
Abstract
The coexistence of asthma and chronic rhinosinusitis with nasal polyposis (CRSwNP) is associated with allergic phenotypes, disease severity and failure of first-line treatment for both asthma and CRSwNP. Recent studies have highlighted shared genetic components for these diseases. To better understand this shared component, we perform genome-wide meta-analyses of asthma (n = 71,481), CRSwNP (n = 9626) and chronic rhinosinusitis without nasal polyposis (CRSsNP, n = 15,448) in FinnGen and UKB (685,602 controls). We detect 131 genomic associations, including 17 novel loci for asthma, 33 novel loci for CRSwNP, and one for CRSsNP. A shared impact on asthma and CRSwNP is observed at 71 loci. A cross-trait meta-analysis using all disorders further implicates 17 loci associated with asthma or asthma and CRSwNP. We also find 17 nonsynonymous associating variants, including a novel TP63 missense variant association with CRSwNP (OR = 1.519 [1.331–1.734]). Gene set analyses confirm enrichment of genes involved with type 2 inflammation, Jak-STAT signaling, and FOXP3 signaling. Our results highlight new shared and separate genetic pathways for CRSwNP and asthma. These provide several avenues of further investigation in functional and epidemiological follow-up, and evidence for immunological and non-immunological mechanisms behind both diseases.
Similar content being viewed by others

Introduction
Chronic rhinosinusitis (CRS) and asthma each affect about 9–12% of the population1. CRS is a heterogenous diseases characterized by persistent inflammation of the nasal and sinus passages, with symptoms lasting over 12 weeks1. Asthma is a heterogenous disease characterized by inflammation, bronchial hyperreactivity and symptoms. They both have a severe impact on quality of life, morbidity and economy1. The overlap between asthma and CRS is ~50%2. Co-morbid asthma and CRS is associated with increased morbidity, suffering and costs3, necessitating studies on the pathogenesis of asthma and CRS to reduce disease burden.
Asthma encompasses various subtypes, with allergic and non-allergic eosinophilic asthma being the most common4. CRS has two main phenotypes: with nasal polyps (CRSwNP) and without nasal polyps (CRSsNP)1. CRSwNP is characterized by the presence of nasal polyps, edematous non-malignant protrusions of the mucosal membranes lining the middle meatal and ethmoidal region of the sinonasal tract. With an estimated prevalence of 1.9–3.5 % from cross-sectional studies1,5,6, about half of CRSwNP patients require specialist intervention due to poor disease control and the need for advanced therapy1,2,7. CRSwNP is underdiagnosed in the general population, and the true prevalence is likely higher1,5. Underdiagnosis is likely due to a lack of disease awareness and poor nasal endoscopy facilities at the primary care level.
CRS and asthma are classified by inflammatory endotypes. T helper cell 2, i.e., Type 2 (T2) high asthma and CRSwNP are linked to eosinophils and cytokines, interleukin (IL)-4, IL-5, IL-13, while Type 2 low endotype involves Th17 pathways and neutrophilic inflammation1. The vast majority of CRSwNP and most asthma cases show Th2-driven inflammation1. Despite the increased knowledge of endotypes of CRSwNP and asthma, there is still lacking knowledge of the genome environmental inteactions leading to the onset of co-morbid CRSwNP and asthma2,8. Hence, it is important to study the shared genetics of CRSwNP and asthma to enlighten the etiopathology of these diseases.
T2-high asthma and CRSwNP share similarities in their risk factors, such as genetic loci related to type 2 inflammation pathways and to IgE overproduction, allergic rhinitis, recurrent respiratory infections, bacterial colonization, autoimmunity responses and eosinophilia9. There are also different risk factors for CRSwNP and type 2-high asthma, which may explain why some individuals get only type 2-high asthma and some only CRSwNP. Shared environmental contributors include epithelium dysbiosis, cigarette smoke and other air pollutants. A shared genetic component between the conditions has been recognized as well10,11, and the shared heritability is greatly enhanced in CRSwNP9,12. Both asthma and CRS have implicated type 2 inflammation pathways of innate immunity, including IL-4, IL-6, IL-13, and the TSLR pathway1,12,13,14.
In asthma, over 212 genomic loci have been identified to date, including loci harboring GSDMB/ORMDL, HLA, TSLP, IL1RL1/Il18R, and IL3310,15,16,17,18, confirming the important role of type 2 inflammatory pathways. For CRSwNP, a genome-wide association study (GWAS) in Icelandic and English populations detected a protective gain-of-function variant in ALOX15, suggesting an important role for LOX-high pathways9. Recent genetic analyses9,12 in Finland and the UK have implicated 17 genomic loci associated with CRSwNP. Importantly, beyond observed clinical overlap, 10 of the 17 CRSwNP loci also associate with asthma. Our previous in silico analyses12 using LD score regression19 established a genetic correlation of 68.7% between asthma and CRSwNP, highlighting both the similarity and potential heterogeneity of heritable factors in one or both diseases. Several of the shared pathways between asthma and CRSwNP have been targeted in biologic therapeutics7,20,21, warranting further study of the shared biological aspects of the diseases.
In this work, we show that the shared genetic findings between asthma and nasal polyposis highlight shared and distinct biological processes and structures relevant to both diseases, offering clues to mechanisms of treatment resistance and disease subtypes. To understand the shared genetic contribution, we conduct genome-wide analyses of asthma, CRSwNP, and CRSsNP in FinnGen and the UK Biobank (UKB). Independent analyses of co-morbid disorders have been showed to have a potential to misrepresent shared etiologies22. Here, we leverage a previously established approach12,17,23 using cross-trait analysis of all three endpoints to emphasize shared genetic risk, instead of limiting analysis to trait-specific associations. A note on the study design is included in the Supplement (Supplementary Note 1).
Results
Genome-wide association analysis
We performed genome-wide association studies (GWASs) of asthma, chronic rhinosinusitis with nasal polyposis (CRSwNP), and chronic rhinosinusitis without nasal polyposis (CRSsNP) in FinnGen and UKB (Table 1, Fig. 1). We identified 87 genome-wide significant (GWS) loci associated with asthma (Fig. 2, Supplementary Data 1), of which 10 have not been previously linked with asthma10,15,17,18,23,24 (Table 2). All novel asthma lead variants had co-directional impact with at least nominal significance (p < 0.05) in both cohorts, although heterogeneity for 1p22.1 (harboring TGFBR3) was high (I2 = 76.3%). Of the asthma-associated loci, 26/87 were GWS associated with CRSwNP, including three (5q22.1, 6p21, and 9p24.1, near genes TSLP, HLA, and IL33, respectively) associated with CRSsNP as well. The asthma locus 16q24.3 near CHMP1A was also associated with CRSsNP (ORCRSsNP = 1.068 [1.043–1.094]). Interestingly, age correction did not appear to have any impact in the FinnGen analysis (Supplementary Fig. 1).

Genome-wide association was carried out on each of the three phenotypes separately, and using an asthma and/or CRS cross-trait phenotype where all participants with asthma or chronic rhinosinusitis (CRS) with nasal polyposis (CRSwNP) or without (CRSsNP) were selected as cases. All four GWASs had the same controls without any of the tested diseases (685,602 controls in total).

Negative log10 p values from genome-wide association meta-analysis of asthma (top, 71,481 cases vs 685,602 controls) and CRSwNP (bottom, 9626 cases vs 685,602 controls). The red dashed line denotes genome-wide significance (p < 5e-8). P values are derived from one-sided χ2-distribution with one degree of freedom.
A total of 50 loci associated with CRSwNP (Fig. 2, Supplementary Data 2), including 26 loci also associating with asthma. Most of the CRSwNP loci (33/50) have not been previously reported in the context of CRSwNP (Table 3). Several new loci are relevant to immunological pathways (e.g., 5p13.2 near IL7R, 10p15.2 near IL2RA, and 22q12.3 near IL2RB), including the allergic asthma locus IL4R (ORCRSwNP = 1.151 [1.104–1.199], p = 2.54e-11). Effect heterogeneity between FinnGen and UKB was notable (I2 > 50 %) for ten loci, although co-directional effects were observed in both cohorts for nine of these loci. The 9q22.3 locus did not reach significance in the UKB (pUKB = 0.17 for lead variant rs7044307-G near SEC61B).
For CRSsNP, we identified five GWS loci (Supplementary Fig. 2, Supplementary Data 3), including one novel association at 9q33.3, near NEK6. The locus, previously linked to allergic rhinitis and vitiligo, associated with CRSsNP (OR = 1.069 [95 % CI 1.044–1.094], p = 2.28e-8). The CRSsNP association for the 5q22.1 locus near TSLP exhibited heterogeneity (I2 = 68.8 %) between FinnGen and UKB, and effect estimates were contra-directional for the lead variant rs79411074-G. Conversely, the remaining four loci demonstrated co-directional and homogeneous effects between the cohorts.
Cross-trait analysis
Given the previously observed9,12 shared loci and genetic correlation between asthma and CRS, we conducted a further GWAS with all participants with asthma, CRSwNP and CRSsNP as cases (n = 88,965). This enhances the analysis of shared genetic risk, at the expense of single phenotype-specific genetic risk, as seen in previous studies involving asthma and allergic diseases17,23. The asthma and/or CRS GWASs were run in FinnGen and UKB separately with the same pipelines as above, and the same controls. The GWAS resulted in 95 GWS loci (Supplementary Data 4), of which 17 were not detected in single-phenotype analyses (Table 4).
In our genome-wide analyses, we observed inflation of test statistics, especially concerning asthma and the cross-trait phenotype, in both the meta-analyses and the original GWASs carried out in FinnGen and UKB as indicated genomic lambda values exceeding 1.1 (Supplementary Table 1). However, as the LD Score Regression intercept values were close to 1, the inflation is likely due to a polygenic signal19,25. Also, the Q-Q plots exhibited a distribution of observed p values commonly observed in genome-wide analyses with significant hits, suggesting no substantial deviation from the expected distribution (Supplementary Figs. 1–3). Together with trait-specific analyses, the cross-trait analysis allowed us to identify 131 genomic loci associating with asthma, CRS, or their combination (Supplementary Data 5).
Bayesian phenotype grouping
To further elucidate the effects of shared genetic variants on specific phenotypes, we implemented a Bayesian phenotype grouping approach employing MetABF26. This method utilizes a Bayesian tree analysis framework to identify the most probable underlying model from a given set while accounting for overlapping cases, in addition to prevalence differences. The models considered were: SHARED, with an equal or highly similar true effect on all tested phenotypes; phenotype-specific models (e.g., CRSwNP), with a true non-zero effect in only one phenotype; and NULL, where there is no true effect for the tested phenotypes.
Through this analysis, we identified that 13 of the 17 loci, which did not appear in single-phenotype GWAS, contributed to both asthma and CRSwNP. Of the remaining 4 loci, three appeared to affect only asthma: 2q24.1 near GPD2, 8p23.1a near SGK223, and 8p23.1d near DEFB136 (Fig. 3). Interestingly, while lacking CRSwNP impact, these three loci showed possible shared effect to CRSsNP (Supplementary Fig. 2), and 2q24.1 in particular. The impact of the 3p14.3 locus near PPA1P16 could not be definitively categorized as either shared or asthma-specific. Among the shared impact loci was 7q22.3, lead by the missense variant rs6967330-G in the viral receptor-encoding CDHR3; this variant has been previously linked to early childhood asthma with severe exacerbations27 and CRS28.

Left panel: Bayesian analysis indicating the most probable model. NULL: Null model. SHARED: the effect is identical or very similar for both asthma and CRSwNP. ASTHMA: there is an effect on asthma but no effect on CRSwNP. CRSwNP: there is an effect on CRSwNP but no effect on asthma. Right panel: Forest plot, presenting natural logarithm of odds ratio, with 95 % confidence intervals (±1.96 standard errors), of lead variants in phenotype-specific analyses.
The Bayesian analysis showed an even greater shared signal between asthma and CRSwNP than initial results revealed. Specifically, the cross-trait asthma and/or CRS GWAS implicated 43 asthma loci that did not appear in the CRSwNP GWAS. Further examination of the phenotype-specific impact of these loci indicated that 26 loci likely exert a shared effect on both asthma and CRSwNP (Fig. 4). Intriguingly, five asthma loci showed a significantly higher effect on CRSwNP: 6q24.2 near PHACTR2 (ORCRSwNP = 1.14 [1.07–1.21]), 5q31.1 near TCF7 (ORCRSwNP = 1.11 [1.05–1.17]), 11q24.3 near ETS1 (ORCRSwNP = 1.12 [1.07–1.17]), 12q13.3 near STAT6 (ORCRSwNP = 1.09 [1.04–1.13]), and 17p12 near TTC19 (ORCRSwNP = 1.07 [1.04–1.11]). Considering CRSsNP as well (Supplementary Fig. 3), a probable asthma-specific effect (PP > 50 % for ASTHMA model) was apparent for only 6 of 43 loci: 4p14, 4q24, 7p21.1, 8p23.1b, 8p23.1c, and 10p12.31.

Left panel: Bayesian analysis indicating the most probable model. SHARED: the effect is identical or very similar in both asthma and CRSwNP. ASTHMA: there is an effect on asthma but no effect on CRSwNP. Right panel: Forest plot of natural logarithm of odds ratios, with 95 % confidence intervals (±1.96 standard errors), of lead variants in phenotype-specific analyses.
To analyze trends of shared and distinct impact on asthma and CRSwNP, we employed linemodels29 for probabilistic clustering of variants based on estimated effect sizes. This method corrects for correlation between cases of different phenotypes specified for the FinnGen and UKB subpopulations. We assessed three grouping models: “ASTHMA,” suggesting a non-zero effect on asthma only; “CRSwNP,” indicating a non-zero effect exclusively on CRSwNP; and “BOTH,” which posits a highly correlated and proportional effect on both conditions.
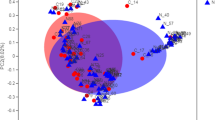
Overall, we identified a total of 131 genomic loci (Supplementary Data 5), of which 71 exhibited proportional associations with both asthma and CRSwNP, 14 were specific to CRSwNP, 20 were unique to asthma, and an additional 26 remained unclassified (Fig. 5). Notably, the unclassified loci displayed co-directional impacts and were presumed to associate with asthma, with some potentially influencing CRSwNP as well. Of the unclassified loci, 1p22.1, 15q22.2, and 17q21.33 were classified as shared in the MetABF analysis (Fig. 4). Importantly, among loci shared by both conditions, the effect on CRSwNP was consistently stronger than that on asthma, with a slope of 1.88 in log-odds space in favor of CRSwNP, even if 95% confidence intervals of individual loci still overlapped with that of asthma. This tendency for a more pronounced effect on CRSwNP was also observed in previously asthma-specific loci, such as 16p13.3 near CLEC16A and 16p12.1 near IL4R. Such findings align with prior observations9 regarding loci like 9p24.1 near IL33, 5q22.1 near TSLP, and 10p14 near GATA3, which were among the first identified as related to CRSwNP17,18,23. In our disease-specific GWASs, 22.9 % (20/87) of asthma loci were determined not to impact CRSwNP. Similarly, 28 % (14/50) of CRSwNP loci exhibited no significant effect on asthma despite the significantly lower case count of CRSwNP, further emphasizing the nuanced genetic landscape shared between these conditions.

Scatterplot of log-odds effect on asthma in FinnGen-UKB meta-analysis (βASTHMA) on the horizontal axis, and log-odds effect on CRSwNP in FinnGen-UKB meta-analysis (βCRSwNP) on the vertical axis. The red line denotes the “Asthma only” model, where the true log-odds effect is non-zero only for asthma. Similarly, the blue line denotes the ’CRSwNP only’ model, with the variant’s true effect is assumed to only exist for CRSwNP. Purple denotes the model for a shared effect (“Both”), where a non-zero effect to both asthma and CRSwNP is assumed to exist and fit or correlate with a fitted slope (k = 1.880129) passing through the origin. All three models have an assumed correlation of 0.999, with dashed lines representing expected deviation with the correlation. Each circle represents a locus lead variant, with coloring according to the model where the linemodels algorithm predicts the variant to belong with ≥90% posterior probability. Variants that do not meet this criterion in any model are represented by circles filled with white and outlined in black.
Fine-mapping of potential causal variant
We performed fine-mapping of non-HLA loci in the FinnGen cohort using SuSIE30. This fine-mapping identified high-quality credible sets for 46 genomic loci (Supplementary Data 6–9). Among credible sets, we selected predicted non-synonymous variants, and tested for their specific impacts in the UKB if they had been omitted in the original analysis due to rarity. These results were meta-analyzed, with GWS variants reported in Table 5. This resulted in known high-impact variants, such as rs144651842-A in IL4R, rs2305479-T in GSDMB, and rs113135335-G in BCL2L11 for asthma, and rs34210653-A in ALOX15 for CRSwNP, and novel variant associations such as the atopic dermatitis-associated31 rs183884396-A in TESPA1 for asthma.
Fine-mapping singled a low frequency (AF = 1.39 %) TP63 variant rs190865056-A that significantly increases risk for CRSwNP (OR = 1.52 [95 % CI 1.331–1.734]), with no significant impact on asthma. This variant is reported to be Finnish-enriched in the gnomAD database32, suggesting ancestry overlap for UKB participants with the variant. In most of the 13 TP63 transcripts, rs190865056-A is predicted to result in an isoleucine-asparagine missense mutation. A phenome-wide association performed for this variant in the FinnGen R9 PheWEB (n = 2272 phenotypes) also detected a co-directional impact on basal cell carcinoma (OR = 1.28 [95% CI 1.17–1.40], p = 5.1e-08). The association with basalioma was investigated in all 130 non-HLA loci, where it was further associated with six loci (1p13.2, 1q21.3, 3p13, 6q15, 12q13.2, and 16q24.3), five of which had opposite direction of effect.
Colocalization analyses
In order to study the differences between loci groupings according to effect on asthma and CRSwNP, we also tested the impact of non-HLA loci credible sets from FinnGen. These were associated and colocalized with FinnGen R9 endpoints selected based on previously observed17,18,23 asthma comorbidities: atopic dermatitis, allergic endpoints, and autoimmune diseases (Supplementary Data 10). Atopic dermatitis had probable colocalization (posterior probability > 0.9) with 7 loci, of which 6 had a shared asthma/CRS impact, and one unassigned. Allergic phenotypes colocalized with 16 loci, mostly having a shared asthma/CRS impact, with the exception of 3p21.3 locus harboring EEFSEC, which our previous analysis labeled as asthma-specific. Autoimmune diseases colocalized with five loci (5p13.2, 11q13.5, 12q13.2, 12q13.3, and 17q21.32), all with shared asthma/CRS impact. For three of these five loci (5p13.2, 12q13.3, and 17q21.32) the nearest genes (IL7R, STAT6, and TBX21, respectively) have previously reported Mendelian immune deficiency variants. In total, 10 loci harbored genes with such reported Mendelian immune deficiency variants33 (Supplementary Data 5). These loci with immune deficiency-linked genes shared impact on both asthma and CRS in 9 of 10 loci and one, 10p15.1, remained unassigned, with GWS impact to asthma (ORasthma = 0.804 [95% CI 0.756–0.855], p = 1.90E-12).
We also investigated asthma subtype association among asthma-associated loci (Supplementary Data 10). Here, we colocalized asthma-associated loci with subtype analyses from FinnGen R9 PheWEB. Specifically, we investigated colocalization with childhood asthma, defined as onset before the age of 16 (n = 5865), and asthma-related infections (n = 16,018). Childhood onset was colocalized (PP > 0.9) with 10 loci, all with shared impact to CRS, and 8 colocalized with allergic diseases. Asthma-related infections were colocalized (PP > 0.9) with 10 shared loci and one unassigned (17q21.33). Interestingly, loci harboring genes with Mendelian immune-deficiency variants did not colocalize with asthma-related general infections at all, suggesting the existence of genetically distinct pathways.
Thirdly, we colocalized (PP > 0.9) credible sets with eQTLs identified in gene expression data in 44 non-gonadal tissues from GTEx34 (Supplementary Data 11). 16 loci colocalized with differing expression of 29 genes among all 44 tissues. In lung tissue samples, three loci colocalized with gene expression of three genes: 4q24 (near NPNT) had codirectional effect with expression of long non-coding RNA (lncRNA) ENSG00000250740; 12q13.2 (near RPS26) had opposite effect on SUOX and RPS26 expression; and 17q21.1 (near GSDMB) had codirectional effect with MED24 expression.
In silico analyses
In order to further study the shared and distinct genes and pathways, we performed Multi-marker Analysis of GenoMic Annotation (MAGMA, v1.10)35 to identify genes and gene sets enriched in each data set. This analysis summarizes GWAS results over gene regions to identify associated genes rather than single alleles. Genes were downloaded from NCBI RefSeq (n = 19526). For gene set analysis, we used canonical gene sets (n = 7561) described at MSigDB 7.036. MAGMA highlighted a total of 330 genes, of which 121 genes associated with CRSwNP, 7 associated with CRSsNP, and 246 associated with asthma (Supplementary Data 12). MAGMA identified 246 genes in the asthma and/or CRS cross-trait analysis, of which 34 were not significant in single-phenotype MAGMA analyses. The gene set analysis identified 49 sets of genes with increased association to phenotypes (Supplementary Data 13), including known asthma-associated pathways such as Jak-STAT signaling37, innate lymphoid cell lines38, and RUNX3 signaling39. CRSwNP was also associated with Jak-STAT and RUNX3 signaling, in addition to the NFAT transcription factor pathway and others. A notable shared signal was also confirmed with genes associated with atopic dermatitis mechanisms and therapies, a corollary to clinical manifestations40,41,42,43.
Immunologically relevant tissues were enriched with asthma (Supplementary Table 2). We performed a tissue-specific enrichment analysis using MAGMA, associating tissue-specific gene expression profiles from 54 tissues from GTEx v834 with disease-gene associations. This linked asthma-associated genes with spleen, whole blood, and EBV-transformed lymphocyte tissues, and spleen and whole blood gene expression with asthma and/or CRS. No tissue was significantly linked with CRSwNP or CRSsNP after multiple testing correction. Interestingly, lung tissue gene expression was less associated with asthma-associated genes than lymphoid cell lines, such as blood and spleen, and lung tissue association was not significant after multiple testing correction. This analysis is limited due to underrepresentation of airway tissues.
Finally, we performed LD score regression to establish estimates of heritability (Supplementary Table 1) and genetic correlation between the phenotypes (Supplementary Table 3). Liability scale SNP-based heritability for asthma was estimated at 17.1 % in UKB and 13.7 % in FinnGen. For CRSwNP we estimate a higher SNP-based heritability of 24.1 % in UKB and 33.2 % in FinnGen, which is also evidenced by similar numbers of genetic loci with relatively few CRSwNP cases. In FinnGen CRSsNP had an estimated SNP-based heritability of 6.6 %. In UKB, CRSsNP heritability was not significantly different from 0 and was excluded from genetic correlation analyses. Genetic correlation between FinnGen and UKB analyses has a confidence interval upper bound of 100% for CRSwNP (p = 1.66E-29), whereas for asthma the correlation between cohorts was high but not complete (rG = 86.5 % [95 % CI 80.6–92.3 %], p = 7.04E-185). Genetic correlation between asthma and CRSwNP was lower in UKB (61.4 %) and FinnGen (52.72 %) than our previous estimate (68.7 %)12. In our previous study, SAIGE was used for genome-wide association, which is based on saddlepoint approximation and has been reported to inflate estimated effects. This has potentially also inflated our previous genetic correlation estimate, and we hold our present estimates based on REGENIE genomic analysis to be more reliable.
Discussion
Asthma and CRS are common diseases with often lifelong burden and about half of CRS cases have asthma44, possibly due to shared pathomechanisms45,46. Recent advances in biomarker research, driven by biological therapies, have identified various CRS clusters with distinct inflammatory profiles, predominantly Th2 high IL-5 levels are associated with a nasal polyp phenotype and increased asthma prevalence47. Studies show that Th2-driven CRSwNP is present in about 80% of European patients, whereas Th1/Th17 inflammation is more common in Asian patients45. Asthma with nasal polyps is more challenging to manage and prone to frequent exacerbations45. Poor symptom control in severe asthma patients correlates with high sinus CT scores48. Biological treatments targeting type 2 immune responses have been effective in improving control of both asthma and CRS and lowering the necessity for further sinus surgeries45.
Here we show for the first time a systematic analysis of the shared genetic contribution between asthma and CRSwNP to elucidate the observed clinical overlap of the conditions. We report the first genome-wide meta-analyses of asthma and CRSwNP in the Finnish and British populations. We detect 17 new loci associating with asthma, 33 new loci associating with CRSwNP, and one locus associating with CRSsNP. Comparing effect estimates allows us to distinguish a total of 131 loci, of which 20 associate only with asthma, 14 only with CRSwNP, and 71 loci with both. We also provide evidence of a trend of shared loci having a higher absolute risk increase for CRSwNP, including many loci traditionally considered asthma-specific, such as 5q22.1 near TSLP, or 9p24.1 near IL33. This is in line with clinical observations, where some biologics targeting asthma pathways have shown impact on CRSwNP as well7,21, and even improved impact in patients with both diseases20. The apparent trend of higher genetic CRSwNP impact may stem at least in part from an expected heterogeneity of asthma diagnoses.
Our results indicate that there exist both shared and distinct genetic effects to asthma and CRSwNP, and that these effects can, in most cases, be clearly separated. Previous studies have shown largely similar genetic signals for subtypes of asthma. Here, we present evidence of genetically distinct signals impacting CRSwNP, suggesting separate pathways and mechanisms yet to be identified. CRSwNP-specific loci differ from asthma loci by a lack of association with allergic diseases. By contrast, allergic diseases and immune deficiency phenotypes seemed to largely correlate with impacting both asthma and CRSwNP. Efforts to recognize what constitutes an asthma-specific genetic effect yielded few consistent clues in biological and clinical comorbidities. The apparently higher CRSwNP impact among shared variants may well be explained by more special care-based diagnoses, in addition to a higher heterogeneity of asthma etiologies. Further work is needed to clarify whether these signals can be distinguished downstream as well, which might imply the existence of endophenotypes in both CRSwNP and asthma.
Several loci previously associated with childhood asthma16,18,49 (e.g., 15q22.33, 16p12.1, and 3q21.3 near genes SMAD3, IL4R, EEFSEC, respectively) were also linked here with a more generally selected asthma. This observation sheds lights on the high but not entirely complete genetic correlation between childhood and adult-onset asthma18. Less clear is the mechanism behind childhood asthma loci such as 6p25.3 near IRF4 and 17q21.32 near TBX21, which we associate here with CRSwNP but not asthma. Interestingly, despite CRSwNP usually manifesting in adulthood, some loci associated with childhood asthma also overlap with CRSwNP. This observation emphasizes the complexities of heritable etiologies shared between CRSwNP and both asthma types. The apparently higher CRSwNP impact among shared variants may well be explained by more special care-based diagnoses, in addition to a higher heterogeneity of asthma etiologies. Additionally, while the shared genetic contributions of atopic dermatitis and asthma have previously been well characterized12,17,23,43, we found no overlap between CRSwNP-specific loci and atopic dermatitis. While epidemiologically expected50, this absence raises the question of the underlying mechanisms driving the impact of atopic dermatitis, asthma, and CRSwNP, suggesting a common immunological contribution related to type 2 inflammation., as further evidenced by type 2 targeting therapeutics impacting all three diseases7,21,40. However, the unique influences of asthma-specific loci remain a matter for exploration, potentially relating to interaction of environmental risk factors – such as obesity51 and smoking52.
Among variants with expected direct impact on the polypeptide chain, we identify a rare, Finnish-enriched missense variant rs190865056-A in TP63 to associate with CRSwNP. Similar to the ALOX15 missense variant rs34210653-A, rs190865056-A has little apparent impact on the risk for asthma9. A member of the p53 family, the p63 protein encoded by TP63 activates bullous pemphigoid antigen 1 (BPAG-1)53 and acts as a cell fate determinant of development of limb and epithelium54,55. Several high-impact variants have been linked with syndromes impacting limb development and skin conditions such as ectodermal dysplasias56. Li and colleagues57 performed immunohistochemistry and quantitative PCR for nasal polyp tissue of 65 CRSwNP patients and inferior turbinate samples of 19 controls, detecting that p63 expression is elevated in patients with CRSwNP, relates to epithelial hyperplasia in nasal polyp tissue, and is reduced in CRSwNP patients after oral glucocorticoid treatment. While not currently considered oncogenic, p63 is elevated in basaliomas, squamous cell carcinomas, thymomas, urothelial carcinomas, and salivary gland neoplasias58. For CRSwNP, the shared impact on basal cell carcinoma of the TP63 variant appears to be more of an exception, as most CRSwNP loci with an impact on basaliomas had an opposite direction of effect.
Limitations of this biobank-scale registry-based study include collider bias (increased chance of diagnosis in the hospital setting) and a smaller proportion of pediatric patients than in the general population. The approach of combining cases and using all three disorders to select controls emphasizes the shared biology, and it is therefore likely that further disease-specific or opposite-effect variants remain to be identified. While the approach used here emphasizes shared genetic impact more than distinct genetic impact, we note that most shared-impact loci appeared in the original asthma or CRSwNP-specific GWASs. While most known types of asthma and CRS cases are represented in this study sample, early childhood asthma is under-represented. The analysis of CRSsNP in the UKB dataset was underpowered due to underrepresentation, and the genetics of CRSsNP requires further study. The observational approach provides strong correlations and the sophisticated measurement tools provide strong evidence, from which establishing causal relationships require further study with potential benefits to treatment. The use of British and Finnish ancestries also limits analysis to variants represented in both ancestries, omitting several ancestry-enriched variants that require further study to determine their validity. This also limits the impact of these results outside of European ancestries59. The genomic lambda values suggest inflation is likely attributable to a polygenic signal, although it may also be partially influenced by residual population stratification not fully controlled by principal component adjustments.
On the whole, this study shows that shared heritable affinity to developing CRSwNP has an even higher overlap with that of asthma than expected. Shared heritability to asthma and CRSwNP is explained in part by type 2 inflammation, as previously noted, but also points to other immunological dimensions, evidenced by overlaps with allergic and atopic phenotypes, immune deficiency, and autoimmunity. CRSwNP-specific heritability, carried in part by rare disease-specific variants such as the TP63 missense variant rs190865056-A, seems to differ from shared heritability, at least in its lack of impact on allergic phenotypes, while asthma-specific heritability shows few differing comorbidities. Given the success of biologic therapeutics in CRSwNP, asthma, and atopic dermatitis, it is likely that further insight can be obtained by future investigations combining other immunologically related endpoints with asthma or CRSwNP.
Methods
Study sample
Finnish participants were obtained from the FinnGen study60, a recently completed nation-wide collection of Finnish genetic samples comprising close to 10% of the population. The dataset used in this study comprised 392,423 participants from FinnGen release 9 (February 2022), including both legacy samples, maintained by the Biobank of the Finnish Institute for Health and Welfare (THL), and recent biobank samples recruited at Finnish university hospitals. The sample population evenly represents both sexes (55.9% females) and all ages. Phenotypic information include disease information from the Care Register for Health Care (THL), the Primary Health Care Register (THL), the Causes of Death Register (Statistics Finland) and the Drug Reimbursement Register (KELA, the Social Insurance Institution of Finland). Asthma cases were established by a diagnosis of J45 (ICD-10) or 493 (ICD-9 or ICD-8) in the Care Register for Health Care or Causes of Death Register. CRSwNP cases were established by a diagnosis of J33 (ICD-10), 471 (ICD-9), or 505 (ICD-8) in these registers. CRSsNP cases were established by a diagnosis of J32 (ICD-10), 473 (ICD-9), or 503 (ICD-8) in these registers in the absence of nasal polyposis diagnosis (J33 in ICD-10, 471 in ICD-9 and 505 in ICD-8).
The co-occurrence of phenotypes is summarized in Supplementary Tables 6 and 7. For comparative effect analyses, we used atopic dermatitis (n = 13,473), allergic phenotypes (allergic rhinitis, allergic conjunctivitis, or allergic asthma; n = 34,614), and autoimmune diseases (n = 96,150). These endpoints are encoded in FinnGen R9 PheWEB as L12_ATOPIC, ALLERG_RHINITIS, H7_ALLERGICCONJUNCTIVITIS, ALLERG_ASTHMA, and AUTOIMMUNE, respectively.
For further sub-phenotype analysis, we selected participants with asthma diagnosed before age 16 (n = 5865, 13.9 %), as defined in the FinnGen R9 PheWEB (ASTHMA_CHILD_EXMORE). Asthma-related infections were defined as specialist-diagnosed upper and respiratory tract infections in asthma cases (n = 16,018, 37.9 %) as defined in FinnGen R9 PheWEB (ASTHMA_INFECTIONS).
Participants in FinnGen provided informed consent for biobank research, based on the Finnish Biobank Act. Alternatively, separate research cohorts, collected prior to the Finnish Biobank Act came into effect (in September 2013) and the start of FinnGen (August 2017), were collected based on study-specific consents and later transferred to the Finnish biobanks after approval by Fimea, the National Supervisory Authority for Welfare and Health. Recruitment protocols followed the biobank protocols approved by Fimea. The Coordinating Ethics Committee of the Hospital District of Helsinki and Uusimaa (HUS) approved the FinnGen study (protocol HUS/990/2017). The FinnGen study approval permits are listed in Supplementary Table 4, and biobank access decisions are listed in Supplementary Table 5.
UK participants were obtained from the UKB61; a prospective cohort with extensive phenotype and genotype data from up to 500,000 individuals aged 40–69 at recruitment. The phenotype definitions were based on ICD10 codes recorded from the hospital inpatient records from the British health registries comprising both primary and secondary diagnoses. These data have been coupled with the UKB and can be found in fields 11634–11846. CRSwNP is defined by ICD-10 code J33 (Nasal polyp). CRSsNP is defined by ICD-10 code J32 (Chronic sinusitis), excluding individuals with the diagnosis code J33. Asthma is defined by ICD-10 codes J45 (Asthma) and J46 (Status asthmaticus).
For controls, we selected all study participants without any of the three conditions.
Genotyping and quality control
As described above, a total of 392,423 FinnGen samples were collected both as legacy cohorts that have been genotyped previously, and recruited samples genotyped after recruitment, using a total of 15 different DNA chips. We excluded variants with a high missingness (>2%), minor allele count <5, and considerable violation of Hardy-Weinberg equilibrium (pHWE < 1e-6). A detailed description of genotyping chips used, quality control, and imputation have been previously described elsewhere60. We excluded samples with ambiguous sex, genotype missingness >0.05, incomplete minimum phenotype information (age, sex, and hospital record data), and non-Finnish ancestry in principal component analysis. For FinnGen release 9, the SISu62 v4 LD reference panel was used in imputation, resulting in 20,167,810 variants analyzed over 377,277 participants.
For UKB analysis, the genotyping and QC pipeline has been described previously. On top of this QC pipeline, for UKB we have chosen to include only variants with MAF > 1% for computational and statistical reasons. This resulting in the inclusion of 7,268,980 variants. Functional variants detected in FinnGen were separately tested for replication in the UKB with no constraint on allele frequency.
Genome-wide association
We analyzed mixed logistic model regression genome-wide using the REGENIE63 software (v2.2.4) on 20,167,811 variants in FinnGen. Covariates used in all FinnGen GWAS were sex, age, genotyping batch, and principal components (PCs) 1–10.
In UKB, the analysis was performed in European unrelated individuals with logistic regression using plink2.064. Covariates in all UKB analyses were sex and PCs 1–15.
Principal components were computed with fastPCA, which is also described in the QC pipeline. For a detailed description of principal component analysis in the FinnGen cohort, please see the pipeline open source pages (https://github.com/FINNGEN/). Only samples with Finnish ancestry in PC analysis merged with 1000 Genomes65 samples were included. The post-QC number of participants included in the analyses was 377,277.
GWAS results from FinnGen and UKB were meta-analyzed for validity. Only meta-analyzed variants with p < 5e-8 were considered significant.
Genetic loci were selected starting from the variant with the lowest p-value (lead) in the chromosome, including variants with LD > 0.1 of the lead variant within a 2 megabase window around the lead variant, then beginning anew until no variants with p < 5e-8 remained. We defined loci as novel if they did not appear in OpenTargets or GWAS Catalog with an association to the associated phenotype. We also manually curated loci according to the phenotypes of asthma10,17,18,23,24, CRSwNP9,12 or CRSsNP12.
Loci were fine-mapped for credible sets of causative variants using the SuSiE30 software v0.9.26 in FinnGen. All credible set variants were annotated for predicted functional consequence. Fine-mapped variants with non-synonumous annotations were considered replicated only if they were GWS in meta-analysis with UKB.
Colocalization of finemapped credible sets were investiged using an in-house pipeline powered by eCAVIAR66. High-quality credible sets from the FinnGen R9 results were colocalized with allergic phenotypes (see above), in addition to eQTLs of gene expression in non-gonadal tissue, as made available by GTEx34. A colocalized signal was considered indicative of a truly shared loci if posterior probability was higher than 90 %.
Cross-trait analysis
In order to increase our yield among shared genetic contributors to both asthma and CRS, we ran a GWAS using all cases with asthma, CRSwNP or CRSsNP as case. Quality control and analysis pipelines were the same as for single-phenotype analyses. Asthma and/or CRS cross-trait analysis was run separately in FinnGen and UKB, and cohort-specific analyses were then meta-analyzed as described below.
Meta-analysis
We conducted four sets of inverse variance-weighted meta-analyses, including one for each single-phenotype GWAS and one for the cross-trait GWAS. The meta-analyses were performed using METAL V.2011-03-2567. We assessed statistical significance using the standard genome-wide significance level (p < 5e-08).
Bayesian analyses
In order to specify whether loci identified in the asthma and/or CRS analysis were truly shared, or whether an association was carried by a single phenotype only, we employed a Bayesian analysis framework (MetABF26). This analysis pipeline employs a covariance matrix to adjust for overlapping case counts, and approximates a Bayesian factor (ABF) for different explanatory models of a given set of effect sizes for a given variant. We ran the Bayesian analysis on asthma and/or CRS cross-trait GWAS lead variants. Analysis was run in FinnGen and UKB separately, since case overlaps could only be adjusted for within cohorts. The tested models in analyses were:
-
NULL, where there is no true effect on the tested phenotypes
-
SHARED, where all phenotypes had one identical or highly similar true effect size;
-
ASTHMA, where the true effect for asthma is non-zero and 0 for all other phenotypes; and
-
CRSwNP, where the true effect for CRSwNP is non-zero and 0 for all other phenotypes.
NULL and phenotype-specific models are calculated using single models. The SHARED model is calculated by evaluating the ABF for one model where the effect is exactly the same in both CRSwNP and Asthma (“fixed”), and another where the effect is highly correlated (at least 90%; “correlated”). Priors for “fixed” and “correlated” are set to half that of NULL and phenotype-specific models to adjust for an otherwise inflated ABF. These two Bayesian factors are then summed to establish an ABF for the SHARED model, which then represents a hypothesized shared effect. A model was considered confirmed if it reached a posterior probability of 80%. No independent signal model was considered, as the choice of using shared controls would deflate signals from variants with opposing effects.
To analyze trends of shared and distinct impact on asthma and CRSwNP, we employed a recently developed approach (linemodels29) to group variants according to estimated effect. Briefly, this approach performs a probabilistic clustering of variants based on effect sizes, correcting for epidemiological overlap between cases of different phenotypes specified for subpopulations (FinnGen and UKB). We tested for grouping according to three models: “ASTHMA”, where variants were assumed to have a non-zero effect on asthma and not CRSwNP; “CRSwNP”, where variants were assumed to have a non-zero effect on CRSwNP and not asthma; and “BOTH”, where variants were assumed to have a highly correlated and proportional effect to both asthma and CRSwNP. The threshold for belonging to a particular cluster was set at posterior probability >90%.
Multi-marker Analysis of GenoMic Annotation
We used the Multi-marker Analysis of GenoMic Annotation (MAGMA35, v1.10) software to run gene, gene set, and tissue enrichment analyses from meta-analyzed summary statistics data from CRSwNP, Asthma, and Asthma and/or CRS analyses. Genes were downloaded from NCBI RefSeq (n = 37358). Significance threshold was set at p < 4.46e-7 for the gene analysis. For gene set analysis, we used canonical gene sets (n = 6490) described at MSigDB (v2023.1.Hs), with a significance threshold of p < 7.70e-6. Tissue enrichment analysis was performed using the publicly available FUMA pipeline68 and leveraged RNA-seq data summaries provided by GTEx v834. This analysis tests for associations between tissue specific gene expression profiles, and disease-gene associations. 54 GTEx tissues were used and three phenotypes tested, setting the significance threshold at p < 3.09e-4.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The summary statistics from genome-wide meta-analysis generated in this study have been deposited in the GWAS Catalog database under accession codes GCST90652530 (asthma), GCST90652531 (CRSwNP), GCST90652532 (CRSsNP) and GCST90652529 (asthma and/or CRS) [https://www.ebi.ac.uk/gwas/]. The summary statistics from specific cohorts are publicly available at locally hosted buckets (FinnGen: https://storage.googleapis.com/fg-publication-green-public/F_2021_032_20250820/ASTCRSNP.tar.gz and UKB: https://storage.googleapis.com/fg-publication-green-public/F_2021_032_20250820/ASTCRSNP_ukb.tar.gz). Based on National and European regulations (GDPR) access to individual-level sensitive health data must be approved by national authorities for specific research projects and for specifically listed and approved researchers. The health data described here was generated and provided by the National Health Register Authorities (Finnish Institute of Health and Welfare, Statistics Finland, KELA, Digital and Population Data Services Agency) and approved, either by the individual authorities or by the Finnish Data Authority, Findata, for use in the FinnGen project. Therefore, we, the authors of this paper, are not in a position to grant access to individual-level data to others. However, any researcher can apply for the health register data from the Finnish Data Authority Findata (https://findata.fi/en/permits/) and for individual-level genotype data from Finnish biobanks via the Fingenious portal (https://site.fingenious.fi/en/) hosted by the Finnish Biobank Cooperative FINBB. All Finnish biobanks can provide access for research projects within the scope regulated by the Finnish Biobank Act, which is research utilizing the biobank samples or data for the purposes of promoting health, understanding the mechanisms of disease or developing products and treatment practices used in health and medical care. You can learn more about accessing other FinnGen data here: https://www.finngen.fi/en/access_results. Access to UKBB data can be obtained upon request [http://www.ukbiobank.ac.uk/resources/]. Variant summary data for the SISu v4.1 reference panel are available at https://sisuproject.fi with information on previous versions available upon request. Gene annotations and gene sets were downloaded from public repositories (NCBI [https://www.ncbi.nlm.nih.gov/refseq/], MSigDB [https://www.gsea-msigdb.org/gsea/msigdb/collections.jsp]). RNA expression data summaries for tissue enrichment analysis are publically available at the GTEx consortium [https://www.gtexportal.org/home/].
References
Fokkens, W. J. et al. European position paper on rhinosinusitis and nasal polyps 2020. Rhinology 58, 1–464 (2020).
Bachert, C., Bhattacharyya, N., Desrosiers, M. & Khan, A. H. Burden of disease in chronic rhinosinusitis with nasal polyps. J. Asthma Allergy 14, 127–134 (2021).
Stevens, W. W. et al. Clinical characteristics of patients with chronic rhinosinusitis with nasal polyps, asthma, and aspirin-exacerbated respiratory disease. J. Allergy Clin. Immunol. Pr. 5, 1061–1070.e3 (2017).
Levy, M. L. et al. Key recommendations for primary care from the 2022 Global Initiative for Asthma (GINA) update. NPJ Prim. Care Respir. Med. 33, 7 (2023).
Johansson, L., Åkerlund, A., Melén, I., Holmberg, K. & Bende, M. Prevalence of nasal polyps in adults: the Skovde population-based study. Ann. Otol., Rhinol. Laryngol. 112, 625–629 (2003).
Dietz de Loos, D. et al. Prevalence of chronic rhinosinusitis in the general population based on sinus radiology and symptomatology. J. Allergy Clin. Immunol. 143, 1207–1214 (2019).
Bachert, C. et al. Biologics for chronic rhinosinusitis with nasal polyps. J. Allergy Clin. Immunol. 145, 725–739 (2020).
Nuutinen, M. et al. The relative proportion of comorbidities among rhinitis and rhinosinusitis patients and their impact on visit burden. Clin. Transl. Allergy 12, e12181 (2022).
Kristjansson, R. P. et al. A loss-of-function variant in ALOX15 protects against nasal polyps and chronic rhinosinusitis. Nat. Genet. 51, 267–276 (2019).
Han, Y. et al. Genome-wide analysis highlights contribution of immune system pathways to the genetic architecture of asthma. Nat. Commun. 11, 1776 (2020).
El-Husseini, Z. W., Gosens, R., Dekker, F. & Koppelman, G. H. The genetics of asthma and the promise of genomics-guided drug target discovery. Lancet Respir. Med. 8, 1045–1056 (2020).
Saarentaus, E. C. et al. Inflammatory and infectious upper respiratory diseases associate with 41 genomic loci and type 2 inflammation. Nat. Commun. 14, 83 (2023).
Saikumar Jayalatha, A. K., Hesse, L., Ketelaar, M. E., Koppelman, G. H. & Nawijn, M. C. The central role of IL-33/IL-1RL1 pathway in asthma: from pathogenesis to intervention. Pharm. Ther. 225, 107847 (2021).
Lambrecht, B. N., Hammad, H. & Fahy, J. V. The cytokines of asthma. Immunity 50, 975–991 (2019).
Buniello, A. et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2018).
Willis-Owen, S. A. G., Cookson, W. O. C. & Moffatt, M. F. The genetics and genomics of asthma. Annu. Rev. Genom. Hum. Genet. 19, 223–246 (2018).
Ferreira, M. A. et al. Shared genetic origin of asthma, hay fever and eczema elucidates allergic disease biology. Nat. Genet. 49, 1752–1757 (2017).
Ferreira, M. A. R. et al. Genetic architectures of childhood- and adult-onset asthma are partly distinct. Am. J. Hum. Genet. 104, 665–684 (2019).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Corren, J. et al. Efficacy of tezepelumab in severe, uncontrolled asthma: pooled analysis of the PATHWAY and NAVIGATOR clinical trials. Am. J. Respir. Crit. Care Med. 208, 13–24 (2023).
Parulekar, A. D., Kao, C. C., Diamant, Z. & Hanania, N. A. Targeting the interleukin-4 and interleukin-13 pathways in severe asthma: current knowledge and future needs. Curr. Opin. Pulm. Med. 24, 50–55 (2018).
Martin, G. P., Sperrin, M., Snell, K. I. E., Buchan, I. & Riley, R. D. Clinical prediction models to predict the risk of multiple binary outcomes: a comparison of approaches. Stat. Med. 40, 498–517 (2021).
Zhu, Z. et al. A genome-wide cross-trait analysis from UK Biobank highlights the shared genetic architecture of asthma and allergic diseases. Nat. Genet. 50, 857–864 (2018).
Torgerson, D. G. et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat. Genet. 43, 887–892 (2011).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Trochet, H. et al. Bayesian meta-analysis across genome-wide association studies of diverse phenotypes. Genet. Epidemiol. 43, 532–547 (2019).
Bønnelykke, K. et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat. Genet. 46, 51–55 (2014).
Chang, E. H. et al. Association between the CDHR3 rs6967330 risk allele and chronic rhinosinusitis. J. Allergy Clin. Immunol. 139, 1990–1992.e2 (2017).
Pirinen, M. linemodels: clustering effects based on linear relationships. Bioinformatics 39, btad115 (2023).
Wang, G., Sarkar, A., Carbonetto, P. & Stephens, M. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. B 82, 1273–1300 (2020).
Sliz, E. et al. Uniting biobank resources reveals novel genetic pathways modulating susceptibility for atopic dermatitis. J. Allergy Clin. Immunol. 149, 1105–1112.e9 (2022).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
McKusick, V. A. Mendelian inheritance in man and its online version, OMIM. Am. J. Hum. Genet. 80, 588–604 (2007).
Aguet, F. et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Georas, S. N., Donohue, P., Connolly, M. & Wechsler, M. E. JAK inhibitors for asthma. J. Allergy Clin. Immunol. 148, 953–963 (2021).
Pivniouk, V., Gimenes Junior, J. A., Honeker, L. K. & Vercelli, D. The role of innate immunity in asthma development and protection: Lessons from the environment. Clin. Exp. Allergy 50, 282–290 (2020).
Yu, Y., Wang, L. & Gu, G. The correlation between Runx3 and bronchial asthma. Clin. Chim. Acta 487, 75–79 (2018).
Agache, I. et al. Efficacy and safety of dupilumab for moderate-to-severe atopic dermatitis: a systematic review for the EAACI biologicals guidelines. Allergy 76, 45–58 (2021).
Castro, M. et al. Dupilumab efficacy and safety in moderate-to-severe uncontrolled asthma. N. Engl. J. Med. 378, 2486–2496 (2018).
Hoy, S. M. Dupilumab: a review in chronic rhinosinusitis with nasal polyps. Drugs 80, 711–717 (2020).
Ravnborg, N. et al. Prevalence of asthma in patients with atopic dermatitis: a systematic review and meta-analysis. J. Am. Acad. Dermatol. 84, 471–478 (2021).
Jarvis, D. et al. Asthma in adults and its association with chronic rhinosinusitis: the GA2LEN survey in Europe. Allergy 67, 91–98 (2012).
Laidlaw, T. M., Mullol, J., Woessner, K. M., Amin, N. & Mannent, L. P. Chronic rhinosinusitis with nasal polyps and asthma. J. Allergy Clin. Immunol. Pract. 9, 1133–1141 (2021).
Dixon, A. E. Rhinosinusitis and asthma: the missing link. Curr. Opin. Pulm. Med. 15, 19–24 (2009).
Seccia, V. et al. Management of patients with severe asthma and chronic rhinosinusitis with nasal polyps: a multidisciplinary shared approach. J. Pers. Med. 12, 1096 (2022).
Castillo, J. A., Plaza, V., Rodrigo, G., Julia, B. & Mullol, J. Chronic rhinosinusitis with and without nasal polyps and rhinitis in adult asthma. Frequency distribution and relationship with asthma control and severity (the IRIS-ASMA study). Eur. Respir. J. 42, P3448 (2014).
Kauppi, P. et al. A second-generation association study of the 5q31 cytokine gene cluster and the interleukin-4 receptor in asthma. Genomics 77, 35–42 (2001).
Halling, A.-S. et al. Association between atopic dermatitis and nasal polyposis: what is the evidence?. J. Eur. Acad. Dermatol. Venereol. 35, e290–e293 (2021).
Bantulà, M., Roca-Ferrer, J., Arismendi, E. & Picado, C. Asthma and obesity: two diseases on the rise and bridged by inflammation. J. Clin. Med. 10, 169 (2021).
Thomson, N. C., Polosa, R. & Sin, D. D. Cigarette smoking and asthma. J. Allergy Clin. Immunol. Pr. 10, 2783–2797 (2022).
Osada, M. et al. p63-specific activation of the BPAG-1e promoter. J. Investig. Dermatol. 125, 52–60 (2005).
Yang, A. et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398, 714–718 (1999).
Mills, A. A. et al. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 398, 708–713 (1999).
Corona-Rivera, J. R. et al. A family with EEC syndrome in the son and ADULT syndrome in his father caused by the c.797G>A (p.Arg266Gln) pathogenic variant in the TP63 gene. Mol. Syndromol. 15, 51–57 (2023).
Li, C. W. et al. Role of p63/p73 in epithelial remodeling and their response to steroid treatment in nasal polyposis. J. Allergy Clin. Immunol. 127, 765–772.e2 (2011).
Steurer, S. et al. p63 expression in human tumors and normal tissues: a tissue microarray study on 10,200 tumors. Biomark. Res. 9, 7 (2021).
Martin, A. R. et al. Human demographic history impacts genetic risk prediction across diverse populations. Am. J. Hum. Genet. 100, 635–649 (2017).
Kurki, M. I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613, 508–518 (2023).
Sudlow, C. et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12, e1001779 (2015).
Institute for Molecular Medicine Finland (FIMM). Sequencing Initiative Suomi project (SISu) v3. http://sisuproject.fi (2020).
Mbatchou, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet 53, 1097–1103 (2021).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 4, 7 (2015).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Hormozdiari, F. et al. Colocalization of GWAS and eQTL signals detects target genes. Am. J. Hum. Genet. 99, 1245–1260 (2016).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Acknowledgements
We want to acknowledge the participants and investigators of the FinnGen study. The FinnGen project is funded by two grants from Business Finland (HUS 4685/31/2016 and UH 4386/31/2016) and the following industry partners: AbbVie Inc., AstraZeneca UK Ltd, Biogen MA Inc., Bristol Myers Squibb Inc. (and Celgene Corporation & Celgene International II Sàrl), Genentech Inc., Merck Sharp & Dohme LCC, Pfizer Inc., GlaxoSmithKline Intellectual Property Development Ltd., Sanofi US Services Inc., Maze Therapeutics Inc., Johnson&Johnson Innovative Medicine Inc., Novartis AG, Boehringer Ingelheim International GmbH and Bayer AG. Following biobanks are acknowledged for delivering biobank samples to FinnGen: Auria Biobank (www.auria.fi/biopankki), THL Biobank (www.thl.fi/biobank), Helsinki Biobank (www.helsinginbiopankki.fi), Biobank Borealis of Northern Finland (https://www.ppshp.fi/Tutkimus-ja-opetus/Biopankki/Pages/Biobank-Borealis-briefly-in-English.aspx), Finnish Clinical Biobank Tampere (www.tays.fi/en-US/Research_and_development/Finnish_Clinical_Biobank_Tampere), Biobank of Eastern Finland (www.ita-suomenbiopankki.fi/en), Central Finland Biobank (www.ksshp.fi/fi-FI/Potilaalle/Biopankki), Finnish Red Cross Blood Service Biobank (www.veripalvelu.fi/verenluovutus/biopankkitoiminta), Terveystalo Biobank (www.terveystalo.com/fi/Yritystietoa/Terveystalo-Biopankki/Biopankki/) and Arctic Biobank (https://www.oulu.fi/en/university/faculties-and-units/faculty-medicine/northern-finland-birth-cohorts-and-arctic-biobank). All Finnish Biobanks are members of BBMRI.fi infrastructure (https://www.bbmri-eric.eu/national-nodes/finland/). Finnish Biobank Cooperative—FINBB (https://finbb.fi/) is the coordinator of BBMRI-ERIC operations in Finland. The Finnish biobank data can be accessed through the Fingenious® services (https://site.fingenious.fi/en/) managed by FINBB. This work was supported by Academy of Finland (grant no. 360387 to E.C.S., grant no. 338229 to E.S.), and the Finnish Medical Foundation (grant no. 6465 to E.C.S.). The Academy of Finland Center of Excellence for Complex Disease Genetics (grant no 312074 and 352793 to A.P.), Sigrid Juselius Foundation, Finland (to A.P.), Instand-NGS4P, EU H2020, grant no 874719, REALMENT, EU H2020, grant no 964874 (to A.P.) We thank the GWAS catalog (https://www.ebi.ac.uk/gwas/) and the GTEx database (https://www.gtexportal.org/) for sharing expression data and summary statistics of GWAS studies. This research has been conducted using the UK Biobank Resource under Application Number 54273. Open access funded by Helsinki University Library.
Author information
Authors and Affiliations
Consortia
Contributions
Each author’s contribution(s) to the paper is listed according to the CRediT model. Conceptualization: E.C.S. Methodology: E.C.S., K.F.R., Ee.S. Validation: E.C.S. Formal analysis: E.C.S., K.F.R., Ee.S. Investigation: E.C.S., K.F.R., Ee.S., FINNGEN. Resources: K.B., FINNGEN. Data Curation: E.C.S., K.F.R., FINNGEN. Visualization: E.C.S. Funding acquisition: E.C.S., A.P., A.M. Project administration: A.P., A.M. Supervision: A.M., A.P., K.B., J.K. Writing—original draft: E.C.S. Writing—review and editing: E.C.S., K.F.R., Ee.S., A.B.Z., T.L., S.T.S., H.K., J.K., K.B., A.P., A.M.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Åsa Torinsson Naluai, Michelle Daya, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saarentaus, E.C., Fischer-Rasmussen, K., Sliz, E. et al. 131 genetic loci highlight immunological pathways and tissues in nasal polyposis and asthma. Nat Commun 16, 9879 (2025). https://doi.org/10.1038/s41467-025-64847-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64847-4
