
Abstract
Polymer nanocomposites (PNCs) benefit from the enhanced properties originating from the synergistic effects of nanoparticles (NPs) and a polymer matrix. However, the incorporation of NPs slows PNC relaxation, and particularly at high-particle-loading, the PNC melts become non-flowing because the interfacially adsorbed polymers bridge the particles, generating a kinetically quasi-permanent particle network. Herein, by introduction of bound polymer loops on the NP surfaces, we molecularly design a relaxation-enhanced PNC, where interfacial polymers adhering to the NP surfaces freely relaxed, enabling formation of a dynamic, loose particle network that facilitates flow of the PNC melts. The resultant molten high-NP-loading composites maintain fluid-like and low-viscosity dynamics, while the corresponding glassy materials possess enhanced toughness and strength. Thus, the demonstrated principle of preparing relaxation-enhanced PNCs by optimizing the way by which polymers are attached to the filler surface represents the state-of-the-art strategy for overcoming the tradeoff between the processability and mechanical performance of PNCs.
Similar content being viewed by others

Introduction
Since the beginning of the polymer industry, the addition of fillers to polymer matrices has been fundamental to enhance materials’ properties1,2,3,4. However, fillers also substantially increase the viscosity and slow the flow of the PNC melts or solutions. Above a certain NP-loading threshold, PNC melts become permanently elastic and stop flowing because of the formation of polymer-chain-bridged NP networks5,6,7,8, wherein the composites are no longer suitable for extrusion or injection molding. Polymer scientists and engineers have long sought to maintain the excellent flowability and processability of high-NP-loading PNC melts while enhancing the mechanical properties of the corresponding materials.
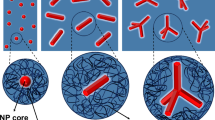
Since the 1990s, the investigation of PNCs has established that the manipulation of polymer–NP interfaces is an effective strategy for altering PNCs’ rheological and mechanical properties9. Near solid interfaces, chain molecules are adsorbed via the enthalpic gain for anchoring chain segments to surfaces10,11,12,13,14,15,16,17. The adsorbed layers possess nanoarchitectures comprising inner flattened chains with high-density-segment–solid contacts and loosely adsorbed polymers extending away from the interface (Fig. 1a)14,15,16,17,18,19. Within the normal experimental timescales, the adsorbed polymers are less likely to desorb because of the high energetic barrier for the simultaneous detachment of numerous adsorbed monomers in the chains10,12,20,21,22,23,24,25,26. On NP surfaces, polymer adsorption connects NPs, generating mesoscopic PNC clusters (Fig. 1a), and above a fair NP-loading (i.e., 10–20 vol %), the NP clusters percolate into a firm NP network, preventing the flow of molten PNCs27,28,29,30,31,32. In the meanwhile, interfacial adsorption increases polymer densities; the high-density interfacial layer, which is a few nanometers in size, possess nearly zero thermal expansion and immobilized segmental dynamics, and are referred to as a dead layer33,34,35,36,37,38. This layers stiffen the composites and increase the moduli of glassy PNCs, and nevertheless energy dissipation is suppressed because of the nullified segmental motion of the interfacial polymers, reducing the toughness and strength of the PNCs. That is, the peculiarly slow and constrained dynamics of the interfacially adsorbed polymers at both the chain and segmental levels39,40,41,42,43,44,45,46 could cause the sluggish flow of the molten PNCs and the embrittlement of the formed glassy materials; see Fig. 1a.

a, b show the structures and dynamics of the conventional and relaxation-enhanced PNCs, respectively. Namely, formation of an immobilized adsorbed layer tightens the particle network and decreases interfacial energy dissipation, eliciting decreased melt flowability and degraded glassy strength of the conventional PNCs. Conversely, the mobile polymer loops on the NP surface serve to loosen the particle network and increase dissipation, thereby facilitating melt flow and strengthening the mechanical properties of the relaxation-enhanced PNCs.
In this work, by introducing an interfacial layer comprising large polymer loops bound to the filler’s surface, we designed a relaxation-enhanced PNC, where the polymers adhering to the filler interface readily relaxed, improving both the PNCs’ processability (e.g., reducing the melts’ viscosities) and mechanical properties (e.g., increasing the glasses’ strength and toughness); see the design principle in Fig. 1b. The free-relaxation bound loops loosened the particle network during melt flow and dissipated energy during mechanical deformation of the glassy polymers. In addition, topological entanglements formed between the bound loops and matrix chains carried substantial loads during deformation, further strengthening the materials47,48,49. Herein, we present the preparation of a relaxation-enhanced PNC comprising silica NPs and polystyrene (PS), characterization of the bound polymer loops, and measurements of the PNCs’ rheological and mechanical properties.
Results
Preparation of loop-covered NPs and relaxation-enhanced PNCs
The preparation process for the relaxation-enhanced PNCs is illustrated in Fig. 2a, wherein the initial step entails the preparation of a polymer loop-covered NPs by coating the NPs surface with a layer of statistical copolymer of poly(styrene-ran−4-hydroxystyrene) [P(S-ran-HS)]. Because HS possess strong affinity with the silica surface because of favorable H-bonding interactions between the HS’s phenolic hydroxyl group and silanol groups on silica (SiOx) surface, it will be predominantly pinned to the surface over the copolymer’s hydrophobic S components19,50, thus providing a means to generate bound loops (BLs) on NP surfaces. Specifically, silica NPs with diameter of 65 ± 10 nm were dispersed in a P(S-ran-HS) matrix by casting composite dispersions of colloidal silica NPs and P(S-ran-HS) in methyl ethyl ketone and subsequently dried to prepare SiOx-NP-filled P(S-ran-HS) composites. The composites were then annealed at 150 °C (i.e., Tg + 50 °C; Tg: the glass transition temperature) for 24 h under vacuum to expedite adsorption of the HS parts in the P(S-ran-HS) chains on the NP surface. The BL–SiOx NPs were obtained via solvent leaching of the composites with chloroform to remove nonattached P(S-ran-HS) chains. The details of the BL–SiOx NP preparation method are included in the section of method and the Supplementary Information (SI).

a Schematic of the preparation of the BL–SiOx NPs and relaxation-enhanced PNCs. b–e Representative transmission electron microscopy (TEM) images of bare SiOx and SiOx NPs bearing 3.4 (fHS = 0.05) and 6.1 nm (fHS = 0.02) thick P(S-ran-HS) BLs. f Thickness of the bound polymer layer derived by TEM, TGA and AFM plotted as a function of fHS, Error bars are standard deviations. The inset shows the equation relating hBL and fHS, assuming a looped chain on the particle surface19; ρpolym is the density (ρPS = 1.05 g cm−3), MS is the molar mass of the S unit, Σ = 1.1 nm−2 is the number of available anchoring points per unit of area on the SiOx surface, and μ is a coefficient (μ = 0.85)19. The gray dashed–dotted curve in (f) represents the prediction using equation in the inset. g Dynamic light-scattering (DLS) traces of the particle size distributions of Ph–SiOx and BL–SiOx NPs in methyl ethyl ketone. h–j TEM images of the BL3–SiOx, BL6–SiOx and Ph–SiOx NPs in the PNCs.
The formation of the BLs on the SiOx NPs was confirmed using transmission electron microscopy (TEM), thermogravimetric analysis (TGA) and atomic force microscopy (AFM); see experimental details in the SI. The TEM images of the BL-coated SiOx NPs showed a clear boundary between the SiOx NPs and the interfacial polymers (Fig. 2c–e) compared to bare SiOx NPs (Fig. 2b). Clearly, the BLs of the P(S-ran-HS) containing higher HS mole fractions in the chain (fHS) were thinner than those containing lower fHS values, i.e., as fHS increased from 0.02 to 0.05, the BL thickness (hBL) decreased from approximately 6 ± 1 nm to 3 ± 1 nm because the reduced fHS values are associated with longer arched sequences (S) between the HS-anchoring points, facilitating the formation of larger loops and thicker BLs. The relation hBL ~ fHS can be quantified using the equation in the inset of Fig. 2f, which was derived based on the proposed nanostructure comprising anchored HS and loop S segments19,50. The adequate fitting confirmed the formation of the NP-surface-bound PS loops, whose height was controlled by altering fHS. In the following sections, the BL–SiOx NPs possessing 3 and 6 nm thick BL layers on average are referred to as BL3– and BL6–SiOx NPs, respectively. Incorporation of the BL–SiOx NPs into the PS matrix formed the proposed relaxation-enhanced PNCs, as shown in Fig. 1b.
The BL–SiOx NPs were dispersed primarily as single NPs in methyl ethyl ketone, as confirmed by the dynamic light-scattering (DLS) curves in Fig. 2g. The NP dispersion was mixed with a toluene solution of PS (weight-averaged molecular weight (Mw) = 370 kg mol−1; polydispersity index (PDI) = 1.07), see the SI for the details of the preparation of the composites. The mixture was naturally dried at room temperature to obtain the composites, which were underwent subsequent thermal annealing at Tg + 30 °C to remove the residual solvents. The TEM images of the composites revealed that the BL–SiOx NPs were well distributed in the PS matrix (Fig. 2h, i) because the SiOx NP-bound loops and PS matrix possessed the same chemistry (e.g., an athermal mixture). However, in the PNCs filled with trimethoxy-phenylsilane-modified silica NPs (i.e., phenyl-modified SiOx; Ph–SiOx), the particles aggregated; see Fig. 2j. In the following sections, the BL3– and BL6–SiOx NP- and Ph–SiOx-filled PNCs are designated as BL3–, BL6–, and Ph–SiOx PNCs, respectively.
Molecular dynamics of the bound polymer loops
Solid-state proton nuclear magnetic resonance (1H-NMR) spectroscopy was employed to directly evaluate the polymer dynamics in the PNCs melts at T = 180 °C. Molecular mobility can be deduced from the free-induction decay (FID) of proton time-domain NMR signals, where fast and slow decays indicate rigid and mobile components, respectively51,52. For the Ph–SiOx PNC melt, the formation of an immobilized adsorption layer on the particle surface elicited the rapid decay of the time-domain FID signal (Fig. 3a). Notably, the FID signal of the BL6–PNC melts underwent a slower decay, similar to that of the pure PS melt (Fig. 3a), suggesting that the PS loops bound to the filler’s surface in the BL–PNCs experienced enhanced relaxation in comparison to the densely adsorbed polymers in Ph–SiOx PNCs, which are difficult to relax thermally. Using a protocol similar to that for preparing the relaxation-enhanced PNCs, we obtained PS films supported by an ~6-nanometer-thick PS BL-covered silicon wafer (see the experimental details in the SI) to investigate the density of the interfacial polymers. The X-ray reflectivity (XRR) measurements of the films revealed that the densities of the bound PS loops were marginally higher than those of the bulk polymers, which contrasts with the 6–8 nm thick high-density interfacially adsorbed layers on the phenyl-modified silicon wafer, see Fig. 3b. The increased thermal relaxation and reduced density of the BLs are consistent with the fabrication of relaxation-enhanced PNCs.

a Proton nuclear magnetic resonance free-induction decay (1H-NMR FID) signals of the PS, Ph–SiOx, and BL6–PNC melts. b XRR profiles for the bilayer films comprising a bottom BL or an adsorbed layer and a top PS layer; insets show schematics of the bilayer films and corresponding density profiles generated in the direction perpendicular to the substrate by fitting the XRR profiles with a bilayer model. c Shifting factors (aT) plotted as functions of temperature for PS/silica PNCs filled with different NPs. For comparison, those for pure PS are also plotted using solid diamonds. The standard deviation values for all the shift factors are less than half an order of magnitude.
To elucidate the dynamics of the PS chains in the BL–PNCs, we compared the rheological shifting factors (aT) recorded at various temperatures for the pure PS and Ph–SiOx and BL–PNCs (Fig. 3c), where the aT values are the time constants used to construct the master G’(ω) and G″(ω) (storage and loss modulus, respectively) curves, as shown in Fig. 4. As shown in Fig. 3c, the temperature-dependent aT values of the pure PS and Ph–PNCs containing low (3 and 8 vol%) NP loadings (φ) followed the Williams–Landel–Ferry (WLF) equation (Supplementary Eq. 3)53, and while that of the Ph–SiOx PNCs changed to an Arrhenius dependence (Supplementary Eq. 4) when the NP loading (φ) reaches to 20 vol%. The Arrhenius-like behavior of PS dynamics in the Ph–PNCs at high NP concentrations indicates the suppression of cooperative polymer dynamics, and instead the localized polymer-desorption-derived relaxation dominates the behavior32,49. In other words, at high NP concentrations, the cooperative dynamics, which promote polymer flow, were hindered because of the irreversible polymer adsorption. However, unlike the Ph–SiOx PNCs, the BL3– and BL6–PNCs showed almost the same temperature-dependent aT values as those of the pure PS, following the WLF equation, implying that the PS dynamics were almost unaffected by incorporation of the BL-NPs. The relaxation and diffusion of the matrix’s PS chains in the BL-PNCs did not have to overcome the high desorption barriers and, therefore, could be accelerated, thereby loosening the particle network, as schematically shown in Fig. 1b.

a, b G’(ω) and G″(ω) master curves plotted as functions of the frequency for PS and Ph–SiOx and BL6–PNCs with various NP loadings (Tref = 160 °C). For the different NP loadings, the curves were vertically shifted for clarity. The solid and dashed lines show the fitting results for G’(ω) and G″(ω), respectively, obtained using the model shown in the SI (Supplementary Eqs. 5–9). c Gter plotted as functions of φ for the PNCs. (d) |η*| plotted as functions of the frequency for pure PS and the PNCs (φ = 6–8 vol%). In (d), the dashed curves were obtained using |η*| = (G’2 + G″2)1/2/ω according to the model fitting shown in (a, b).
Linear viscoelasticity (LVE) of PNCs
The LVE of the PS and the PNCs was characterized based on the low-amplitude oscillatory shear measured using a rotational rheometer. The G’(ω) and G″(ω) of an entangled PS (Mw = 370 kg/mol) are plotted using gray symbols in Fig. 4a. At the intermediate timescale, the rubbery plateau in the G’(ω) represents the elastic response of a transient entanglement network54. Before and after the entanglement modulus plateau (Ge), G″ exceeded G’, and the crossover frequencies were identified as the Rouse and terminal relaxation times (τe and τter) of an entanglement strand and the entire chain, respectively. At t > τter (i.e., the terminal zone), free diffusion of a chain was excited, and the polymers transitioned to a liquid state, where both G’ and G″ decreased with decreasing frequency; however, G’ decreased much faster than G″ with decreasing ω2 (i.e., G’ ≈ ω2 and G″ ≈ ω1). The addition of the PS–SiOx NPs resulted in a sluggish flow, see Fig. 4a. For the PNCs filled with 8 vol% Ph–SiOx NPs, an additional terminal plateau with a moduli (Gter) of ≈ 2 kPa appeared at ω < 10−4 s−1, indicating the cessation of the PNCs’ terminal flow because of the formation of an NP network27,28,29. With increasing NP fraction, the terminal plateau occurs at higher ω and with larger Gter values. Notably, the addition of Ph–SiOx NPs up to 19 vol% results in the emergence of a terminal plateau situated immediately following the rubbery plateau. That is, the terminal relaxation of the PNCs was entirely suppressed, suggesting the generation of a dense particle network, where the interparticle distance is close to the tube’s diameter for polymer reptation.
In contrast to the φ-sensitive LVE spectra of the Ph–SiOx PNCs (Fig. 4a), the spectra of the BL–PNCs (Fig. 4b and Supplementary Fig. 7) were less dependent on the NP content. Specifically, for the BL6–PNCs, possessing large loops and thick BLs (Fig. 4b), the low-frequency G’ value of the φ = 3 vol% PNCs decayed with decreasing ω in a manner similar to that by which the low-frequency G’ of the pure PS decayed, meaning that a trace of BL6–SiOx NPs did not change the PNC flow. For the φ = 8 vol% PNCs, G’ relaxed slightly slower than that of the PS melt but much faster than that of the Ph–SiOx PNCs at the same NP concentration. With increasing φ to 20 vol%, where terminal flow is prohibited for Ph–SiOx PNCs, G’ relaxed for BL6–PNCs, and no obvious terminal plateau appeared down to 10−4 rad s−1. The results for mechanical creep of the pure PS, Ph–SiOx PNCs and BL6–PNCs align well with those from oscillatory shear experiments; see Supplementary Fig. 8; i.e., a terminal plateau appears in Ph-SiOx PNC, and however, such plateau disappears in BL6-PNC.
To quantify the effect of NPs on the PNCs’ dynamics, we used a simple analytical model proposed by Chen and Colby et al.41 to describe the PNCs’ linear rheology. The dashed and solid curves in Fig. 4a, b represent the fitting results; the modeling details are presented in the SI. From the fittings, Gter, characterizing the network density, can be derived. Clearly, for all the PNCs, Gter increased with increasing φ and were the lowest for the BL6–PNCs (Fig. 4c), suggesting that the attachment of the BLs to the NP surface facilitated the PNCs’ terminal relaxation, i.e., one of the essential features of the relaxation-enhanced PNCs.
To further evaluate the ease of the BL–PNCs’ flow, the complex viscosity [|η*| = (G’2 + G″2)1/2/ω] was calculated for the PS and φ = 6–8 vol% PNCs, see Fig. 4d. For the pure PS, |η*| increased with decreasing frequency and plateaued to a constant value at low frequencies, where the polymers flowed freely under low stress. In contrast, for the Ph–SiOx PNCs, |η*| increased monotonically with decreasing frequency without plateauing at the zero-shear viscosity, indicating sluggish flow because of the Ph–SiOx blending. For the BL3– and BL6–PNCs, |η*|’s low-frequency plateau reappeared, and the plateau viscosity gradually decreased to that of the pure PS with increasing hBL, confirming the accelerated terminal relaxation of the PNCs possessing larger BLs on the NP surface. Notably, the low-frequency viscosity of the BL6-PNCs decreased by over 10 times compared to the Ph-SiOx PNCs; Fig. 4d. Nonetheless, the BL strategy was less effective for the low-molecular-weight PS characteristic of intrinsically weak particle networks55, see Supplementary Fig. 9.
Mechanical properties of the glassy PNCs
The mechanical properties of the relaxation-enhanced PNCs were assessed using tensile measurements and dynamic mechanical analysis (DMA). The glassy BL–PNCs possessed a Young’s modulus and a tensile strength of 4.1 GPa and 51 MPa, respectively, which were 46% and 42% higher than those of the pure PS, see Fig. 5a, b. In contrast, at the same nanofiller–polymer ratio, the Ph–SiOx PNCs possessed both decreased tensile strength and elongation at break because of NP aggregation and the stiffening of the interfacial polymers absorbed on the NPs. Furthermore, an additional loss modulus peak, arising from the internal friction induced by the relaxation of polymer near the NP interface, appeared in the DMA dissipation spectra of the PNCs (Fig. 5c). The interfacial dissipation peak for the BL–PNCs was much more intense than that for the Ph–SiOx PNCs and was even comparable to the intensity of the α-relaxation peak of the pure PS. Therefore, the enhanced relaxation of the BL–PNC interfacial polymers increased the dissipation factor of the composites and helped to enhance the PS-based BL–PNCs56,57, see Figs. 1b and 5c.

a Stress–strain curves, b storage (E’) and c loss modulus (E”), as well as the deconvoluted peaks, of the glassy PS and Ph–SiOx (φ = 19 vol%) and BL3– (φ = 25 vol%) and BL6– (φ = 20 vol%) PNCs, respectively. The corresponding scanning electron microscopy (SEM) images of the fractured cross-sections of the glassy (d, d1) PS and (e, e1) Ph–SiOx and (f, f1) BL6–PNCs.
The analysis of the fractured cross-sectional morphologies further confirmed that BL–SiOx reinforced the PNCs. Figure 5d–f shows the scanning electron microscopy (SEM) images of the fractured surfaces of the films after tensile testing. Because PS is intrinsically brittle, it exhibited relatively smooth cleavage fracture behavior. Such fractography is typical for brittle polymers and reveals that the crack propagated rapidly and catastrophically. At the tensile-fractured surface of the Ph–SiOx PNCs, NP aggregation was evident, and the SiOx NPs appeared to separate from the polymer matrix during fracturing, indicating weak polymer–filler interactions. Although the fractured surface of the BL–SiOx PNCs was still brittle, it was rougher because of some locally plastic deformation. Meanwhile, no SiOx particles were exposed on the fractured surface, suggesting exceptionally strong polymer–filler interactions that could not be destroyed during fracturing. Such amplified interactions could be explained by the formation of topological entanglements between the BLs and matrix chains, which function as a load-bearing structure and further toughen the materials, see Fig. 1b. Additionally, we used MD simulation to construct the same BL-PNCs as in experiments, and direct visualize the loop-chain entanglements on particle surface; see Supplementary Fig. 10. These results revealed that the BL–SiOx PNCs possessed higher tensile strength and increased toughness compared with those of the pure PS films and Ph–SiOx composites.
Discussion
Our work demonstrates that the design of a relaxation-enhanced PNC, where polymers near NPs freely relax, is a feasible strategy for modulating PNCs’ melt flows, processability, and mechanical properties. Our strategy involves the attachment of large polymer loops to NP surfaces by coating the NPs with a nanolayer of a well-designed random copolymer. The free sliding of the matrix chains through the mobile loops generated a loosened and dynamic NP network, reducing the viscosity and improving the flowability of the melts (i.e., the BL-PNC melt’s viscosity was reduced by more than tenfold compared with that of the loop-free PNC melt’s viscosity). Moreover, during deformation, the active dynamics of the bound polymer loops generated additional dissipation, and the entanglements between the BLs and matrix chains enhanced the polymer–filler interactions and toughened, strengthened the PNC glasses (i.e., the PNCs’ mechanical strength increased by 42%). Clearly, the proposed BL strategy reduces the melts’ viscosity while strengthening the mechanical properties of glassy PNCs—a feat that is difficult to accomplish using traditional particle modification techniques, such as grafting chains or surfactants onto NP surfaces.
Therefore, relaxation-enhanced PNCs solve the longstanding problem of the tradeoff between improving the processability and mechanical properties of polymer nanocomposites. This practical endeavor also suggests that the PNCs’ rheological and mechanical properties can be effectively controlled by tuning the dynamics and structure at polymer–NP interfaces, opening new doors for understanding and manipulating nanoparticle-filled composites. We anticipate that the proposed BL strategy, which does not require sophisticated synthetic or complex processing procedures, will be applied to the large-scale manufacturing of polymer composites exhibiting diverse structures and functions.
Methods
Preparation and characterization of P(S-ran-HS)
Poly(styrene-ran-4-hydroxystyrene) copolymer P(S-ran-HS) was prepared by radical copolymerization of styrene (S) and 4-acetoxystyrene (AS) at 60 °C using 2,2’-azobis(2-methylpropionitrile) as initiator and followed by hydrolysis of the acetoxy group (Supplementary Figs. 1, 2), as previously reported19,50. Copolymerization between S and AS with the reactivity ratio of rS = 0.8 and rAS = 1.04 (rS·rAS → 1), respectively, produces a typically statistic copolymer with S and AS components randomly distributed in the P(S-ran-AS) chains. The hydrolyzation of the acetoxy group in P(S-ran-AS) results in the formation of a statistic copolymer of P(S-ran-HS)58,59. The nuclear magnetic resonance (NMR) spectra and gel permeation chromatography (GPC) were employed for the confirmation of the chemical structure and Mw of P(S-ran-HS) (Supplementary Figs. 3, 4); see SI for the details.
Preparation and characterization of BL-SiOx NPs
The copolymer, P(S-ran-HS), was dissolved in methyl ethyl ketone (MEK) to form a polymer solution with a concentration of 10 wt%. The solution was then mixed with colloidal suspension of SiOx NPs (grade: MEK-ST, diameter of 65 ± 10 nm, supplied by Nissan Chemical) dispersed in MEK. The mixture was stirred thoroughly and dried naturally for 2 days to obtain the P(S-ran-HS)/silica composite materials, which was underwent subsequent annealing at 150 °C for 24 h under vacuum, to expedite adsorption of P(S-ran-HS) chain onto the SiOx NPs surface and to form interfacial bound loops. The annealed composites were then solvent leached thoroughly to isolate nanoparticles bearing bound loops from an unadsorbed polymer matrix, see SI for the experimental details. After drying the nanoparticle at 50 °C for 4 h, SiOx particles covered with a bound polymer layer of P(S-ran-HS) (i.e., BL-SiOx NPs) obtain readily. As a control, trimethoxy-phenylsilane-modified silica NPs (i.e., phenyl-modified SiOx; Ph–SiOx) were prepared through an interfacial self-assembly process; see SI for the preparation details. Transmission electron microscopy (TEM), thermogravimetric analysis (TGA) (Supplementary Fig. 5) and atomic force microscope (AFM) (Supplementary Fig. 6) were employed to characterize the thickness of bound loops layer absorbed on the surface of SiOx NPs (hBL); see SI for the details.
Preparation of relaxation-enhanced PNCs (BL-SiOx)
PS polystyrene (PS) (Mw = 370 or 80 kg/mol, Ð = 1.07, purchased from Polymer Source, Canada) was dissolved in 4 ml toluene and stirring continuously for ∼4 h to obtain a PS solution with concentration of 10 wt%. The NPs powders were dispersed in the MEK and sonicated 1 h to form NPs dispersions. The nanoparticles dispersion was mixed with toluene solution of PS, sonicated and stirred overnight for effective mixing. The resulting nanoparticle/PS dispersion was dried at room temperature for 24 h. Afterwards, the composite powders were underwent subsequently annealing at Tg + 30 °C in a convection oven for 48 h to remove the residual solvents, yielding the PNCs. A series of PNCs with different volume fractions (φ vol%) of NPs were prepared.
Characterization of dispersion of nanoparticles in PNCs
The dispersion of nanoparticles in PNCs was characterized by transmission electron microscopy (TEM) using an FEI Tecnai F20 ST microscope (field-emission gun operated at 3.8 kV extraction voltage) operating at 200 kV. The composite specimens were cut into 100 nm-thick thin sections by ultramicrotomy using a Leica Ultracut UCT microtome with a diamond knife, and the thin films were transferred onto the 200-mesh copper grids at room temperature.
Characterization of rheology, dynamics and mechanical properties of PNCs
The linear viscoelasticity of PS and PNCs was characterized by the small amplitude oscillatory shear (SAOS) on an Anton Paar MCR702 (Austria) rheometer with 8 mm parallel plates. Frequency sweep tests were conducted at constant stresses ranging from 200 to 1000 Pa within the linear viscoelastic region at various temperatures (from 130 to 270 °C). Subsequently, time-temperature superposition (TTS) was employed to construct master curves for each sample at the reference temperature (Tref = 160 °C).
Solid-state 1H-NMR measurements were performed on a Bruker Minispec mq20 low-field spectrometer at 20 MHz proton resonance frequency. The sample temperature was controlled with a BVT3000 heater working with a flow of heated air. In order to avoid O2-related sample degradation, the NMR tube (10 mm outer diameter) was first flushed with argon gas. The Minispec has a typical π/2 pulse length of about 3 μs and a receiver dead time of about 10 μs.
The density of the interfacial polymers was characterized by X-ray reflectivity (XRR) based on a bilayer model consist of a PS film supported by a P(S-ran-HS) bound layer on a silicon wafer. Specifically, an 80 nm thick films were prepared by spin-coating the P(St-ran-HS) (fHS = 0.02) toluene solution onto a silicon surface, and were annealed at 150 °C for 24 h under vacuum to expedite the formation of the interfacial bound loops. Next, the unbound chains were removed via solvent leaching in chloroform to obtain an exposed interfacial bound layer with a thickness of ~6 nm on silicon surface. The interfacial bound layer was dried under vacuum at 100 °C for 1 h, after which a 20 nm thick PS film was spin-coated on top. The bilayer films were annealed at 150 °C under vacuum overnight to dry the film and equilibrate the structure of the interfacial polymer chains. For comparison, we prepared PS films with an adsorbed layer on a Ph-modified silicon surface. Specifically, a PS film with thickness of ~20 nm was prepared by spin-coating a PS toluene solution onto the Ph-modified silicon surface, followed by annealing at 150 °C for 24 h under vacuum conditions to accelerate the adsorption of PS chains at the interface. XRR experiments were performed on the films using a Bruker D8 Advance diffractometer equipped with a reflectometry with the X-ray source (Cu Kα radiation, λ = 1.54056 Å). The reflectivity data were analyzed using the Motofit program of the IGOR software.
Tensile tests were performed at a constant speed of 5 mm/min at room temperature on a MOXIN MX-0350 tester to measure the mechanical properties of the glassy PNCs. BL6-SiOx and Ph-SiOx PNC samples were mold and cut into dumbbell-shape films with the gauge size of 20 × 2 × 0.2 mm for the tests.
Dynamic mechanical analysis was performed on the Mettler Toledo DMA1 in the dual cantilever bending modes under a 0.03 % strain at a frequency of 1 Hz. Temperature scans were performed from 20 to 160 °C at a rate of 5 °C /min at air atmosphere. The samples were cut into 30 × 6 × 0.2 mm strips for the tests.
The micromorphology of the fracture surfaces of the composites was directly visualized using scanning electron microscopy (SEM, Ultra55, Zeiss, Germany). The PNCs films after tensile testing were bonded directly to the conductive adhesive and sprayed with gold for 300 s using a Quorum SC7620 sputter coater at 10 mA and then characterized by SEM.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. All data are available from the corresponding author upon request.
References
Cheng, S. et al. Focus: structure and dynamics of the interfacial layer in polymer nanocomposites with attractive interactions. J. Chem. Phys. 146, 203201 (2017).
Kumar, S. K., Benicewicz, B. C., Vaia, R. A. & Winey, K. I. 50th anniversary perspective: Are polymer nanocomposites practical for applications?. Macromolecules 50, 714–731 (2017).
Steck, J., Kim, J., Kutsovsky, Y. & Suo, Z. Multiscale stress deconcentration amplifies fatigue resistance of rubber. Nature 624, 303–308 (2023).
Zou, H., Wu, S. & Shen, J. Polymer/silica nanocomposites: preparation, characterization, properties, and applications. Chem. Rev. 108, 3893–3957 (2008).
Bailey, E. J. & Winey, K. I. Dynamics of polymer segments, polymer chains, and nanoparticles in polymer nanocomposite melts: a review. Prog. Polym. Sci. 105, 101242 (2020).
Akcora, P. et al. “Gel-Like” mechanical reinforcement in polymer nanocomposite melts. Macromolecules 43, 1003–1010 (2010).
Choi, J. et al. Universal scaling of polymer diffusion in nanocomposites. ACS Macro Lett. 2, 485–490 (2013).
Venkatesh, R. B. & Lee, D. Conflicting effects of extreme nanoconfinement on the translational and segmental motion of entangled polymers. Macromolecules 55, 4492–4501 (2022).
Nepal, D., Haines, J. & Vaia, R. A. Polymer nanocomposites: 35 years on. MRS Bull. 49, 236–246 (2024).
Napolitano, S. Irreversible adsorption of polymer melts and nanoconfinement effects. Soft Matter 16, 5348–5365 (2020).
Tian, H. et al. Intramolecular dynamic coupling slows surface relaxation of polymer glasses. Nat. Commun. 15, 6082 (2024).
Panagopoulou, A. & Napolitano, S. Irreversible adsorption governs the equilibration of thin polymer films. Phys. Rev. Lett. 119, 097801 (2017).
Nieto Simavilla, D., Huang, W., Housmans, C., Sferrazza, M. & Napolitano, S. Taming the strength of interfacial interactions via nanoconfinement. ACS Cent. Sci. 4, 755–759 (2018).
Gin, P. et al. Revealed architectures of adsorbed polymer chains at solid-polymer melt Interfaces. Phys. Rev. Lett. 109, 265501 (2012).
Luo, J., Zha, H., Tian, H. & Zuo, B. Physical adsorption and glass transition of thin polystyrene films at a graphene interface. Chin. J. Polym. Sci. 43, 1163–1169 (2025).
Jiang, N., Shang, J., Di, X., Endoh, M. K. & Koga, T. Formation mechanism of high-density, flattened polymer nanolayers adsorbed on planar solids. Macromolecules 47, 2682–2689 (2014).
Koga, T. et al. Impact of an Irreversibly Adsorbed Layer on Local Viscosity of Nanoconfined Polymer Melts. Phys. Rev. Lett. 107, 225901 (2011).
Cheng, S. et al. Unexpected molecular weight effect in polymer nanocomposites. Phys. Rev. Lett. 116, 038302 (2016).
Zuo, B., Zhou, H., Davis, M. J. B., Wang, X. & Priestley, R. D. Effect of local chain conformation in adsorbed nanolayers on confined polymer molecular mobility. Phys. Rev. Lett. 122, 217801 (2019).
Frantz, P. & Granick, S. Kinetics of polymer adsorption and desorption. Phys. Rev. Lett. 66, 899–902 (1991).
Schneider, H. M., Frantz, P. & Granick, S. The bimodal energy landscape when polymers adsorb. Langmuir 12, 994–996 (1996).
Tian, H., Bi, C., Li, Z., Wang, C. & Zuo, B. Metastable polymer adsorption dictates the dynamical gradients at interfaces. Macromolecules 56, 4346–4353 (2023).
Monnier, X., Napolitano, S. & Cangialosi, D. Direct observation of desorption of a melt of long polymer chains. Nat. Commun. 11, 4354 (2020).
Napolitano, S. & Wübbenhorst, M. The lifetime of the deviations from bulk behaviour in polymers confined at the nanoscale. Nat. Commun. 2, 260 (2011).
Guiselin, O. Irreversible adsorption of a concentrated polymer solution. Europhys. Lett. 17, 225 (1992).
Johnson, H. E. & Granick, S. New mechanism of nonequilibrium polymer adsorption. Science 255, 966–968 (1992).
Sternstein, S. S. & Zhu, A.-J. Reinforcement mechanism of nanofilled polymer melts as elucidated by nonlinear viscoelastic behavior. Macromolecules 35, 7262–7273 (2002).
Long, D. & Sotta, P. Stress relaxation of large amplitudes and long timescales in soft thermoplastic and filled elastomers. Rheol. Acta 46, 1029–1044 (2007).
Chen, Q. et al. Mechanical reinforcement of polymer nanocomposites from percolation of a nanoparticle network. ACS Macro Lett. 4, 398–402 (2015).
Cui, W., You, W. & Yu, W. Mechanism of mechanical reinforcement for weakly attractive nanocomposites in glassy and rubbery states. Macromolecules 54, 824–834 (2021).
Wang, Y. et al. Quantitative correlation between hierarchical nanofiller structure and rheology of polymer/fumed silica nanocomposites. Macromolecules 56, 934–946 (2023).
Baeza, G. P. et al. Network dynamics in nanofilled polymers. Nat. Commun. 7, 11368 (2016).
DeMaggio, G. B. et al. Interface and surface effects on the glass transition in thin polystyrene films. Phys. Rev. Lett. 78, 1524–1527 (1997).
Napolitano, S. & Wübbenhorst, M. Dielectric signature of a dead layer in ultrathin films of a nonpolar polymer. J. Phys. Chem. B 111, 9197–9199 (2007).
Scheutjens, J. M. H. M. & Fleer, G. J. Statistical theory of the adsorption of interacting chain molecules. 2. Train, loop, and tail size distribution. J. Phys. Chem. 84, 178–190 (1980).
Bitsanis, I. A. & ten Brinke, G. A lattice Monte Carlo study of long chain conformations at solid–polymer melt interfaces. J. Chem. Phys. 99, 3100–3111 (1993).
O’Shaughnessy, B. & Vavylonis, D. Irreversibility and polymer adsorption. Phys. Rev. Lett. 90, 056103 (2003).
O’Shaughnessy, B. & Vavylonis, D. Irreversible adsorption from dilute polymer solutions. Eur. Phys. J. E 11, 213–230 (2003).
Tanaka, K. et al. Interfacial mobility of polymers on inorganic solids. J. Phys. Chem. B 113, 4571–4577 (2009).
Tsuruta, H. et al. Local conformation and relaxation of polystyrene at substrate interface. Macromolecules 45, 4643–4649 (2012).
Midya, J. et al. Disentangling the role of chain conformation on the mechanics of polymer tethered particle materials. Nano Lett. 19, 2715–2722 (2019).
Hao, Z. et al. Mobility gradients yield rubbery surfaces on top of polymer glasses. Nature 596, 372–376 (2021).
Randazzo, K. et al. Direct visualization and characterization of interfacially adsorbed polymer atop nanoparticles and within nanocomposites. Macromolecules 54, 10224–10234 (2021).
Yang, Y., Tian, H., Napolitano, S. & Zuo, B. Crystallization in thin films of polymer glasses: the role of free surfaces, solid interfaces and their competition. Prog. Polym. Sci. 144, 101725 (2023).
Perez-de-Eulate, N. G., Sferrazza, M., Cangialosi, D. & Napolitano, S. Irreversible adsorption erases the free surface effect on the Tg of supported films of poly(4-tert-butylstyrene). ACS Macro Lett. 6, 354–358 (2017).
Burroughs, M. J., Napolitano, S., Cangialosi, D. & Priestley, R. D. Direct measurement of glass transition temperature in exposed and buried adsorbed polymer nanolayers. Macromolecules 49, 4647–4655 (2016).
Moll, J. F. et al. Mechanical reinforcement in polymer melts filled with polymer grafted nanoparticles. Macromolecules 44, 7473–7477 (2011).
Zhao, D. et al. Role of filler shape and connectivity on the viscoelastic behavior in polymer nanocomposites. Macromolecules 48, 5433–5438 (2015).
Yang, J., Melton, M., Sun, R., Yang, W. & Cheng, S. Decoupling the polymer dynamics and the nanoparticle network dynamics of polymer nanocomposites through dielectric spectroscopy and rheology. Macromolecules 53, 302–311 (2020).
Luo, J. et al. Effect of conformation of interfacial adsorbed chains on physical aging of polymer nanocomposites. J. Chem. Phys. 160, 064906 (2024).
Zhang, R. et al. Reversible cross-linking, microdomain structure, and heterogeneous dynamics in thermally reversible cross-linked polyurethane as revealed by solid-state NMR. J. Phys. Chem. B 118, 1126–1137 (2014).
Gao, Y. et al. Critical effect of segmental dynamics in polybutadiene/clay nanocomposites characterized by solid state 1h NMR spectroscopy. J. Phys. Chem. C. 118, 5606–5614 (2014).
Williams, M. L., Landel, R. F. & Ferry, J. D. The temperature dependence of relaxation mechanisms in amorphous polymers and other glass-forming liquids. J. Am. Chem. Soc. 77, 3701–3707 (1955).
Rubinstein, M. & Colby, R. H. Polymer Physics. (Springer Berlin Heidelberg, 2003).
Huang, J., Zhou, J. & Liu, M. Interphase in polymer nanocomposites. JACS Au 2, 280–291 (2022).
Huang, J. et al. Ultrahigh energy-dissipation elastomers by precisely tailoring the relaxation of confined polymer fluids. Nat. Commun. 12, 3610 (2021).
Huang, J. et al. Full-scale polymer relaxation induced by single-chain confinement enhances mechanical stability of nanocomposites. Nat. Commun. 15, 6747 (2024).
Zhu, K. J., Chen, S. F., Ho, T., Pearce, E. M. & Kwei, T. K. Miscibility of copolymer blends. Macromolecules 23, 150–154 (1990).
Woo, D. et al. Styrene/4-hydroxystyrene random, block and gradient copolymers modified with an organic dye: synthesis by controlled radical polymerization and characterization of electrorheological properties. Polymer 47, 3287–3291 (2006).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant nos. 22525303, 22303084, and 52373025). J.L. further acknowledges the Natural Science Foundation of Zhejiang Province (Grant no. LQ24E030010).
Author information
Authors and Affiliations
Contributions
B.Z. and J.L. conceived and supervised the experiments; Q.X. and H.Q. performed experiments; Z.W. performed and analyzed the simulations; S.G. and Z.Z. contributed to data analysis.; Q.X. contributed to original draft writing. B.Z. and J.L. contributed to review and editing. All authors participated in discussions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Humberto Almeida and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, Q., Qu, H., Wu, Z. et al. Relaxation-enhanced polymer nanocomposites induced by bound polymer loops on the particle surfaces. Nat Commun 16, 9870 (2025). https://doi.org/10.1038/s41467-025-64841-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64841-w
