
Abstract
Local adaptation may facilitate range expansion during invasions, but the mechanisms underlying successful invasions remain unclear. Cheatgrass (Bromus tectorum), native to Eurasia and Africa, has invaded globally, with severe impacts in western North America. We aim to identify mechanisms and consequences of local adaptation in the North American cheatgrass invasion. We sequence 307 range-wide genotypes and conduct controlled experiments. We find that diverse lineages invaded North America, where long-distance gene flow is common. Nearly half of North American cheatgrass comprises a mosaic of ~19 locally adapted, near-clonal genotypes, each seemingly very successful in a different part of North America. Additionally, ancestry, phenotype, and allele frequency-environment clines in the native range predict those in the invaded range, indicating pre-adapted genotypes colonized different regions. Common gardens show directional selection on flowering time that reverse between warm and cold sites, potentially maintaining clines. In the USA Great Basin, genomic predictions of strong local adaptation identify sites where cheatgrass is most dominant. Our results indicate that multiple introductions and migration within the invaded range fuel local adaptation and success of cheatgrass in western North America. Understanding how environment and gene flow shape adaptation and invasion is critical for managing ongoing invasions.
Similar content being viewed by others

Introduction
Biological invasions are a major cause of global biodiversity decline and ecosystem disruption, but the mechanisms driving ongoing invasions remain poorly understood1,2. In particular, the role of adaptive evolution in enabling invasive species to succeed is poorly understood. Is invasion success primarily determined by the susceptibility of invaded ecosystems3, or are the worst invaders adapted to spread and dominate4? For example, local adaptation to invaded environments could increase fitness and abundance5, while ongoing gene flow into the invaded range could swamp or reshape local adaptation6,7. If colonizing propagules are diverse, new populations may quickly adapt to local environments, facilitating invasive spread4,8. However, colonizing genotypes may reach new environments to which they are maladapted, swamping local adaptation and potentially hindering further spread6,7,9,10. Furthermore, if the diversity of colonizing propagules is low, new populations may be unable to adapt to new conditions11, phenotypic plasticity of invasive genotypes may counteract local adaptation1, and/or colonization bottlenecks may increase the frequency of deleterious mutations12. Testing these hypotheses requires a rare combination of genomic, fitness, and abundance data13.
Multiple mechanisms could contribute to local adaptation during invasions, generating distinct patterns of genomic and phenotypic variation14,15. In general, selection may change along environmental gradients and promote genotypic and phenotypic clines16. If environmental gradients are similar in native versus invaded regions, clines may be similar between native and invaded regions, indicating niche conservatism of different lineages17,18,19. Alternatively, if selective pressures are novel in the invaded range, clines may be distinct between native and invaded regions, suggesting niche shift of some lineages19,20,21,22. Furthermore, invasive genotypes may closely match the genetic diversity of the native range23, represent newly admixed populations24, or form novel genotypes via introgression from congeners25. Understanding successful invasions thus requires dissecting global patterns of genomic and phenotypic variation, which has seldom been accomplished. Although some studies have examined genomic and phenotypic differences between native and invasive populations, sampled populations between ranges are hard to compare because they often differ in spatial scale and/or do not incorporate enough environmental variation26.
Bromus tectorum L. (cheatgrass) is a grass native to Eurasia and northern Africa that spread across North America by the 1890s27,28, heavily influencing ecological dynamics of arid and semi-arid ecosystems of the North American Intermountain West29. At least some introductions likely came via contamination in grain shipments27,28. Cheatgrass occurs in high abundance across an estimated 31% (210,000 km2) of this region30, displacing native perennials via rapid reproduction and shortened fire return intervals29, reducing biodiversity and degrading wildlife habitat31. It is highly selfing, typically winter annual, with a high-quality rerence genome (~2.5 Gb)28,32. Existing genetic studies, while limited to small numbers of markers or populations, suggest that multiple introductions from different regions in Europe might have occurred in North America28,33,34,35,36,37,38. Studies have shown evidence for local adaptation in phenology at small scales39,40,41,42,43, and substantial genetic differentiation in phenology between populations from different regions32,44, although range-wide patterns of local adaptation remain elusive. Due to cheatgrass’s high rate of selfing, novel recombinant genotypes are expected to be rare, limiting novel genomic diversity in the invaded range. However, repeated introductions into North America and post-introduction dispersal could have promoted the adaptative potential and invasive spread of cheatgrass populations.
Here, we aim to identify mechanisms and consequences of local adaptation in the North American cheatgrass invasion. We hypothesize that for cheatgrass, as a self-pollinating species with few constraints on dispersal, local adaptation in the invaded range would be likely due to rapid spread of pre-adapted genotypes to suitable environments (as opposed to adaptation by de novo mutation or novel admixtures), provided there were multiple diverse introductions. We sequence whole genomes of a global panel of 307 genotypes from the native and invaded ranges and measure phenotypes and performance in one growth chamber experiment and two field common gardens. We ask whether there were multiple and diverse introductions to North America and examine genetic consequences of the invasion. We evaluate how geography, environment, and phenotype shape genomic diversity. We test whether ancestry, trait, and allele frequency-environment clines were repeated in native and invasive genotypes, and if selection maintains clines. Finally, we integrate field surveys of cheatgrass abundance in the USA Great Basin30 to assess whether genomic matching to local climates facilitated invasive dominance. Our results reveal that multiple introductions and migration within the invaded range fueled local adaptation and success of cheatgrass in western North America.
Results and discussion
Diverse native range ancestries invaded North America
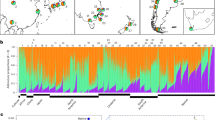
Cheatgrass populations in North America stem from multiple, diverse introductions. Using ~267k unlinked single-nucleotide polymorphisms (SNPs), different clustering analyses of global genomic variation showed that population genetic structure largely followed geography in the native range and to a lesser degree in North America (Fig. 1 showing K = 4 ancestral genetic clusters/ancestries, Supplementary Figs. 1, 2). In the native range, west Asian, Mediterranean, and Atlantic genotypes primarily fell in a single ancestry, while central and eastern European genotypes were mostly assigned to two ancestries differentiated by latitude and were overall more intermediate (i.e., composed of multiple ancestries). In the invaded range, genotypes were assigned to all four ancestries in western North America (WNA, west of the Rocky Mountains), but only to two ancestries in eastern North America (ENA, east of the Rocky Mountains) (Fig. 1a–c, Supplementary Fig. 3). The majority of invasive genotypes were similar to genotypes from north, central, or eastern Europe (Fig. 1d–f). In WNA, however, warm desert genotypes in southern California and Nevada were similar to genotypes from Iran and Afghanistan. The warm Mojave and the cool Pacific Northwest also harbored genotypes similar to those from the western Mediterranean (Fig. 1e, f). For regions with less extensive invasions, results showed that: Argentines are similar to Spanish genotypes, an Australian is similar to western Mediterranean genotypes and a widespread lineage from WNA, New Zealand genotypes are similar to northeastern European genotypes, and a Korean genotype is similar to central eastern European genotypes and a widespread lineage from ENA.

a Admixture proportions for K = 4 ancestral genetic clusters (colors) for invasive and native genotypes in different regions; WNA: western North America (n = 107), ENA: eastern North America (n = 67), out: not in North America (n = 8), MD: Mediterranean (n = 24), NCE EU: north-central-east Europe (n = 53), WA: west Asia (n = 28). Geographic distribution of b invasive (n = 194, North American only) and c native (n = 105) genotypes. d Genetic differentiation (FST) between native and invaded regions, with notations following a. e Principal components analysis showing PC1 (y-axis) and PC2 (x-axis) explaining 20.6% of genomic variation. Axes are shifted to better reflect the latitudinal distribution of genotypes. Gray letters denote geographic origin in the native range and stars represent genotypes in the invaded range. f Neighbor-joining tree annotated with native (gray letters) and invaded locations (black numbers and stars). Native notations follow the ISO alpha-3 country code or their cardinal direction in Europe (EU). Black numbers mark groups of 2–14 near-clonal, and often widely distributed, invasive genotypes. Stars mark branches with invasive genotypes. Source data are provided in Supplementary Data 1, and the genetic data for e, f is publicly available via Figshare at https://doi.org/10.6084/m9.figshare.29367845.
The diversity of genotypes found in WNA reflects colonization by propagules from different native regions, while patterns in ENA reflect reduced genetic diversity. Accordingly, population-specific FST (i.e., the degree of relatedness among individuals) was higher in the invaded compared to the native range (0.2 and 0.03, respectively), especially for ENA (0.39) compared to WNA (0.18). Pairwise FST values were lowest between European and North American genotypes, while other pairs of regions were more diverged (e.g., those involving the Mediterranean and west Asia, Fig. 1d). The native and invaded range were moderately genetically differentiated (FST = 0.11), comparable to the differentiation between genotypes from ENA and WNA (FST = 0.12). In the native range, pairwise FST values were larger (Fig. 1d), showing strong divergence between European and west Asian genotypes (FST = 0.25).
WNA and ENA: different patterns of diversity
Much of North America harbors great genomic diversity with little evidence of elevated genetic load and inbreeding compared to the native range (Supplementary Figs. 4–6 using ~15.1 M SNPs). In WNA, nucleotide diversity (π, Supplementary Fig. 4a, b) was comparable to the most diverse native region, north-central-eastern Europe (0.0016 ± 4.5 × 10–6 se vs. 0.0018 ± 4.6 × 10–6 se, respectively), followed by the Mediterranean (0.0015 ± 3.8 × 10–6 se) and west Asia (0.0011 ± 3.1 × 10–6 se). Nucleotide diversity was much lower in ENA (0.0009 ± 4.5 × 10–6 se). In WNA, the skew in the site frequency spectrum (Tajima’s D, Supplementary Fig. 4c, d) was positively shifted (mean = 2.8 ± 0.006 se), indicating an excess of intermediate-frequency SNPs, consistent with strong population structure and heterogeneous ancestry across the region (see also ref. 38). In ENA, Tajima’s D was low (mean = 0.5 ± 0.009 se), indicating more rare variants and suggesting recent population expansion. In the Mediterranean and north-central-eastern Europe, Tajima’s D was positively shifted (Mediterranean mean = 1.6 ± 0.005, north-central-eastern Europe mean = 2.2 ± 0.007), reflecting substantial population structure within these regions. The Mediterranean comprises multiple distinct eastern and western lineages (see also ref. 45), while north-central-eastern Europe comprises multiple lineages with some intermediate genotypes. In contrast, West Asian genotypes appeared more closely related to each other (Tajima’s D mean = 0.3 ± 0.006).
To understand the effects of potential bottlenecks and drift in North America, we first examined deleterious mutation load using ~15.1 M SNPs, under the hypothesis that most protein changing mutations are deleterious. Estimated mutation load was not different between native versus invaded range genotypes of the same ancestry (two-way ANOVA: range F(1290) = 57.8, p = 4 × 10–13, ancestry F(4290) = 46.7, p < 2 × 10–16, interaction F(3290) = 4.4, p = 0.005; Tukey HSD range p = 0.2, Supplementary Fig. 5). The central-eastern European ancestry (teal in Supplementary Fig. 5), widespread in North America, showed the lowest load in both ranges, suggesting large effective population size at some point in the past. In contrast, the West Asian and Mediterranean ancestry (pink in Supplementary Fig. 5) was associated with higher load in both ranges.
Next, we examined runs of homozygosity (ROH46) using a panel of 101 closely related native and invasive genotypes sequenced directly from field collections (Supplementary Fig. 6). Native and invasive genotypes (grouped by range) had similar Tajima’s D, thus similar skew in the site frequency spectrum. The native group, however, had lower counts of ROH and a much higher FROH (the proportion of the genome with ROH), resulting in inference of a strong selfing rate (Supplementary Fig. 6a–c). Selfing rates were significantly different between the native and invasive groups (two-tailed t-test t = 3.84, df = 11.90, p = 0.002) (Supplementary Fig. 6c). Moreover, selfing rates appeared significantly higher in ENA compared to WNA (two-tailed t-test t = 2.81, df = 7.37, p = 0.025) (Supplementary Table 1). This could reflect relaxed selection for reproductive assurance in invasive genotypes from specific environments. For example, although selfing is more common, some highly inbred desert lineages appeared overrepresented among parents of heterozygotes in a previous common garden experiment47. Our results suggest that the North American cheatgrass invasion is not associated with higher inbreeding due to selfing compared to the native range (Supplementary Fig. 6c, d).
Taken together, the high diversity in WNA indicates great potential for adaptation in this heavily invaded region. In contrast, the lower diversity in ENA reflects colonization by a few closely related lineages (see also ref. 34) that persist as ruderal plants in urban and agricultural environments.
Strong isolation-by-environment in North America
Both geography and environment shape genomic diversity in the native range, but geography plays a weak role in North America. Isolation-by-distance (based on ~267k SNPs) was strong in the native range (geographic vs. genetic distance Mantel p = 10–4, Fig. 2a) but very weak in North America (Mantel p = 0.03, Fig. 2b). At 0–100 km distance, pairs of distantly related genotypes were common in WNA, but not in ENA or the native range (Supplementary Fig. 7a, b). Even at the smallest scales (0–25 km), isolation-by-distance appeared weaker in the invaded compared to the native range (Supplementary Fig. 7c, d). Moreover, several groups in North America (“1–19” in Fig. 1f) composed of 2–14 near-clonal genotypes ( > 98% SNPs identity) were found across distances of >3000 km (Fig. 2b, Supplementary Fig. 7a, 8a). In contrast, such widely distributed, near-clonal genotypes were absent in the native range (Fig. 2a, Supplementary Fig. 7b). These patterns suggest long-distance dispersal within North America by lineages descended from distinct native range populations. Furthermore, groups of near-clonal genotypes occupied significantly different environments (PERMANOVA of multivariate environment predicted by clonal group: p = 0.0001, R2 = 0.65) that together encompass the extent of climate space in North America (Supplementary Fig. 8b). This suggests that although genotypes might not be dispersal limited in North America, their spread may be limited by different environmental constraints, suggesting local adaptation. The weak spatial patterns in North America may also reflect genotype sorting along the steep, heterogeneous climatic gradients that are common in WNA. Pairwise climatic distance (based on Euclidean distance in Supplementary Fig. 7e) significantly increased with spatial distance in both native and invaded ranges (Mantel p = 10–4 in both ranges, native Mantel Spearman r = 0.7, invaded Mantel Spearman r = 0.6; Supplementary Fig. 7f, g), but this relationship was weak in WNA (Mantel Spearman r = 0.3 WNA vs. 0.7 ENA), reflecting the fine-scale climatic heterogeneity of this region.

Strong isolation-by-distance in the a native (**Mantel p = 10-4) but weak in the b invaded range (*Mantel p = 0.03); plots show raw pair-wise data with a spline. Euler Plots show genomic variation is best explained by both the abiotic environment and spatial distance in c the native range, but only by the abiotic environment in d the invaded range. Fields of squares represent total genomic variation, circles represent genomic variation explained by a particular group of variables calculated using variance partitioning with RDA ordination (native n = 105, invaded n = 194). e Native and f invasive genotypes projected on the first two canonical axes of RDA (x-axis: RDA1, y-axis: RDA2). Arrows represent environmental predictors that strongly correlate with a maximal proportion of variation in linear combinations of SNPs. ELV: elevation, PET: potential evapotranspiration, PRC: total annual precipitation, PSE: precipitation seasonality, TAR: temperature annual range, TDR: temperature diurnal range, TMP: annual mean temperature. Genotypes are colored by K = 4 ancestral clusters. Geographic annotations are depicted in bolded black; N EU: north Europe, E EU: east Europe, C EU: central Europe, MD: Mediterranean, WA: west Asia, W coast: west coast, InterM. W: intermountain West, ENA: Eastern North America. Source data are provided in Supplementary Data 1; genetic and geographic distances are publicly available via Figshare at https://doi.org/10.6084/m9.figshare.29367845.
To further examine genomic differentiation along climate gradients, we performed redundancy analysis (RDA) with variance partitioning, comparing the role of climate and spatial variables in explaining genomic variation. SNP variation was better explained by these predictors in the native than in the invaded range (native R2adj = 0.25, invaded R2adj = 0.10; Fig. 2c, d). Spatial variables explained little SNP variation in North America (native R2adj = 0.07, invaded R2adj = 0.005), confirming low isolation-by-distance. In both ranges the abiotic environment explained the largest portion of SNP variation (native R2adj = 0.13, invaded R2adj = 0.09; Fig. 2e, f), highlighting the importance of isolation-by-environment in both the native and invaded range and consistent with invasive local adaptation via native pre-adaptation19.
Repeated ancestry-climate clines
Ancestry-environment clines were remarkably similar in the native and invaded ranges, suggesting environmental filtering of pre-adapted genotypes that could disperse long distances or via directed gene flow (as opposed to local adaptation by novel genotypes). We focused on aridity and temperature gradients representative of global climatic variation in the cheatgrass range (see Supplementary Fig. 7e) and used generalized-additive-models (GAMs) to detect significant ancestry-climate trends between ranges (Supplementary Fig. 9). In native and invasive genotypes, the west Asian and Mediterranean genetic cluster (pink) was more frequent in drier regions (GAM p = 0.0004, pseudo-R2 = 0.5), the northern Europe cluster (blue) was more frequent in humid regions (GAM p = 0.007, pseudo-R2 = 0.08), the central Europe cluster (teal) was more frequent in regions with little precipitation seasonality (GAM p = 10–5, pseudo-R2 = 0.2), and the presumably northeast Europe ancestry (green) was more frequent in regions with colder winters (GAM p = 0.002, pseudo-R2 = 0.1).
Repeated phenotype-climate clines
Consistent with the hypothesis that pre-adaptation to local climate facilitated the cheatgrass invasion, we found similar phenotype-environment clines in the invaded and native ranges. A principal components (PC) analysis on genetic variation among 169 native and invasive genotypes for eleven growth chamber phenotypes (Supplementary Data 1, Supplementary Table 2) detected multi-trait axes of variation (Fig. 3a). PC1 explained 35.4% of the variation and suggested a life history axis of delayed flowering and high vegetative investment (more tillers and leaves) versus rapid flowering and high reproductive investment (taller, more fecund inflorescences). PC2 explained 22.2% of the variation and indicated an axis associated with larger plants with greater growth after vernalization versus shorter plants with little growth after vernalization. Native genotypes had on average earlier flowering (two-tailed t-test t = 4.09, df = 41.36, p = 0.0002) and higher reproductive investment (two-tailed t-test t = –1.92, df = 43.69, p = 0.06) than invasive genotypes (PC1 two-tailed t-test t = 3.02, df = 38.14, p = 0.004) which may be due to different ancestry proportions in the native range. We found no significant native versus invasive trait differences after accounting for relatedness (p > 0.4), thus no evidence for evolution of increased competitive ability by invasive cheatgrass48. Additionally, near-clonal groups were significantly different in multivariate phenotypes (PERMANOVA of multivariate phenotype predicted by clonal group: p = 0.03, R2 = 0.39), yet the remaining non-clonal genotypes were also diverse (Supplementary Fig. 8c). These results highlight how North America hosts diverse life histories.

a Eigenvector plot with loadings of eleven phenotypes onto PC1 (x-axis) and PC2 (y-axis) describing axes of life history variation of 169 genotypes in a growth chamber; fl: Flowering, n: Number, inflor: Inflorescence. b Growth chamber phenotype-environment associations for invasive (left; n = 138–145) and native genotypes (right; n = 31–36). Coefficients of determination (R2), trends (gray lines), and 95% confidence intervals (gray shades) come from two-sided linear regressions. Significance comes from two-sided linear-mixed kinship models (i.e., accounting for relatedness among genotypes) of trait in response to environment: invaded PC1 *p = 0.02, PC2 *p = 0.03, days to flower *p = 0.03; native PC1 ***p = 9.3 × 10−6, PC2 *p = 0.01, days to flower ***p = 1.8 × 10−8 (only the two most relevant climate variables were tested, thus p-values are not adjusted for multiple comparisons). c Fitness advantage of early flowering genotypes at a warm site/common garden (WI Wild Cat, gray crosses, n = 93) and of late flowering genotypes at a cool site/common garden (SS Sheep Station, gray open circles, n = 82) in two consecutive years (top: 2022 Spring harvest and bottom: 2023 Spring harvest). Trends (gray lines) and 95% confidence intervals (gray shades) come from linear regressions. Significance comes from linear-mixed kinship models of fitness (seed count for 2022 and inflorescence mass for 2023) in response to mean first day of flowering (fl), site, and their interaction (int): Spring 2022 fl ***p = 7.4 × 10−6, site ***p = 5.3 × 10−6, int ***p = 3.7 × 10−7; Spring 2023 fl *p = 0.006, site **p = 0.0006, int **p = 0.0003. In all panels genotypes are colored by K = 4 ancestral clusters. Source data are provided in Supplementary Data 1.
Multiple trait-climate clines potentially maintained by selection were mirrored between the native and invaded range (Fig. 3b), indicating sorting of genotypes along humidity and temperature gradients. We focused on two climate variables that we hypothesized would capture distinct climatic stressors: maximum vapor pressure deficit (Pa), for drought adaptation, and mean winter temperature (°C), for cold adaptation (Supplementary Fig. 7e). To test for evidence of selection maintaining clines, we used linear-mixed models that accounted for genomic similarity (lmkin below), similar to QST–FST tests49. When significant, these models suggest selection is driving trait-climate clines, because the cline is stronger than expected by the genome-wide patterns of variation. In native and invasive genotypes, earlier flowering was associated with higher aridity (native lmkin-p = 2e–8, R2 = 0.5; invaded lmkin-p = 0.03, R2 = 0.3), suggesting a locally adaptive cline of rapid phenology/early reproductive investment in arid regions versus delayed phenology/early vegetative investment in humid regions. Also, clines showed evidence of selection specifically within WNA, but not in ENA (Supplementary Fig. 10). These patterns suggest local adaptation via life history clines in WNA. In contrast, life history in ENA is not associated with climate, potentially because a single generalist ruderal strategy is adaptive throughout ENA.
Selection on flowering time along a temperature gradient in WNA
In field common gardens, selection on flowering time changed direction between sites that differed in temperature. To test whether phenotypic clines in WNA were promoted by selection, we conducted two common garden experiments in different climates in Idaho with fall plantings across two years (2021 and 2022). One site was cooler (Sheep Station, ID, USA 44.2456°N, 112.2144°W, annual mean temperature 6 °C) and the other warmer (Wildcat, ID, USA 43.4744°N, 116.9018°W, annual mean temperature 12 °C). We planted 95 diverse genotypes from across WNA, for a total of 14,800 plants50. We measured flowering time, survival, and fecundity. In both years (Fig. 3c), selection favored later flowering at the cool site, with late flowering genotypes having ~3× the fitness of earlier flowering genotypes ( ~ 300 vs. ~100 seeds produced per original sown seed). By contrast, at the warm site, selection favored earlier flowering, with the earliest flowering genotypes having ~10× the fitness of the later flowering genotypes ( ~ 170 vs. ~17 seeds produced per original sown seed). This suggests that late flowering genotypes have an extreme disadvantage in warm climates. This strong selection is consistent with our finding that the hottest sites in WNA were almost exclusively comprised of west Asian-like genotypes. We saw no clear admixture from distantly related, but geographically proximate European-like genotypes inhabiting cooler and wetter higher elevations in WNA (Fig. 1a), suggesting a barrier to dispersal of maladapted genotypes. Thus, climate gradients in WNA appear to impose changes in selection, maintaining a strong phenotypic cline.
Repeated allele frequency-climate clines
Putative quantitative trait loci (QTL) for traits under selection showed similar allele frequency-climate clines in the invaded and native ranges (Fig. 4, Supplementary Figs. 11–12). Using genome-wide association studies (GWAS), we identified several genetic loci associated with variation in flowering time and number of tillers (Supplementary Data 2). We implemented two methods that accounted for population genetic structure: univariate mixed model LMM and multilocus mixed model MLMM. Of the 400 top GWAS SNPs (100 top SNPs × 2 phenotypes × 2 GWAS methods), just one SNP was segregating exclusively within the invaded range. The other 399 SNPs segregated in both native and invaded ranges, supporting the hypothesis that de novo mutations have not been major drivers of local adaptation in North America.

a, d Geographic distribution of QTL SNP alleles in the native (top) and invaded (bottom) range; black crosses represent the reference/major allele, and red open circles the alternate/minor allele. b, e Zoomed-in Manhattan plots showing two-sided Wald-test p-values (plotted as –log10) from GWAS and genomic location of top SNP (marked in green), with respective false-discovery-rate (FDR) and minor allele frequency (MAF). c, f Phenotypic (boxplots to the left) and environmental variation (boxplots to the right) of flowering time QTL SNP alleles identified with GWAS. Boxplots indicate median (middle line), 25th, 75th percentile (box), and whiskers cover the data extent. Max VPD: Maximum vapor pressure deficit in kPa. A, G, T, C on maps and x-axis of boxplots indicate nucleotides. Listed p-values come from two-sided t-tests comparing GWAS SNP alleles/nucleotides: c native days to flower **p = 0.002, max VPD ***p = 0.0004, invaded days to flower ***p = 5.5 × 10−6, max VPD ***p = 0.0002; f native days to flower ***p = 4.4 × 10−7, max VPD ***p = 3.4 × 10−8, invaded days to flower ***p = 2.2 × 10−16, max VPD ***p = 0.0004. On the x-axis of boxplots n individuals denotes the exact number of homozygous individuals/genotypes carrying each nucleotide (flowering time: native n = 32, invaded n = 138–139; max VPD: native n = 99–104, invaded n = 193–194). Source data are provided in Supplementary Data 1, 2.
We annotated SNPs based on genome-wide linkage-disequilibrium (LD, Supplementary Fig. 13a). We found that at 194.5 kb, LD decayed to ~80% of the background LD (here taken as 5 Mb). A similar LD decay pattern was observed across chromosomes (Supplementary Fig. 13b), while between chromosomes average R2 ~ 0.1. Below, we thus highlight QTL based on the position of the closest gene within a 200 kb window centered at the GWAS SNP. We focus on flowering time because this was the only trait for which we found gene functions clearly related to phenotype.
The top flowering time QTL (detected with LMM) contained multiple SNPs along a haploblock of ~28 Mb (chromosome 1: 56–84 Mb, allele frequency (AF) ~ 0.9) containing ~64 genes with annotations based on homology to Oryza sativa and Arabidopsis thaliana. These genes were enriched for gene ontology terms describing developmental processes involving reproductive structure/system, embryo, embryo ending in seed dormancy, post-embryonic, fruit, and seed (8 O. sativa genes and 14 A. thaliana genes, p < 0.0003, FDR = 0.01). Such a large haploblock could indicate a structural variant, a potential driver of local adaptation51,52. This locus thus merits further investigation.
The top SNP of the haploblock (chromosome 1: 71007448 bp, AF = 0.91; top in LMM and 2nd top in MLMM) was 25 kb downstream of a O. sativa homolog, the DnaJ protein Erdj3b. Expression of Erdj3b in O. sativa is critical for heat stress tolerance during seed development53. Late flowering alleles were more frequent in humid/colder regions of the native (two-tailed t-test t = –3.66, df = 83.61, p = 0.0004) and invaded range (two-tailed t-test t = –4.31, df = 26.41, p = 0.0002) (Fig. 4a–c), suggesting cheatgrass adaptation to temperature gradients may be linked to seed sensitivity to temperature stress.
The fourth top flowering time QTL (only found with LMM) comprised three SNPs (chromosome 1: 236616590 bp, 236616999 bp, 236617691 bp, AF = 0.82) 0.5 kb upstream (putative promoter region) of the A. thaliana homolog ATE1 (AT5G05700). ATE1 regulates seed maturation, seedling metabolism, and abscisic acid germination sensitivity54. Early flowering alleles were found in drier regions of the native (two-tailed t-test t = 6.73, df = 42.19, p = 3.4e–8) and invaded range (two-tailed t-test t = 4.82, df = 11.67, p = 0.0004), specifically the Mediterranean and west Asia in the native range, and the Mojave and Lahontan Basin in the invaded range, but also reaching Mediterranean climates of coastal WNA (Fig. 4d–f). These patterns suggest that even the specific QTL underlying local adaptation in the native range have been similarly reused for local adaptation in the invaded range.
We compared our GWAS results with a published study that performed a GWAS for flowering time using a smaller and much less diverse genotype panel32. There was no overlap among the 200 top GWAS SNPs we found (100 top SNPs × 2 GWAS methods) and the SNPs detected in that study (Table 2 in ref. 32), likely due to our larger and more diverse panel of genotypes.
Cheatgrass dominates where local adaptation is predicted to be stronger
Whole genome-environment associations in the native range predicted local adaptation in the invaded range, especially where cheatgrass is most dominant. To further evaluate whether invasive genotypes matched local climates as in the native range, we used a predictive genome-environment model. Using the native range RDA model of genotype as a function of climate (Fig. 2e), we first predicted invasive genotypes for locations of our sequenced samples. Next, we calculated the genetic distance between predicted and observed genotypes, similar to metrics sometimes referred to as ‘genomic offset’55. Genotype-environment matching (i.e., low genetic distance, or offset, between predicted and observed genotypes) was strongest at northern latitudes across North America, particularly in WNA. Putative maladaptation (i.e., high genetic distance between predicted and observed genotypes) was strongest in the southeast USA (Fig. 5a). By comparing mean genetic distance to means of 1000 null permutations, we found the mean genetic distance was significantly lower than the null expectation in WNA (p < 0.002), but not in ENA (p = 0.5, Fig. 5b). This finding is consistent with the hypothesis that local adaptation to climate in WNA reflects the patterns observed in the native range, while cheatgrass in ENA has a novel strategy or is not locally adapted. Unlike WNA, cheatgrass populations in ENA are more restricted to highly disturbed urban and agricultural sites, rarely forming large monospecific stands56.

a Geographic distribution of the genomic offset estimated for each invasive genotype (closed circles, n = 194). The genomic offset or maladaptation is the genetic distance between observed invasive genotypes and the genotype-environment predictions in the invaded range based on the native range genotype-environment association. Colors in the map indicate the degree of maladaptation, from low (purple) to high (yellow). b Histograms of the mean genetic distance (offset) of 1000 null permutations in western North America (WNA, n = 127) and eastern North America (ENA, n = 67), relative to their estimated mean genetic distance (red lines). c Within the Great Basin (polygon in a, n = 55), the mean genetic distance (offset) is significantly lower in areas where cheatgrass occurs in high (i.e., representing >15% vegetation cover) vs. low abundance; two-sided t-test p = 0.006 and two-sided permutation test p = 0.01. Boxplots indicate median (middle line), 25th, 75th percentile (box), and whiskers cover the data extent. Source data are provided in Supplementary Data 1.
To assess whether matching of specific genotypes to local environments promotes cheatgrass invasion, we compared the strength of genotype-environment correlations with variation in cheatgrass abundance from 11,307 field surveys across the Great Basin (Fig. 5a), the region where the invasion has its worst impacts30. Locations where cheatgrass occurs in high abundance showed significantly high genotype-environment matching based on the native range model compared to sites where cheatgrass does not dominate (n = 55, two-tailed t-test t = 2.89, df = 52.3, p = 0.006, Fig. 5c), suggesting local adaptation promotes cheatgrass dominance. This pattern was consistent when comparing genotype-environment matching of high-abundance sites to 1000 null permutations of genotypes within the Great Basin (p = 0.01), evidence that this pattern was not merely due to environmental characteristics of the low-abundance sites but reflects the match of genotypes to their local environments.
Synthesis
Biological invasions pose a major environmental threat, but the roles of genomic diversity, repeated introductions, and adaptation are poorly understood. Our results have important implications for understanding the evolution of local adaptation in invasive species that have greatly expanded their range. Past studies suggest that selfing species with large native ranges—like cheatgrass—are more likely to establish self-sustaining populations in new regions23,57,58, but the mechanisms promoting successful establishment have been less explored.
We show that multiple diverse introductions and long-distance dispersal post-introduction likely increased the chances of cheatgrass genotypes arriving to favorable North American environments. Across Eurasia, cheatgrass shows clines indicative of local adaptation and continuous isolation by distance, likely shaped by multiple migrations and subsequent isolation after the Last Glacial Maximum45. In the invaded range, many genotypes represent a mosaic of near-clonal lineages across sometimes widely spread locations but similar environments, and non-clonal genotypes sorted along the steep climate gradients of western North America. Despite differences in genomic diversity and likely demographic history between the native and invaded ranges, western North American genotypes closely matched the native signatures of local adaptation along temperature/aridity gradients. Thus, in North America, environmental filtering of pre-adapted genotypes likely led to reuse of native diversity and facilitated range expansion18,19,59. Accordingly, common gardens revealed that changing selection likely maintains a major life history cline in the USA Intermountain West. Furthermore, genomic signatures of local adaptation also predicted cheatgrass ecological dominance across the USA Great Basin, indicating that factors supporting local adaptation, such as high genetic diversity from repeated introductions, fuel the invasion.
Our findings emphasize that any sources of genetic diversity could continue to reshape adaptation in established invasive species60. With high genomic diversity and no dispersal limitation, range-wide adaptation could persist over time even under shifting environments. Limiting ongoing introductions and intra-continental dispersal of genotypes (e.g., by limiting seed contaminants in grains) could likely help minimize rapid adaptation of invasive plants. For annual selfers like cheatgrass, this strategy might limit local adaptation via pre-adaptation, but also via de novo variation from uncommon but potentially important outcrossing events28,39,47.
Methods
Plant material
Natural inbred lines of Bromus tectorum were obtained from 1) the Genome Resources Information Network (GRIN), 2) Greenhouse inbred/selfed plants (S1 or S2) of field samples in western North America collected 2019–2020, 3) field samples from the native and invaded range collected 2020–2022, and 4) DNA extractions of frozen seedlings (contributed by Brian Rector, USDA–ARS) for 29, 111, 155, and 12 samples, respectively. With this panel of genotypes, we targeted sites with distinct environmental conditions, favoring environmental variation over intra-population sampling. Sites were ~1–6600 km apart within the native or invaded ranges: 194 North American, 105 Eurasian, and 8 from regions with less extensive invasions: 2 from Argentina, 1 from Australia, 3 from New Zealand, and 2 from South Korea. Material was imported to the USA under USDA APHIS permits P37-17-01651 and P37-18-00230.
Environments of origin and regional classification
GPS coordinates of genotypes were taken directly from collection sites and used to extract data from raster files downloaded from the CHELSA v2.1 climate repository61,62, and to create an elevation raster layer with the get_elev_raster function in the R63 package elevtr (v.0.99.0)64. Coordinates were transformed to spatial points in the WGS84 Coordinate System (same as.tif raster files) with the SpatialPoints function in the R package sp (v.2.1-3)65, and data from rasters was obtained using the extract function in the R package raster (v.3.6-26)66. The final environmental dataset included 52 variables (Supplementary Data 1) and environmental gradients across the cheatgrass distribution were identified with PCA (R function prcomp, variables scaled and centered) (see Supplementary Fig. 7e). Furthermore, a shapefile of Level I Ecological Regions of North America67 was downloaded from the USA Environmental Protection Agency. Data from this shapefile were extracted with the over function in the R package sp using the spatial points obtained above (Supplementary Data 1) to assign genotypes into ecological regions. North American genotypes were assigned to eastern North America (ENA) or western North America (WNA) based on ecological region at location of origin. WNA: marine west coast forest, Mediterranean California, North American deserts, northwestern forested mountains, and temperate Sierras. ENA: eastern temperate forests, Great Plains, and northern forests (Supplementary Fig. 3). Native genotypes were assigned to central-north-east Europe, Mediterranean, or west Asia based on geographic location.
Growth conditions
Genotypes with available seeds (295) were germinated in a growth chamber at 20 °C (80% humidity, 12 h light/12 h dark, 200 μmol m2 s−1 light intensity) to increase seed, verify identification, measure phenotypes (in the first seed bulking only), and obtain tissue for whole genome sequencing and genotyping. The first seed bulking was performed in 2020–2021 (193 genotypes) and the second in 2022 (102 genotypes). Three replicates were grown in a 1:1 mix of commercial grade sand and growing medium (PGX PRO-MIX, Premier Tech Manufacturer) to obtain seeds. Two replicates were grown in 100% growing medium to obtain tissue for DNA. Plants were grown in conetainers (1.5-inch diameter, 164 ml) in a RL98 rack (Stuewe & Sons, Inc.). Cotton balls were placed at the bottom of each conetainer to prevent soil loss through drainage holes. Plants selected for DNA extraction were kept under constant temperature until tissue collection. Seedlings ( ~ 5–15 day-old plants) of the grow out sets were vernalized in a cold room at 4 °C (30% humidity, 8 h light/16 h dark, ~75 μmol m2 s−1 light intensity) for 10 weeks, then transferred back to the growth chamber at 20 °C day/15 °C night (50% humidity, 14 h light/10 h dark, 200 μmol m2 s−1 light intensity) and kept well-watered until harvesting. Plants were randomized within trays, with every other position left empty within racks (for 49 pots/tray), and once plants flowered, surrounded with a hard plastic transparent cylinder (with air-flow holes) attached to the base of pots to avoid any outcrossing. Trays were periodically rotated (twice per week) and moved around the growth chamber (twice per month) to mitigate positional effects.
DNA purification
Total genomic DNA for each genotype was extracted from ~100 mg of fresh tissue collected from one healthy individual in the DNA plant set, using the Viogene Plant Genomic DNA Extraction System (Viogene BioTek, Corp.). To ensure purity and high molecular weight, DNA was subsequently cleaned with 0.9X AMPure XP magnetic beads (Beckman Coulter, Inc.). DNA purity was assessed with a Thermo Scientific NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Inc.) and quantified with an Invitrogen Qubit 2.0 fluorometer (Thermo Fisher Scientific, Inc.).
Next generation whole-genome sequencing
A set of 303 genotypes was sequenced at an average coverage of ~1.2–4.8X per genotype (genome size ~2.5 Gb) at the Texas A&M AgriLife Research (Genomics and Bioinformatics Service). At least 500 ng of genomic DNA was used to construct paired-end sequencing libraries (PerkinElmer NEXTFLEX Rapid XP DNA-Seq Kit HT) which were sequenced on an Illumina NovaSeq 6000 S4 platform −2 × 150 v.1.5 (Illumina, Inc.). Sequence cluster identification, quality prefiltering, base calling and uncertainty assessment were done in real time using Illumina’s NCS 1.0.2 and RFV 1.0.2 software with default parameter settings. Sequencer.cbcl basecall files were demultiplexed and formatted into.fastq files using bcl2fastq 2 2.19.0 script configureBclToFastq.pl. Raw reads were processed with FastQC (v.0.11.8)68 and filtered for low quality and adapter regions using Trimmomatic (v.0.39)69. Filtered.fastq files contained ~20–80 × 106 reads totaling ~3–12 Gb per genotype.
A separate set of four genotypes was sequenced at an average of 70–120X per genotype at the Joint Genome Institute for genome size estimation. At least 10 µg of genomic DNA was used to construct libraries for Illumina sequencing as above. Duplicate reads were removed based on paired sequence matching using Clumpify70. BBDuk (v.38.90)70 was used to trim reads that contained adapter sequences and homopolymers of G’s of size ≥5 at the ends of reads. BBDuk was also used to remove reads that contained one or more ‘N’ bases, had an average quality score <6, or had a minimum length ≤49 bp or 33% of the full read length. Filtered.fastq files contained ~1.16–2.04 M reads totaling ~174–308 Gb per genotype.
Mapping reads to the reference genome
Paired-end reads from each genotype were mapped to the Bromus tectorum genome using BWA-MEM (v.0.7.15)71 with default parameter settings. Then in SAMtools (v.1.18)72 read alignments were converted into BAM format (with: view -bhS), sorted by read names (sort -n) to check and update mate coordinates (fixmate -rpcm), sorted by genomic coordinates (sort) to mark and remove PCR duplicates (markdup -Srs), and filtered for incomplete and poor quality alignments (view -b -m 30 -q 30).
Initial variant calling
Analysis of Next Generation Sequencing Data (ANGSD v. 0.938)73 software was used to detect SNPs and calculate genotype likelihoods across the 307 samples, thus allowing to account for genotype uncertainty in downstream analyses. An initial variant calling step was performed on all samples, separately for each chromosome, using base call and mapping quality filters (-uniqueOnly 1 -minMapQ 30 -C 50 -baq 1 -minQ 20 -remove_bads 1 -only_proper_pairs 1) with the SAMtools genotype likelihood framework (GL -1) and the output written to Beagle format (-doGlf 2). To avoid potential biases arising from sequencing errors and excessive repetitive regions, we kept biallelic SNPs with a sequencing coverage of minimum 5× and maximum 25× in 50% of all samples and with an allele frequency above 0.05 (-SNP_pval 1e-6 -doCounts 1 -setMinDepth 1535 -setMaxDepth 7675 -minInd 154 -minMaf 0.05). For each site, the major and minor allele were inferred from genotype likelihoods. Allele frequencies were estimated both while assuming known major and minor alleles but also while taking the uncertainty of the minor allele inference into account (-doMajorMinor 1 -doMaf 3). Estimated variant calls for each chromosome were then used in a second step to produce a.bcf file with genotype likelihoods and posterior probabilities (-sites -doBcf 1 -doMajorMinor 3 -doMaf 1 -doPost 1 -GL 1) that was converted into.vcf with view in BCFtools (v.1.18)74.
SNP dataset
Haplotype-phasing was performed in Beagle (v.4.1)75 with default parameter settings, using genotype likelihoods to remove uncertainty in the initial.vcf file and add precision based on similarities between pairs of individuals. SNP imputation was performed in Beagle (v.5.2)76 with default parameter settings, which are considered appropriate for a global population. Resulting per chromosome.vcf files were concatenated and indexed in BCFtools. To remove potential paralogs, sites with excess heterozygosity (flagged ‘ExcHet<1’) and with >5% heterozygote genotypes were filtered out. The resulting.vcf dataset contained 15,101,725 SNPs and was subsequently reformatted to.gds with the snpgdsVCF2GDS function (method = “biallelic.only”) in the R package SNPRelate (v.0.9.19)77.
Population genetic structure
We used multiple methods to infer population genetic structure that we interpret collectively. Using the dataset of 15,101,725 SNPs, sites in high linkage-disequilibrium were detected in PLINK (v.1.9)78 with a window size of 150 kb, a step size of 1, and a pairwise R2 threshold of 0.5, resulting in 266,504 unlinked sites. Unlinked sites were used in ANGSD to estimate genotype likelihoods in the SAMtools framework, with the output written to Beagle (-GL 1 -doGlf 2 -doMajorMinor 1 -doMaf 3). Unlinked sites were also used to produce a.vcf of SNPs using BCFtools view and subsequently reformatted to.gds.
First, we estimated individual admixture proportions in NGSadmix (v.33)79 and inferred population genetic structure with PCA in PCAngsd (v.1.11)80, which work directly with genotype likelihoods that contain all relevant information of unobserved genotypes. Individual admixture proportions were estimated with maximum likelihood in 12 replicates, for K = 2–12 genetic clusters, on sites with minor allele frequency (MAF) > 0.05, at least in 50% individuals, and <75% missing data. Cross-validation of number of clusters was determined from log-likelihoods of the NGSadmix output across all replicates81, supporting K = 4. Because NGSadmix assumes Hardy-Weinberg equilibrium in the ancestral populations and this assumption might be violated in a selfer like cheatgrass, we compared our NGSadmix results to admixture proportions based on sparse nonnegative matrix factorization algorithms implemented in sNMF82. Using the unlinked SNPs dataset and a regularization parameter of alpha = 100, inferred admixture proportions were almost identical between both methods (Supplementary Fig. 2). We thus use results from NGSadmix, the recommended method for datasets with low to medium sequencing coverage like ours79. For the PCA in PCAngsd, individual allele frequencies were estimated on all sites in an iterative approach using a truncated singular value decomposition model, and the covariance matrix was estimated using the inferred individual allele frequencies from prior information for the unobserved genotypes.
Then, we computed an unrooted phylogenetic tree with the Neighbor-Joining (NJ) algorithm65 based on a genetic dissimilarity matrix using the unlinked SNPs. We inferred genetic dissimilarity with the snpgdsDiss function in the R package SNPRelate (v.0.9.19)83. The tree was plotted with the plotnj function in the R package phyclust (v.0.1-33)84 and tips were colored according to their admixture proportions using the tiplabels function and the pies option in the R package ape (v.5.7-1)85. We also calculated Nei’s86 pairwise FST and Weir & Goudet’s87 population-specific FST with functions pairwise.fst.dosage and fs.dosage in the R package hierfstat (v.0.5-11)88. For this, a genotype matrix with dosage data for all sites was estimated with the function snpgdsGetGeno in the R package SNPRelate. Pairwise FST measures population genetic differentiation between regions, whereas population-specific FST measures regional deviations from the ancestral population. High values of population specific-FST indicate high within-group allele sharing and potentially greater divergence from ancestral populations, while low values indicate possible ancestral populations.
Genomic diversity and Tajima’s D
We compared nucleotide diversity (π89) between ranges and regions in the native and invaded range using our dataset of 15,101,725 sites. Regional.vcf datasets containing all sites were generated with BCFtools (v.1.18)74 (view -S), and genome-wide estimates of π were obtained with VCFtools (v.0.1.15)90 using 50 kb sampling windows. Deviations from neutral evolution between geographic regions were examined with the Tajima’s D statistic91, which compares the mean number of pairwise differences against the number of segregating sites observed in a set of sequences. Tajima’s D for each region was calculated in 50 kb sampling windows for shared SNPs in VCFtools using --TajimaD.
Genetic load
Genotype mutation load (under the hypothesis that most protein changing mutations are deleterious) was estimated separately from the high impact and missense variants, both normalized by (divided by) the number of synonymous variants. To this end, we performed variant effect annotations with SnpEff (v 5.1 f)92. First, we constructed a SnpEff database for B. tectorum. The reference genome and gene annotation files were downloaded from the Comparative Genomics platform CoGe (https://genomevolution.org/coge/), genome ID: id6435626. A coding sequence file was produced using gffread (v 0.12.8)93 and the SnpEff annotation pipeline was applied to the.vcf of 307 genotypes and ~15 M SNPs. Variants categorized as high-impact (chromosome large deletion, chromosome large duplication, chromosome large inversion, exon deleted, exon deleted partial, exon duplication, exon duplication partial, exon inversion, exon inversion partial, frame shift, gene deleted, gene fusion, gene fusion half, gene fusion reverse, gene rearrangement, protein-protein interaction locus, protein structural interaction locus, rare amino acid, splice site acceptor, splice, site donor, stop lost, stop gained, start lost, start gained, transcript deleted) or missense (non-synonymous) were subsequently identified and counted per genotype, along with synonymous variants. We then used 2-way ANOVA and Tukey HSD tests to examine differences in genetic loads between native and invaded range genotypes of the same ancestry. Genotypes were assigned to a cluster based on having >0.55 ancestry proportion for the NGSadmix K = 4 ancestral genetic clusters. If no K = 4 ancestry was >0.55, genotypes were designated as intermediate.
Self-fertilization rates
We examined the causes of inbreeding between native and invaded genotypes with several statistics. We used a subset of 101 closely related native and invasive genotypes that were sequenced from seedlings of plants collected directly from the field (as opposed to a greenhouse bulking or GRIN). Based on the ~15 M SNPs dataset, we first computed runs of homozygosity46,94 (with BCFtools roh), Tajima’s D (with VCFtools --TajimaD in 100 kb windows), and heterozygosity (with PLINK (v.1.9) --het) for one lineage represented in the native and North American invaded range (native n = 27, invaded n = 74 [ENA = 47, WNA = 27]). Then we implemented random forests in R to analyze these genetic statistics together and estimate selfing rates for each group using a recently published model (“sequential model”46). We compared group means with a two-tailed t-test.
Isolation-by-distance
We examined isolation-by-distance in native and invasive genotypes, and in genotypes from WNA and ENA, with the same LD-filtered SNP dataset. Genome-wide pairwise genetic dissimilarity matrices were obtained as described above. Pairwise geographic distances were calculated in kilometers from genotype coordinates with the spDists function in the R package sp (v.2.1-3), using the WGS84 ellipsoid projection. Simple Mantel tests95 were used to test if geographic distance predicts genetic distance with the Mantel function in the R package vegan (v.2.6-4)96, using 9999 permutations and method = “spearman”. We conducted linear regressions (with the R function lm) to assess the proportion of genetic variance explained by geographic distance. We also used the Mantel R function in vegan to assess how climatic distance changed with geographic distance using the same parameters as before.
Environmental differentiation between clonal groups
We examined if the 19 genetically different groups of 2–14 nearly clonal genotypes (detected based on >99% SNP similarity) were differentiated by environment using climate data at their location of origin. We used clonal group identity as the predictor of 52 CHELSA climate variables (Supplementary Data 1, Supplementary Fig. 8b) with PERMANOVA using function adonis2 (9999 permutations, Euclidean distances) in vegan.
Variance partitioning of genomic diversity
Redundancy analyses (RDA) were used to model how sets of variables explained SNP variation and for identifying abiotic gradients explaining the most genome-wide SNP variation97. To model geographic patterns in the RDA, a distance matrix obtained from coordinates was converted into a spatial weighting matrix to get a reduced-dimension set of orthogonal variables (Moran’s eigenvector maps, MEMs98). MEMs are eigenvectors of the pairwise spatial weighting matrix among samples. Weighting matrices among unique sample locations were generated using the listw.candidates function in the adespatial (v.0.3-23) R package99. Two algorithms were implemented, Gabriel graph and distance-based graph, to generate three candidate connectivity matrices. The Gabriel graph results primarily in connections among neighboring sites. A distance-based graph connects sites closer than a given threshold, for which we used two values: minimum distance required to connect all points (i.e., the largest distance of a minimum spanning tree) and infinity (resulting in a fully connected graph). With each of these three connectivity matrices, two spatial weighting matrices were generated using two distance-decay functions: linear (weight between two sites = 1 − D/Dmax, where D is distance between sites and Dmax is maximum distance among all sites) or concave up (weight between two sites = D − 0.01). Then, the forward selection of MEM eigenvectors algorithm was used to optimize the number of eigenvectors (restricted to those with positive eigenvalues) included in RDA for each MEM set100. Optimization was based on adjusted R2, and the MEM set with greatest adjusted R2 was defined as the optimal set. These eigenvectors were included in the RDA below on native and invaded whole-SNP datasets filtered for MAF > 0.05 (native: 234,122 SNPs, invaded: 212,292 SNPs).
RDA was then conducted with variance partitioning97 to quantify proportion of genome-wide SNP variation explained by each of two categories of covariates: abiotic variables and geographic MEMs. We selected abiotic variables that were informative and non-colinear based on the PCA explaining range-wide environmental variation (Supplementary Fig. 7e). Variance partitioning estimates proportion of SNP variation that is explained by the collection of variables in each category and by collinearity among variables. To identify environmental gradients associated with genome-wide divergence, RDA was also conducted using only abiotic variables for native and invasive genotypes. We computed RDA and performed variance partitioning with functions rda and varpart, respectively, in vegan. Finally, to examine spatial environmental heterogeneity in the native range and North America, climatic distance was estimated for each region (native, ENA, WNA) based on pairwise Euclidean distances in the environmental PCA produced above, using the function vegdist in vegan.
Ancestry-environment associations
To assess environmental filtering of pre-adapted genotypes in North America, we examined ancestry-climate associations in invasive versus native genotypes using generalized additive models (GAMs). GAMs allow us to account for nonlinear patterns between predictors and the response variable101. Environmental predictors were the same aridity and temperature gradients used for trait-environment clines, in addition to precipitation seasonality (all representative of climatic variation in cheatgrass genotypes; see Supplementary Fig. 7e). For each NGSadmix ancestral cluster (K = 4), GAMs were implemented with the function gam in the R package mgcv (v.1.9-1)102 with a logit link function and beta-distributed residuals. Genotypes were assigned to a cluster based on having >0.55 ancestry proportion for the NGSadmix K = 4 ancestral genetic clusters. Intermediate genotypes (i.e., composed of multiple ancestries) were excluded from this analysis.
Phenotypes
During the grow out in 2020, we measured phenotypes on up to 184 genotypes with 2–3 replicates that emerged within ~9–18 days of planting and survived until harvesting. Plants were monitored every two days until phenotyping was terminated (~250 days after germination when >90% of plants had flowered), and then once a week until the last plants were harvested. Eleven phenotypes were recorded: seedling and adult (i.e., reproductive) height (used to get spring growth), number of leaves, number of tillers, days to flower, inflorescence height, dry biomass, total seed mass (i.e., fecundity), individual seed mass (i.e., seed mass), total seed length, and awn length. No phenotypes were recorded after termination, but seed data were recorded for 11 extra genotypes that had not flowered prior to termination.
Seedling and adult height were measured in centimeters from the base of the plant to the tip of the longest leaf. Seedling height was measured the day plants were taken out of vernalization, i.e., 12 weeks after planting. Adult height was taken from reproductive plants, close to or at harvesting. Spring growth (cm) was the growth of plants after vernalization, taken as the difference between adult and seedling height. Number of leaves and tillers was counted when adult height was measured in reproductive plants. Days to flower were counted from the day of individual germination to the day awn tips appeared through the boot and was recorded until termination. Harvesting was performed when >75% of the main inflorescence was matured; at this point inflorescence height was measured in centimeters from the base of the plants to the tip of the tallest panicle. Uncleaned seeds were collected, stored in coin envelopes, kept under dry conditions for ~30 days, and weighed in milligrams in a tared balance. Vegetative tissue was cut at the soil surface, collected in paper bags, oven-dried for 48 h at 38 °C, and weighed in milligrams in a tared balance. The sum of these two weights constituted total dry biomass. Fecundity and seed mass were determined for a single replicate per genotype due to the time intensity involved in this task. Viable seeds (i.e., filled) were cleaned and weighed in milligrams in a tared balance, constituting fecundity, then 20 random seeds were weighted to get an average seed mass. Seed and awn length were measured with a caliper in five seeds per replicate. Seed length was measured from the base of the raquilla to the tip of the awn, and awn length was measured from the tip of the palea to the tip of the awn.
As mentioned in Plant material, for 155 genotypes planted seeds were directly collected from the field, thus maternal environment effects could be a possible source of phenotypic variation. However, we found a strong correlation between growth chamber and common garden flowering time (common garden—explained in Field common gardens- planted in 2021 from S1 seeds). Flowering time was averaged at the genotype level from the raw data for each study (i.e., growth chamber and common garden). The Pearson and Spearman correlation coefficients were 0.65 and 0.58, respectively, suggesting little maternal effects in growth chamber data.
After quality/error checking, the best linear unbiased estimate (BLUE) of phenotypes was calculated per genotype with the BLUE function in the R package polyqtlR (v.0.1.1)103, using genotype as the predictor of trait measurements across 2–3 replicates and tray as a random effect. We then calculated broad sense heritability (H2) of traits as the proportion of phenotypic variance explained by genotype in a linear model. The total set of phenotyped genotypes included: 184 with vegetative height/growth/count data, 173 with flowering/inflorescence data, 178 with dry biomass data, and 182 with seed data, for a total of 169 genotypes with no missing phenotypes.
Trait variation and environmental associations
To detect axes of life history variation, we summarized the natural genetic variation in our growth chamber phenotypes with PCA (function prcomp, variables scaled and centered) in R. PC1, PC2, and flowering time were then used as response variables for investigating trait differences between ranges and environmental gradients in phenotypes. To assess differences in trait means between native and invasive genotypes, we implemented two-tailed t-tests as well as linear mixed models that accounted for kinship between genotypes. To assess phenotypic differentiation between groups of nearly clonal genotypes, we performed PERMANOVA with group identity as the predictor and eleven phenotypes as response with function adonis2 in the R package vegan (9999 permutations, Euclidean distances). To assess trait-environment clines we used kinship linear-mixed models, which when significant (i.e., p ≤ 0.05), they provide evidence of selection (vs. population genetic structure/drift) explaining variation, similar to QST–FST tests49. A kinship matrix was estimated with identity-by-state (IBS, i.e., allele sharing between pairs of genotypes) using the dataset of 15,101,725 SNPs and function snpgdsIBS in the R package SNPRelate. This kinship matrix was used to fit linear mixed models with random genotype effects using function lmekin in the R package coxme (v.2.2-20)104. We focused on maximum monthly vapor pressure deficit (Pa), describing aridity, and mean air temperature of the coldest quarter (°C), describing winter temperature. These two climate variables showed the highest loads on PC1 and PC2, respectively, on a PCA of 52 climate variables for the 307 native and invasive genotypes (Supplementary Fig. 7e). To test if clines were repeated, absent, or shifted in the invaded relative to the native range, we also tested for an interaction between ranges in the models.
Field common gardens
We conducted a replicated common garden experiment in the 2022 and 2023 growing seasons, across two sites in the Intermountain West that varied in their regional climatic conditions: a cool site with little temperature seasonality (Sheep Station, ID [44.2456°N, 112.2144°W]) and a warm site with pronounced temperature seasonality (Wildcat, ID [43.4744°N, 116.9018°W]). We grew replicates of 95 genotypes from Fall 2021 to Spring 2022 and 93 genotypes from Fall 2022 to Spring 2023 at two different densities (low = 100 seeds/1 m2; high = 100 seeds/0.04 m2) and under two different temperature treatments (low = white gravel; high = black gravel) in a factorial design at both sites50. For each growing season, we planted seeds directly on the ground in the fall of the year preceding the growing season. To track individual plants, seeds were glued to a toothpick (detailed protocol in Vahsen et al. 50). For the 2022 growing season, we planted 100 plants each per plot in a randomized blocked design, with each density and temperature treatment combination represented once across 10 total blocks per site, for a total of 8000 plants (2 density treatments × 2 gravel treatments × 10 blocks × 100 plants × 2 common garden sites = 8000 total plants50). For the 2023 growing season, we reduced the replication at the plot level, such that at the cold, less seasonal site, there were 80 plants per plot (3200 total plants) and at the warm, seasonal site, there were 90 plants per plot (3600 total plants). Plants were not irrigated throughout the experiment. We recorded emergence starting in early winter the year preceding the growing season and recorded individual plant growing stage on a roughly biweekly basis starting in the spring of the growing season. We considered plants to be flowering for this analysis when their florets were first observed to have emerged and were green in color.
At the end of the growing season, we opportunistically harvested the aboveground biomass of plants so that florets had a chance to mature (shift from green to purple in color), while avoiding plant senescence and the dropping of seeds. Plant harvesting occurred within the following date ranges for each common garden site and year: cold, less seasonal site = June 23–July 8, 2022, and June 14–August 4, 2023; warm, seasonal site = February 24–June 28, 2022, and March 4 – June 11, 2023. We stored the aboveground biomass of each individual plant in a separate paper envelope at room temperature in the lab prior to processing seed mass and weight. For processing in 2022, we hand-separated each plant into vegetative and reproductive biomass. Depending on the total number of seeds, we then either separated, counted, and weighed all individual seeds (i.e., if n < 50), or we took a subset of 50 seeds from the total number of seeds and recorded their weight. Then, for those plants for which only a subset of seeds was counted and weighed, we estimated the total seed count given the weight of the seed subset and the total reproductive biomass. For processing in 2023, we only hand-separated each plant into vegetative and reproductive biomass and did not count the seeds. Thus, the fitness metric reported for the 2022 data is total seed count and the fitness metric reported for the 2023 data is reproductive biomass. Plants that were infested with smut or were noted to have dropped seeds prior to or during harvest were not included in the analyses.
We compared the direction and magnitude of selection on flowering phenology between the cold, less seasonal site and the warm, seasonal site for both the 2022 and 2023 growing seasons. For each year and common garden site combination, we calculated the average fitness (2022 = seed count; 2023 = reproductive biomass) and average first flowering day for each genotype, across all treatments. In the calculations of average first flowering day, if a plant did not flower (i.e., fitness = 0), it was assigned the average first flowering day for all plants of that genotype that did flower at some point during the growing season. For each growing season year, we regressed the mean fitness data on the mean flowering time data across genotypes and compared the slopes between the cold, less seasonal site and the warm, seasonal site. Positive slopes on this graph indicate that flowering later is selected, while negative slopes indicate that flowering earlier is selected. Models that included a random intercept for each genotype, with a correlation structure specified by a kinship matrix, also provided evidence of selection.
Genome wide association studies (GWAS)
To identify QTL for growth chamber phenotypes, we implemented GWAS that controlled for kinship on our BLUEs dataset (n = 173–184 genotypes, 14.6–14.7 M SNPs excluding SNPs with MAF < 0.05) using two methods: a univariate linear mixed model (LMM) and a multilocus mixed model (MLMM). SNP genotype data were generated with function snpgdsGetGeno in the R package SNPRelate. The univariate LMM was fit with GEMMA (v.0.98.5)105 with an IBS matrix as the random effect accounting for relatedness between genotypes. The MLMM was implemented with FarmCPUpp106, which computes a restricted kinship matrix based on pseudo-quantitative trait nucleotides (QTN) selected from a preliminary GWAS step. FarmCPUpp also takes principal components of genome-wide SNP variation as covariates, providing a stronger control of population genetic structure. Principal components were obtained with function snpgdsPCA in the R package SNPRelate. We chose PC1–PC3, which together explain ~49% of genomic variation (potentially capturing most neutral variation) and after which the variance explained by each successive PC rapidly levels off. Moreover, while GEMMA tests for an association with each SNP individually, FarmCPUpp performs additional steps that detect pseudo-QTNs based on LD and uses model selection and multiple regression to retain the best set of pseudo-QTNs. Thus, while GEMMA might reveal large blocks of significantly associated SNPs (which might correlate with chromosomal rearrangements), this signal should be lost with FarmCPUpp (which might be better at detecting causal SNPs). To detect statistical significance of GWAS SNPs, we used a false-discovery-rate (FDR) threshold of 0.05 on output p-values. Associations were inspected with Manhattan plots and model fits were assessed with quantile-quantile (Q-Q) plots.
Linkage disequilibrium (LD) and SNP annotation
To assign GWAS SNPs to annotated cheatgrass genes, we first investigated linkage disequilibrium (LD) decay with genomic distance in our sequenced panel of 307 genotypes. We used PopLDdecay107, which takes a.vcf with samples and computes the square of Pearson correlations (R2) between pairs of SNPs genome-wide (using the ~15 M SNPs dataset) or per chromosome. We excluded SNPs with MAF < 0.05 and >5% heterozygote individuals and calculated R2 between pairs of SNPs at a minimum of 10 bp apart and a maximum of 5 Mb apart. We detected the pattern of LD decay by plotting mean LD values in 100 bp bins from 0–5 Mb genomic distance. We observed substantial long-range LD with mean R2 ~ 0.3 even at 5 Mb (Supplementary Fig. 13a), in line with strong population structure. However, there was a clear decay in LD with genomic distance. The initial mean LD (R2 = 0.45) at 10 bp decayed halfway (R2 = 0.376) of the minimum observed LD (R2 = 0.3) by 194.5 kb. Because mean LD was high even at 5 Mb, we also assessed inter-chromosomal LD. To this end, we randomly sampled ~15 K genome-wide SNPs and used the program GWLD108 to get an R2 between all pairs of SNPs (with MAF > 0.05) in different chromosomes. Using the Bromus tectorum CoGe annotations, top GWAS SNPs (those with the lowest 100 p-values for each GWAS) were assigned to candidate genes using a 200 kb window based on the observed LD decay. Given the range-wide LD we observed, we also used a 1 Mb window, but no other clear QTL were revealed. We thus report QTL using a 200 kb window.
QTL-environment clines and enrichment analysis
Environmental variation of allele frequency in QTL detected with GWAS was examined with two-tailed t-tests and kinship linear-mixed models. QTL-environment tests were performed separately for the native and invaded range. To find significantly over-represented GO terms or parents of these terms in the haploblock detected for flowering time, we uploaded a gene set to the PlantRegMap GO Term Enrichment tool109, based on A. thaliana and O. sativa homologs obtained from CoGe annotations.
Genome-environment matching of invasive genotypes
We further tested for evidence of pre-adaptation by quantifying how well a native range genotype-environment association (GEA) model predicted genetic composition in the invaded range. Our approach is like the genomic offset statistics used for predicting climate change impacts on maladaptation55,97. We used the RDA-generated native range GEA to predict maladaptation of invaded range genotypes, separately for WNA and ENA, with the function predict in the R package vegan. The Euclidean distance between predicted and observed allele frequencies for each invasive genotype was then estimated, representing the genetic maladaptation to the invaded range site. To evaluate if the mean genetic maladaptation WNA and ENA was different from random, predictions and genetic (i.e., Euclidean) distances were recalculated in 1000 reshuffled environments. Genetic maladaptation was considered significantly lower than expected by chance if it fell in the lower 0.025 tail of the random distribution of mean genomic distances in a two-tailed test.
Genome-environment matching and invasive spread
We examined if cheatgrass dominance was correlated with the strength of local adaptation using available cheatgrass abundance data. Using random forest models, Bradley et al. 30 created a regional classification of cheatgrass presence across the Great Basin based on 11,307 surveyed sites. Their classification differentiates between cheatgrass present at high abundance ( ≥ 15%) and cheatgrass absent or at low abundance ( < 15%). Genetic maladaptation/offset was compared between sites where cheatgrass is in high abundance to sites where cheatgrass is in low abundance (high vs. low in Fig. 5c, respectively) with a two-tailed t-test (n = 54 sites or Great Basin genotypes). To assess if this pattern was driven by the match of local genotypes to local environments (as opposed to the environmental characteristics of the low abundance cheatgrass sites), we recalculated genomic offset in 1000 reshuffled Great Basin environments and compared offset of regions where cheatgrass dominates to the 1000 null permutations of genotypes within the Great Basin.
Plotting
For plots depicting ancestral clusters from NGSadmix (i.e., Figs. 1b, c, e, 2e, f, 3), samples were colored according to their admixture proportions using the function geom_scatterpie in the R package scatterpie (v.0.2.1)110, implemented in ggplot2111. Maps (i.e., Figs. 1b, c, 4a, d, 5a, Supplementary Figs. 3, 5, 8a, 12a, d) were also plotted with ggplot2 using the natural earth world country polygons obtained with the function ne_countries(scale = “medium”, returnclass = “sf”) in the R package rnaturalearth (version 1.1.0)112. For North America, we used the function ne_states(country=c(“united states of america”, “canada”, “mexico”), returnclass = “sf”) in the same package.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The sequence data of 307 genotypes generated in this study have been deposited in the European Nucleotide Archive under accession code PRJEB97687. The beagle (genotype likelihoods), vcf (SNP calls), raw phenotype data, and genetic/geographic/climatic distances are publicly available via Figshare [https://doi.org/10.6084/m9.figshare.29367845]113. Source Data with the complete genotype-level dataset assembled here and used for analyses and to generate figures is provided in Supplementary Data 1. Source Data with GWAS results used in Fig. 4 are provided in Supplementary Data 2. Herbarium vouchers of genotypes are deposited in The Pennsylvania State University Herbarium (PAC). Due to limited availability, seeds of genotypes used in this study are available from the lead contact upon request.
Code availability
The code used to analyze NGS data is publicly available via Figshare [https://doi.org/10.6084/m9.figshare.29367845]113.
References
Daly, E. Z. et al. A synthesis of biological invasion hypotheses associated with the introduction–naturalization–invasion continuum. Oikos 2023, e09645 (2023).
Catford, J. A., Jansson, R. & Nilsson, C. Reducing redundancy in invasion ecology by integrating hypotheses into a single theoretical framework. Diversity Distrib. 15, 22–40 (2009).
Liu, D. et al. Regional invasion history and land use shape the prevalence of non-native species in local assemblages. Glob. Chang. Biol. 30, e17426 (2024).
Colautti, R. I. & Barrett, S. C. H. Rapid adaptation to climate facilitates range expansion of an invasive plant. Science 342, 364–366 (2013).
Urban, M. C. et al. Evolutionary origins for ecological patterns in space. Proc. Natl. Acad. Sci. USA 117, 17482–17490 (2020).
Tigano, A. & Friesen, V. L. Genomics of local adaptation with gene flow. Mol. Ecol. 25, 2144–2164 (2016).
Bridle, J. R. & Vines, T. H. Limits to evolution at range margins: When and why does adaptation fail?. Trends Ecol. Evol. 22, 140–147 (2007).
Simón-Porcar, V. I., Silva, J. L. & Vallejo-Marín, M. Rapid local adaptation in both sexual and asexual invasive populations of monkeyflowers (Mimulus spp.). Ann. Bot. 127, 655–668 (2021).
Kirkpatrick, M. & Barton, N. H. Evolution of a species’ range. Am. Nat. 150, 1–23 (1997).
Eckert, C. G., Samis, K. E. & Lougheed, S. C. Genetic variation across species’ geographical ranges: the central–marginal hypothesis and beyond. Mol. Ecol. 17, 1170–1188 (2008).
Mayr, E. Animal Species and Evolution (Harvard University Press, 1963).
Robinson, J., Kyriazis, C. C., Yuan, S. C. & Lohmueller, K. E. Deleterious variation in natural populations and implications for conservation genetics. Annu. Rev. Anim. Biosci. 11, 93–114 (2023).
Bay, R. A. et al. Genomic signals of selection predict climate-driven population declines in a migratory bird. Science 359, 83–86 (2018).
Nota, A., Bertolino, S., Tiralongo, F. & Santovito, A. Adaptation to bioinvasions: When does it occur?. Glob. Chang. Biol. 30, e17362 (2024).
Rosche, C. et al. Climate outweighs native vs. nonnative range-effects for genetics and common garden performance of a cosmopolitan weed. Ecol. Monogr. 89, e01386 (2019).
Santangelo, J. S. et al. Global urban environmental change drives adaptation in white clover. Science 375, 1275–1281 (2022).
Leger, E. A. & Rice, K. J. Assessing the speed and predictability of local adaptation in invasive California poppies (Eschscholzia californica). J. Evol. Biol. 20, 1090–1103 (2007).
Liu, C., Wolter, C., Xian, W. & Jeschke, J. M. Most invasive species largely conserve their climatic niche. Proc. Natl. Acad. Sci. USA 117, 23643–23651 (2020).
Sherpa, S. & Deprés, L. The evolutionary dynamics of biological invasions: a multi-approach Perspective. Evolut. Appl. 14, 1463–1484 (2021).
Turner, K. G., Ostevik, K. L., Grassa, C. J. & Rieseberg, L. H. Genomic analyses of phenotypic differences between native and invasive populations of diffuse knapweed (Centaurea diffusa). Front. Ecol. Evol. 8, 577635 (2021).
Kreiner, J. M., Caballero, A., Wright, S. I. & Stinchcombe, J. R. Selective ancestral sorting and de novo evolution in the agricultural invasion of Amaranthus tuberculatus. Evolution 76, 70–85 (2022).
Endriss, S. B., Alba, C., Norton, A. P., Pyšek, P. & Hufbauer, R. A. Breakdown of a geographic cline explains high performance of introduced populations of a weedy invader. J. Ecol. 106, 699–713 (2018).
Gioria, M., Hulme, P. E., Richardson, D. M. & Pyšek, P. Why are invasive plants successful?. Annu. Rev. Plant Biol. 74, 635–670 (2023).
Vallejo-Marín, M. et al. Population genomic and historical analysis suggests a global invasion by bridgehead processes in Mimulus guttatus. Commun. Biol. 4, 327 (2021).
Bieker, V. C. et al. Uncovering the genomic basis of an extraordinary plant invasion. Sci. Adv. 8, eabo5115 (2022).
Lucas, M. S. et al. Re-focusing sampling, design and experimental methods to assess rapid evolution by non-native plant species. Biol. Invasions 26, 1327–1343 (2024).
Mack, R. N. Invasion of Bromus tectorum L. into western North America: an ecological chronicle. Agro Ecosyst. 7, 145–165 (1981).
Novak, S. J. & Mack, R. N. “Chapter 4. Mating system, introduction and genetic diversity of Bromus tectorum in North America, the most notorious product of evolution within Bromus section Genea” in Exotic Brome-Grasses in Arid and Semiarid Ecosystems of the Western US: Causes, Consequences, and Management Implications, Germino, M. J., Chambers, J. C. & Brown, C. S. Eds. (Springer International Publishing, 2016), pp. 99–132.
Germino, M. J., Belnap, J., Stark, J. M., Allen, E. B. & Rau, B. M. “Chapter 3. Ecosystem impacts of exotic annual invaders in the genus Bromus” in Exotic Brome-Grasses in Arid and Semiarid Ecosystems of the Western U. S.: Causes, Consequences, and Management Implications, Germino, M. J., Chambers, J. C. & Brown, C. S. Eds. (Springer International Publishing, 2016), pp. 61–95.
Bradley, B. A. et al. Cheatgrass (Bromus tectorum) distribution in the intermountain western United States and its relationship to fire frequency, seasonality, and ignitions. Biol. Invasions 20, 1493–1506 (2018).
Porensky, L. M. & Blumenthal, D. M. Historical wildfires do not promote cheatgrass invasion in a western Great Plains steppe. Biol. Invasions 18, 3333–3349 (2016).
Revolinski, S. R., Maughan, P. J., Coleman, C. E. & Burke, I. C. Preadapted to adapt: Underpinnings of adaptive plasticity revealed by the downy brome genome. Commun. Biol. 6, 326 (2023).
Novak, S. J. & Mack, R. N. Genetic variation in Bromus tectorum (Poaceae): comparison between native and introduced populations. Heredity 71, 167–176 (1993).
Barlett, E., Novak, S. J. & Mack, R. N. Genetic variation in Bromus tectorum (Poaceae): differentiation in the eastern United States. Am. J. Bot. 89, 602–612 (2002).
Valliant, M. T., Mack, R. N. & Novak, S. J. Introduction history and population genetics of the invasive grass Bromus tectorum (Poaceae) in Canada. Am. J. Bot. 94, 1156–1169 (2007).
Schachner, L. J., Mack, R. N. & Novak, S. J. Bromus tectorum (Poaceae) in midcontinental United States: Population genetic analysis of an ongoing invasion. Am. J. Bot. 95, 1584–1595 (2008).
Huttanus, T. D., Mack, R. N. & Novak, S. J. Propagule pressure and introduction pathways of Bromus tectorum (cheatgrass; Poaceae) in the central United States. Int. J. Plant Sci. 172, 783–794 (2011).
Pawlak, A. R., Mack, R. N., Busch, J. W. & Novak, S. J. Invasion of Bromus tectorum (L.) into California and the American southwest: Rapid, multi-directional and genetically diverse. Biol. Invasions 17, 287–306 (2015).
Leger, E. A., Espeland, E. K., Merrill, K. R. & Meyer, S. E. Genetic variation and local adaptation at a cheatgrass (Bromus tectorum) invasion edge in western Nevada. Mol. Ecol. 18, 4366–4379 (2009).
Hufft, R. A. & Zelikova, T. J. “Chapter 5. Ecological genetics, local adaptation, and phenotypic plasticity in Bromus tectorum in the context of a changing climate” in Exotic Brome-Grasses in Arid and Semiarid Ecosystems of the Western U. S.: Causes, Consequences, and Management Implications, Germino, M. J., Chambers, J. C. & Brown, C. S. Eds. (Springer International Publishing, 2016), pp. 133–154.
Meyer, S. E. & Allen, P. S. Ecological genetics of seed germination regulation in Bromus tectorum L. I. Phenotypic variance among and within populations. Oecologia 120, 27–34 (1999).
Meyer, S. E. & Allen, P. S. Ecological genetics of seed germination regulation in Bromus tectorum L. II. Reaction norms in response to a water stress gradient imposed during seed maturation. Oecologia 120, 35–43 (1999).
Meyer, S. E., Nelson, D. L. & Carlson, S. L. Ecological genetics of vernalization response in Bromus tectorum L. (Poaceae). Ann. Bot. 93, 653–663 (2004).
Meyer, S. E., Leger, E. A., Eldon, D. R. & Craig, C. E. Strong genetic differentiation in the invasive annual grass Bromus tectorum across the Mojave–Great Basin ecological transition zone. Biol. Invasions 18, 1611–1628 (2016).
Kelly, L. J., Mack, R. N. & Novak, S. J. Genetic analysis of Bromus tectorum (Poaceae) in the Mediterranean region: biogeographical pattern of native populations. Heredity 126, 178–193 (2021).
Zeitler, L. & Gilbert, K. J. Using runs of homozygosity and machine learning to disentangle sources of inbreeding and infer self-fertilization rates. Genome Biol. Evol. 16, evae139 (2024).
Meyer, S. E., Ghimire, S., Decker, S., Merrill, K. R. & Coleman, C. E. The ghost of outcrossing past in downy brome, an inbreeding annual grass. J. Hered. 104, 476–490 (2013).
Hierro, J. L., Eren, Ö, Čuda, J. & Meyerson, L. A. Evolution of increased competitive ability may explain dominance of introduced species in ruderal communities. Ecol. Monogr. 92, e1524 (2022).
McKay, J. K. & Latta, R. G. Adaptive population divergence: markers, QTL and traits. Trends Ecol. Evol. 17, 285–291 (2002).
Vahsen, M. L. et al. Phenological sensitivity of Bromus tectorum genotypes depends on current and source environments. Ecology 106, e70025 (2025).
Todesco, M. et al. Massive haplotypes underlie ecotypic differentiation in sunflowers. Nature 584, 602–607 (2020).
Battlay, P. et al. Large haploblocks underlie rapid adaptation in the invasive weed Ambrosia artemisiifolia. Nat. Commun. 14, 1717 (2023).
Resentini, F., Orozco-Arroyo, G., Cucinotta, M. & Mendes, M. A. The impact of heat stress in plant reproduction. Front. Plant Sci. 14, 1271644 (2023).
Holman, T. J. et al. The N-end rule pathway promotes seed germination and establishment through removal of ABA sensitivity in Arabidopsis. Proc. Natl. Acad. Sci. USA 106, 4549–4554 (2009).
Gain, C. et al. A quantitative theory for genomic offset statistics. Mol. Biol. Evol. 40, msad140 (2023).
Morrow, L. A. & Stahlman, P. W. The history and distribution of Downy Brome (Bromus tectorum) in North America. Weed Sci. 32, 2–6 (1984).
Razanajatovo, M. et al. Plants capable of selfing are more likely to become naturalized. Nat. Commun. 7, 13313 (2016).
van Kleunen, M., Manning, J. C., Pasqualetto, V. & Johnson, S. D. Phylogenetically independent associations between autonomous self-fertilization and plant invasiveness. Am. Nat. 171, 195–201 (2008).
NA, A. et al. Niche shift in invasive species: is it a case of “home away from home” or finding a “new home”?. Biodivers. Conserv. 31, 2625–2638 (2022).
Smith, A. L. et al. Global gene flow releases invasive plants from environmental constraints on genetic diversity. Proc. Natl. Acad. Sci. 117, 4218–4227 (2020).
Brun, P., Zimmermann, N. E., Hari, C., Pellissier, L. & Karger, D. N. Global climate-related predictors at kilometer resolution for the past and future. Earth Syst. Sci. Data 14, 5573–5603 (2022).
Brun, P., Zimmermann, N. E., Hari, C., Pellissier, L. & Karger, D. N. Data from: CHELSA-BIOCLIM+ A novel set of global climate-related predictors at kilometer-resolution. EnviDat; https://doi.org/10.16904/envidat.332. (2022).
R. Core Team, R: A language and environment for statistical computing, R Foundation for Statistical Computing; https://www.R-project.org/. (2021).
Hollister, J. & Shah, T. elevatr: Access elevation data from various APIs, version 0.99.0; http://github.com/usepa/elevatr. (2017).
Bivand, R. S., Pebesma, E. J., Gómez-Rubio, V. & Pebesma, E. J. Applied Spatial Data Analysis with R (Springer, 2008).
Hijmans, R. J. raster: Geographic data analysis and modeling, version 3. 6-26; https://rspatial.org/raster. (2023).
Commission for Environmental Cooperation Working Group, Ecological Regions of North America – toward a common perspective (Commission for Environmental Cooperation, 71 Map (scale 1:12,500,000). Montreal, Canada, Revised 2006).
Andrews, S. FastQC: A quality control tool for high throughput sequence data [Online], version 0.11.8; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Bushnell, B. BBTools package, version 38.90; https://sourceforge.net/projects/bbmap/. (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Korneliussen, T. S., Albrechtsen, A. & Nielsen, R. ANGSD: Analysis of next generation sequencing data. BMC Bioinforma. 15, 1–13 (2014).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Browning, S. R. & Browning, B. L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 81, 1084–1097 (2007).
Browning, B. L., Zhou, Y. & Browning, S. R. A one-penny imputed genome from next-generation reference panels. Am. J. Hum. Genet. 103, 338–348 (2018).
Zheng, X. et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–3328 (2012).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Skotte, L., Korneliussen, T. S. & Albrechtsen, A. Estimating individual admixture proportions from next generation sequencing data. Genetics 195, 693–702 (2013).
Meisner, J. & Albrechtsen, A. Inferring population structure and admixture proportions in low-depth NGS data. Genetics 210, 719–731 (2018).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Frichot, E., Mathieu, F., Trouillon, T., Bouchard, G. & François, O. Fast and efficient estimation of individual ancestry coefficients. Genetics 194, 973–983 (2014).
Saitou, N. & Nei, M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425 (1987).
Chen, W.-C. “Overlapping codon model, phylogenetic clustering, and alternative partial expectation conditional maximization algorithm,” thesis, Iowa State University (2011).
Paradis, E. & Schliep, K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2019).
Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. 70, 3321–3323 (1973).
Weir, B. S. & Goudet, J. A unified characterization of population structure and relatedness. Genetics 206, 2085–2103 (2017).
Goudet, J. Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 5, 184–186 (2005).
Nei, M. & Li, W.-H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 76, 5269–5273 (1979).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly. (Austin) 6, 80–92 (2012).
Pertea, G. & Pertea, M. GFF utilities: GffRead and GffCompare. F1000Res 9, PMC7222033 (2020).
Ceballos, F. C., Joshi, P. K., Clark, D. W., Ramsay, M. & Wilson, J. F. Runs of homozygosity: windows into population history and trait architecture. Nat. Rev. Genet. 19, 220–234 (2018).
Diniz-Filho, J. A. F. et al. Mantel test in population genetics. Genet. Mol. Biol. 36, 475–485 (2013).
Oksanen, J. et al. vegan: Community ecology package, version 2.6-4; https://CRAN.R-project.org/package=vegan (2022).
Capblancq, T. & Forester, B. R. Redundancy analysis: a Swiss army knife for landscape genomics. Methods Ecol. Evol. 12, 2298–2309 (2021).
Dray, S., Legendre, P. & Peres-Neto, P. R. Spatial modelling: a comprehensive framework for principal coordinate analysis of neighbour matrices (PCNM). Ecol. Model. 196, 483–493 (2006).
Dray, S. et al., adespatial: Multivariate Multiscale Spatial Analysis, version 0.3-23; https://github.com/sdray/adespatial (2018).
Bauman, D., Drouet, T., Fortin, M.-J. & Dray, S. Optimizing the choice of a spatial weighting matrix in eigenvector-based methods. Ecology 99, 2159–2166 (2018).
Hastie, T. J. & Tibshirani, R. J. Generalized Additive Models. (Chapman and Hall, 1990).
Wood, S. N. Fast stable restricted maximum likelihood and marginal likelihood estimation of semiparametric generalized linear models. J. R. Stat. Soc. Ser. B Stat. Methodol. 73, 3–36 (2011).
Bourke, P. M. et al. Detecting quantitative trait loci and exploring chromosomal pairing in autopolyploids using polyqtlR. Bioinformatics 37, 3822–3829 (2021).
Therneau, T. M. coxme: Mixed effects cox models, version 2.2-20; https://CRAN.R-project.org/package=coxme (2022).
Zhou, X. & Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 44, 821–824 (2012).
Kusmec, A. & Schnable, P. S. FarmCPUpp: efficient large-scale genomewide association studies. Plant Direct 2, e00053 (2018).
Zhang, C., Dong, S.-S., Xu, J.-Y., He, W.-M. & Yang, T.-L. PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35, 1786–1788 (2019).
Zhang, R. et al. GWLD: an R package for genome-wide linkage disequilibrium analysis. G3 Genes|Genomes|Genet. 13, jkad154 (2023).
Tian, F., Yang, D.-C., Meng, Y.-Q., Jin, J. & Gao, G. PlantRegMap: charting functional regulatory maps in plants. Nucleic Acids Res. 2019, gkz1020 (2019).
Guangchuang, Y. scatterpie: Scatter Pie Plot (Version 0.2.1). [R package]. CRAN. https://cran.r-project.org/web/packages/scatterpie (2016).
Wickham, H., Chang, W. & Wickham, M. H. Package ‘ggplot2.’ Create Elegant Data Vis. Using Gramm. Graph. Version 2, 1–189 (2016).
South, A., Michael, S. & Massicotte, P. rnaturalearthdata, World vector map data from natural Earth used in ‘rnaturalearth’ (Version 1.0.0) [R package]. CRAN. https://cran.r-project.org/package=rnaturalearthdata (2024).
Gamba, D. et al. Local adaptation to climate has facilitated the global invasion of cheatgrass Datasets. Figshare https://doi.org/10.6084/m9.figshare.29367845 (2025).
Acknowledgements
Any opinions, findings, conclusions, or recommendations expressed in the material are those of the authors and should not be construed to represent any official USDA determination or policy. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government. Xavier Mack, Carlos Rodríguez-Gonzalez, Yuxin Luo, and Katherine Blocklove helped with measuring phenotypes, harvesting, and performing DNA extractions. Ian Burke, Samuel Revolinski, Peter Maughan, and Craig Coleman granted us access to the reference genome prior to publication. The USGS FIREss team contributed to common garden establishment and data collection on the Wildcat site. iNaturalist was a key resource for identifying seed collectors, among those were also Dave Barnett, David Board, Chalon Boesel, John Bradford, Jaime Braschi, Howard Bruner, Victoria Bustamante, Charles Campbell, Jeanne Chambers, Mike Chen, Mark Chynoweth, Jason Cooper, Massimo Cristofaro, Melodie Cunningham, Kirk Davies, Janelle Downs, Torsten Eriksson, Maggie Eshleman, Erica Fleishman, Carol Kadonsky & Bill Foreman, Berit Gehrke, Tom Getts, Richard Gill, Dana Hartel, Nate Hartley, Patricia Hollins, Alex Hood, Tayla Hook, Parker Hopkins, Becky Hufft, Jennifer Kalt, Lorri Kendrick, Molly Ladd, Matt Lavin, Steven Lee, Jonathan Levine, Marisa Mancillas, John Maron, Grace McCartha, Randal Mindell, Chandra Moffat, Brooke Moore, James Nagler, Yael Orgad, Matthew Pedrotti, David Pyke, Sasha Reed, Matt Rinella, Viktoria Ropak, Håkan Rydin, Geno Schupp, Adam Searcy, Tim Springer, Amy Symstad, Tracy Thomas, Trudy Trevarthen, Samantha vanDeurs, Biljana Vidovic, Viktoria Wagner, Gretchen Whetham, David Wilderman, Eileen Wyza, and Pauline & Jon Zweck. Funding: National Science Foundation grant DEB-1927282 (PBA), DEB-1927009 (JRL), DEB-1927177 (MBH); Joint Genome Institute of the U.S. Department of Energy grant New Investigator Award-506608 (JRL); National Institutes of Health grant R35GM138300 (JRL).
Author information
Authors and Affiliations
Contributions
Conceptualization: D.G., C.S.B., E.A.L., D.M.B., M.J.G., L.M.P., M.B.H., P.B.A., J.R.L. Methodology: D.G., T.M.M., N.P., S.R., A.P., C.S.B., E.A.L., D.M.B., M.J.G., L.M.P., M.B.H., P.B.A., J.R.L. Investigation: D.G., M.L.V., J.J.V.E., A. Das, R.B.-Z., E.A.L., D.M.B., M.J.G., L.M.P., P.B.A., J.R.L., O.B., C.S.B., R.B., S.B.-M., S.M.C., S.L.D.-F., A. Diamond, R.C.D., P.W.D., D.J.E., T.A.V., H.H., M.C.H., R.A.H., S.J., J.M.K., E.K., S.K., M.K., A.K., M.L., S.L., D.M., C.L.M., J.W.M., D.U.N., J.P.O., R.P., L.A.P., L.R., B.G.R., C.R., M.S., R.K.S., A.S., B.M.S., R.L.S., K.G.T., A.K.U., A.V.W., C.-A.W., J.Z. Funding acquisition: P.B.A., M.B.H., J.R.L., M.J.G. Project administration: P.B.A., M.B.H., C.S.B., D.M.B., M.J.G., L.M.P., J.R.L. Writing—original draft: D.G., J.R.L. Writing—review and editing: all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Fernando Lattanzi, Samuel Revolinski, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Unaffiliated: Randy Bodkins, .Holly Hoitink, Michel Langeveld, Daniel Martin, Marina Sadykova.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gamba, D., Vahsen, M.L., Maxwell, T.M. et al. Local adaptation to climate has facilitated the global invasion of cheatgrass. Nat Commun 16, 10203 (2025). https://doi.org/10.1038/s41467-025-64799-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64799-9
