
Abstract
The global adoption of hydrogen and biofuels, though central to decarbonization, risks aggravating NO2 pollution as their combustion drives up NOx emissions through hotter flames and excess oxygen. Effective ambient NO2 removal is critical for protecting human health, yet existing adsorbents often release toxic NO, limiting practical deployment. Here, we overcome this grand challenge by developing divalent metal cation-exchanged zeolites that achieve complete NO2 capture with “zero” NO emission. Exemplified by Ca2⁺- and Mn2⁺-exchanged zeolites, these materials operate under both dry and humid (70% RH) conditions, even at NO2 concentrations up to 500 ppm. Mechanistic studies reveal that divalent cations, acting as surface active sites, can suppress NO formation via stabilizing its intermediate. We further demonstrate real-world applicability by integrating these zeolites into a wearable air-purifying respirator, enabling a “zero-NOx shield” for personal protection. This work establishes NO2 adsorption chemistry and offers a scalable pathway to clean air solutions.
Similar content being viewed by others


Introduction
The global transition from conventional fossil fuels to clean alternatives, such as hydrogen (H2) and biofuels, advances vital sustainability goals, including decarbonization and reductions in carbon monoxide, hydrocarbons, and particulate emissions1,2. However, the combustion of these clean fuels can lead to increased nitrogen oxide (NOx) emissions due to their higher flame temperatures and, in the case of biofuels, elevated intrinsic oxygen content3,4. Since hard-to-abate combustion-based industries still account for approximately 30% of global energy consumption5,6,7,8, adopting clean fuels in these sectors could exacerbate ambient NOx pollution, predominantly in nitrogen dioxide (NO2). NO2 is a major environmental and public health hazard, contributing to respiratory diseases and driving the formation of fine particulate matter and ground-level ozone, which were linked to an estimated 38,000 premature deaths worldwide in 20159. Consequently, combustion-related NO2 emissions will remain a significant environmental challenge, underscoring the urgent need for effective mitigation strategies.
Existing NOx abatement technologies, such as selective catalytic reduction (SCR), operate effectively only at elevated temperatures (above 250 °C)10, leaving ambient NO2 largely unaddressed, precisely where human exposure is most acute. Adsorption-based capture using solid porous materials has emerged as a promising solution for ambient-temperature NO2 removal11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27. However, a critical challenge persists: all known adsorbents invariably generate and release toxic nitric oxide (NO) as a by-product during NO2 adsorption, a phenomenon widely attributed to dissociative chemisorption11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27. This “NO release”, often accounting for 20–30% of the NO2 input11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27, poses additional respiratory health risks and can be rapidly re-oxidized to NO2, thereby undermining the goal of zero-emission NO2 capture for air purification28,29. This issue is especially problematic in occupational settings (e.g., steelmaking, cement production, power generation, welding, and mining) and enclosed or densely populated environments (e.g., enclosed parking facilities, transit hubs, ferry terminals, and residential spaces using biofuels)30.
Attempts to mitigate NO release, such as co-adsorbing NO by designing NO-retaining adsorbents31, have been constrained by the inherently stronger binding affinity of NO2 compared to NO, and the prevalence of surface active sites that promote NO formation. Most NO2 adsorbents were reported to have surface active sites that spontaneously facilitate substantial NO formation15,16,17,18,19,20,21,22,23,24,25,26. These active sites, although lacking definitive identification, are generally understood to be surface radical sites (e.g., -C*15,16,17,18,19, -O*20,21,22,23, and -N*24 arising from surface defect) and basic groups25,26, which trigger redox reactions during NO2 adsorption, thereby forming NO. As a result, intrinsically suppressing NO formation through material-level design should be a way out. However, this approach remains largely underexplored and poorly realized.
Zeolites represent a compelling but overlooked platform in this context. Their crystalline, inorganic frameworks, composed of Si-O and Al-O tetrahedra, appear to lack the reactive surface sites commonly associated with NO formation. Preliminary experimental evidence suggests that zeolites, especially when modified, can achieve low levels of NO release32,33,34,35. Yet, systematic efforts to engineer zeolites for true zero-emission NO2 capture remain absent from the literature, largely due to the prevailing notion that NO formation during NO2 adsorption is unavoidable.
Here, we overcome this long-standing challenge by incorporating divalent metal cations into LTA zeolites via ion exchange, yielding materials that achieve complete NO2 capture with “zero” NO emission under both dry and humid ambient conditions. Through combined experimental and theoretical investigation, including dynamic adsorption/desorption experiments, temperature-programmed desorption (TPD), in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), and density functional theory (DFT) calculations—we establish that divalent metal cations suppress NO formation by stabilizing a key intermediate (nitrous acid—HNO2) and preventing its decomposition. We further demonstrate the generality of this strategy across multiple zeolite frameworks and highlight its practical relevance by integrating these materials into wearable air-purifying respirators, thereby establishing a “zero-NOx shield” technology. A schematic overview of this study is presented in Fig. 1. This work not only redefines the fundamental chemistry of NO2 adsorption in porous materials but also introduces a scalable pathway toward emission-free air purification technologies critical for public health.

NOx emissions from fuel combustion primarily exist as NO2 in the atmosphere, motivating targeted removal strategies. Existing adsorbents typically release NO during NO2 capture, limiting their effectiveness. The adsorbents developed in this work achieve complete NO2 capture with “zero” NO emission by incorporating divalent metal cations into the zeolite framework. This approach enables emission-free air purification and demonstrates potential for practical deployment.
Results
Synthesis and characterization of metal-exchanged LTA zeolites
LTA zeolites with silicon-to-aluminum (Si/Al) ratios of 3 and 6 were hydrothermally synthesized36 and converted to their ammonium form (as an intermediate for further ion exchange) and proton (H+) form (as the benchmark). Ion exchange of metal cations featuring different charge-to-size ratios and/or electronic configurations (Na+, K+, Mg2+, Ca2+, Mn2+, Co2+, Ni2+, Cu2+, Zn2+, Y3+, La3+, Ce3+, Eu3+, Tb3+, and Yb3+) was performed via aqueous salt solutions to tailor the adsorbents for optimal NO2 removal with minimal NO release. The detailed procedures for sample preparation are provided in the Supplementary Information (Section 1.1). Powder X-ray diffraction (PXRD), energy dispersive spectroscopy (EDS), X-ray photoelectron spectroscopy (XPS), inductively coupled plasma optical emission spectroscopy (ICP‒OES), and N2 adsorption‒desorption analyses at −196.15 °C confirmed high phase purity, successful cation exchange, and preserved porosity. Detailed explanations are provided in the Supplementary Information (Section 2.1, Supplementary Figs. 4–8 and Tables 1, 2).
Elimination of NO release during NO2 adsorption
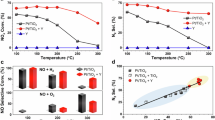
Column breakthrough dynamic adsorption tests at approximately 25 °C and 101.325 kPa using a 500 ppm NO2/Air mixture revealed that our studied LTA zeolites exhibit exceptionally low NO release, ranging from 0.06% to 4.11% of the NO2 input (Fig. 2a and Supplementary Table 3), which is vastly lower than the > 20% typically reported for conventional NO2 adsorbents (Fig. 2b, Table 1, and Supplementary Table 4)9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40. More importantly, unlike those materials that release NO at the outset of NO2 adsorption9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40, our di-/trivalent metal cation-exchanged LTA zeolites release NO only at or slightly before NO2 breakthrough (Fig. 2c and Supplementary Fig. 9), enabling “zero-emission” NO2 capture. Two prominent examples, Ca-LTA-3 and Mn-LTA-3 (Si/Al ratio = 3), maintained “zero” NO release until NO2 breakthrough (Fig. 2a–c and Table 1), even at a reduced feed NO2 concentration of 25 ppm (Fig. 2d and Supplementary Table 5), a condition that typically promotes NO release due to the prolonged adsorption duration. This performance positions di-/trivalent metal cation-exchanged LTA zeolites as uniquely capable of delivering “zero-emission” NO2 capture under ambient conditions.

a Amounts of NO2 adsorbed and NO released, and percentages of released NO relative to NO2 input in H+-, Na+-, Ca2+-, Mn2+-, Ni2+-, and La3+-exchanged LTA-3 zeolites in NO2/Air (500 ppm NO2 balanced with air). b Comparison of NO2 uptake and the percentage of released NO relative to NO2 input in Na+-, Ca2+-, and Mn2+-exchanged LTA-3 zeolites with benchmark adsorbents under similar conditions12,13,20,24,26,53,54. c NO2 breakthrough and NO release curves of H+-, Na+-, Ca2+-, Mn2+-, Ni2+-, and La3+-exchanged LTA-3 zeolites in NO2/Air. d NO2 breakthrough and NO release curves of Na+-, Ca2+-, and Mn2+-exchanged LTA-3 zeolites in 25 ppm NO2/Air (25 ppm NO2 balanced with air). e Cyclic adsorption performance of Na+-, Ca2+-, and Mn2+-exchanged LTA-3 zeolites in NO2/Air. f Comparison of NO2 adsorption regenerability and the percentage of released NO relative to NO2 input in Na+-, Ca2+-, and Mn2+-exchanged LTA-3 zeolites with benchmark adsorbents under similar conditions12,20,24.
In addition to this breakthrough in eliminating NO release, our adsorbents also exhibit high NO2 uptake—up to 3.9 mmol g-1 (Figs. 2a–b, Table 1, and Supplementary Tables 3–4), rapid kinetics—as evidenced by achieving up to 86.43% of the saturating NO2 uptake before NO2 breakthrough (Fig. 2d and Supplementary Table 5), and excellent regenerability—approximately 100% (Figs. 2e–f and Table 1); further details are provided in the Supplementary Information (Section 2.2, Supplementary Figs. 9–11 and Tables 3–8).
Identified factors for eliminating NO release
The exceptionally low NO release observed on our LTA zeolites can stem from two potential factors: minimal NO formation and adsorptive NO retention31,33. To identify the contributions of these factors, we compared representative Na+-, Ca2+-, Mn2+-, and Ni2+-exchanged LTA-3 zeolites (detailed in the Supplementary Information—Section 2.3) via binary adsorption tests: one with 500 ppm NO2 alone (NO2/N2) and the other with 500 ppm NO2 plus 50 ppm NO (NO2/NO/N2). The difference in outlet NO concentration (∆NO) served as a metric for NO retention.
For Na-LTA-3, ∆NO remained at approximately 50 ppm throughout (Fig. 3a), indicating negligible NO retention. This suggests that the low NO release observed (Fig. 2b, Table 1, and Supplementary Table 4) is due mainly to minimal NO formation. In contrast, Ca-LTA-3 had an initial ∆NO of 20 ppm, which increased to 76 ppm before NO2 breakthrough (Fig. 3b). This indicates modest NO retention, constrained by competitive adsorption, with the virtually eliminated NO release (Fig. 2a–d, Table 1, and Supplementary Tables 3, 4) being attributed primarily to minimal NO formation, coupled with limited retention.

NO2 breakthrough and NO release/breakthrough curves of a Na-LTA-3, b Ca-LTA-3, c Mn-LTA-3, and d Ni-LTA-3 adsorbents activated at 300 °C.
On the other hand, Ni-LTA-3 and Mn-LTA-3 maintained ∆NO values below 50 ppm (i.e., ranging from 20 to 36 ppm for Mn-LTA-3 and 0 to 40 ppm for Ni-LTA-3) until NO2 breakthrough (Fig. 3c and 3d), demonstrating superior NO retention. These findings align with the DFT-calculated NO binding energies and experimentally measured NO uptakes (Ni-LTA > Mn-LTA > Ca-LTA > Na-LTA, Supplementary Figs. 12, 13 and Table 9, with detailed explanations presented in the Supplementary Information—Section 2.3). Despite their superior NO retention capabilities, Ni-LTA-3 and Mn-LTA-3 exhibited significantly decreased NO2 uptake in the presence of NO (i.e., the NO2 uptake in NO2/NO/N2 was 1-1.5 mmol g-1 lower than that in NO2/N2, Fig. 3c, d and Supplementary Table 10), underscoring the adverse effect of strong NO retention on NO2 uptake. Therefore, we conclude that the virtually eliminated NO release observed for Mn-LTA-3 and Ni-LTA-3 (Figs. 2a–d, Table 1, and Supplementary Tables 3, 4) is attributed primarily to their minimal NO formation, which is further reinforced by their superior NO retention capabilities.
In summary, our data reveal that the exceptionally low NO release in our LTA zeolites primarily arises from their minimal NO formation. This result indicates that LTA zeolites inherently lack the active sites that trigger significant NO formation—a key distinction from conventional adsorbents—resulting in a unique profile that enables “zero-emission” NO2 capture. Detailed mechanistic analyses of the NO formation in LTA zeolites and its suppression by divalent metal cations are provided in the following sections.
Mechanisms of NO formation during NO2 adsorption
To understand the mechanism of NO formation in our LTA zeolites, we first identified the species formed during NO2 adsorption via TPD (Supplementary Table 11 and Figs. 14, 15) and in situ DRIFTS (Fig. 4a–d and Supplementary Fig. 16). The results revealed the formation of nitric acid (HNO3) and HNO2 in addition to physically adsorbed NO2, indicating that, alongside NO2 physisorption, dissociative chemisorption of NO2 occurs via NO2‒H2O reactions. The presence of residual H2O within our LTA zeolites after activation at 300 °C was identified by the thermalgravimetric analysis (TGA, Supplementary Fig. 17) and differential thermogravimetric analysis (DTG, Fig. 4e) for Na+-, Ca2+-, Mn2+-, and Ni2+-exchanged LTA-3 zeolites37.

In situ DRIFTS results of a Na-LTA-3, b Ca-LTA-3, c Mn-LTA-3, and d Ni-LTA-3 activated at 300 °C upon NO2 adsorption in NO2/Air (500 ppm NO2 balanced with Air) at approximately 25 °C and 101.325 kPa. e Differential thermogravimetric analysis (DTG) results of Na+-, Ca2+-, Mn2+-, and Ni2+-exchanged LTA-3 zeolites. f Comparative study of NO2 adsorption and accompanying NO release in Na-LTA-3 with different amounts of retained H2O. NO2 breakthrough and NO release/breakthrough curves (activated at 300 and 450 °C, respectively) upon column breakthrough dynamic adsorption tests using NO2/N2 (500 ppm NO2 balanced with N2) and NO2/NO/N2 (500 ppm NO2 and 50 ppm NO balanced with N2), respectively, at approximately 25 °C and 101.325 kPa.
Further NO2 adsorption experiments on samples with distinct H2O content confirmed that lower H2O content correlates with reduced NO formation. Na-LTA-3 was specifically selected for this study due to its negligible NO retention capability (Figs. 3a and 4f), which allows its NO release curve in NO2/N2 to accurately reflect NO formation. As demonstrated in Fig. 4f, Na-LTA-3 activated at 450 °C (with lower H2O content than the 300 °C-activated sample, as evidenced by Fig. 4e) exhibited reduced NO release (3% of NO2 input, Supplementary Table 12) compared to its 300 °C-activated counterpart (6.3%, Supplementary Table 12). This trend was consistently observed across Ca2+-, Mn2+-, and Ni2+-exchanged LTA-3 zeolites (Supplementary Table 12 and Fig. 18). This finding indicates that NO formation during NO2 adsorption on LTA zeolites is primarily driven by the NO2‒H2O reaction, which is further supported by cyclic adsorption-desorption results (Supplementary Fig. 19).
Based on the identified dissociatively chemisorbed species (i.e., HNO3, HNO2, nitronium—NO2+, and nitrosonium—NO+) and the established reaction pathways (i.e., NO2–H2O reactions), the mechanism of NO formation in LTA zeolites can be described as follows: NO2 reacts with residual H2O to form HNO3 and the less stable HNO2, with the latter subsequently decomposing to form NO, as presented in Eq. (1).
Similarly, the reaction between dinitrogen tetroxide (N2O4, the dimer of NO2, as shown in Eq. (2)) and H2O also produces HNO2 and NO, along with the formation of NO2+, as depicted in Eq. (3).
Additionally, the presence of O2 in the feed gas can reduce NO formation by oxidizing the transient-formed NO into more favorable adsorbates such as NO2 and NO⁺ within zeolite pores (Supplementary Figs. 20, 21 and Table 13), as illustrated by the mechanisms proposed in Eq. (4) and Eq. (5).
These experimental observations and analyses, together with the proposed mechanisms for NO formation, underpin our subsequent analysis of the suppressed NO formation in divalent cation-exchanged LTA zeolites.
Divalent metal cations as key suppressors of NO formation
Our comprehensive adsorption experiments and in situ DRIFTS analyses reveal that divalent metal cations (e.g., Ca2+, Mn2+, and Ni2+) are crucial for suppressing NO formation during NO2 adsorption, whereas monovalent metal cations (e.g., Na+) lack this ability. Specifically, Na-LTA-3 exhibits a low-level NO release throughout NO2 adsorption, while Ca-LTA-3, Mn-LTA-3, and Ni-LTA-3 eliminate NO release (Fig. 2c, d). Besides, increasing the degree of ion exchange of divalent metal cations (e.g., Ca2+, Mn2+, and Ni2+) within Na-LTA-3 constantly reduces NO release (Supplementary Figs. 22, 23 and Tables 14–16). Additionally, in situ DRIFTS show that Ca2+-, Mn2+-, and Ni2+-exchanged LTA-3 zeolites form greater amounts of HNO3 and HNO2 (the intermediate of NO formation) via NO2–H2O reactions than Na-LTA-3 does, as evidenced by the more prominent peaks observed in the range of 1100–1300 cm-1 (Figs. 4a–d and Supplementary Fig. 16)38,39,40,41,42. These results demonstrate that divalent metal cation-exchanged zeolites consistently exhibit suppressed decomposition of HNO2 into NO. This contrasts with the monovalent metal cation-exchanged ones, which form less HNO2 but exhibit more significant NO release, suggesting that monovalent metal cations lack the capability to suppress HNO2 decomposition.
DFT calculations support this observation, revealing that HNO2 binds significantly more strongly to divalent metal cations (i.e., Ni2+: 1.17 eV, Mn2+: 1.13 eV, and Ca2+: 0.71 eV) than to Na+ (0.42 eV), owing to their higher charge‒to‒size ratios and resultant stronger electrostatic interactions (Fig. 5b)37,43,44. Although trivalent cation-exchanged zeolites could theoretically offer even stronger binding37, their low degree of ion exchange (below 40%, with Na+ remaining the majority cation, Supplementary Table 1) limits their practical effectiveness (Supplementary Fig. 9 and Table 3). In summary, the strong binding of HNO2 by divalent metal cations redirects the pathway of the NO2–H2O reaction away from NO formation (schematically illustrated in Fig. 5c), which is critical for eliminating NO release and achieving “zero-emission” NO2 capture.

a Locations of cations in Na+ (cyan), Ca2+ (red), Mn2+ (green), and Ni2+ (purple)-exchanged LTA zeolites determined by synchrotron PXRD (Supplementary Fig. 24) analysis. b DFT-calculated binding energies of HNO2 in LTA models containing a single cation of each metal, along with the corresponding optimized adsorption configurations. c Schematic illustration summarizing the mechanism of NO formation in zeolites and its suppression facilitated by ion‑exchanged divalent metal cations.
Moreover, the enhanced formation of NO⁺ in Mn-LTA-3 and Ni-LTA-3, as evidenced by more pronounced peaks at approximately 2100 cm-1 (Fig. 4a–d and Supplementary Fig. 16), suggests that these cations may promote the self-disproportionation of NO2 to yield NO3- and NO+ (Eq. (6)).
This pathway assists in diverting NO2 away from the NO2–H2O reaction and facilitates effective electrostatic stabilization of produced NO3- and NO+ on positively-charged cations and negatively-charged zeolite frameworks, respectively, thereby mitigating HNO2 and NO formation (schematically illustrated in Fig. 5c)40,42,45,46,47,48. This desirable property of Mn2+ and Ni2+ may be attributed to their unfilled outer shell d-orbitals and greater Lewis acidity than Na+ and Ca2+, enabling them to act as stronger electron acceptors47,49,50.
Broad applicability of divalent metal cations in zeolites to achieve “Zero” NO release
Our strategy of using divalent metal cations (e.g., Ca2+ and Mn2+) to suppress NO formation and thus eliminate NO release demonstrated broad applicability across various zeolite types. Besides its effectiveness on LTA zeolites, exchanging these cations into commercially available CHA, MOR, and FAU zeolites also results in reduced NO release during NO2 adsorption, achieving “zero-emission” NO2 capture (Fig. 6a, Supplementary Figs. 25–31 and Tables 17–25). These findings indicate that the NO formation/release behavior in zeolites is primarily governed by the type of the metal cation, with the influence of zeolite topology being comparatively limited. This broad applicability underscores the scalability and practical viability of our strategy for advanced NOx removal technologies.

a NO2 breakthrough and NO release curves for Na+-, Ca2+-, and Mn2+-exchanged LTA, CHA, MOR, and FAU zeolites (Si/Al ratio = 6, activated at 300 °C) in NO2/Air (500 ppm NO2 balanced with air) at approximately 25 °C and 101.325 kPa. Correlation between the quantity of retained water (after 300 °C activation) and released NO, along with differential thermogravimetric analysis (DTG) for studied b Na+-, c Ca2+-, and d Mn2+-exchanged LTA, CHA, MOR, and FAU zeolites.
Different zeolite topologies slightly influence the amount of NO released, primarily due to differences in retained H2O content after identical activation processes. Specifically, for LTA, CHA, MOR, and FAU zeolites exchanged with the same metal cation, the amount of NO released positively correlates with the amount of water retained after activation at 300 °C (Fig. 6b–d). This finding reaffirms that the NO2-H2O reactions constitute the dominant pathway for NO formation.
Real-world application potential and demonstrations
Our adsorbents capable of “zero-emission” NO2 capture were rigorously evaluated under conditions that closely mimic real-world challenges. First, we confirmed that our LTA zeolites operate effectively in humid environments—scenarios known to hinder conventional adsorbents by inducing substantial NO release (up to 45% of NO2 input) via NO2-H2O reactions27. Even in gas streams containing ~2 vol% H2O (~70% relative humidity at ambient temperature, representative of tropical and subtropical regions) and various NO2 concentrations—500 ppm (commonly used for evaluating NO2 adsorbents) and 4 ppm (representative of typical indoor NO2 pollution scenarios51)—our di-/tri-valent metal cation-exchanged LTA-3 zeolites consistently achieved “zero-emission” NO2 capture, demonstrating their practical viability for ambient air purification (Fig. 7a–b and Supplementary Table 26). The representative Ca-LTA-3 exhibited excellent recyclability under humid conditions, retaining up to 97% of its NO2 uptake and maintaining “zero-emission” NO2 capture over 10 cycles (Fig. 7c–e). This performance surpasses the previously reported maximum retained NO2 uptake (64%) in cyclic adsorption tests under humid conditions24.

a NO2 breakthrough and NO release curves for Na+-, Ca2+-, Mn2+-, and Ni2+-exchanged LTA-3 zeolites (activated at 450 oC) in humid NO2/Air (500 ppm NO2 balanced with air, containing ~2 vol% of H2O, at ~70% relative humidity, 25 °C, and 101.325 kPa). b NO2 breakthrough and NO release curves for Ca-LTA-3 (activated at 450 oC) in humid 4 ppm NO2/Air (4 ppm NO2 balanced with air, containing ~2 vol% of H2O, at ~70% relative humidity, 25 °C, and 101.325 kPa). c NO2 breakthrough and NO release curves and d corresponding NO2 uptake and percentages of released NO relative to NO2 input for Ca-LTA-3 (activated/regenerated at 450 oC) during cyclic NO2 adsorption in humid NO2/Air. e NO2 breakthrough and NO release curves for Ca-LTA-3 zeolites (activated/regenerated at 450 oC) during cyclic NO2 adsorption in humid 4 ppm NO2/Air. f Schematic illustration of our designed filter for wearable air‑purifying respirators intended for occupational personal protection. g Photograph of the fabricated prototype of our designed filter. h Photograph of the filter integrated into a commercially available respirator (3 M 6200 Half Mask; this product is solely utilized for demonstrating the integration and does not imply endorsement, affiliation, or evaluation by the manufacturer). i NO2 breakthrough and NO release curves, along with inhalation resistance of the filter during the standardized NO2 service-life test, performed using humid 500 ppm NO2 polluted air (containing ~2 vol% of H2O, at ~70% relative humidity, 25 °C, and 101.325 kPa) at a flow rate of 32 L min-1.
Afterward, we integrated our adsorbents into the filter of wearable air-purifying respirators, engineered as a “zero-NOx shield” for occupational protection. This prototype (Fig. 7f–h) not only met but also exceeded the standardized criteria for NO2 removal (outlined by the National Institute for Occupational Safety and Health (NIOSH), procedure codes: RCT-APR-STP-0062 and TEB-APR-STP-0007). Specifically, the filter effectively captured 500 ppm NO2 from humid air with “zero” NOx emissions for 132 minutes (Fig. 7i), which is significantly beyond the required 30-minute service life claimed in RCT-APR-STP-0062. Additionally, the inhalation resistance is 282.3 Pa (Fig. 7i), which falls well within the acceptable range (i.e., <400 Pa, claimed in RCT-APR-STP-0062 and TEB-APR-STP-0007). These results provide a compelling demonstration of how our fundamental discovery can translate into a robust, real-world protective device.
Furthermore, desktop estimations indicate that our adsorbents hold significant promise as a “zero-NOx shield” for indoor air purification through integration into filters of flow-through air purifiers. Calculations based on NO2 uptake suggest that only a small amount of material is required to clean confined spaces effectively. Specifically, 112.5 g of Ca-LTA-3 (corresponding to approximately 100 cm3 in volume) would be sufficient for complete NO2 removal from a workshop of 60 m2 area and 4 m height, contaminated with 25 ppm NO2. These results further highlight the potential of our approach to address long-standing air quality challenges.
Moreover, leveraging the excellent regenerability of our LTA zeolites, the spent materials used for NO2 capture can be regenerated via heating and gas purging, releasing the adsorbed NO2 for catalytic reduction (e.g., by SCR). This approach enables a complete cycle for NO2 adsorption and reduction.
Cost analysis of large-scale production
To assess the practical viability of our advanced NO2 adsorbents, an industrial-scale cost analysis was conducted. The estimated production cost of our zeolite adsorbents is approximately USD 1.93 per kilogram (Supplementary Tables 28, 29) and could be further reduced through process scale‑up. Given that our zeolite adsorbents can be nearly fully regenerated following NO2 adsorption under practical conditions (Fig. 7c–e), the spent materials can be collected, regenerated, and reused—paving another way for cost reduction. Compared with commercial porous adsorbents such as activated carbon, silica gel, and alumina, our zeolite-based adsorbents offer a highly competitive cost-performance profile (Detailed in Supplementary Table 30), underscoring their promise for widespread deployment in air purification technologies.
Discussion
The global transition to clean fuels (e.g., H2 and biofuels) can drive increased NOx emissions across indispensable combustion-based industries. Adsorptive removal of NO2 at ambient temperature is a promising strategy for mitigating air pollution, but conventional porous materials are fundamentally limited by the formation and release of NO, a toxic by-product. In this work, we report a class of adsorbents capable of achieving complete NO2 capture with “zero” NO release at ambient temperature. This realizes true “zero-emission” NO2 capture under ambient conditions. This performance was enabled by precisely tuning the adsorption chemistry through ion-exchanging divalent metal cations (e.g., Ca2+ and Mn2+) into zeolites.
Mechanistic studies combining dynamic adsorption/desorption experiments, in situ spectroscopy, and DFT calculations reveal that these cations suppress NO formation by strongly stabilizing nitrous acid (HNO2), a key intermediate, and, in some cases, promoting the self-disproportionation of NO2 into NO3- and NO+ without NO release. The superior electronegativity, Lewis acidity, and ion exchange capacity of divalent cations are central to this effect, altering the adsorption landscape in a way that prevents redox pathways leading to NO formation.
The breakthrough not only advances the fundamental understanding of NO2 adsorption chemistry in zeolitic frameworks but also opens a paradigm for ambient NO2 abatement technologies. Our materials exhibit robust performance under both dry and humid conditions (up to 70% relative humidity) across a wide NO2 concentration range (e.g., 4–500 ppm), confirming their practical relevance for real-world air purification.
To demonstrate applicability, we engineered these zeolites into functional devices, establishing a “zero-NOx shield” for human protection. When incorporated into wearable air-purifying respirators, the adsorbents substantially reduce inhalation exposure to NO2, enhancing occupational safety in high-risk environments such as firefighting, welding, traffic control, mining, agriculture, military operations, and healthcare. Additionally, integration into flow-through air purification systems could effectively mitigate NO2 levels in urban microenvironments, including transit terminals, (semi-)enclosed parking facilities, tunnels, and roadside booths, as well as confined spaces such as workshops, indoor fireplaces, and vehicles. These deployment routes directly address the pressing public health need to reduce the NOₓ-related respiratory and cardiovascular risks.
Beyond air purification, this study reveals additional opportunities for zeolite-based systems in NOx conversion, storage, and utilization. First, ambient adsorption of NO2 in the presence of water yields HNO3 within zeolite pores, presenting a low-energy pathway for nitric acid production, a sustainable alternative to the energy-intensive Ostwald process. Second, the stabilization of HNO2 and NO+ within divalent metal cation-exchanged zeolites enables their function as solid-state NO reservoirs, with controlled NO release achievable via mild thermal or moisture triggers. This controlled release supports applications in biomedical NO delivery for wound healing and vasodilation, as well as in antimicrobial surface coatings. Third, the high NO2 uptake and concurrent formation of reactive nitrogen species within confined zeolite environments suggest the feasibility of solid-phase nitration, offering a greener, solvent-free route for producing nitrated organics used in pharmaceuticals, dyes, agrochemicals, and energetic materials.
In summary, our work not only overcomes a critical limitation in ambient NO2 adsorption—eliminating the release of toxic NO by-product—but also establishes a versatile platform for advanced air purification and broader chemical engineering applications involving nitrogen oxides and their derivatives.
Methods
Preparation of adsorbents
LTA and CHA zeolites with Si/Al ratios of 3 or 6 were synthesized hydrothermally following established procedures36,52. MOR and FAU zeolites with the Si/Al ratio of 6 were purchased from Zeolyst Co. (CBV10A and CBV712, respectively). Ammonium (NH4+) from zeolites and metal cation (Na+-, K+-, Mg2+-, Ca2+-, Mn2+-, Co2+-, Ni2+-, Cu2+-, Zn2+-, Y3+-, La3+-, Ce3+-, Eu3+-, Tb3+-, and Yb3+-)-exchanged zeolites were prepared via liquid-state ion exchange according to our previously reported methods37,43. Proton (H+) from LTA zeolite was obtained by calcinating the NH4+ form in the airflow. Detailed procedures for sample preparation are provided in the Supplementary Information (Section 1.1).
Characterization
The crystallinity and phase purity of our studied zeolites were evaluated via PXRD patterns in the 2θ range of 5–50° with a step size of 0.026°. PXRD patterns were recorded on a PANalytical X’Pert3 powder diffractometer equipped with a Cu Kα radiation source (λ = 0.15406 nm) operated at 40 V and 40 mA. The crystallinity, phase purity, and cation locations of our studied representative LTA zeolites were further confirmed by synchrotron PXRD (PD beamline, Australian Synchrotron, ANSTO) at wavelengths of 0.5903 Å and 0.6887 Å. The Si/Al ratios and ion exchange degrees of the studied zeolites were determined from their elemental compositions, which were obtained from EDS using an Oxford Aztec Energy X-MAX 50 system in conjunction with field emission gun-scanning electron microscopy (FEG-SEM, FEI Quanta 450 FEG), as well as from ICP‒OES using a Agilent 720ES. These parameters were calculated using Eqs. (7) and (8), respectively:
The valence states of the ion-exchanged metal cations were determined via XPS using Thermo Scientific ESCALAB Xi + . The porous properties of the studied zeolites were characterized by N2 adsorption isotherms at −196.15 °C using a Micromeritics 3Flex gas adsorption analyzer. Prior to the measurements, the samples were activated at 300 °C under high vacuum (10-6 Pa) for 10 hours.
In situ DRIFTS during NO2 adsorption were collected using a Thermo Nicolet i550 spectrometer. Prior to spectral acquisition, the samples were activated at 300 °C for 4 hours under continuous argon (Ar) flow at a rate of 40 mL min-1. After activation, the background spectrum was recorded once the sample cooled to ambient temperature (~25 °C). Subsequently, in situ spectra were collected during NO2 adsorption using a feed gas containing 500 ppm NO2 (balance with either air or N2—designated as NO2/Air and NO2/N2, respectively) at a total flow rate of 50 mL min-1. All spectra were recorded with 64 scans at a resolution of 4 cm⁻¹.
TGA was conducted using an SDT 650 thermal analyzer. The samples were first heated to 300 °C at a rate of 2 or 5 °C min⁻1 and then held for 2 hours, followed by heating to 450 or 600 °C at 5 °C min⁻1 and holding for an additional 1 hour. All measurements were performed under a continuous 200 mL min-1 N2 flow. DTG analysis was obtained by calculating the first derivative of the TGA result.
NO2 adsorption experiments
NO2 adsorption was measured at ambient temperature ( ~ 25 °C) and atmospheric pressure ( ~ 101.325 kPa) by column breakthrough dynamic adsorption experiments, with a description of the experimental setup provided in the Supplementary Information (Section 1.2.1). Prior to the adsorption test, adsorbents (0.2–0.3 g, particle size ~0.4 mm) were activated at 300 or 450 °C under a 40 mL min−1 continuous Ar flow, with a heating rate of 2 °C min⁻1 to the target temperature, followed by holding for 10 hours. During the adsorption tests, a low-concentration NO2 feed (i.e., 500 ppm, 25 ppm, or 4 ppm NO2 balanced with air or N2, respectively) was introduced into the sample column at a flow rate of 200 mL min⁻1. The outlet concentrations of NO2 and NO were continuously monitored using a gas analyzer (MultiRAE Lite and/or MKS Multigas 6030). Data acquisition was terminated once the outlet NO2 concentration reached 20 ppm.
The amount of NO2 adsorbed (also referred to as NO2 uptake, in mmol g-1) was calculated using Eq. (9):
where NNO2 denotes the NO2 uptake in mmol g-1; Q is the feed‑gas flow rate (200 mL min-1); Vm is the molar volume of an ideal gas under ambient conditions; m is the mass of the activated adsorbent in g; C is the NO2 concentration in the feed (500 ppm, 25 ppm, or 4 ppm); Cd is the detected outlet NO2 concentration in ppm at time t; and Ts is the breakthrough time (the time at which Cd reaches 20 ppm) in minutes.
The amount of NO released (in mmol g-1) during NO2 adsorption was calculated via Eq. (10):
where RNO denotes the amount of NO released in mmol g-1, and Cd is the detected outlet NO concentration in ppm at time t. All other parameters are defined as in Eq. (9). To facilitate comparison across different adsorbents, the amount of NO released was normalized as a percentage of the NO2 input.
Binary adsorption experiments of NO2 and NO were conducted under similar conditions using a feed gas of 500 ppm NO2 and 50 ppm NO balanced with N2. The amounts of NO2 adsorbed and NO released were calculated using the methods described in Eqs. (9) and (10).
DFT calculations
DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP 5.4.4) on the Australian Synchrotron Compute Infrastructure (ASCI). The generalized gradient approximation (GGA) and GGA + U methods were applied, employing the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional. Interactions between ionic cores and valence electrons were treated using ultrasoft pseudopotentials, and van der Waals (VdW) interactions were included via the zero-damping DFT-D3 dispersion correction method of Grimme. A plane-wave cutoff energy of 600 eV was used for all calculations, and a Monkhorst-Pack k-point mesh was employed to ensure total‑energy convergence within 1 meV per atom. The LTA zeolite framework used in the DFT calculations was obtained from the Rietveld refinement of the synchrotron PXRD data (Supplementary Fig. 24). The adsorption binding energy (Eads) was calculated via Eq. (11):
Demonstration of zeolite adsorbents in wearable air-purifying respirators
We conducted both the standardized NO2 service-life test and inhalation-resistance test in accordance with the National Institute for Occupational Safety and Health (NIOSH) procedure (code: RCT-APR-STP-0062 and TEB-APR-STP-0007). A customized tubing system was used to perform these experiments, as detailed in the Supplementary Information (Section 1.3). Prior to testing, the adsorbents (60 g of Ca-LTA-3 and 40 g of Ni-LTA-3) were pelleted into 0.7 mm particles to balance high NO2 uptake, minimal NO release, and low breathing resistance (Supplementary Fig. 32 and Table 27) and subsequently packed into a customized simulated filter. The assembled device was activated by heating at 2 °C min-1 to 450 °C under a 10 L min-1 Ar flow and held at this temperature for 3 hours. During the NO2 service-life test, a feed gas consisting of 500 ppm NO2 balanced with air and containing ~2 vol% of H2O at ~70% relative humidity, 25 °C, and 101.325 kPa was supplied at a flow rate of 32 L min-1. The outlet concentrations of NO2 and NO were continuously monitored using a flue gas analyzer (Betoo Instrument, CN), and data acquisition was terminated once the outlet NO2 concentration reached 20 ppm. The inhalation resistance was measured simultaneously by recording the pressure drop across the device. A blank test (without adsorbent loading) was also conducted for background subtraction.
Evaluation of zeolite adsorbents for indoor air purification
The evaluation was performed through desktop calculations based on the NO2 uptake obtained from single breakthrough dynamic adsorption results under dry conditions, assuming operation in effectively dehumidified environments achieved through a desiccant pre‑layer or a dehumidifier.
The specific air purification capacity Vs (in m3) was calculated via Eq. (12):
where Vs denotes the specific air purification capacity in m3, CNO2 is the initial NO2 concentration in the atmosphere, and the other parameters are defined as in Eq. (9).
Data availability
The processed data generated in this study are included in the main text, the Supplementary Information, and Source Data files. Additional data are available from the corresponding author upon request. Source data are provided with this paper.
References
Babaniyi, B. R. et al. in Microbial Biotechnology for Bioenergy (eds Naga Raju Maddela et al.) 81-96 (Elsevier, 2024).–
Dincer, I. & Aydin, M. I. New paradigms in sustainable energy systems with hydrogen. Energy Convers. Manag. 283, 116950 (2023).
Lewis, A. C. Optimising air quality co-benefits in a hydrogen economy: a case for hydrogen-specific standards for NOx emissions. Environ. Sci.: Atmos. 1, 201–207 (2021).
Thangaraja, J., Anand, K. & Mehta, P. S. Biodiesel NOx penalty and control measures - a review. Renew. Sustain. Energy Rev. 61, 1–24 (2016).
IEA (2020), Iron and Steel Technology Roadmap. (2020).
Fatahi, R., Nasiri, H., Dadfar, E. & Chehreh Chelgani, S. Modeling of energy consumption factors for an industrial cement vertical roller mill by SHAP-XGBoost: a “conscious lab” approach. Sci. Rep. 12, 7543 (2022).
A deep dive into hard-to-abate sectors: types, characteristics and challenges, <https://www.iberdrola.com/sustainability/energy-transition/decarbonized-economy-principles-regulatory-actions/hard-to-abate-sectors> (2025).
IEA (2024), Renewables. (2024, Paris, 2024).
Anenberg, S. C. et al. Impacts and mitigation of excess diesel-related NOx emissions in 11 major vehicle markets. Nature 545, 467–471 (2017).
Gu, Y. & Epling, W. S. Passive NOx adsorber: An overview of catalyst performance and reaction chemistry. Appl. Catal. A: Gen. 570, 1–14 (2019).
Bashkova, S., Deoki, D. & Bandosz, T. J. Effect of silver nanoparticles deposited on micro/mesoporous activated carbons on retention of NOx at room temperature. J. Colloid Interface Sci. 354, 331–340 (2011).
Shang, S. et al. Transition-Metal-Containing Porphyrin Metal–Organic Frameworks as π-Backbonding Adsorbents for NO2 Removal. Angew. Chem. Int. Ed. 59, 19680–19683 (2020).
Levasseur, B., Petit, C. & Bandosz, T. J. Reactive adsorption of NO2 on copper-based metal−organic framework and graphite oxide/metal−organic framework composites. ACS Appl. Mater. Interfaces 2, 3606–3613 (2010).
Ebrahim, A. M. & Bandosz, T. J. Carbon coated silica doped with Cerium/Zirconium Mixed Oxides as NO2 Adsorbent at Ambient Conditions. J. Phys. Chem. C. 118, 8982–8992 (2014).
Jeguirim, M., Belhachemi, M., Limousy, L. & Bennici, S. Adsorption/reduction of nitrogen dioxide on activated carbons: Textural properties versus surface chemistry – A review. Chem. Eng. J. 347, 493–504 (2018).
Kante, K., Deliyanni, E. & Bandosz, T. J. Interactions of NO2 with activated carbons modified with cerium, lanthanum and sodium chlorides. J. Hazard. Mater. 165, 704–713 (2009).
Ghouma, I. et al. Activated carbon prepared by physical activation of olive stones for the removal of NO2 at ambient temperature. Comptes Rendus Chim. 18, 63–74 (2015).
Levasseur, B., Gonzalez-Lopez, E., Rossin, J. A. & Bandosz, T. J. Effect of reduction treatment on copper modified activated carbons on NOx adsorption at room temperature. Langmuir 27, 5354–5365 (2011).
Florent, M., Tocci, M. & Bandosz, T. J. NO2 adsorption at ambient temperature on urea-modified ordered mesoporous carbon. Carbon 63, 283–293 (2013).
Shang, S. et al. NO2 removal by adsorption on transition-metal-based layered double hydroxides. ACS EST Eng. 1, 375–384 (2021).
Shang, S. et al. The low-temperature NO2 removal by tailoring metal node in porphyrin-based metal-organic frameworks. Sci. Total Environ. 801, 149710 (2021).
Sivachandiran, L., Thevenet, F., Gravejat, P. & Rousseau, A. Investigation of NO and NO2 adsorption mechanisms on TiO2 at room temperature. Appl. Cata. B: Environ. 142-143, 196-204 (2013).
Yu, J. J., Jiang, Z., Zhu, L., Hao, Z. P. & Xu, Z. P. Adsorption/desorption studies of NOx on well-mixed oxides derived from Co−Mg/Al Hydrotalcite-like compounds. J. Phys. Chem. B 110, 4291–4300 (2006).
Tian, Y. et al. Efficient adsorption removal of NO2 by covalent triazine frameworks with fine-tuned binding sites. J. Hazard. Mater. 441, 129962 (2023).
Ebrahim, A. M. & Bandosz, T. J. Effect of amine modification on the properties of zirconium–carboxylic acid based materials and their applications as NO2 adsorbents at ambient conditions. Microporous Mesoporous Mater. 188, 149–162 (2014).
Deliyanni, E. & Bandosz, T. J. Effect of carbon surface modification with dimethylamine on reactive adsorption of NOx. Langmuir 27, 1837–1843 (2011).
Wang, Y., Wang, T., Gu, Q. & Shang, J. Adsorption removal of NO2 under low-temperature and low-concentration conditions: a review of adsorbents and adsorption mechanisms. Adv. Mater. 37, 2401623 (2024).
Petit, P. C. et al. The Pathophysiology of Nitrogen Dioxide During Inhaled Nitric Oxide Therapy. ASAIO J. 63, 7–13 (2017).
Weinberger, B., Laskin, D. L., Heck, D. E. & Laskin, J. D. The Toxicology of Inhaled Nitric Oxide. Toxicol. Sci. 59, 5–16 (2001).
Pârvulescu, V. I., Grange, P. & Delmon, B. Catalytic removal of NO. Catal. Today 46, 233–316 (1998).
Shang, S. et al. Designing multivariate porphyrin-based metal-organic frameworks with Ni/Co dual-metal atom sites for cooperative NO2 capture and NO retention. Sep. Purif. Technol. 320, 124080 (2023).
Delachaux, F., Vallières, C., Monnier, H. & Lecler, M. T. Experimental study of NO and NO2 adsorption on a fresh or dried NaY zeolite: influence of the gas composition by breakthrough curves measurements. Adsorption 25, 95–103 (2019).
Sun, M. et al. Ambient temperature NO2 removal by adsorption on transition metal ion-exchanged chabazite zeolites. Results Eng. 18, 101134 (2023).
Mosca, A., Öhrman, O., Hedlund, J., Perdana, I. & Creaser, D. NO2 and N2 sorption in MFI films with varying Si/Al and Na/Al ratios. Microporous Mesoporous Mater. 120, 195–205 (2009).
Sun, M. et al. Regulating NO2 adsorption at ambient temperature by manipulating copper species as binding sites in copper-modified SSZ-13 zeolites. J. Mater. Chem. A 12, 30329–30339 (2024).
Palomino, M., Corma, A., Rey, F. & Valencia, S. New Insights on CO2−methane separation using LTA Zeolites with different Si/Al ratios and a first comparison with MOFs. Langmuir 26, 1910–1917 (2010).
Tao, Z., Tian, Y., Ou, S. Y., Gu, Q. & Shang, J. Direct air capture of CO2 by metal cation-exchanged LTA zeolites: Effect of the charge-to-size ratio of cations. AIChE J. e18139 (2023).
Wang, X. et al. Enhanced adsorption and mass transfer of hierarchically porous Zr-MOF nanoarchitectures toward toxic chemical removal. ACS Appl. Mater. Interfaces 13, 58848–58861 (2021).
Wang, X. et al. NO2 Removal under ambient conditions by nanoporous multivariate zirconium-based metal–organic framework. ACS Appl. Nano Mater. 3, 11442–11454 (2020).
Szanyi, J., Kwak, J. H. & Peden, C. H. F. The effect of water on the adsorption of NO2 in Na− and Ba−Y, FAU Zeolites: A combined FTIR and TPD investigation. J. Phys. Chem. B 108, 3746–3753 (2004).
Szanyi, J. & Paffett, M. T. The adsorption of NO and reaction of NO with O2 on H-, NaH-, CuH-, and Cu-ZSM-5: An in situ FTIR investigation. J. Catal. 164, 232–245 (1996).
Szanyi, J., Kwak, J. H., Burton, S., Rodriguez, J. A. & Peden, C. H. F. Characterization of NOx species in dehydrated and hydrated Na- and Ba-Y, FAU zeolites formed in NO2 adsorption. J. Electron Spectrosc. Relat. Phenom. 150, 164–170 (2006).
Tao, Z. et al. Metal cation-exchanged LTA zeolites for CO2/N2 and CO2/CH4 separation: The roles of gas-framework and gas-cation interactions. Carbon Capt. Sci. Technol. 8, 100126 (2023).
Tao, Z. et al. Development of zeolite adsorbents for CO2 separation in achieving carbon neutrality. npj Mater. Sustain. 2, 20 (2024).
Sedlmair, C., Gil, B., Seshan, K., Jentys, A. & Lercher, J. A. An in situ IR study of the NOx adsorption/reduction mechanism on modified Y zeolites. Phys. Chem. Chem. Phys. 5, 1897–1905 (2003).
Ahrens, M., Marie, O., Bazin, P. & Daturi, M. Fe-H-BEA and Fe-H-ZSM-5 for NO2 removal from ambient air – A detailed in situ and operando FTIR study revealing an unexpected positive water-effect. J. Catal. 271, 1–11 (2010).
Mignon, P., Pidko, E. A., Van Santen, R. A., Geerlings, P. & Schoonheydt, R. A. Understanding the reactivity and basicity of zeolites: A periodic DFT study of the disproportionation of N2O4 on alkali-cation-exchanged zeolite Y. Chem. – A Eur. J. 14, 5168–5177 (2008).
Schoonheydt, R. A., Geerlings, P., Pidko, E. A. & van Santen, R. A. The framework basicity of zeolites. J. Mater. Chem. 22, 18705–18717 (2012).
Pidko, E. A., Mignon, P., Geerlings, P., Schoonheydt, R. A. & van Santen, R. A. A periodic DFT study of N2O4 disproportionation on alkali-exchanged zeolites X. J. Phys. Chem. C. 112, 5510–5519 (2008).
Gil, B., Mierzyńska, K., Szczerbińska, M. & Datka, J. Basic sites in zeolites followed by IR studies of NO+. Appl. Catal. A: Gen. 319, 64–71 (2007).
Pennanen, A. S. O., S. R., Sari, A., J., J. M. & and Pasanen, P. Characterization of air quality problems in five Finnish indoor ice arenas. J. Air Waste Manag. Assoc. 47, 1079–1086 (1997).
Yun, J. H. & Lobo, R. F. Effects of temperature pretreatment on propane cracking over H-SSZ-13 zeolites. Catal. Sci. Technol. 5, 264–273 (2015).
Levasseur, B., Ebrahim, A. M. & Bandosz, T. J. Interactions of NO2 with Amine-functionalized SBA-15: Effects of synthesis route. Langmuir 28, 5703–5714 (2012).
Ebrahim, A. M., Levasseur, B. & Bandosz, T. J. Interactions of NO2 with Zr-based MOF: effects of the size of organic linkers on NO2 adsorption at ambient conditions. Langmuir 29, 168–174 (2013).
Acknowledgements
This work was financially supported by the Science and Technology Innovation Commission of Shenzhen Municipality (Ref: JCYJ20210324134006019 to J.S.) and the Research Grants Council of Hong Kong (Ref: CityU 11317722 and 11310223 to J.S. and Q.G.). The funders played no role in the study design, data collection, analysis, interpretation, or manuscript writing. The authors acknowledge the research undertaken at the PD beamline and Australian Synchrotron Compute Infrastructure (ASCI) at the Australian Synchrotron, ANSTO. The authors express their gratitude for the technical assistance provided by Shanshan Shang, Mingzhe Sun, Aamir Hanif, Nuo Chen, Yuan Lai, Yee Lam Lo, and Junyi Chen.
Author information
Authors and Affiliations
Contributions
Z. T. and Y. T. conceived and designed the experiments, performed the majority of the experimental work, analyzed the data, and drafted the manuscript. R. W. contributed to experimental design and manuscript preparation. T. Z. contributed to sample preparation and manuscript preparation. T. W. contributed to the manuscript revision. H. Z., M. L., and J. Q. contributed to data collection. M. K., S. F., and Z. N. provided partial experimental resources and contributed to the manuscript revision. H.W. and Q. G. provided critical intellectual input, supervised the project, contributed experimental resources, and played a key role in shaping the manuscript presentation. J. S. conceived the overall research idea, provided the majority of experimental resources, led manuscript revision, and supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xiaoqin Zou and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tao, Z., Tian, Y., Wang, R. et al. Zero-emission NO2 capture using divalent metal cation-exchanged zeolites for air purification. Nat Commun 16, 10102 (2025). https://doi.org/10.1038/s41467-025-65052-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65052-z
This article is cited by
-
Tailoring Cation Charge-to-Size Ratios in Zeolite Y for High-performance Methane/Nitrogen Separation
Chemical Research in Chinese Universities (2026)
