
Abstract
The Plasmodium falciparum sodium efflux pump PfATP4 is a leading antimalarial target, but suffers from a lack of high-resolution structural information needed to identify functionally important features in conserved regions and guide rational design of next generation inhibitors. Here, we determine a 3.7 Å cryoEM structure of PfATP4 purified from CRISPR-engineered P. falciparum parasites, revealing a previously unknown, apicomplexan-specific binding partner, PfABP, which forms a conserved, likely modulatory interaction with PfATP4. The discovery of PfABP presents an unexplored avenue for designing PfATP4 inhibitors.
Similar content being viewed by others
Introduction
The malaria-causing parasite Plasmodium falciparum traverses two hosts, encountering diverse cellular environments across multiple cell types during its life cycle. Upon invading a host red blood cell, the parasite establishes new permeability pathways (NPPs) to enable nutrient acquisition across the red cell membrane and a parasite-encasing parasitophorous vacuole membrane1,2,3 (Fig. 1a). As the membranes become more permeable, Na+ concentrations in the host cytosol and parasite vacuolar lumen equalize with the bloodstream (~135 mM) (Fig. 1a)4,5. This presents a challenge for the parasites, which, like most living cells, require low intracellular [Na+] (~10 mM) and a Na+ gradient across its plasma membrane to survive. To facilitate this, parasites actively extrude Na+ via the P. falciparum ATP-dependent Na+ efflux pump, PfATP44,6,7. PfATP4 is a type 2 cation pump8,9 and, like other members of the P2-type ATPase family10,11, features five conserved domains. These include an extracellular loop (ECL) domain, the transmembrane domain (TMD) which mediates ion binding and transport, the intracellular nucleotide binding (N) and phosphorylation (P) domains which bind and hydrolyze ATP to drive conformational changes of the TMD during Na+ transport10, and the actuator (A) domain which coordinates these processes by translating ATP binding and hydrolysis into TMD movements10,11.

a Schematic overview showing the dynamics of [Na+] in parasite-infected RBC. After invading RBCs, the parasite experiences a shift in [Na+] as it moves from the bloodstream to the host cytosol. In the host cytosol, [Na+] goes up to ~135 mM due to leakage from the new permeability pathways (NPP/PSAC). Nevertheless, the parasite maintains an intra-parasitic [Na+] of ~10 mM through PfATP4 action. b Bar graph of ATPase assay showing ATP hydrolyzing activity in 100 mM assay buffer (Control), in low [Na+] (10 mM) buffer, with PfATP4 alone and in the presence of known PfATP4 inhibitors, a spiroindolone (Cipargamin; 10 nM) and a pyrazolamide (PA21A092; 100 nM) (Experiment was performed in triplicate, and data is plotted as mean values with standard error of mean. 2 tailed Student t-test analysis was performed for statistical validation, **p = 0.02). All conditions except low [Na+] were performed at high [Na+]. c, d CryoEM map of PfATP4. ECL, extracellular loop (green); P-domain, phosphorylation domain (dark blue); N-domain, nucleotide binding domain (purple); A-domain, actuator domain (light violet); TM, transmembrane domain (colored based on TM clusters). See also Supplementary Movie 1. e The domain architecture of PfATP4 and PfABP is colored as in (c).
The continual rise of drug resistance in the malaria parasite P. falciparum hinders efforts toward sustained control and mitigation. As the target of structurally diverse antimalarial compounds, PfATP4 is a promising antimalarial target12,13,14,15, and its inhibition induces rapid parasite clearance in vivo16. However, resistance mutations in PfATP4 have emerged under drug pressure in vitro12,13,14,17,18, and in clinical isolates19,20. Our understanding of the molecular mechanisms of inhibitory compounds and the mutations conferring resistance against them remains limited in the absence of high-resolution structural information. Unfortunately, attempts to express PfATP4 in heterologous systems have been unsuccessful, thwarting structural studies. Furthermore, while the general P2-type ATPase domain organization is predicted to be conserved in PfATP4, the spatial arrangement of mutations and drug binding sites in relation to key functional and structural components of the transporter remains unclear.
Here, we determine a 3.7 Å resolution cryoEM structure of endogenously purified PfATP4 isolated from parasite-infected human red blood cells. Our structure reveals the molecular details of ATP and ion-binding sites in PfATP4 and provides a structural framework for understanding the spatial organization of resistance-conferring mutations. Most notably, isolating PfATP4 directly from parasite-infected human red blood cells enabled the discovery of a previously unknown conserved companion protein, which may play a key role in modulating PfATP4 activity. These insights will inform the design of drugs with the potential to overcome P. falciparum resistance mechanisms.
Results
Structure of P. falciparum cation ATPase, PfATP4
To obtain PfATP4 protein for structural studies, we used CRISPR-Cas9 to insert a 3×FLAG epitope tag at the C-terminus of PfATP4 in Dd2 P. falciparum parasites (Supplementary Fig. 1a). We show that PfATP4 affinity purified from parasites cultured in human red blood cells exhibit Na+-dependent ATPase activity that is inhibited by established PfATP4 inhibitors, PA21A092 and Cipargamin (Fig. 1b). We then determined a 3.7 Å resolution structure of endogenously purified PfATP4 using single particle cryoEM (Fig. 1c, Supplementary Fig. 1d–f). All five canonical P-type ATPase domains are visible in our cryoEM map and, aside from the flexible A-domain, at sufficient resolution to enable de novo modeling (Fig. 1c, d).
Our atomic model contains 982 residues of the total 1264 residues of PfATP4 (Fig. 2a) and differs significantly (RMSDs of 10.3–22.9 Å) from previous predictions based on homology modeling8 (Supplementary Fig. 2). The P-domain is composed of two segments situated between the TM4 and N domains. The first segment of the P-domain comprises two β-sheets and a short helix, which sits between TM4 and the N-domain. The rest of the P-domain, located between the N-domain and TM5, consists of five β-sheets connected by several short loops and helices (Fig. 1e). The N-domain consists of several β-sheets connected by short helices and long loops and is positioned below the P-domain. The extracellular loop region, which juts into the lumen of the parasitophorous vacuole, is composed of four long β-sheets connected by long loops and flanked by two small helices.

a, Atomic model of PfATP4. ECL, extracellular loop (green); P-domain, phosphorylation domain (dark blue); N-domain, nucleotide binding domain (purple); A-domain, actuator domain (light violet); TM, transmembrane domain (colored based on TM clusters). b Top and side views of transmembrane helices shown as tubes, colored as in (a). c Detailed top view of Na+ coordination site within the TMD with residues of interest indicated. TM helices colored as in (a). d Detailed ribbon side view of TM1 within the TMD, highlighting the Proline at the canonical kink. e Detailed top view of ATP-binding site between the N- and P-domains with residues of interest indicated. Domains colored as in (c) and (d). See also Supplementary Movie 1. f Detailed view of Na+ coordination site of PfATP4 with resistance-conferring mutations indicated. In green are Na+ coordinating residues in the ion-binding pocket within the TMD, as in (c). In cyan are sites of mutations conferring resistance, with A211V and G358S mutated and highlighted. Orange shading indicates the proposed binding site of antimalarial, Cipargamin.
The TMD (Fig. 2b) of PfATP4 contains 10 helices arranged similarly to the α-subunit TMDs of other P2-type ATPases, the two best studied of which are Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA)21 and the α-subunit of Na+/K+ ATPase (NKA)22,23. The PfATP4 TMD consists of three clusters (TM1-2, TM3-4, TM5-10) (Fig. 2b). The ion-binding site within the TMD is located between TM4, TM5, TM6 and TM8, similar to that of SERCA24,25(Fig. 2c). Although our map is not of sufficient resolution to observe density for Na+ in the binding site, all of the Na+ coordinating sidechains are conserved and positioned near-identically to their corresponding residues in SERCA in its Ca2+-bound state (E1-2Ca2+) (RMSD = 0.86–1.83)(Supplementary Fig. 3a)24,25,26. Furthermore, analysis with pyKVFinder27 detects no cavity at the ion-binding site in our model or E1-2Ca2+ SERCA, whereas a cavity is detected at the corresponding site in unbound SERCA (E2-BeF3−) (Supplementary Fig. 3a)26. Beyond the ion-binding pocket, we observe a kink in PfATP4-TM1, matching the ion-bound states of both SERCA28 and NKA29. A Phe positioned at this kink in NKA has been shown to mediate gate closure10,30. In PfATP4, this key residue is replaced by a proline (P176), which could play a similar role (Fig. 2d). Taken together, our structure of PfATP4 is consistent with a Na+ bound state.
The overall architecture of the ATP-binding site between the N- and P-domains of PfATP4 is conserved with those of other P2-type ATPases (Fig. 2e, Supplementary Fig. 3b, Supplementary Movie 1). E557, F614, K652, R703, K846, D865, and N868 as well as the P-domain phosphorylation site D451 maintain a conformation similar to that of SERCA E1.2Ca2+ in its ATP-free state (E442, F482, K514, R559, K683, D702, N705, D351 respectively) (Supplementary Fig. 3b, Supplementary Movie 1), and we do not observe any density corresponding to ATP in the ATP-binding site. However, we observe key differences in the sidechain arrangements at M620, R618, and R840. Specifically, the M620 sidechain flips into the ATP-binding pocket (Supplementary Fig. 3b). Conversely, the sidechains for R618 and R840 both swivel away from the ATP-binding pocket, and the hydrogen bonds formed by the corresponding residues in SERCA are missing (R489-V678 and R677-K492).
Mapping resistance-conferring mutations in PfATP4
Mutations in PfATP4 are associated with resistance against several promising chemical classes of antimalarial drug candidates, suggesting that PfATP4 is the target of these compounds. Recent work looking at ortholog replacement in P. knowlesi has also shown that drug sensitivity in P. falciparum is a function of PfATP4 primary sequence31. Mutations conferring resistance to the spiroindolone Cipargamin mainly localize around the proposed Na+ binding site within the TMD (Supplementary Fig. 3d). Among these, G358S/A, found in recrudescent parasites that arose during Cipargamin Phase2b clinical trials19, were shown to confer high-level resistance against both Cipargamin and (+)−SJ733, a dihydroisoquinolone with a similar parasite-killing phenotype20. Mapping G358S onto our model of PfATP4 reveals that the residue is located on TM3, adjacent to the proposed Na+ coordination site. Notably, the mutation could potentially block Cipargamin binding by introducing a serine or alanine sidechain into the proposed Cipargamin binding pocket (Fig. 2f). Another resistance mutation of note is A211V, which arose against PA21A092 - a pyrazoleamide - under continuous drug pressure17,32. A211V is found within TM2 adjacent to the ion-binding site of PfATP4 and the proposed binding site of Cipargamin. Interestingly, parasite lines with the A211V mutation showed increased susceptibility to Cipargamin17,32.
Discovery of a PfATP4-binding protein (PfABP)
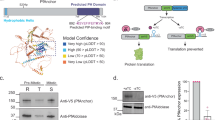
We were surprised to note an additional helix interacting with TM9 of PfATP4 in the TMD (Figs. 1d, 2a, b), which could not be assigned to any of the remaining unmodelled residues at the N-terminus of PfATP4. Sequence-independent modeling of this unidentified helix backbone with ModelAngelo33 followed by a search in findMySequence34 returned a single hit: the C-terminus of PF3D7_1315500, a conserved P. falciparum protein of unknown function35, which we named PfATP4-Binding Protein (PfABP) (Fig. 3). De novo modeling of the PfABP C-terminal sequence into the unmodeled density confirmed the assignment. PfABP is the third most abundant protein in tryptic digest mass spectrometry of our purified sample, further supporting the findMySequence assignment (Fig. 3, Supplementary Table 3, Supplementary Fig. 6).

Dd2-PfATP4-3×FLAG parasites are expanded and harvested, which is followed by affinity purification, cryoEM data collection, and structure determination. Final density revealed an additional TM helix, which could not be assigned to PfATP4. Sequence-independent modeling identified this helix as the C-terminus of PF3D7_1315500, a conserved protein we term PfATP4-Binding Protein (PfABP). Fitting of PfABP confirmed the assignment, supported by mass spectrometry, where PfABP was the third most abundant protein.
PfABP is essential for PfATP4 stability and parasite growth
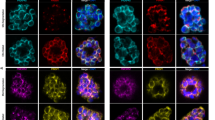
To explore the importance of PfABP, we generated a parasite line conditionally expressing HA-tagged PfABP using the TetR-DOZI system on a Dd2-PfATP4-3×FLAG background. Western blot analysis indicates a >96% reduction in PfABP expression in parasites grown in the absence of anhydrotetracycline (aTc) within the first 24 h (Fig. 4a, Supplementary Fig. 4a), and immunofluorescence assays reveal PfABP localization at the parasite plasma membrane, similar to PfATP4 (Fig. 4b). Removal of aTc led to a severe fitness defect in the parasites, with a drastic reduction in parasite growth after just one cycle of replication (Fig. 4c, d), demonstrating PfABP is indispensable for parasite survival. Interestingly, PfABP knockdown also led to a marked reduction in PfATP4 protein levels within 24 h, highlighting a strict dependency of PfATP4 stability on its association with PfABP (Fig. 4d, Supplementary Fig. 4b, c). Finally, reciprocal pulldown of PfABP-HA and PfATP4-FLAG using either anti-HA or anti-FLAG beads was confirmed by Western blot and further corroborated by tryptic digest mass spectrometry (Fig. 4e, f, Supplementary Table 5).

a Western blot of saponin-lysed parasite pellets collected every 48 h over 8 days, probed with anti-HA for PfABP. Cytosolic aldolase served as a loading control. The reduction in aldolase signal reflects leakage of cytosolic content due to saponin sensitivity of parasite plasma membrane8 as well as parasite death upon PfABP knockdown. b Immunofluorescence microscopy showing PfABP localization. Parasites were fixed and stained with DAPI (blue) for parasite nuclei and anti-HA (green) for PfABP. c Growth assay of PfABP conditional knockdown parasites in the presence and absence of aTc. Parasites were fixed, stained with SyBr Green, and parasitemia quantified by flow cytometry (data plotted as mean of 3 replicates with standard error of mean values). d Western blot of saponin-lysed parasite pellets collected at 24 h and 72 h, probed with anti-HA for PfABP and anti-FLAG for PfATP4. PfEXP2, a parasite vacuolar membrane protein, was used as a loading control. Unlike aldolase leakage, PfEXP2 remains stable at 24 h, demonstrating specific PfABP knockdown together with reduced PfATP4 expression, while its loss at 72 h reflects parasite death. e Reciprocal co-immunoprecipitation under denaturing conditions probed with anti-HA and anti-FLAG. Detection of PfABP-HA in PfATP4-FLAG pulldown and vice versa demonstrates their association. f Blue native PAGE of reciprocal PfATP4-FLAG and PfABP-HA pulldowns, showing co-migration of the complex under both conditions. Each Western blot and micrograph image is representative of three biological experiments. Uncropped and unprocessed versions of all gels are provided as a Source Data file.
PfABP is an apicomplexan-specific P-type ATPase modulator
From our structure of PfATP4-PfABP, we see that the C-terminal domain of PfABP comprises two short intermembrane helices (TM1-2PfABP) packed against one long, single-span transmembrane helix (TM3PfABP) and ending in a short loop within the lumen of the parasitophorous vacuole (Fig. 1c, d, Supplementary Fig. 3e). Extra density extending from the N-terminal end of TM1PfABP can be seen reaching into the parasite cytosol toward the N-domain at higher thresholds but is not of sufficient resolution to be modeled (Supplementary Fig. 3e). PfABP-TM3 runs parallel to PfATP4-TM9, forming significant interactions with the outward face of TM9 as well as parts of the ECL domain (Fig. 5b). The interactions forming the interface between PfABP and PfATP4-TM9 are comprised primarily of van der Waal’s interactions (Fig. 5c) with the exception of a pi-pi stacking interaction between PfABP-T183 and PfATP4-W1089 (Supplementary Fig. 3c), which is surrounded by a cluster of aromatic and positively charged residues that pack against the ECL domain. A cluster of negatively charged residues at the C-terminal end of the PfABP-TMD coincides with the surface of the detergent belt (Supplementary Fig. 3f).

Transmembrane domain of PfATP4 and PfABP (orange) overlayed with TM9-FXYD of NKA (blue) (a) and TM9-sarcolipin (TM9-SLN) of SERCA (purple) (d) with TM9 of PfATP4, NKA, and SERCA aligned to each other. See also Supplementary Movie 2. b Inset from (a) showing the YXYD motif in PfABP and tight packing with ECL. c Inset from (d) showing interaction between TM3-PfABP and TM9-PfATP4. Serine on TM9 (S1204) and Serine on PfABP-TM3 (S155) indicated.
There are several known examples of single TM proteins that bind and regulate P-type ATPases, including the γ-subunit of NKA30,36,37, and the SERCA binding peptides, sarcolipin (SLN)38 and phospholamban (PLB)39. Structural alignment of our PfATP4-PfABP model against the NKA-γ-subunit structure reveals that the interface formed between PfABP-TMD and PfATP4-TM9 closely resembles the interface between NKA-γ-subunit and NKA-TM9 (Fig. 5a, Supplementary Movie 2). A canonical FXYD motif at the C-terminus of the γ-subunit, as well as a Glu-Glu motif within the TM9, are the hallmarks of Na+ affinity modulation in NKA11. Although PfABP does not contain a FXYD motif, there is a similar aromatic charged loop–YXYD–in the corresponding location (Fig. 5b). Together, these similarities suggest PfABP may regulate PfATP4 activity. Interestingly, PfATP4-TM9 also lacks the highly conserved Glu-Glu motif but instead has Ser-Ile-Ser residues in the same region (Fig. 5c). These serine substitutions have previously been implicated in inhibiting interaction of NKA-TM9 with the γ-subunit in fish40, suggesting similar evolutionary adaptations in PfATP4.
Conversely, alignment against SERCA and its binding partners reveals that SLN and PLB bind to a different face of the corresponding helix in the SERCA TMD (Fig. 5d, Supplementary Movie 2). Some of the key residues on SERCA TM9 mediating the binding of SLN to SERCA–hydrogen bonds between T5-T932 and a hydrophobic pocket (V19, I22, L953)38 are conserved in PfATP4-TM9 and Plasmodia (T1198, L1223), raising the exciting possibility for designing analogous single TM peptides that bind to and inhibit PfATP4 by regulating Na+ efflux.
Discussion
In this study, we determine the cryoEM structure of the P. falciparum P-type ATPase PfATP4, endogenously purified from P. falciparum parasites cultured in human erythrocytes. By mapping onto our structure key mutations in PfATP4 that confer resistance to Cipargamin, a promising antimalarial candidate undergoing clinical trials, we find that most major resistance-conferring mutations in PfATP4 cluster around the Na+ coordination site within the TMD (Supplementary Fig. 3d). Compared to other P2-type ATPases, our structure also reveals key differences at the sidechain level in predicted active sites. Of note, while M494, R489 and R677 form hydrogen bonds that reduce the size of the cavity during ATP binding25 in ATP-free SERCA, the corresponding residues in PfATP4 (M620, R618, R840) are positioned such that hydrogen bonds would not be possible (Supplementary Fig. 3b). Further functional studies can leverage these differences to specifically target PfATP4 in a selective manner.
Intriguingly, we discovered a previously unknown interaction between PfATP4 and PfABP, an essential protein of unknown function. Conditional knockdown of PfABP led to a rapid reduction in PfATP4 levels and a dramatic loss of parasite viability, demonstrating that PfABP is essential for the stability of PfATP4 and consequently for maintaining the levels of PfATP4 required at the parasite plasma membrane for parasite survival (in blood stages). Furthermore, structural similarities between the observed interaction between PfATP4 and PfABP in our structure and that of similar single-span TM binding partners of homologs SERCA and NKA suggest that PfABP may modulate the Na+ efflux activity of PfATP4. The ability to modulate the efficiency of Na+ pumping by PfATP4 could help the parasite accommodate the extreme changes in extracellular Na+ concentration it encounters as it traverses different environments. This would be of particular importance for combating the steady rise of Na+ concentration inside the red blood cell during intraerythrocytic growth of the parasite.
Compensatory adaptations in other ATPases have previously been observed through differential expression of α-subunits, as well as situational binding of tissue-specific β- and γ-subunits41. One acclimatization of note, found in rainbow trout, is the “reciprocal expression” of α1a- and α1b- subunits based on fresh versus salt water conditions40,42. A second example exists in human renal tissue, where different FXYD isoforms (γ-subunits) interact with TM9 of NKA ATPases to alter Na+ sensitivity. Given the similarity of the interface between NKA-γ-subunit and PfATP4-PfABP, PfABP-TMD may play a similar role in modulating the ATPase activity of PfATP4. While FXYD-family proteins lack homologs in P. falciparum and other apicomplexans, multiple sequence alignment reveals that PfABP is highly conserved in apicomplexan parasites, particularly the transmembrane helices at the C-terminus of the protein (Supplementary Fig. 5a, b). Moreover, absence of similarly conserved PfABP-related proteins in Dinoflagellates and other organisms expressing ATP4-related P-type ATPases (ENA-pumps43;) is consistent with the observation that Myzozoan ATP4 homologs may comprise a subtype distinct from the type 2D ATPases8,44.
Taken together, these observations suggest that PfABP may act as both a stabilizer of PfATP4 and a modulator of PfATP4 activity. In fact, a recently published study of the mammalian Plasma Membrane Calcium ATPase (PMCA) Ca2+ pump with its accessory protein, neuroplastin (NPTN)45, revealed that NPTN is essential both for PMCA2 stability and for reshaping a lipid-binding cavity that modulates Ca2+ transport. It is possible that PfABP might similarly fulfill two roles – stabilizing PfATP4 and potentially modulating Na+ efflux as needed.
More broadly, it appears that PfABP may represent a distinct class of FXYD-like Type 2 ATPase modulators specific to apicomplexans. Despite the structural diversity of compounds developed against PfATP4 so far, all have given rise to resistance-conferring mutations. As such, the PfATP4-PfABP interaction represents an unexplored avenue for therapeutic intervention, which, due to the highly conserved interface between PfATP4 and PfABP, may provide a higher barrier to resistance.
Methods
Plasmid construction for endogenous tagging of PfATP4
To endogenously tag PfATP4 with a 3×FLAG epitope at the C-terminus, we implemented a CRISPR-Cas9-based gene modification strategy. We designed a guide RNA targeting the C-terminal region of the PfATP4 gene and cloned its DNA into a pCas9 plasmid using an NEB Hi-Fi DNA assembly kit as per the manufacturer’s protocol. We PCR amplified the 3′-UTR region using primers (see Supplementary Table. 1) flanked by restriction enzyme sites for XhoI/BstEII. For the 5′-homologous regions, a gene block of 600 bp for amino acid 1115–1264 with a modified region for the CRISPR target site amino acid 1246–1264 was synthesized with sites for BstEII/AvrII flanking the fragment. These homologous fragments were inserted into the p2MG-hDHFR-3×-Flag plasmid via T4 ligation and verified by whole plasmid sequencing.
Parasite culture and generation of parasites with FLAG-tagged PfATP4
Asexual P. falciparum Dd2 wild-type parasites (Gift from Prof. David Fidock, Columbia University, New York, USA) were cultured and continuously maintained in commercially available human O+ erythrocytes (Purchased from BioIVT, Hicksville, NY) at 2.5% hematocrit in HEPES (15 mM, Millipore-Sigma) and Sodium bicarbonate (2.1 g/L, Thermo Fisher Scientific) buffered RPMI-1640, supplemented with 0.5% Albumax (Gibco), hypoxanthine (10 mg/Liter, Fisher Scientific) and 50 mg/Liter gentamycin (VWR). The culture was maintained at a 37 °C incubator under 90% N2, 5% CO2, and 5% O2 atmosphere. The parasites were maintained at 5% parasitemia unless otherwise specified. To generate C-terminally tagged PfATP4 parasites, the Dd2 wild-type parasites were transfected with 50 µg P2MG-hDHFR-3×FLAG and ATP4-mCas9 plasmid. Transgenic parasites were selected based on drug resistance to (5 nM) WR99210. The integration was confirmed by Western blot analysis of the tagged protein.
The transgenic parasites expressing PfATP4-FLAG were evaluated for any fitness defect due to the modification of the endogenous locus by a comparative growth assay with the parent Dd2 by assessing parasitemia over an 8-day period. The tagged PfATP4-FLAG parasites showed no growth defects in comparison to wild-type parasites (Supplementary Fig. 1b). The expression of the FLAG-tagged PfATP4 was evaluated by Western blotting as described previously8. Briefly, trophozoite stage parasites were saponin lysed and solubilized using Laemmli buffer. The samples were heated at 65°C for 10 min and run on a 10% SDS-PAGE gel in denaturing conditions. The samples were transferred to a PVDF membrane and probed with a mouse anti-FLAG primary antibody overnight at 4 °C (Sigma Cat# F1804, 1:7500 v/v). The blots were washed 3× with 0.01% Tween-20 in 1× TBS buffer and probed with goat anti-mouse-HRP antibodies (Abcam Cat# AB97040,1:10,000 v/v) and Blots were imaged in a Bio-Rad ChemiDoc™MP Imaging System. All the cell lines were checked and tested negative for mycoplasma contamination.
Immuno-purification of FLAG-tagged PfATP4
Synchronized large-scale cultures at 10% parasitemia of Dd2-PfATP4-FLAG parasites were set up, and trophozoite stage parasites were freed from host RBCs by saponin lysis8. A modified protocol from Ho et al.46 was used for immuno-purification of the FLAG-tagged PfATP4 in its native condition for structural studies. The saponin-lysed parasite pellets were washed 3× with PBS, snap-frozen in liquid nitrogen and stored in a −80 °C freezer until used. The frozen pellets were solubilized overnight in 200 mM 6-amino caproic acid, 50 mM Bis-Tris (pH-7), 1 mM EDTA, 1 mM AEBSF, 10% Glycerol, 2% digitonin in the presence of a 1:1000 dilution of a fungal cocktail of protease inhibitors. The solubilized sample was centrifuged (3000 × g) to remove the debris, and the supernatant was batch-bound overnight with M2 FLAG affinity resin (Sigma Aldrich) at 4 °C. The FLAG resin was washed with 25 mM HEPES buffer with 10 mM MgCl2, 50 mM KCL, 100 mM NaCl, and 0.02% digitonin, and the bound PfATP4 eluted with 300 µg/mL 3×FLAG peptide in the above-mentioned wash buffer and reserved for biochemical and structural studies.
ATPase assay
We performed an ATPase assay to confirm that the purified PfATP4 was active. We used an ATP determination kit (Invitrogen Cat # A22066, Lot-1967043) to measure ATP consumption and assess ATPase activity of PfATP4. We incubated purified PfATP4 with 100 µM ATP for 30 min at room temperature in a buffer containing either 100 mM NaCl, 10 mM NaCl, or PfATP4 inhibitors. Elution buffer, wash buffer, and manufacturer-provided reaction buffers were used as a control. After incubation, the reactions were developed as per the manufacturer’s protocol, and the luminescence was measured using a Tecan Infinite Plex spectroscope. All experiments were conducted in biological and technical triplicate. 10× EC50 of PfATP4 inhibitors Cipargamin (10 nM) and pyrazolamide (100 nM) were used in the assay.
Single particle sample freezing and data collection
Purified PfATP4 was applied to glow-discharged R1.2/1.3, 300 mesh Cu Quantifoil EM grids (Quantifoil Micro Tools) and vitrified in liquid ethane on an FEI Vitrobot Mark IV. Grids were screened on a Glacios TEM at 200 kV with a Gatan K3 camera. High-resolution data was collected on an FEI Titan Krios at 300 kV with a Gatan K3 camera and Quantum energy filter (Gatan K3-BioQuantum). A total of 41,434 movies were acquired at a magnification of 105,000× and a defocus range of −1.5 to −2.5 in electron counting mode (0.83 Å/px), with a total dose of 58 e− per Å2.
Single particle data processing, modeling, and analysis
Movies were motion corrected and CTF estimated using Patch-Motion Correction and CTF in CryoSPARC v447,48. Particles were picked using a combination of CryoSPARC Blob and Template picker. A total of 1,817,437 particles were extracted with a 320-pixel box, unbinned (0.823 Å/px), and iteratively cleaned up through rounds of 2D classification. 668,394 promising particles were used for ab initio reconstruction into two classes and cleaned up further by heterogeneous refinement to remove particles containing empty detergent micelles. Further iterative refinement was performed with NU- and local refinement. Reference-based motion correction (RBMC) was then performed, yielding a 0.2 Å improvement in the map.
After RBMC, to improve the P, N, and A domains, we re-centered the particles at the P-domain and re-extracted in a 380-pixel box. Following re-extraction, NU-Refinement yielded improvement in both the P and N domains. To further improve the N-domain, we performed focused 3D classification followed by 3D refinement around the P and N domains, which improved the backbone connectivity in the N-domain. None of these strategies yielded improvements in the highly flexible A-domain. All half-maps were then postprocessed in RELION49 to yield the final sharpened maps that were used for atomic modeling. Local resolution estimation was performed in RELION.
Model building and refinement were performed using ModelAngelo33, Coot50, UCSF ChimeraX51, and ISOLDE52. The protein sequence was obtained from PlasmoDB35. An initial atomic model of the transmembrane domain was generated using ModelAngelo. We then combined predicted fragments and corrected problem areas in Coot. All remaining segments were then built de novo in Coot and iteratively refined in Isolde52. Three different maps, one centered near the detergent belt, a second re-centered near the P-domain, and a third local refined map of the re-centered map, were used for de novo model building. Final refinement and validation were performed using Phenix.
For identification of the additional helical density that remained unaccounted for after manual atomic model building (PfABP), we used ModelAngelo without an input sequence, but found the predicted sequence did not produce any plausible hits in NCBI BLAST against the plasmodium, human, and mosquito proteomes. The chain trace of helix built by ModelAngelo was used for protein prediction with FindMySequence against the P. falciparum proteome, which identified UniProt ID Q8IEF99. Q8IEF9 contains a putative transmembrane region, and BLAST suggests that the protein is highly conserved and exclusive to Plasmodium. AlphaFold253 prediction of Q8IEF9 yields a transmembrane helix that fits the additional helical density in our cryoEM map. AlphaFold354 multimer prediction of ATP4 with Q8IEF9 shows identical binding of their transmembrane helices, further supporting the identity.
The final model and maps were deposited to the PDB (9N10) and EMDB (48800 [https://www.ebi.ac.uk/emdb/EMD-48800] and 48801), respectively. Mutations in PfATP4 implicated in resistance to various antimalarials were then mapped onto the final model and analyzed. Figures were generated using Adobe Illustrator and ChimeraX.
Generation of PfABP conditional knockdown parasite line
We designed a 1061 bp synthetic gene block to generate a parasite line with conditional knockdown of PfABP expression. We selected 550 bp of the C-terminal genomic region of the PfABP gene with a unique CRISPR guide RNA target, as well as 500 bp from the 3′UTR region of the gene. The gene block was synthesized with flanking BssHII and BstEII restriction sites and an EcoRV linearization site to clone into the pMG75-3HA plasmid with a blasticidin selection marker. We electroporated the plasmid constructs into Dd2-PfATP4-3×FLAG parasites and selected the parasites with 250 nM anhydrous tetracycline (aTC) and 2.5 µg/mL blasticidin (Supplementary Table 1).
Growth assay and localization of PfABP
The PfABP conditional knockdown parasites were tested for functional essentiality of the PfABP gene. We performed a 72 h and 8-day continuous growth assay in the presence or absence of 250 nM aTC. Parasites were synchronized with 0.5 M alanine and split at 0.5–1% starting parasitemia in fresh RBC. Samples were collected every 48 h, fixed with 4% formaldehyde, and stored at 4 °C. All the fixed samples were washed with PBS, incubated with 2× SyBr Green (Thermo Fisher S7563) in PBS for 30 min, and washed with PBS to remove extra stain. 100,000 cells were counted for each replicate in the BD Acuri C6 flow cytometer and analyzed for percent parasitemia with the inbuilt software. For Western blot analysis, parasites were washed with PBS and divided equally into two flasks at time zero (2% parasitemia, 5% hematocrit, 20 mL culture with fresh RBCs). One flask was maintained in the presence of aTc, and the other without. At 24 h and 72 h, 10 mL of culture from each condition was harvested and subjected to saponin lysis. Parasite pellets were solubilized in 50 μL lysis buffer, and 20 μL of each lysate was resolved by SDS-PAGE for Western blot analysis. In parallel, we collected saponin-lysed parasite samples every two days and analyzed them by Western blot to assess PfABP knockdown. Localization of PfABP was determined by immunofluorescence microscopy in trophozoite stage parasites fixed with 4% formaldehyde and 0.0075% glutaraldehyde. Fixed parasites were permeabilized with 0.025% Triton X-100 and then blocked with 3% w/v BSA. These parasites were then incubated overnight with mouse anti-HA antibodies (Santa Cruz Cat# SC-7392, 1:300 v/v) and anti-mouse IgG H + L Alexa Fluor™Plus 488 (Invitrogen, A32723, 1:500 v/v). Images were taken in a Nikon Ti microscope and processed using Nikon NIS Elements (5.30.02) Imaging Software.
Reciprocal pulldown of PfATP4-3×FLAG, PfABP-3×HA and proteomics analysis
We performed reciprocal pulldowns to ascertain complex formation between PfATP4 and PfABP. Briefly, two aliquots of 200 µl saponin-free parasite pellets were solubilized following the same protocol used for the PfATP4-3×FLAG pulldown experiments. The solubilized samples were incubated with either anti-FLAG resin (Sigma, Cat# A2220) or anti-HA resin (Pierce, Cat # 88837) overnight at 4 °C, followed by elution with either FLAG or HA peptide. The eluted samples were electrophoresed using SDS-PAGE or Blue Native PAGE gels and transferred to PVDF membranes. The membranes were probed with mouse anti-HA antibodies (Santa Cruz Cat# SC-7392, 1:10,000 v/v) or mouse anti-FLAG M2 antibodies (Sigma Cat# F1804, 1:7500 v/v) to assess reciprocal pulldown of PfATP4 and PfABP. To further confirm that the PfABP-HA and PfATP4 exist as a complex, the pulled-down samples were subjected to proteomic analysis by the University of South Florida proteomics facility.
Tryptic digest mass spectrometry
Protein samples were prepared for LC-MS/MS using s-traps (Protifi). 5% SDS in 50 mM triethylammonium bicarbonate (TEAB) was added to the IP beads. Protein samples were reduced with 20 mM dithiothreitol (DTT) at 95 °C for 10 min and alkylated using 40 mM iodoacetamide (IAA) in the dark at room temperature for 30 min. The samples were acidified with 12% phosphoric acid (final concentration 1.2%) before adding 6× volumes of s-trap buffer (90% Methanol, 100 mM TEAB). The protein solution was loaded onto micro s-traps and centrifuged at 4000 × g for 30 s. Three washes with 150 μL s-trap buffer were performed. 0.5 μg of Trypsin/LysC (Promega) was added to the s-trap filter and digested overnight (~16 h) at 37 °C. Peptides were eluted with sequential additions of 50 mM TEAB, water+0.1% formic acid, and 50/50 acetonitrile/water+0.1% formic acid, centrifuged at 4000 × g for 1 min each. Peptides were completely dried in a vacuum-centrifuge concentrator before resuspension in water+0.1% formic acid.
Peptides were separated using a Vanquish Neo UHPLC (Thermo) with a 50 cm EASY-Spray PepMap Neo C18 column (Thermo) and analyzed on a Q Exactive Plus hybrid quadrupole mass spectrometer (Thermo). Peptides were separated over a 1 h gradient (Mobile Phase A: water+0.1% Formic Acid, Mobile Phase B: Acetonitrile+0.1% Formic Acid; 2% B–32% B) and data was acquired using data-dependent acquisition (DDA) with a Top 30 method. Full MS scans were acquired at 70k resolution, and MS/MS scans were acquired at 17.5k resolution.
Raw data files were searched using MaxQuant (v2.4.9) against the current Plasmodium Falciparum protein sequence downloaded from PlasmoDB.org, as well as MaxQuant-generated databases of reverse protein sequences and common contaminant protein sequences. Digestion settings specified Trypsin/P specific enzyme digestion, with a maximum of 2 missed cleavages and a minimum peptide length of 7. Both PSM (peptide spectral match) and protein FDR cutoffs were set at 1%. Cysteine carbamidomethylation was set as a fixed modification. Methionine oxidation and acetyl (protein n-term) were set as variable modifications. The MS/MS match mass tolerance was set at 20 ppm.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The EM maps generated in this study have been deposited in the Electron Microscopy Data Bank (EMDB) under accession numbers 48800 and 48801. The atomic model has been deposited in the Protein Data Bank (PDB) under accession number: 9N10. Source Data are provided as a Source Data file. Source data are provided with this paper.
References
Boddey, J. A. & Cowman, A. F. Plasmodium nesting: remaking the erythrocyte from the inside out. Annu. Rev. Microbiol. 67, 243–269 (2013).
Desai, S. A. Unique properties of nutrient channels on Plasmodium-infected erythrocytes. Pathogens 12, 1211 (2023).
Kirk, K., Staines, H. M., Martin, R. E. & Saliba, K. J. Transport properties of the host cell membrane. Novartis Found. Symp. 226, 55–66; discussion 66–73 (1999).
Kirk, K. Ion regulation in the malaria parasite. Annu. Rev. Microbiol. 69, 341–359 (2015).
Staines, H. M., Ellory, J. C. & Kirk, K. Perturbation of the pump-leak balance for Na(+) and K(+) in malaria-infected erythrocytes. Am. J. Physiol. Cell Physiol. 280, C1576–1587 (2001).
Spillman, N. J. & Kirk, K. The malaria parasite cation ATPase PfATP4 and its role in the mechanism of action of a new arsenal of antimalarial drugs. Int. J. Parasitol. Drugs Drug Resist. 5, 149–162 (2015).
Spillman, N. J. et al. Na(+) regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the spiroindolone antimalarials. Cell Host Microbe 13, 227–237 (2013).
Rachuri, S. et al. Mutational analysis of an antimalarial drug target, PfATP4. Proc. Natl. Acad. Sci. USA 122, e2403689122 (2025).
Macha, A. M. & Becker, W. A. Comparison of predicted with actual body weight selection gains of Coturnix coturnix japonica. Züchter. Genet. Breed. Res. 47, 251–255 (1976).
Stock, C. et al. Fast-forward on P-type ATPases: recent advances on structure and function. Biochem. Soc. Trans. 51, 1347–1360 (2023).
Dyla, M., Kjærgaard, M., Poulsen, H. & Nissen, P. Structure and mechanism of P-type ATPase ion pumps. Annu. Rev. Biochem. 89, 583–603 (2020).
Vaidya, A. B. et al. Pyrazoleamide compounds are potent antimalarials that target Na+ homeostasis in intraerythrocytic Plasmodium falciparum. Nat. Commun. 5, 5521 (2014).
Jiménez-Díaz, M. B. et al. +)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium. Proc. Natl. Acad. Sci. USA 111, E5455–E5462 (2014).
Rottmann, M. et al. Spiroindolones, a potent compound class for the treatment of malaria. Science 329, 1175–1180 (2010).
Lehane, A. M., Ridgway, M. C., Baker, E. & Kirk, K. Diverse chemotypes disrupt ion homeostasis in the malaria parasite. Mol. Microbiol. 94, 327–339 (2014).
White, N. J. et al. Spiroindolone KAE609 for falciparum and vivax malaria. N. Engl. J. Med. 371, 403–410 (2014).
Flannery, E. L. et al. Mutations in the P-type cation-transporter ATPase 4, PfATP4, mediate resistance to both aminopyrazole and spiroindolone antimalarials. ACS Chem. Biol. 10, 413–420 (2015).
Lee, A. H. & Fidock, D. A. Evidence of a mild mutator phenotype in Cambodian Plasmodium falciparum malaria parasites. PLoS ONE 11, e0154166 (2016).
Schmitt, E. K. et al. Efficacy of cipargamin (KAE609) in a randomized, phase II dose-escalation study in adults in sub-Saharan Africa with uncomplicated Plasmodium falciparum malaria. Clin. Infect. Dis. 74, 1831–1839 (2022).
Qiu, D. et al. A G358S mutation in the Plasmodium falciparum Na+ pump PfATP4 confers clinically-relevant resistance to cipargamin. Nat. Commun. 13, 5746 (2022).
Toyoshima, C. & Inesi, G. Structural basis of ion pumping by Ca2+-ATPase of the sarcoplasmic reticulum. Annu. Rev. Biochem. 73, 269–292 (2004).
Jorgensen, P. L., Hakansson, K. O. & Karlish, S. J. D. Structure and mechanism of Na,K-ATPase: functional sites and their interactions. Annu. Rev. Physiol. 65, 817–849 (2003).
Kaplan, J. H. Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 71, 511–535 (2002).
Toyoshima, C., Nakasako, M., Nomura, H. & Ogawa, H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature 405, 647–655 (2000).
Zhang, Y. et al. Cryo-EM analysis provides new mechanistic insight into ATP binding to Ca2+ -ATPase SERCA2b. EMBO J. 40, e108482 (2021).
Chen, Z. et al. Cryo-EM structures of human SPCA1a reveal the mechanism of Ca2+/Mn2+ transport into the Golgi apparatus. Sci. Adv. 9, eadd9742 (2023).
Guerra, J. V. daS. et al. pyKVFinder: an efficient and integrable Python package for biomolecular cavity detection and characterization in data science. BMC Bioinform. 22, 607 (2021).
Zhang, M. et al. Plasma membrane H+-ATPase overexpression increases rice yield via simultaneous enhancement of nutrient uptake and photosynthesis. Nat. Commun. 12, 735 (2021).
Guo, Y. et al. Cryo-EM structures of recombinant human sodium-potassium pump determined in three different states. Nat. Commun. 13, 3957 (2022).
Nguyen, P. T. et al. Structural basis for gating mechanism of the human sodium-potassium pump. Nat. Commun. 13, 5293 (2022).
Mohring, F. et al. Cation ATPase (ATP4) orthologue replacement in the malaria parasite Plasmodium knowlesi reveals species-specific responses to ATP4-targeting drugs. mBio 13, e0117822 (2022).
Tewari, S. G. et al. Metabolic responses in blood-stage malaria parasites associated with increased and decreased sensitivity to PfATP4 inhibitors. Malar. J. 22, 56 (2023).
Jamali, K. et al. Automated model building and protein identification in cryo-EM maps. Nature 628, 450–457 (2024).
Chojnowski, G. et al. findMySequence: a neural-network-based approach for identification of unknown proteins in X-ray crystallography and cryo-EM. IUCrJ 9, 86–97 (2022).
Amos, B. et al. VEuPathDB: the eukaryotic pathogen, vector and host bioinformatics resource center. Nucleic Acids Res. 50, D898–D911 (2022).
Minor, N. T., Sha, Q., Nichols, C. G. & Mercer, R. W. The gamma subunit of the Na,K-ATPase induces cation channel activity. Proc. Natl. Acad. Sci. USA 95, 6521–6525 (1998).
Rivard, C. J., Almeida, N. E., Berl, T. & Capasso, J. M. The gamma subunit of Na/K-ATPase: an exceptional, small transmembrane protein. Front. Biosci. 10, 2604–2610 (2005).
Winther, A.-M. L. et al. The sarcolipin-bound calcium pump stabilizes calcium sites exposed to the cytoplasm. Nature 495, 265–269 (2013).
Akin, B. L., Hurley, T. D., Chen, Z. & Jones, L. R. The structural basis for phospholamban inhibition of the calcium pump in sarcoplasmic reticulum. J. Biol. Chem. 288, 30181–30191 (2013).
Jorgensen, P. L. Importance for absorption of Na+ from freshwater of lysine, valine and serine substitutions in the alpha1a-isoform of Na,K-ATPase in the gills of rainbow trout (Oncorhynchus mykiss) and Atlantic salmon (Salmo salar). J. Membr. Biol. 223, 37–47 (2008).
Blanco, G. Na,K-ATPase subunit heterogeneity as a mechanism for tissue-specific ion regulation. Semin. Nephrol. 25, 292–303 (2005).
Tipsmark, C. K. Identification of FXYD protein genes in a teleost: tissue-specific expression and response to salinity change. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R1367–R1378 (2008).
Rodríguez-Navarro, A. & Benito, B. Sodium or potassium efflux ATPase a fungal, bryophyte, and protozoal ATPase. Biochim. Biophys. Acta 1798, 1841–1853 (2010).
Lehane, A. M. et al. Characterization of the ATP4 ion pump in Toxoplasma gondii. J. Biol. Chem. 294, 5720–5734 (2019).
Vinayagam, D. et al. Molecular mechanism of ultrafast transport by plasma membrane Ca2+-ATPases. Nature https://doi.org/10.1038/s41586-025-09402-3 (2025).
Ho, C.-M. et al. Malaria parasite translocon structure and mechanism of effector export. Nature 561, 70–75 (2018).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Kimanius, D., Dong, L., Sharov, G., Nakane, T. & Scheres, S. H. W. New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem. J. 478, 4169–4185 (2021).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D Struct. Biol. 74, 519–530 (2018).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Acknowledgements
We thank Rebecca Lees for helpful discussions and suggestions. We thank Aarti Ramanathan for her initial enthusiasm and contribution to this study. We thank the Weill Cornell Medicine Proteomics and Metabolomics Core Facility and the University of South Florida Advanced Research Core for Mass Spectrometry for assistance in mass spectrometry. CryoEM data was collected on Titan Krios instruments in the Columbia Electron Microscopy Center. We acknowledge support from the National Institutes of Health (DP5OD029613 to CMH; and R01AI132508 and R01AI154499 to ABV).
Author information
Authors and Affiliations
Contributions
Conceptualization: C.M.H., A.B.V., A.S., and M.T.H. Methodology: C.M.H., A.B.V., A.S., and M.T.H. Investigation: parasite and biochemistry work: A.S. and J.M.M.; Plasmid construct designing: S.B. and A.S.; Phylogenetic analysis: M.W.M.; single particle cryoEM work: M.T.H., J.Z., and Z.Z.; Formal analysis: M.T.H., J.Z., A.S., C.M.H., and A.B.V. Resources: A.B.V. and C.M.H. Data curation: M.T.H. and C.M.H. Writing - original draft: M.T.H. and C.M.H. Writing - review & editing: all authors. Visualization: M.T.H. and C.M.H. Supervision: C.M.H. and A.B.V. Project administration: C.M.H. and A.B.V. Funding acquisition: C.M.H. and A.B.V.
Corresponding authors
Ethics declarations
Competing interests
M.T.H., A.S., J.Z., M.W.M., S.B., J.M.M., Z.Z., and C.M.H. report no competing interests. A.B.V. is listed as an inventor on US Patent 9,464,057. Applicants: MMV Medicines for Malaria Venture; University of Washington; Drexel University; Inventors: Jeremy Burrows, Matthew Wyvratt, Akhil Vaidya, Sandhya Kortagere, Erkang Fan, Arnab Chatterjee, Advait Nagle, Tomoyo Kato. “New Anti-Malarial Agents” (US Patent number 9,464,057). Patent granted: October 11, 2016. This patent covers compound PA21A092 used as an inhibitor for PfATP4.
Peer review
Peer review information
Nature Communications thanks Slavica Pavlovic-Djuranovic, Wai-Hong Tham, and the other anonymous reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Haile, M.T., Shukla, A., Zhen, J. et al. Endogenous structure of antimalarial target PfATP4 reveals an apicomplexan-specific P-type ATPase modulator. Nat Commun 16, 9092 (2025). https://doi.org/10.1038/s41467-025-64815-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64815-y
