysts, based on our experience of multiple failures before finally obtaining a high-quality mass spectrometry signal, we provide a protocol in the form of a logical flow chart to help readers better design experiments, detect errors, and modify reactors (Fig. 4h).
In addition, to reinforce the TDME effect suggested by operando TC-DEMS, we conducted Raman spectroscopy measurements under isotope labeling conditions. RhRu3Ox was reacted in electrolytes prepared with H218O at room temperature and 60 °C. As shown in Supplementary Fig. 34, the sample before labeling exhibits two characteristic vibrational modes at 523 and 643 cm−1, corresponding to the Eg and A1g modes of Ru–O bonding32,33, respectively. Upon labeling at room temperature, these peaks remained unchanged, indicating negligible participation of lattice oxygen. In contrast, the sample labeled at 60 °C displayed clear redshifts to 518 and 636 cm−1, respectively, confirming that 18O was incorporated into the lattice of RhRu3Ox34,35. Nevertheless, these redshifts remained below the theoretical maxima for oxygen isotope substitution, which could be attributed to the surface-localized 18O exchange and the partially involved LOM pathway during the catalytic process (Supplementary Note 5). The above temperature-dependent isotopic results provide compelling spectroscopic evidence that lattice oxygen involvement becomes significant at elevated temperatures, further supporting the TDME hypothesis.
To further rationalize the TDME behavior, it is essential to address a seemingly paradoxical observation: the onset of the mechanistic shift is not accompanied by a significant change in the Tafel slope. (Fig. 2e). This phenomenon, however, is not inherently paradoxical. As reported by Shinagawa et al. in alkaline OER36, the Tafel slope is jointly governed by the electron transfer characteristics of the rate-determining step (RDS) and the surface coverage of intermediates formed in preceding steps. Even if the RDS changes, the Tafel slope may remain largely unaffected if the coverage of key intermediates does not vary notably37. Our microkinetic modeling under acidic OER conditions (Supplementary Note 6 and Supplementary Fig. 35) further supports this notion, demonstrating that distinct RDS scenarios can produce similar Tafel behavior due to the dominant influence of intermediate coverage. These results suggest that relying solely on Tafel slopes may overlook subtle mechanistic changes, particularly in complex multistep acid OER electrocatalytic systems. In addition to the microkinetic analysis, the intrinsic nature of the LOM pathway also helps explain this apparent inconsistency. Specifically, the RDS in the AEM mechanism (M–O + H2O → M–OOH + H+ + e−) involves proton-coupled electron transfer and is thus governed by electrochemical kinetic. In contrast, the typical RDS in the LOM mechanism (M–O* + M–Olat → O2 + M* + Mvac) does not involve electron transfer and is primarily controlled by thermodynamic Brønsted–Evans–Polanyi (BEP) relationships13,38. Such an electron-transfer-independent step falls beyond the descriptive capacity of electrochemical kinetic parameters, such as the Tafel slope. Therefore, even though the underlying mechanism begins to involve LOM at elevated temperatures, the Tafel slope may remain largely unchanged. Based on the microkinetic analysis and the electron-transfer-independent nature of the RDS in the LOM pathway, the occurrence of a mechanistic evolution does not necessarily lead to a significant change in the Tafel slope; the two are neither sufficient nor necessary conditions for each other.
After having a certain understanding of the TDME effect, the next critical task is to develop effective strategies for the controllable regulation of TDME behavior. Typically, the activation of the LOM pathway is triggered by an upward shift of the O 2p band toward or even above the Fermi level (Ef), rendering lattice oxygen oxidizable and thus capable of participating in the reaction1,39. It is worth noting that an increase in temperature will lead to thermal broadening of the Fermi-Dirac distribution40. As a result, O 2p states that are fully occupied and lie below Ef at room temperature may develop partial hole character under thermal excitation, thereby electronically activating lattice oxygen and enabling the LOM pathway. To avoid thermally induced LOM activation, it is critical to ensure that the O 2p band remains below the Fermi level at the intended operating temperature. However, the position of the O 2p band should not be lowered arbitrarily. If it becomes too deep, the hybridization between O 2p and Ru 4d orbitals will be weakened, leading to reduced Ru–O covalency and a lower Ru vacancy formation energy, which in turn increases the risk of metal dissolution and structural degradation41,42. This trade-off highlights a seesaw relationship between Ru–O covalency and the O 2p band position: while elevating the O 2p band enhances covalency and bonding stability, an excessively high O 2p level can cross the Fermi level at the operating temperature, thereby triggering the LOM pathway and accelerating structural degradation.
Based on this understanding, we propose an electronic structure design strategy guided by the target operating temperature. Specifically, the suitable O 2p band position should be determined in reverse from the desired working temperature range, such that even under thermal excitation, the O 2p states remain below Ef, effectively suppressing or delaying the onset of LOM. In practice, this can be achieved by incorporating dopants to moderately downshift the O 2p band, thereby increasing the activation energy required for lattice oxygen oxidation. Additionally, selecting dopants with appropriate ionic radii can induce local lattice contraction or strain, enhance structural compactness, and suppress oxygen diffusion and O–O coupling processes, thereby further delaying the onset of the LOM mechanism from a structural perspective.
Theoretical analysis
To provide theoretical insights into the mechanisms of the switch of the RhRu3Ox, DFT computations were performed. The detailed computational methods are available in the Methods section. Firstly, it is assumed that incorporating a minor quantity of Rh would negligibly affect the surface state of the parental RuO2(110). The Rh-doped models are constructed based on this surface with a distinguished surface state. Hence, we first conduct the surface state verification on RuO2(110), and the results are presented in Fig. 5a, b. Obviously, under the potential for OER, RuO2(110) will be pre-covered by 1 ML O*. Comparative analysis of various Rh doping sites within the RuO2 matrix demonstrates that incorporation of Rh at the coordination unsaturated site (CUS) represents the most energetically favorable configuration, as depicted in Fig. 5c. With this derived structure, it is now feasible to explore the OER mechanism of the RhRu3Ox.

a, b Calculated 1D surface Pourbaix diagram as a function of potential vs. RHE (pH 1; temperature=298.15 K) (a) and 2D surface Pourbaix diagram as a function of potential vs. SHE and pH (temperature: 298.15 K) (b) of RuO2 (110). c, d AEM and LOM OER mechanisms illustration (c) and relevant free energy diagrams of these two mechanisms on RhRu3Ox and undoped RuO2 (d).
The reaction pathways for the two distinct OER mechanisms are illustrated in Fig. 5c. Steps 1 through 4 delineate the associative AEM, while steps 1, 2, 5, and 6 outline the LOM. Subsequently, the free energy diagrams for these two mechanisms are computed upon RhRu3Ox and undoped RuO2. The results are presented in Fig. 5d. The Gibbs free energy (ΔG) of the rate-determination step (RDS) for AEM and LOM on RhRu3Ox is 0.32 eV (HOO*) and 0.27 eV (O*), respectively. Clearly, RhRu3Ox is supposed to exhibit an increased propensity for LOM. However, the operando TC-DEMS experiments reveal that, under the room temperature conditions, RhRu3Ox predominantly employs AEM, rather than LOM. This preference is attributed to the kinetic barrier associated with the activation of lattice oxygen in the LOM pathway, which becomes a significant factor at lower temperatures. As indicated by the green peak in Fig. 5d, the activation barrier for this process is approximately 0.47 eV, which is much higher than ΔG for both AEM and LOM. In standard conditions, the presence of this activation barrier renders the LOM difficult to initiate. However, this barrier can be effectively surmounted at elevated temperatures, as implied by the Ab initio molecular dynamics (AIMD) simulations in Supplementary Fig. 36. Specifically, when the temperature is increased to 360 K, the average activation barrier for the LOM pathway decreases to 0.34 eV, indicating that higher temperatures facilitate a more favorable LOM process.
Discussion
In summary, we synthesized a RhRu3Ox catalyst with notable activity and stability in acidic OER. The notable performance of RhRu3Ox operating in the PEM-WE device also shows its application value in practical hydrogen production. More importantly, we discovered a so-called temperature-dependent mechanism evolution (TDME) effect for RhRu3Ox during the OER process through the operando TC-DEMS technology. The reaction path follows AEM at low temperatures, while LOM is involved at high temperatures. This discovery further enriches the toolbox for regulating OER reaction pathways. We believe that the design principles established in this study for modulating TDME behavior, along with the TDME concept itself, may potentially be extended to the understanding and optimization of a broader range of transition metal–based OER catalysts.
Methods
Materials
Rhodium (III) Chloride Hydrate (RhCl3 Rh 38.5–42.5%), Ruthenium (III) chloride hydrate (RuCl3 Ru 35.0–42.0%), Palladium (II) chloride (PdCl2 Pd 59–60%), Chloroplatinic (IV) acid hydrate (H2PtCl6 Pt 37.5%), Hexachloroiridium (IV) Acid Hydrate (H2IrCl6 Ir 36.0–44.0%) and perchloric acid (HClO4 70.0–72.0%) were purchased from Sigma–Aldrich Co., Ltd. Hydrochloric acid (HCl 36.0~38.0%) and isopropyl alcohol ( ≥99.95%) were purchased from Sinopharm Chemical Reagent Co., Ltd. Carbon black (BP2000) was purchased from Cabot Carbon Ltd. HiSPEC 40% Pt on Vulcan XC-72R (Pt/C 40 wt%) was purchased from Johnson Matthey Catalyst Co., Ruthenium dioxide (RuO2) was purchased from Premetek Co., Ltd. Nafion solution (5 wt%) was purchased from DuPont Co. Heavy-oxygen water (97.5% 18O) was purchased from Henan Herunsheng Isotope Technology Co., Ltd. All reagents were purchased without further purification. Ultrapure Milli-Q water (18.2 MΩcm−1) was used to prepare samples and electrolyte solutions.
Synthesis of catalysts
In a typical synthesis procedure, 0.2 mmol of RhCl3 was added to 20 ml of HCl, dissolved until clear, then 130 ml of deionized water (DIW) was added, followed by 0.6 mmol of RuCl3. The mixture was then ultrasonic treated for 2 h. Add 325 mg carbon black (Cabot, BP2000), the weight of which is 4 times the weight of the total metal in the precursor, and stir for 18 h to disperse to ensure uniform dispersion. Next, a rotary evaporator is used to dry the mixture at 80 °C, 100 rpm, 8 × 103 Pa to collect the remaining powder. The dried powder was placed in H2/Ar (5% H2) flowing at 900 °C for 2 h, and annealed in air at 450 °C for 3 h, and the heating rate was 5 °C/min. Take 50 mg of the sample and put it into 20 ml, 1 M HCl for acid leaching for 12 h, centrifuge, and then centrifuged and washed with deionized water for 3 times. Finally, the samples were dried overnight in an oven at 60 °C to obtain RhRu3Ox catalyst. Other samples of different binary metal oxides MRu3Ox (M = Ir, Pd, Pt, Ru) are prepared in the same way, but with different precursors.
Characterizations
X-Ray powder diffraction (XRD) was carried out on a X‘Pert MPD X-ray diffractometer with Cu Ka radiation (λ = 1.5406 Å). The morphology of the samples was analyzed by a Hitachi 7700 transmission electron microscope (TEM). High-resolution TEM (HRTEM) and corresponding energy dispersive X-ray (EDX) spectral element mapping were performed on JEM-F200 with an accelerated voltage of 20–200 KV and beam current ≥ 2.5 nA @ 0.7 nm Probe Size. High-angle annular dark-field scanning TEM (HAADF-STEM) images were recorded on the FEI Titan cube Themis G2 300 with a 200 kV probe corrector. The surface area was calculated by nitrogen adsorption-desorption isotherm using the Brunauer-Emmett-Teller (BET) method on Micromeritics ASAP 2460. The X-ray photoelectron spectra (XPS) spectra was collected on Thermo Scientific ESCALAB 250Xi, the monochromatic X-ray source is Al kα 150 W with a beam spot of 500 μm. The X-ray absorption edge spectra were collected on the BL14W1 beamline of Shanghai Synchrotron Radiation Facility (SSRF) and analyzed with the software of Ifeffit Athena. The Q1 edge position is defined as the energy at normalized absorption of 0.5. ICP data were obtained by Thermo Fisher iCAP RQ, and all powder samples were dissolved in boiling aqua regia. The Ru: Rh molar ratio of our sample is about 3.12:1, so we noted it as RhRu3Ox. Raman spectra were captured using a LabRAM HR Evolution spectrometer with an excitation wavelength of 633 nm.
Electrochemical measurements
Electrochemical measurements were carried out at room temperature in a water-jacketed five-neck cell (C011, Tianjin Aida) using a standard three-electrode configuration connected to a Multipotentiostat (IM6ex, ZAHNER Elektrik, Germany) in an O2-saturated 0.1 M HClO4 (pH = 1 ± 0.02) electrolyte, which was freshly prepared by diluting 1.66 mL of HClO4 (Sigma–Aldrich) with deionized water to a final volume of 200 mL. A Pt foil (Pt 210, Tianjin Aida) and Hg/Hg2SO4 (Saturated K2SO4, Tianjin Aida) were employed as the counter and reference electrode, respectively. In a typical test, 5 mg of catalyst was added to 1 ml of isopropyl alcohol and 20 µl of Nafion solution (5 wt%; Sigma-Aldrich) and ultrasonicated 1 h to yield a homogeneous catalyst ink. To avoid the impact of the carbon oxidation current generated by the glass carbon electrode or the contact resistance generated by the passivation of other electrode materials on the stability test, we chose the Au electrode to test our samples. Before electrode preparation, the Au electrode (area, 0.19625 cm2, Tianjin Aida) surface was cleaned by Plasma Cleaner (Harrick PDC-002) at 740 V DC, 40 mA DC, 29.6 W for 2 min. After the cleaning procedure, 16 µl catalyst ink was cast on the Au electrode in four drops, and dried in vacuum at room temperature after each drop (catalyst loading: 0.4 mg cm−2). The prepared Au electrode was assembled with the RDE assembly (Pine Instruments) as the working electrode at a rotation rate of 2500 r.p.m. For the OER experiment, LSV tests were recorded with a scanning rate of 10 mV s−1 in the potential range of 1.1–1.8 V versus RHE. All potentials were measured against the MMS reference electrode and converted to the reversible hydrogen electrode (RHE) scale by using E (vs RHE) = E (versus MMS) + 0.645 + 0.0591 × pH43,44. Electrochemical impedance spectroscopy (EIS) data were collected from 0.1 Hz to 100 kHz, at an amplitude of 5 mV at 1.45 V (versus RHE). The current density-time (i-t) curves were measured by chronopotentiometry testing at 10 mA cm−2. ECSA is calculated by: ECSA = Cdl/Cs, where Cdl is the double-layer capacitance and Cs is the specific capacitance. According to previous reports6,43, Cs = 0.035 mF cm−2 was selected for calculation. Cdl is obtained from Cdl = ic/ν, where ic is the charging current and ν is the scanning rate. To obtain ic and ν, we performed CV tests at several different scan rates (2.5, 5.0, 7.5, 10, 15, and 20 mV s−1) in the non-Faraday potential region of 1.18–1.28 V (versus RHE). S number is calculated by the following formula: \(S={n}_{{O}_{2}}/{n}_{{Ru}({{\rm{Dissolved}}})}\), where \({n}_{{O}_{2}}\) is the evolved oxygen (calculated from the total charge Q, \({n}_{{O}_{2}}\) = it/4eNA) and \({n}_{{Ru}({{\rm{Dissolved}}})}\) is the amount of dissolved total Ru (\({n}_{{Ru}({{\rm{Dissolved}}})}={c}_{{Ru}\left({{\rm{Dissolved}}}\right)*{V}_{{Solution}}}/{M}_{{Ru}}\)) measured by inductively coupled plasma-mass spectrometry (ICP-MS). To correct the ohmic drop, all potentials measured in RDE are calibrated using 100% iR compensation.
PEM–WE measurements
The membrane electrode assembly (MEA) was prepared by the catalyst coating (CCM) method. The ionomer of 30 wt% of the total anode catalyst was uniformly mixed with the anode catalyst (40 wt% for the cathode) in a solvent with a volume ratio of isopropyl alcohol to water of 1:1. A 3 mg cm−2 anode catalyst layer (RhRu3Ox, Hom-RuO2, Com-RuO2) and a 0.6 mg cm−2 Pt/C layer were formed by vacuum adsorption air brush at 40 °C on a coffee-colored Teflon high temperature cloth (Jiangsu Aokai New Material Technology Co., Ltd.). Hot press transfer was then performed on a blank membrane electrode (Nafion 115, thickness: 127μm, Anhui Contango New Energy Technology Co., Ltd.) of 2*2 cm2 at 135 °C, 11 T, 5 min (Model 4386, Carver, Inc.). The assembly of PEM-WE is shown in (Fig. 3a, b). In order to reduce the contact resistance, platinum-plated titanium alloy material is selected to make the anode polar plate, the anode PTL is platinized titanium fiber felt (0.25 mm, SCI Materials Hub), the gaskets of specific thickness on both sides are made of polytetrafluoroethylene (PTFE), the cathode PTL is carbon paper, and the cathode polar plate is made of graphite. All PEM-WE tests were performed on an integrated electrolysis system (Scribner 600 PEM Electrolysis Test System). The electrolyte we used was 18 MΩ DIW, and the flow rate was 100 ml/min. The cathode carrier gas was N2, and the flow rate was 40 ml/min. I–V curves were measured with scan current mode at 50–2200 mA cm−2 at room temperature and ambient pressure. The stability test was carried out with constant current mode at 2000 mA cm−2 at room temperature (or 60 °C) and ambient pressure.
Operando TC-DEMS
The operando TC-DEMS measurements were carried out in a homemade electrochemical reactor configuration (Yu-Qu configuration) coupling with a differential electrochemical mass spectrometer (Linglu QMG 250) and connected by a quartz capillary tube. In order to control the temperature more accurately, we also added a water-cooling system around the heating jacket to ensure the control temperature accuracy of ±0.1 °C. The working electrode used is to air-spray the catalyst on a 1*1 cm2 glass carbon sheet with a load of 0.4 mg cm−2, the counter electrode is a carbon rod, and the reference electrode is an Ag/AgCl solid electrode. In the 18O labeling step, we flushed the Ar flow to remove air from the reactor for 15 min, then we ran the reactor and heat up to target temperature. Next, we maintain a constant current mode of 10 mA cm−2, and if the required voltage is close to the RDE test results, we label it in H218O/0.1 M HClO4 for 20 min (Raman spectra were collected under the same electrochemical labeling conditions); If inconsistent, the experiment can be corrected by referring to the reasons for possible failure summarized in our protocol.
The reactor and electrodes were then washed with a large amount of 16O water and soaked with 16O water three times for 5 min each time to remove any 18O species adsorbed on the catalyst by van der Waals force. Then, reassembling the operando TC-DEMS equipment and flushed Ar flow to the reactor for 15 min. The baseline signal of all mass-selected products (m/z = 32 and 34) needs to be stabilized before heating up to the target temperature, which usually takes about 20 min. The mass signal was smoothed by averaging every twenty data points. If the signal baseline is still unstable after 30 min, check the air tightness of the reactor itself and the connection of the quartz capillary ends. After heating to the target temperature, a triangular wave potential of 1.1–1.7 V versus RHE is applied at a sweep rate of 2 mV s−1. If the signal peak can be seen to be notably higher than the baseline, the test is completed. The signal peak usually has a delay of about 400 s after the application of the angular wave potential due to capillary injection. If there is no signal peak notably higher than the baseline, please refer to our protocol again to check.
The Tafel slopes of RhRu3Ox and Hom-RuO2 at 30, 40, 50, 60 and 70 °C were measured in the same reactor. The apparent activation energy (Ea, app) is derived from the following formula:
Where j0 is the exchange current density, which can be calculated from the slope and intercept of the Tafel diagram, the formula is as follows:
Computational details
Spin-polarized density functional theory (DFT) computations were conducted via the Vienna ab initio simulation package (VASP)45, in which the projector augmented wave (PAW) method46,47 was employed. Plane-wave basis set47 was chosen to expand the Kohn-Sham48 wave functions to consider the valence electrons. The relevant kinetic energy cutoff was set at 520 eV. The revised Perdew–Burke–Ernzerhof (RPBE) functional49 was employed as a parametrization to the general gradient approximation (GGA)50 to compute the electron exchange and correlation interactions. All geometrical structures were allowed to be relaxed until the force subject to each atom less than 0.05 eV/Å. The Brillouin zone was sampled by a (3×3×1) k-point mesh51. To separate two periodic surfaces along the z-direction, a 15 Å vacuum slab was employed. The two bottommost layers were held stationary at their bulk lattice positions, while the remaining layers were permitted to undergo relaxation. The Python Materials Genomics (pymatgen)52 and Atomic Simulation Environment (ASE) libraries53 were employed to handle crystal structure manipulation, input generation, and the evaluation of surface stabilities. The computational parameters for bulk structures were derived from the Materials Project database54. Prior to the study, a series of more rigorous computational methodologies were assessed, encompassing increased kinetic energy cutoffs, expanded k-point mesh grids, reduced convergence thresholds for forces, augmented layer thickness, and expanded unit cell dimensions. Nevertheless, the variations in binding energies and the optimized adsorption geometries were found to be negligible. The methodologies for the surface Pourbaix diagrams computations were referred to the work by Hansen et al.55, with the thermodynamics calculated by employing the computational hydrogen electrode model, introduced by Nørskov et al.56, as a function of pH and electrochemical potential. Corrections for zero-point energy (ZPE) and entropy were referenced from previous investigations57 at standard conditions (298.15 K). Moreover, solvation corrections were specifically applied to the HO* intermediates, considering the pronounced stabilization effect attributable to hydrogen bonding interactions, with correction values cited from the literature58. To further probe the temperature-induced effect on the mechanism evolution from AEM to LOM, ab initio molecular dynamics (AIMD) simulations59,60 were performed using the VASP code, employing the RPBE exchange–correlation functional. The simulations utilized a plane-wave basis set with an energy cutoff of 400 eV and applied Gaussian smearing with a width of 0.1 eV. All simulations were carried out at the Γ-point with a time step of 1 fs, and the system temperature was maintained at 360 K using a Nosé thermostat over the course of 10,000 simulation steps. The structures obtained after 5000 steps were used for further analysis. To note, the relevant structures after 5000 steps are presented in GitHub in an interval of 100 steps.
Data availability
The source data generated in this study are provided in the Source Data file. MD simulation structures generated in this study have been deposited in Figshare under accession code https://doi.org/10.6084/m9.figshare.2955424761. Source data are provided with this paper.
References
Shi, Z. P. et al. Customized reaction route for ruthenium oxide towards stabilized water oxidation in high-performance PEM electrolyzers. Nat. Commun. 14, 14 (2023).
Hao, S. Y. et al. Dopants fixation of Ruthenium for boosting acidic oxygen evolution stability and activity. Nat. Commun. 11, 11 (2020).
Wang, J. et al. Single-site Pt-doped RuO2 hollow nanospheres with interstitial C for high-performance acidic overall water splitting. Sci. Adv. 8, 11 (2022).
Liu, H. et al. Eliminating over-oxidation of ruthenium oxides by niobium for highly stable electrocatalytic oxygen evolution in acidic media. Joule 7, 558–573 (2023).
Tang, J. Y., Zhong, Y. J., Su, C. & Shao, Z. P. Silver compositing boosts water electrolysis activity and durability of RuO2 in a proton-exchange-membrane water electrolyzer. Small Sci. 3, 9 (2023).
Wu, Z. Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100–108 (2023).
Wen, Y. Z. et al. Stabilizing highly active Ru sites by suppressing lattice oxygen participation in acidic water oxidation. J. Am. Chem. Soc. 143, 6482–6490 (2021).
Kim, D. et al. Electronic structure modulation of MnO2 by Ru and F incorporation for efficient proton exchange membrane water electrolysis. J. Mater. Chem. A 10, 21985–21994 (2022).
Wang, X. J. et al. Electronic structure modulation of RuO2 by TiO2 enriched with oxygen vacancies to boost acidic O2 evolution. ACS Catal. 12, 9437–9445 (2022).
Deng, L. et al. Valence oscillation of Ru active sites for efficient and robust acidic water oxidation. Advanced materials (Deerfield Beach, Fla.), e2305939 (2023).
Chen, F. Y., Wu, Z. Y., Adler, Z. & Wang, H. T. Stability challenges of electrocatalytic oxygen evolution reaction: from mechanistic understanding to reactor design. Joule 5, 1704–1731 (2021).
Goderis, S. et al. Reevaluation of siderophile element abundances and ratios across the Cretaceous-Paleogene (K-Pg) boundary: implications for the nature of the projectile. Geochim. Cosmochim. Acta 120, 417–446 (2013).
Nong, H. N. et al. Key role of chemistry versus bias in electrocatalytic oxygen evolution. Nature 587, 408–413 (2020).
Nong, H. N. et al. A unique oxygen ligand environment facilitates water oxidation in hole-doped IrNiOx core-shell electrocatalysts. Nat. Catal. 1, 841–851 (2018).
Zheng, Y. R. et al. Monitoring oxygen production on mass-selected iridium-tantalum oxide electrocatalysts. Nat. Energy 7, 55–64 (2022).
Hubert, M. A. et al. Acidic oxygen evolution reaction activity-stability relationships in Ru-based pyrochlores. ACS Catal. 10, 12182–12196 (2020).
An, L. et al. Recent development of oxygen evolution electrocatalysts in acidic environment. Adv. Mater. 33, 31 (2021).
Zhao, B. H. et al. Economically viable electrocatalytic ethylene production with high yield and selectivity. Nat. Sustain. 6, 827–837 (2023).
Mayyas., A., Ruth., M., Pivovar., B., Bender., G. & Wipke., K. Manufacturing cost analysis for proton exchange membrane water electrolyzers. (National Renewable Energy Laboratory, 2019).
Wang, Q. L. et al. Long-term stability challenges and opportunities in acidic oxygen evolution electrocatalysis. Angew. Chem. -Int. Ed. 62, 22 (2023).
Geiger, S. et al. The stability number as a metric for electrocatalyst stability benchmarking. Nat. Catal. 1, 508–515 (2018).
Wolter, O. & Heitbaum, J. Differential electrochemical mass-spectroscopy (dems) - a new method for the study of electrode processes. Ber. Bunsen-Ges. Phys. Chem. Chem. Phys. 88, 2–6 (1984).
Bruckenstein, S. & Raogadde, R. Use of a porous electrode for in-situ mass spectrometric determination of volatile electrode reaction products. J. Am. Chem. Soc. 93, 793–794 (1971).
Deka, N. et al. On the operando structure of ruthenium oxides during the oxygen evolution reaction in acidic media. ACS Catal. 13, 7488–7498 (2023).
Feng, S. et al. Intrinsic room-temperature ferromagnetism in a two-dimensional semiconducting metal-organic framework. Nat. Commun. 14, 7063 (2023).
Ribeiro, J., Tremiliosi, G., Olivi, P. & de Andrade, A. R. XAS characterization of the RuO2-Ta2O5 system local (crystal) structure. Mater. Chem. Phys. 125, 449–460 (2011).
Lazaridis, T., Stühmeier, B. M., Gasteiger, H. A. & El-Sayed, H. A. Capabilities and limitations of rotating disk electrodes versus membrane electrode assemblies in the investigation of electrocatalysts. Nat. Catal. 5, 363–373 (2022).
Lagadec, M. F. & Grimaud, A. Water electrolysers with closed and open electrochemical systems. Nat. Mater. 19, 1140–1150 (2020).
Lum, Y. et al. Tuning OH binding energy enables selective electrochemical oxidation of ethylene to ethylene glycol. Nat. Catal. 3, 14–22 (2020).
Hao, S. Y. et al. Torsion-strained iridium oxide for efficient acidic water oxidation in proton exchange membrane electrolyzers. Nat. Nanotechnol. 16, 1371–U1376 (2021).
Grimaud, A. et al. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution (vol 9, pg 457, 2017). Nat. Chem. 9, 1 (2017).
Zhang, J. H. et al. Tantalum-stabilized ruthenium oxide electrocatalysts for industrial water electrolysis. Science 387, 48–55 (2025).
He, Z. Y. et al. Activating lattice oxygen in NiFe-based (oxy)hydroxide for water electrolysis. Nat. Commun. 13, 12 (2022).
Abdullah, M. I. et al. Tackling the activity-stability paradox of reconstructed NiIrOx electrocatalysts by bridged W-O moiety. Nat. Commun. 15, 18 (2024).
Pavlovic, Z., Ranjan, C., van Gastel, M. & Schlögl, R. The active site for the water oxidising anodic iridium oxide probed through in situ Raman spectroscopy. Chem. Commun. 53, 12414–12417 (2017).
Shinagawa, T., Garcia-Esparza, A. T. & Takanabe, K. Insight on Tafel slopes from a microkinetic analysis of aqueous electrocatalysis for energy conversion. Sci. Rep. 5, 21 (2015).
Qu, M. R. et al. Ordered interfacial domain expansion catalysis enhances hydrogen evolution for proton exchange membrane electrolysis. Energy Environ. Sci. 13, (2025).
Grimaud, A. et al. Activation of surface oxygen sites on an iridium-based model catalyst for the oxygen evolution reaction. Nat. Energy 2, 10 (2017).
Grimaud, A. et al. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 9, 457–465 (2017).
Peljo, P., Villevieille, C. & Girault, H. H. The redox aspects of lithium-ion batteries. Energy Environ. Sci. 18, 1658–1672 (2025).
Jin, H. et al. Safeguarding the RuO2 phase against lattice oxygen oxidation during acidic water electrooxidation. Energy Environ. Sci. 15, 1119–1130 (2022).
Shen, Y. et al. Cr dopant mediates hydroxyl spillover on RuO2 for high-efficiency proton exchange membrane electrolysis. Nat. Commun. 15, 12 (2024).
Lin, Y. C. et al. Chromium-ruthenium oxide solid solution electrocatalyst for highly efficient oxygen evolution reaction in acidic media. Nat. Commun. 10, 13 (2019).
Su, J. W. et al. Assembling ultrasmall copper-doped ruthenium oxide nanocrystals into hollow porous polyhedra: highly robust electrocatalysts for oxygen evolution in acidic media. Adv. Mater. 30, 8 (2018).
Hafner, J. Ab-initio simulations of materials using VASP: density-functional theory and beyond. J. Comput. Chem. 29, 2044–2078 (2008).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133 (1965).
Hammer, B., Hansen, L. B. & Nørskov, J. K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 59, 7413 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188 (1976).
Ong, S. P. et al. Python Materials Genomics (pymatgen): A robust, open-source Python library for materials analysis. Comput. Mater. Sci. 68, 314–319 (2013).
Larsen, A. H. et al. The atomic simulation environment—a Python library for working with atoms. J. Phys.: Condens. Matter 29, 273002 (2017).
Jain, A. Commentary: the materials project: a materials genome approach to accelerating materials innovation. APL mater 1, 011002 (2013).
Hansen, H. A., Rossmeisl, J. & Nørskov, J. K. Surface Pourbaix diagrams and oxygen reduction activity of Pt, Ag and Ni (111) surfaces studied by DFT. Phys. Chem. Chem. Phys. 10, 3722–3730 (2008).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886–17892 (2004).
Bajdich, M., García-Mota, M., Vojvodic, A., Nørskov, J. K. & Bell, A. T. Theoretical investigation of the activity of cobalt oxides for the electrochemical oxidation of water. J. Am. Chem. Soc. 135, 13521–13530 (2013).
Gauthier, J. A., Dickens, C. F., Chen, L. D., Doyle, A. D. & Nørskov, J. K. Solvation effects for oxygen evolution reaction catalysis on IrO2 (110). J. Phys. Chem. C. 121, 11455–11463 (2017).
Marx, D. & Hutter, J. Ab initio molecular dynamics: Theory and implementation. Mod. Methods Algorithms Quantum Chem. 1, 141 (2000).
Zhu, A. et al. Rational design of precatalysts and controlled evolution of catalyst-electrolyte interface for efficient hydrogen production. Nat. Commun. 16, 1880 (2025).
Qu, M. R., AIMD simulation data supporting Temperature-dependent mechanism evolution on RhRu3Ox catalyst for acidic water oxidation, Figshare, https://doi.org/10.6084/m9.figshare.29554247 (2025).
Acknowledgments
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grants XDB0450402, S.-H.Y.), the National Key Research and Development Program of China (2021YFA0715700, S.-H.Y.), the National Natural Science Foundation of China (Grants 22293044, 22205223, S.-H.Y.), the Major Basic Research Project of Anhui Province (2023z04020009, S.-H.Y.), the New Cornerstone Investigator Program, the JSPS KAKENHI (No. JP25K01737 and JP24K23069, S.-H.Y.). We acknowledge the MASAMUNE-IMR at the Center for Computational Materials Science (Institute for Materials Research, Tohoku University) (No. 202412-SCKXX-0204 and 202412-SCKXX-0211, H.L. (Hao Li)), the JSPS KAKENHI (No. JP25K01737 and JP24K23069), and the Institute for Solid State Physics (ISSP) at the University of Tokyo for providing supercomputer resources. This work was partially carried out at the USTC Center for Micro- and Nanoscale Research and Fabrication. We acknowledge SCI Materials Hub, Beijing Scistar Technology Co., Ltd., and Shanghai Linglu Instrument Equipment Co., Ltd. for their assistance with the PEM-WE and DEMS tests.
Author information
Authors and Affiliations
Contributions
S.-H.Y. conceived and supervised the project. M.-R.Q., S.Z., and R.W. performed the experiments, collected, and analyzed the data. M.-R.Q. independently conducted PEM-WE testing and developed TC-DEMS technology. H.L. (Heng Liu) and H.L. (Hao Li) carried out the DFT calculations. J.X. performed the HAADF-STEM measurements. Y.-Y.Q. helped with the synthesis of the materials. S.-H.F., X.-Z.S., H.-L.D., R.-Q.L., and W.-S.Y. performed the XANES and EXAFS experiments and analyzed the data. S.-H.Y., M.-R.Q., S.Z., and R.W. co-wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Javier Brey and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qu, MR., Liu, H., Feng, SH. et al. Temperature-dependent mechanism evolution on RhRu3Ox for acidic water oxidation. Nat Commun 16, 9261 (2025). https://doi.org/10.1038/s41467-025-64286-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64286-1
