
Abstract
The human genome contains a sophisticated array of elements that regulate gene activity and organismal functions. Developing a large window foundation model capable of efficiently processing long sequence inputs is essential yet challenging for decoding the multi-layered and complex landscape of the cis-regulatory elements. Here, we introduce OmniReg-GPT, a generative foundation model designed for the low-resource pretraining of long genomic sequences by optimized attention mechanism. During pretraining, OmniReg-GPT captures the complete distribution of regulatory elements across nucleotide to megabase scales with efficient training speed and memory usage. We demonstrate exceptional performance in downstream regulotary applications spanning the entire spectrum of genomic scales, including various cis-regulatory elements identification, context dependent gene expression prediction, single-cell chromatin accessibility analysis, and 3D chromatin contact modeling. As a generative model, OmniReg-GPT also holds the potential to generate candidate cell-type-specific enhancers through prompt engineering. Overall, OmniReg-GPT extends the boundaries of foundation models in the genomic field, and provides a valuable pretraining model resource which can be extensively applied for genomic researches.
Similar content being viewed by others


Introduction
A long-standing goal in genomics is to understand the language of genome sequence, especially for the non-coding regulatory sequence1,2,3. As the major biological outputs of the genome, protein coding genes comprise only a tiny fraction of human genome, whereas most of the human genome consists of non-coding sequence, which might be functionally involved in gene regulation in a highly complex style. For example, cis-regulatory elements underlying non-coding DNA sequences could dynamically and cooperatively regulate the gene transcription, by spatially, temporally, and combinatorially modulating the recruitment and activity of RNA polymerase.
Using various functional genomic sequencing technologies, large-scale consortia such as ENCODE4 and Roadmap Epigenomics5,6 have experimentally profiled the regulatory genome across diverse cellular contexts, and systematically identified a vast amount of non-coding regulatory elements, such as promoters, enhancers, silencers, and insulators. Based on these foundational resources, supervised deep learning have emerged as powerful tools in genomics, demonstrating its ability to build predictive mapping from paired DNA sequence and functional genomic data7,8,9,10,11. Those models have facilitated our understanding the language of genome sequence and enable broad predictive applications, such as cis-regulatory elements annotation, gene expression modeling, and variant effect prediction. However, the reliance on specific functional data has led to the proliferation specialized models, each tailored for a single predictive goal. We still lack a comprehensive mapping with global representations of regulatory sequences empowered with multi-scale understanding and predictive capabilities.
In recent years, foundation models have demonstrated the ability to learn universal representations from unlabeled data and swiftly adapt to specific downstream tasks through the pretraining and finetuning paradigm12,13,14. Meanwhile, several DNA pre-trained models have been developed for the genome sequence language understanding, such as DNABERT215, GENA16, Nucleotide Transformer17, and Species-Specific Models18. Those models have shown wide applicability in diverse down-stream tasks, including predicting various types of regulatory elements, chromatin profiles, and transcription factor binding using the embedding representations of sequences. However, current models typically pretrained on short genomic sequence, largely because the quadratic computation and memory complexity of the self-attention mechanism in Transformer-based architectures hinders their ability to process long genomic contexts efficiently. The understanding of complex regulatory sequences and grammar necessitates models with sufficient capacity and extensive receptive fields to capture interactions across long genomic contexts. From the perspective of model architecture design, a highly efficient foundation model capable of integrating long-range genomic signals is therefore necessary to decode the complex regulatory landscape.
Furthermore, the evaluation of current DNA foundation models has largely been confined to a narrow set of downstream tasks, such as identifying isolated regulatory elements. The focus on specific regions, while precise, might ignore the breadth and complexities of genomic regulatory grammar that involve interactions across larger genomic contexts. For instance, while specialized supervised models have been applied in context-dependent challenges like modeling gene expression, chromatin accessibility and 3D chromatin contacts, these tasks remain significant hurdles for most existing pre-trained models. Additionally, the lack of generative capabilities in current models restricts their utility in generating functional sequences.
Here, we present OmniReg-GPT, a foundation generative model that leverages a hybrid attention structure composed of local and global attention mechanisms. This architecture reduces the quadratic complexity of attention layers to linear complexity, enabling a comprehensive receptive field with high computational efficiency. Capitalizing on this efficiency, we pretrained OmniReg-GPT on the human reference genome using 20 kb sequence windows. To verify its ability, we conducted multiple downstream experiments on genome understanding tasks, including predicting cis-regulatory elements, predicting cell-type agnostic and specific gene expression. By pretraining on large genomic windows, OmniReg-GPT broadens the capabilities of DNA foundation models, yielding promising results across these tasks and enabling analyses that capture broader regulatory contexts. Notably, we applied OmniReg-GPT to predict single-cell chromatin accessibility and to measure 3D chromatin contact over a 2-megabase sequence window, illustrating its potential to capture complex regulatory networks that control gene expression. Finally, we deployed OmniReg-GPT in a zero-shot setting to predict enhancer effects from enhancer-promoter compatibility data, and generated computational hypothesis for diverse cell-type-specific enhancers. Overall, OmniReg-GPT will extend the boundaries of foundation models in the genomic field, and provide a valuable pretraining model resource which can be extensively applied for genomic research.
Results
Overview of OmniReg-GPT: advancing long-sequence modeling through high-efficiency pretraining
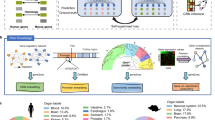
To address the computational challenges of quadratic time and space complexities in long genomic sequence pretraining using Transformer architecture, we develop OmniReg-GPT, a model designed to efficiently integrate both local and global genomic sequence information through a hybrid attention mechanism (Fig. 1A and Supplementary Fig. 1). OmniReg-GPT retains the generative pretraining transformer architecture and next-token-prediction task, while utilizing a Byte Pair Encoding tokenizer for efficient sequence processing from the genome (Fig. 1A and Supplementary Table 1). As the overall pipeline of application, we aimed to pretrain OmniReg-GPT model with 20 kb length on sequences from the human reference genome, and then use it for a wide range of downstream tasks, such as predicting chromatin feature profiling, local rules of genome regulation and topology level 3D chromatin interaction. Notably, this model can also generate context-dependent enhancers by leveraging multi-scale regulatory grammar embedded within its parameters (Fig. 1B).

A OmniReg-GPT Workflow: The model undergoes generative pretraining on randomly extracted sequences from the human genome. The core of OmniReg-GPT features a hybrid attention module that combines local and global attention mechanisms, optimizing resource efficiency during training. Local attention divides query into windows and concatenates each query window with the previous window to form key-value pairs, then merges window dimensions into batch dimension for efficient attention computation. OmniReg-GPT is then applied to a range of predictive tasks through simple transfer learning, and is also used for generating functional regulatory elements. B The pretrained embeddings can be leveraged to predict multi-scale gene regulation profiles, including chromatin feature profiling, local genomic regulation rules, and chromatin topology and interactions. By incorporating regulatory knowledge within the model parameters, OmniReg-GPT is capable of in silico generating cell type-specific, high-activity enhancers through its reasoning abilities. Schematics in A and B were created using BioRender. (Wang, A. (https://BioRender.com/gnepe2b) and (https://BioRender.com/q9xdysb).
At the core of model architectures, OmniReg-GPT consists 12 local blocks for generating contextual embeddings and 2 global blocks for constructing comprehensive sequence representations, accumulating 270 million parameters (Supplementary Fig. 1). Within the local blocks of OmniReg-GPT, the sequence is segmented into portions by a defined window size. Then a local window attention is employed, allowing attention to be performed with both the preceding window and the sequence itself. This approach reduces the complexity of attention from O(L2) to O(L), while maintaining information aggregation and processing (Methods). To further enhance representation, we implement a token shift strategy along the hidden dimension. The resulting outputs are concatenated and processed through a feedforward network layer to generate the layer output. In the global blocks, global attention is utilized to enhance the capture of long-range interactions, further improving the model’s capacity to identify dependencies across extended genomic sequences. In both types of blocks, we have incorporated computational efficiency optimization. Each attention module employs Flash attention for accelerated computation19 and adopts Rotary Position Embedding to facilitate length extrapolation20.
To validate the effectiveness of our architectural design choices, we performed several comparison studies. Firstly, we compared three attention variants of the hybrid attention mechanism during pretraining, including 1) 14 layers of local attention only, 2) 14 layers of full attention only, and 3) hybrid architecture with 12 layers of local attention and 2 layers of global attention. The pretraining results on 5 kb and 20 kb input genomic sequence revealed that the hybrid architecture consistently achieved the lowest training loss, outperforming both pure local and full attention variants (Supplementary Fig. 2 a,b). Secondly, we evaluated the model’s time and space complexity during long-sequence pretraining, by comparing OmniReg-GPT to various Transformer-based DNA foundation models across different sequence lengths. As expected, OmniReg-GPT exhibited remarkable efficiency in GPU memory usage during pretraining. On a single NVIDIA Tesla V100 with 32GB memory, OmniReg-GPT architecture could accommodate DNA sequence inputs up to 200 kb, despite its parameter size twice larger than other methods (Fig. 2A, and Supplementary Table 2). In contrast, Gena-bigbird16, which employed sparse attention, was restricted to 100 kb inputs on the same hardware. Additionally, to keep the parameter sizes comparable across models, we used a reduced hidden dimension for OmniReg-GPT when comparing training speeds. OmniReg-GPT achieved superior training throughput (Fig. 2B, and Supplementary Table 3), whereas Gena-bigbird traded off speed to increase window size.

A Line plot showing the relationship between GPU memory usage and input sequence length for OmniReg-GPT and other two latest transformer-based models. The 32GB V100 GPU memory limit is indicated by the dashed line. The 138 million parameter size for OmniReg-GPT refers to the variant with a hidden dimension of 768. B Bar plot depicting the relative computation time ratios of three models across different sequence lengths, with the slowest model at each length set as the baseline (1.0). Note that DNABERT-2 is absent at 50k due to memory exhaustion. C Comparative performance of OmniReg-GPT against DNA foundation models trained on human genome (top panel) and multi-species genome (bottom panel) in predicting histone marks, promoters and enhancers, measured by Matthew’s correlation coefficient (MCC). D, E AUROC scores for fine-tuned multi-classification models (OmniReg-GPT, DNABERT2 and Gena-LM, NT-V2-multispecies(500 M), HyenaDNA-1k) of CpG methylation (n = 7) and histone modification tasks (n = 18) from Bend Benchmark. For each model, individual dots represent AUROC scores for each specific task, while the boxplot height indicates the mean AUROC across all tasks. Specifically, for the CpG methylation tasks, the mean AUROC scores for OmniReg-GPT, NT-V2-multispecies(500 M), DNABERT2, Gena-LM and HyenaDNA-1k were 0.872, 0.858, 0.810, 0.743 and 0.800 respectively; for the histone modification tasks, mean AUROC scores were 0.764, 0.780, 0.739, 0.701 and 0.726, respectively. Statistical significance was assessed by a one-sided paired Wilcoxon test (per-task AUROC pairs, alternative: OmniReg-GPT > baseline; *p < 0.05, **p < 0.01, ***p < 0.001). For the CpG methylation task, the exact p values for the comparisons are: vs DNABERT2 (p = 0.011), vs GenaLM (p = 0.010), vs NT-V2-multispecies (p = 0.011), vs Hyena-1k (p = 0.011). For the CpG methylation task, the exact p values for the comparisons are: vs DNABERT2 (p = 0.0001), vs GenaLM (p = 0.0001), vs NT-V2-multispecies (p = 0.0001), vs Hyena-1k (p = 0.0001). F Bar plot comparing the PR-AUC values of OmniReg-GPT, NT-V2-multispecies (500 M), DNABERT2, Gena-bigbird and HyenaDNA-32k for eQTL variants binary classification tasks across sequence lengths of 2k, 6k, and 10k base pairs. G Precision-Recall Curves plot of OmniReg-GPT, NT-V2-multispecies (500 M), DNABERT2, Gena-bigbird and HyenaDNA-32k for the pathogenic variants binary classification task with length of 10k base pairs. Source data are provided as a Source Data file.
Such enhanced efficiency directly benefits the handling of long genomic sequences input at pretraining stage, which could be widely adopted to the modern genomic language model, especially in resource-constrained environments for most of the biological applications. To further accelerate research progress in this field, we have made OmniReg-GPT fully open-access, providing both the source code and pretrained model weights to the community.
Benchmark of genomics foundational models
To evaluate and highlight the capacity of OmniReg-GPT, we benchmarked latest genomic foundational models in various genome understanding scenarios. Specifically, we assessed four leading genomic foundational models, including DNABERT215, HyenaDNA-1kb21, Gena-lm16, and Nucleotide Transformer17, along with their long-sequence variants HyenaDNA-32kb, Gena-bigbird and Nucleotide Transformer V2-multispecies, which were particular suited for tasks requiring extended genomic context. The evaluation spanned thirteen representative regulatory sequence understanding tasks from the Nucleotide transformer benchmark, encompassing ten histone modification datasets (each 1000 bp in length), two promoter classification datasets (each 300 bp in length), and one enhancer classification dataset (400 bp in length). Among models pretrained exclusively on the human genome, OmniReg-GPT demonstrated superior Matthews Correlation Coefficient (MCC) in the majority of genome understanding scenarios (9 out of 13), and received the highest aggregated scores on both histone and regulatory elements prediction tasks. To further evaluate the robustness of model performance across different sequence context length, we extended the input sequences to 1 kb, 2 kb, and 4 kb, and compared the predictive performance for proximal Promoter and distal Enhancer understanding tasks (Methods). The results showed that performance of Promoter task decreased slightly with larger window sizes, while the Enhancer task performance remained stable across different window sizes (Supplementary Fig. 3). Focusing specifically on distal enhancers, OmniReg-GPT showed improved F1 scores and recall with increasing window size (Supplementary Fig. 4a), indicating classification of distal enhancers could be enhanced with broader input sequence context. NT-v2-multispecies-500M showed a similar trend but lower overall performance (Supplementary Fig. 4b).
To thoroughly evaluate the practical utility of OmniReg-GPT, we conducted further benchmark inspecting the complex regulatory sequences, using datasets derived from diverse functional genomic sequencing experiments. Firstly, we leveraged broader epigenetic markers from the Bend benchmark, which collected biologically meaningful tasks for DNA language model benchmarking22. The benchmark showed that OmniReg-GPT outperformed in CpG methylation (Fig. 2D) and histone modification predicting scenarios (Fig. 2E), with AUROC metrics significantly higher than other foundation models on each of the 7 methylation tasks (p < 0.02, Wilcoxon signed-rank test) and 18 histone markers (p < 2e-4, Wilcoxon signed-rank test). Secondly, we benchmark on the extensive genomic datasets from DeepSEA23, which compiled a diverse compendium of genome-wide chromatin profiles in various cell types from ENCODE and Roadmap projects. OmniReg-GPT exhibited performance comparable to DNABERT-2 in predicting various epigenetic profiles, including TF binding, Histone marks (HMS), DNase I-hypersensitive sites (DHS) prediction tasks (Supplementary Fig. 5), with AUROC metric slightly lower than DNABERT-2. Notably, OmniReg-GPT has demonstrated enhanced predictive performance in several specific TF binding tasks. This was particularly evident in the prediction of binding sites for the NRSF motif and the BRF1 motif, where DNABERT-2 achieved AUROC scores below 0.8.
As a genomic sequence-based foundation model, we further evaluated the OmniReg-GPT’s ability to assess the impact of genetic variants on the predictions. Specifically, we benchmarked OmniReg-GPT on predicting the effects of expression quantitative trait loci (eQTLs) on gene expression and distinguishing pathogenic SNPs from common SNPs (Methods). For the eQTL prediction task, OmniReg-GPT consistently outperformed other state-of-the-art sequence-based models across varying sequence lengths (2k, 6k, and 10k base pairs), achieving the highest AUROC scores of 0.724, 0.719, and 0.701, respectively (Fig. 2F). In the pathogenic variant classification task, OmniReg-GPT also demonstrated superior performance, attaining an AUROC score of 0.679, significantly higher than Nucleotide Transformer V2-multi-species (0.622), DNABERT-2 (0.608), Gena-bigbird (0.633), and HyenaDNA-32kb (0.497) (Fig. 2G).
In summary, our comprehensive benchmark of genomics foundation models on diverse genomic sequence understanding tasks, demonstrated that OmniReg-GPT held the capacity to understand various regulatory sequences, and discern the functional impacts of regulatory and pathogenic variants. Although it did not outperform every other model in all tasks due to variations in model parameter sizes and pretraining dataset scales, the results highlight OmniReg-GPT’s robustness and effectiveness in a broad spectrum of downstream genomic scenarios.
OmniReg-GPT improves context-dependent gene expression prediction
Gene expression, the principal biological output of the genome, is spatial-temporally controlled by the regulatory genome sequences using the context-dependent grammar24. Genomic foundational models hold the promise to model the gene transcription regulation, and further reveal the complex context-dependent grammar. Here, we adopted OmniReg-GPT in predicting and modeling gene expression across broader sequence spans (Fig. 3A, Methods). To achieve this, we designed two comprehensive downstream experiments. The first focused on cell-type-agnostic and expression-modeling scenarios, while the second was specifically designed to investigate cell-type-specific scenarios at single cell resolution. This evaluation also served as a validation of OmniReg-GPT, which demonstrated on long-range and context-dependent genomic understanding, extending the local sequence and epigenomic benchmarks for genomic foundation models.

A Schematic illustration of predicting cell gene expression at cell-type-agnostic gene-expression and single cell level gene activity. B Bar plot comparing OmniReg-GPT’s performance to four foundational models on cell-type-agnostic datasets across human genome. C The per-cell AUROC values of OmniReg-GPT, DNABERT2, Gena-bigbird, HyenaDNA-32k on human (n = 97), mouse (n = 98) and fly (n = 59) across different cell types. Mean AUROC values for each model: OmniReg-GPT (human: 0.84, mouse: 0.79, fly: 0.76), DNABERT2 (human: 0.75, mouse: 0.67, fly: 0.53), Gena-bigbird (human: 0.52, mouse: 0.65, fly: 0.69), HyenaDNA-32k (human: 0.52, mouse: 0.72, fly: 0.53). Statistical significance was assessed by one-sided Wilcoxon signed-rank test (paired comparisons) between OmniReg-GPT and each baseline model across multi-task AUROC scores, with the alternative hypothesis that OmniReg-GPT performs better than the baseline (*p < 0.05, **p < 0.01, ***p < 0.001). The exact p values for the comparisons are: for human, vs DNABERT2 (p = 1.22 × 10−17), vs Gena-bigbird (p = 1.22 × 10−17), and vs HyenaDNA-32k (p = 1.22 × 10−17); for mouse, vs DNABERT2 (p = 8.33 × 10−18), vs Gena-bigbird (p = 8.33 × 10−18), and vs HyenaDNA-32k (p = 8.33 × 10−18); and for fly, vs DNABERT2 (p = 2.39 × 10−11), vs Gena-bigbird (p = 2.39 × 10−11), and vs HyenaDNA-32k (p = 2.39 × 10−11).Data are presented as half-violin plots (right side) showing kernel density estimation, box plots (center) displaying the median (center line), upper and lower quartiles (box limits), and 1.5× interquartile range (whiskers), and individual data points (left side). Violin plot width represents data density at each AUROC value. D The per-gene AUROC values of OmniReg-GPT, DNABERT2, Gena-bigbird, HyenaDNA-32k on human (n = 784), mouse (n = 1411) and fly (n = 946) across different genes. Mean AUROC values for each model: OmniReg-GPT (human: 0.69, mouse: 0.73, fly: 0.69), DNABERT2 (human: 0.64, mouse: 0.67, fly: 0.65), Gena-bigbird (human: 0.66, mouse: 0.66, fly: 0.60), HyenaDNA-32k (human: 0.65, mouse: 0.64, fly: 0.65). Statistical significance was assessed by one-sided Wilcoxon signed-rank test (paired comparisons) between OmniReg-GPT and each baseline model across multi-task AUROC scores, with the alternative hypothesis that OmniReg-GPT performs better than the baseline (*p < 0.05, **p < 0.01, ***p < 0.001). The exact p values for the comparisons are: for human, vs DNABERT2 (p = 1.11 × 10−47), vs Gena-bigbird (p = 1.36 × 10−25), and vs HyenaDNA-32k (p = 1.48 × 10−30); for mouse, vs DNABERT2 (p = 5.21 × 10−35), vs Gena-bigbird (p = 9.85 × 10−45), and vs HyenaDNA-32k (p = 4.23 × 10−46); and for fly, vs DNABERT2 (p = 1.11 × 10−8), vs Gena-bigbird (p = 3.51 × 10−48), and vs HyenaDNA-32k (p = 3.00 × 10−8). Data are presented as half-violin plots (right side) showing kernel density estimation overlaid with box plots. Box plots display the median at the center line, upper and lower quartiles as box limits, and 1.5× interquartile range as whiskers. Violin plot width represents data density at each AUROC value. E t-SNE visualization of cell relationships learned by OmniReg-GPT based on the weights of the classifier. The clusters were identified using the Louvain algorithm. The adjusted mutual information (AMI) score of 0.95 indicates a high level of similarity between the observed cell clusters and those predicted by OmniReg-GPT, demonstrating the model’s accuracy in capturing the underlying cell cluster structure. Source data are provided as a Source Data file.
We first evaluated the gene expression prediction performance in a cell-type-agnostic dataset. Specifically, cell-type agnostic gene expression level was obtained from the Xpresso human and mouse dataset25, defining the cell-type-agnostic expression level for each gene as its median value across numerous tissues and cell lines. Five DNA foundation models were fine-tuned to predict gene expression using 20 kb regulatory region around the Transcription Start Site (Fig. 3A, Methods). After finetuning, OmniReg-GPT achieved R2 score of 0.55 in predicting held-out gene expression for human (Fig. 3B). The performance of OmniReg-GPT outperformed DNABERT2(0.11 R2 score), Gena-bigbird (0.23 R2 score), Nucleotide-multispecies (0.27 R2 score), and HyenaDNA-32k (0.18 R2 score), respectively (Fig. 3B). We also analyzed Xpresso mouse dataset for further multi-species validation, where OmniReg-GPT achieved an R2 score of 0.65 for mouse, surpassing other methods (Supplementary Fig. 6). OmniReg-GPT achieved a performance nearly double that of other DNA foundation models, underscoring its efficacy in capturing the contextual sequence grammar underlying gene expression regulation.
Next, we extended OmniReg-GPT to cell-type-specific gene regulation by predicting gene expression at single-cell level. Single-cell atlas have characterized comprehensive intercellular heterogeneity, where genes are differently regulated and expressed in specific cell types. Following a binarized single cell gene expression dataset of Nvwa26, we evaluated multiple baseline methods on human cell atlas with 134,557 cells and 97 cell types through a single multitask model trained on all cells. OmniReg-GPT consistently achieved the highest average per-cell AUROC scores of 0.83 across human atlas (Fig. 3C, Supplementary Fig. 7). In contrast, the performance of other pretrained models fluctuated considerably, with DNABERT2 received average AUROC scores of 0.75, Gena-bigbird and HyenaDNA-32k failing to yield effective results (Fig. 3C). We extended this analysis to mouse and fly single cell atlases, where OmniReg-GPT similarly demonstrated superior per-cell AUROC performance across all three species (human: p < 1e-16; mouse: p < 1e-17; fly: p < 1e-12, Wilcoxon rank test; Fig. 3C). Additionally, OmniReg-GPT exhibited exceptional performance in per-gene AUROC analysis across human, mouse, and fly genomes, achieving a mean score of 0.70 for human genes and comparable excellence in other species, significantly outperforming other models (human: p < 1e-24; mouse: p < 1e-34; fly: p < 1e-7, Wilcoxon rank test; Fig. 3D). By directly comparing between predicted and observed human cell atlas, we found OmniReg-GPT captured gene expression patterns among major cell types (Supplementary Fig. 8). To further quantify the model’s predictive fidelity on cell type specificity, we projected and visualized the weights of the single cell classifiers by OmniReg-GPT using t-distributed stochastic neighbor embedding(t-SNE). The t-SNE embeddings showed that OmniReg-GPT learned distinct cell relationships from the human cell atlas, with cells from the same cell type identically grouped together (Fig. 3E). The calculated adjusted mutual information (AMI) score between re-clustering of single cell classifier weights and the biologically defined cell types had further confirmed OmniReg-GPT’s ability to accurately capture cell-type-specific gene expression patterns (Fig. 3E).
These results indicated that genomic foundational models possessed the capability to predict the gene expression from regulatory sequences. OmniReg-GPT improved both cell-type-agnostic and cell-type-specific gene expression prediction by effectively modeling context-dependent regulatory grammar of gene transcription. Notably, evaluation on the cell-type-specific gene expression patterns also showed that the regulatory sequence embedding derived from OmniReg-GPT could robustly adapt to diverse cellular conditions.
OmniReg-GPT accurately predicts single-cell chromatin accessibility and transcription factor activity
The functional regulatory genome is activated in a spatial-temporal manner, orchestrating a complex symphony of cis-regulatory element activity across diverse organs, tissues, cell types, and cellular states. Single-cell Assay for Transposase-Accessible Chromatin sequencing (scATAC-seq) provides unprecedented single-cell resolution to characterize inter-cellular heterogeneity of chromatin accessibility27,28,29 and reveals the driver sequence features, such as transcription factor binding sites, underlying cell-type specific chromatin accessibility30. Here, we adopted OmniReg-GPT to model chromatin accessibility of regulatory genome at the single cell level, and identify the underlying cell-type specific cis-regulatory elements (Fig. 4A).

A Schematic illustration of predicting single cell chromatin accessibility under peak level sequence. B OmniReg-GPT prediction performance on held-out peaks evaluated by AUROC per peak (top) and AUROC per cell (bottom) of the Buenrostro2018 dataset. C t-SNE of classifier-weight–derived cell embeddings (bottom), colored by annotated cell type. Top right: hematopoietic differentiation schematic from Buenrostro2018. Cell types: hematopoietic stem cell (HSC), multipotent progenitor (MPP), lymphoid-primed MPP (LMPP), common lymphoid progenitor (CLP), plasmacytoid dendritic cell (pDC), common myeloid progenitor (CMP), granulocyte–macrophage progenitor (GMP), megakaryocyte–erythroid progenitor (MEP), monocyte (Mono), and unknown (UNK). D Distributions of per-cell TF PWM-ISM scores for GATA1 and KLF1 for cells in HSC (n = 75), MPP (n = 76), CMP (n = 54) and MEP (n = 80) cell types. The PWM-ISM score is the dot product of the PWM and ISM measurements at sites of motif matches (GATA1 at chr11:5297906 and KLF1 at chr11:5297940). A one-sided Wilcoxon rank-sum test was performed to test for significance. *P < 0.01; Exact P values are P = 2.12 × 10−4 for MPP versus HSC, P = 1.63 × 10−6 for CMP versus MPP, and P = 4.83 × 10−67 for MEP versus CMP for GATA1; P = 1.91× 10 − 16 for MPP versus HSC, P = 2.04 × 10−31 for CMP versus MPP and P = 7.37 × 10−61 for MEP versus CMP for KLF1. Data are presented as violin plots showing kernel density estimation overlaid with box plots displaying the median (center line), upper and lower quartiles (box limits), 1.5× interquartile range (whiskers), and individual outliers (points beyond whiskers). Schematic in A was created using BioRender. (Wang, A. (https://BioRender.com/kzkgyyw).). Source data are provided as a Source Data file.
Based on a well-established and widely-benchmarked scATAC dataset of FACS-sorted hematopoietic differentiation31, OmniReg-GPT successfully predicts single-cell chromatin accessibility by leveraging the genomic foundational model and the scATAC-seq constructed epigenetic landscape (Fig. 4A). Specifically, we froze the parameters of OmniReg-GPT, and trained a classification layer for predicting single-cell peak accessibility with the input of 1344 bp DNA sequences underlying the corresponding peak (Fig. 4A). OmniReg-GPT achieved the mean AUROC score of 0.717 when evaluated across peaks for each cell (‘per cell’) (Fig. 4B). Meanwhile, the model achieved the average AUROC score of 0.717 when evaluated across cells for each peak (‘per peak’) (Fig. 4B). By comparing between predicted and observed chromatin accessibility, we observed that the predicted accessibility pattern showed substantial cell-to-cell variability, closely mirroring the experimental data (Supplementary Fig. 9). To evaluate the predicted cell-type specificity, we interpreted the classifier weight matrix as cell embeddings, where each column represents a cell-type-specific embedding vector. In the t-SNE visualization, cells with the same original cell type labels naturally clustered together (Fig. 4C), demonstrating that our learned embeddings successfully captured the inherent cell-type-specific patterns. To evaluate the quality of learned cell embeddings, we performed Louvain clustering on the classifier’s weight matrix and assessed cluster agreement with ground-truth cell-type labels using three standard metrics: Adjusted Rand Index (ARI), Adjusted Mutual Information (AMI), and Homogeneity. OmniReg-GPT achieved superior performance across all metrics compared to baseline methods including scBasset32, SnapATAC33, ArchR34 and Chromvar35 (Supplementary Fig. 10).
We further extended OmniReg-GPT to deduce TF binding activity at single-cell resolution. Leveraging the classification layer trained on the Buenrostro2018 dataset, we conducted in silico motif insertion to measure the cell-type specific activity of all human TFBSs (Methods). The anticipated activity patterns of inserted motifs revealed the same cell-type specificity as canonical TFBS. For example, OmniReg-GPT demonstrated the transcription factors CEBPB, GATA1 and HOXA9 activities in monocytes, megakaryocyte-erythroid (MEP) cells, and hematopoietic stem cells (HSC) respectively, which aligned perfectly with their canonical regulatory roles in the corresponding cell types31 (Supplementary Fig. 11).
Then, we adapted the OmniReg-GPT to infer TFBS activity in gene regulation at a per-cell, per-nucleotide resolution. We employed in silico saturation mutagenesis (ISM) on a 100-bp enhancer sequence, which is pivotal for erythroid-specific β-globin expression36,37. OmniReg-GPT could predict changes of chromatin accessibility upon nucleotide variation in each cell, and further identify the cell-type specific TFBS activity (Methods). OmniReg-GPT successfully identified GATA1 and KLF1 motifs as key active regulatory TFBS that bind to the enhancer region and regulate β-globin expression38 (Supplementary Fig. 12). Furthermore, equipped with the trained classification layer at a single cell resolution, OmniReg-GPT could correctly identify that the contribution of GATA1 and KLF1 to accessibility intensifies as cells differentiate within the erythroid lineage (Fig. 4D, and Supplementary Figs. 13,14).
Our results demonstrated the broad applications of OmniReg-GPT in analyzing the regulatory genome using scATAC-seq data, including predicting single-cell chromatin states and identifying the underlying cell-type specific regulatory programs. OmniReg-GPT represented a pioneering application of genomic sequence foundation models in single-cell chromatin accessibility data, which could facilitate the practical use of genomic sequence foundation models for single-cell analyses.
OmniReg-GPT predicts 3D chromatin organization
In the cell nucleus of eukaryotes, chromosomes are physically folded to form hierarchical three-dimensional (3D) structure at megabase scale. The 3D-organization of the genome directly mediates physical contacts between promoters and distant cis-regulatory elements, and further functionally facilitates TF regulation and gene transcription activities in contact domains39,40. High-throughput Chromatin conformation capture technologies, such as Hi-C, have comprehensively measured 3D chromatin organization of genome sequence41. To learn how megabase-scale genomic sequence influences 3D genome architecture, we leveraged the capacity of OmniReg-GPT to accommodate extensive sequences and predict chromatin organization (Fig. 5A, Methods).

A OmniReg-GPT generates embeddings of 2-megabase window, and then a specialist model is employed to predict Hi-C interaction matrix. B The training loss curve of OmniReg-GPT and C.Origami with different input features. C Experimental and OmniReg-GPT predicted Hi-C matrices of IMR-90 on training (chr2), validation (chr10) and test (chr15) chromosomes. D Insulation scores calculated from experimental (solid line) and OmniReg-GPT-predicted (dotted line) Hi-C matrices. Pearson correlation coefficients between prediction and target insulation scores are presented. E The CTCF-binding and chromatin accessibility profiles. Schematic in A was created using BioRender. (Wang, A. (https://BioRender.com/d5b184s).). Source data are provided as a Source Data file.
Trained on the same Hi-C interaction dataset of human IMR-90 cell line in C.Origami42, a specialist model predicting chromatin organization using DNA sequence and genomic features, OmniReg-GPT showed its ability to predict Hi-C contact frequency maps from sequence information alone. We compared the performance of OmniReg-GPT with two configurations of C.Origami: one utilizing solely the DNA sequence information, and the other incorporating additional data modalities, namely CTCF binding and chromatin accessibility signals. The training process showed that the rich representations provided by OmniReg-GPT embeddings were instrumental in achieving better model convergence (Fig. 5B).
To systematically evaluate the performance of OmniReg-GPT, we expanded our analysis to chromosome-wide predictions. At this broader scale, OmniReg-GPT maintained robust performance with median insulation score correlations (Pearson) of 0.52 and 0.49 on validation and test chromosomes, respectively (Supplementary Fig. 15). Furthermore, we conducted distance-stratified correlation analysis against experimental Hi-C data to evaluate how prediction accuracy varies with genomic distance (Methods). Notably, our model achieved correlations above 0.6 within 1-Mb regions on chromosome 10 (validation) and above 0.6 within 0.5-Mb regions on chromosome 15 (test), demonstrating strong predictive power at biologically relevant distances despite the increased complexity of chromosome-wide predictions (Supplementary Fig. 16).
To provide a more detailed assessment, we further analyzed the 3D-organization prediction at 2-Mb local organization scale and 8192-bp bin resolution. We found that OmniReg-GPT accurately identified topological domains and chromatin loops in the contact matrices on training (chr2), validation (chr10), and test (chr15) chromosomes (Fig. 5C). We then calculated insulation scores as the average of interaction frequencies crossing over each bin for both the experimental and predicted Hi-C matrices. The correlation of insulation scores between experimental and predicted Hi-C matrices also validated the chromatin organization similarity with pearson correlation coefficients of 0.82(p < 3e-70), 0.85(p < 3e-58), and 0.80 (p < 2e-53) for training, validation and test chromosomes respectively (Fig. 5D). Moreover, we also observed that 3D-organization could be mainly explained with CTCF and ATAC signals, consistent with the central role of CTCF in TAD-level structural organization (Fig. 5E).
Our experiments in 3D chromatin organization showed that OmniReg-GPT learned chromatin interactions at base-pair resolution and could transfer this knowledge to capture regulatory elements over megabase distances. Equipped with high computational efficiency and the ability to generalize to large genomic sequences, OmniReg-GPT extended the application of genomic sequence foundation models in 3D chromatin organization modeling at megabase scale.
OmniReg-GPT generates cell-type specific enhancers
Generating desired regulatory enhancer sequences with cell-type specific function is essential in synthetic biology43,44,45. However, the vast search space and complex cell-type specific activities of all possible DNA sequences have made it impractical to summarize the explicit design rules of native regulatory enhancer elements. Given that generative pretraining models, such as GPTs, have shown promise in zero-shot generating meaningful samples, we explore OmniReg-GPT’s generative potential in the strategic optimization of highly efficient cell-type specific enhancer sequences design.
Firstly, we verified OmniReg-GPT’s ability to accurately predict the importance of enhancers beyond native genome sequences using Self-transcribing active regulatory region sequencing (STARR-seq) (Fig. 6A). The synthetic enhancer-promoter compatibility data from Bergman et al. 46 have prioritized transcriptional activities by assessing a wide range of combinations of 1000 enhancer and 1000 promoter fragments in human K562 cells. We compared OmniReg-GPT’s zero-shot scores and Enformer’s K562 CAGE (Cap analysis gene expression sequencing) track predictions to derive the activity levels of enhancers and promoters against the STARR-seq experimental dataset47. Both models explained a substantial portion of the enhancer and promoter activity variance in their predictions (97%). Notably, OmniReg-GPT explained a greater proportion of this variance through enhancer strength (20%) compared to Enformer (6%) (Fig. 6B). Additionally, OmniReg-GPT demonstrated a higher correlation between the predicted enhancer effect and the reported enhancer strength compared to Enformer (Supplementary Fig. 17).

A Schematic illustration of using OmniReg-GPT’s likelihood as a metric of STARR-seq and measuring the activities of enhancer and promoter sequence (sequences 264 bp in length were selected and cloned in all pairwise combinations into the promoter and enhancer positions of a plasmid vector together). B Correlation of activity oracle from experiment, OmniReg-GPT prediction and Enformer prediction, with intrinsic promoter strength and the combination of promoter and enhancer strength. C OmniReg-GPT designs cell-type-specific and high activity enhancers with the help of the score model and a progressive prompt setting. D OmniReg-GPT designs synthetic sequences that achieve higher cell-type specific enhancer activity compared to natural sequences in K562 cell line (n = 2000 sequences for each source in CODA, n = 4000 for each source in Round0, n = 6000 for each source in Round1). Data are shown as violin plots (kernel density estimation) with centered box plots displaying median, quartiles, and 1.5× IQR whiskers. The ‘native’ refers to sequences derived from DHS-natural sequences through two rounds of enhancer generation. E Heatmap of alignment scores between the generated sequences in the first round(left), and second round(right) compared with the initial CODA sequence from four different sources. F Distribution of MinGap scores between generated sequences and DHS-natural sequences (n = 4000 sequences for each source). Data are presented as half-violin plots (right side) showing kernel density estimation overlaid with box plots. Box plots display the median at the center line, upper and lower quartiles as box limits, and 1.5× interquartile range as whiskers. Violin plot width represents data density at each AUROC value. Schematic in A was created using BioRender. (Wang, A. (https://BioRender.com/e49fdgh).). Source data are provided as a Source Data file.
For generating cell-type specific enhancers, we then adopted a zero-shot sequence design framework based on OmniReg-GPT (Fig. 6C). Equipped with CODA, a platform included predictive models and optimization algorithms43, we employed a progressive prompt setting and selected the top 10% of sequences for each source in two rounds of generation, with counts of 4000 and 6000 sequences respectively. Specifically, in the first round, we selected the top 2000 sequences with the highest MinGap scores from four sources provided by CODA, including DHS-natural sources and three optimization algorithms—AdaLead, FastSeqProp, and Simulated Annealing. These top sequences served as seed prompts for the subsequent generation of additional sequences. During the second round, we first scored the activity of generated sequences using the expert model from CODA, and then selected the top 10% of sequences based on MinGap scores for further analysis (Methods).
We iteratively generated and optimized sequences in silico with improved enhancer activity and diversity for the K562, HepG2, and SK-N-SH cell lines. In K562 cells (Fig. 6D), during the first round, enhancers generated by OmniReg-GPT from three optimization algorithm sources exhibited activity comparable to the most effective sequence in the CODA dataset, with an average improvement of 1.3%. In the second round, a significant concentration of activity was observed, with an average enhancement of 30.5%. Sequences derived from DHS-natural sources demonstrated even more substantial enhancement, showing increases of 23.1% in the first round and 81.7% in the second round. These findings indicated that high-activity enhancers could be designed effectively even without relying on pre-optimized sequences as initial inputs. Sequence alignment further revealed that OmniReg-GPT was capable of highly unique enhancer generation. Specifically, sequences from the first and second rounds in K562 cells showed average identities of 40.75% and 23.76%, respectively, compared to the original source sequences from the CODA dataset (Fig. 6E). For the other two cell types, final round sequences from optimization algorithm sources exhibited average activity increases of 33.8% in HepG2 and 5.4% in SK-N-SH. Sequences from DHS-natural sources showed even greater improvements, with increases of 53.4% in HepG2 and 93.3% in SK-N-SH (Supplementary Fig. 18). Across all target cell types, synthetic sequences generated from all sources consistently exhibited significantly higher MinGap values, surpassing DHS-natural sequences from the CODA platform by a substantial margin, confirming our strategy’s effectiveness across different cell types (Fig. 6F).
To assess the TF-binding potential of synthetic enhancers, we performed motif enrichment analysis using STREME48 and TOMTOM against the JASPAR CORE (2022) database49 (Methods). Synthetic enhancers retained significant TF-binding motifs, with notable enrichment of motifs not present in the original sequences. For example, in K562, 16.80% of motifs in round 1 and 10.24% in round 2 were newly identified; similar trends were observed in HepG2 and SK-N-SH, suggesting that synthetic enhancers can reveal previously uncharacterized TF-binding potential (Supplementary Fig. 19). To identify functional motifs informed by predicted activity contributions, we performed in silico mutagenesis (ISM), using OmniReg-GPT to calculate nucleotide contribution scores based on zero-shot embedding distances. Positive motif patterns were extracted using TF-MoDISco Lite50 and matched to known human TF-binding motifs. In total, we discovered 60, 51, and 62 TF motifs in K562, HepG2, and SK-N-SH cells, respectively. Notably, well-known activators such as GATA—an essential TF predicted by OmniReg-GPT to drive activity exclusively in K562 cells—were identified in K562, whereas HNF1B and HNF4A, master regulators of hepatocyte development, showed positive contribution scores exclusively in HepG2 (Supplementary Tables 4, 5, 6). In SK-N-SH cells, we also detected ASCL1, a key transcription factor in neuroblastoma.
Discussion
In this study, we developed a generative pretraining approach, OmniReg-GPT, with high computational efficiency and robust adaptive performance for multi-scale genome understanding and generation using a hybrid attention mechanism. This architecture supports up to 200 kb of genomic sequence pretraining on a standard 32GB memory GPU, enabling the model to learn multi-layer gene regulation across long-range genomic scales. Our hybrid attention architecture employs local attention that restricts each token to attend only to a fixed-size context window during pretraining, maintaining consistent attention patterns when scaling to longer sequences. This is complemented by strategic global attention layers that efficiently aggregate local features to capture long-range dependencies. Additionally, our implementation of Rotary Position Embedding (RoPE) encodes positional information through sinusoidal functions at multiple frequencies, providing an implicit hierarchical positional encoding that effectively distinguishes between short-range and long-range relationships. This architectural design enables OmniReg-GPT to generalize effectively to longer genomic contexts, a capability not achieved by alternative approaches such as state space models, sparse attention, or position extrapolation techniques. Importantly, this research lays the groundwork for a resource-efficient and high-performance training framework that can serve as a valuable foundation model for future DNA sequence models, promising to accelerate advancements in genomic research by optimizing both computational efficiency and predictive accuracy.
OmniReg-GPT’s rich latent representation provides enhanced transfer capabilities for diverse regulatory genome understanding and predictive tasks across varying sequence lengths. OmniReg-GPT demonstrates strong performance when benchmarked against advanced genomic foundational models on standard and real-world datasets, showcasing its effectiveness in various regulatory genomic sequence understanding tasks. Importantly, we extended genomic foundation models in decoding the intricate DNA sequence syntax for spatio-temporal and long-range regulation. To our knowledge, OmniReg-GPT was the first sequence foundation model applied in spatio-temporal regulation at single-cell resolution. For the gene expression regulation, OmniReg-GPT improves both cell-type-agnostic and cell-type-specific gene expression prediction by effectively modeling context-dependent regulatory grammar of gene transcription. Using single-cell chromatin accessibility data, we demonstrated the broad applications of OmniReg-GPT in analyzing the regulatory genome, including predicting single-cell chromatin states and identifying cell-type specific regulatory programs. As for the long-range regulation, our experiments in 3D chromatin organization showed that OmniReg-GPT learned chromatin interactions at kilobase-scale resolution. Across these diverse downstream genomic tasks, OmniReg-GPT markedly outperforms existing pretraining techniques. Finally, our pilot applications of OmniReg-GPT model on sequence generation, demonstrated its ability to precisely pinpoint enhancer activity and diversity in an expansive search space. Additionally, the results highlight the model’s robust capability to efficiently design cell-type specific sequences, underscoring the powerful generative potential of genome foundation models.
While the advent of OmniReg-GPT marks considerable progress in regulatory genomic analysis, there remain several opportunities for further refinement and expansion. First, our framework optimizes pretraining on the human reference genome solely, streamlining computational resources and reducing experimental costs. While our current hyperparameter choices and architecture design have demonstrated robust performance, future work could leverage scaling laws to systematically optimize the hyperparameters such as learning rate, batch size and the ratio of local and global attention layers as model scales and diversity of training data expand, particularly when incorporating cross-species genomes. Second, despite robust generalizability across gene regulation tasks, our exploration of regulatory syntax is still constrained by a focus on the single modality and limited cell types. With the advent of new assays and sequencing technologies, we now have the potential to incorporate additional data modalities, such as protein-based data, into our analyses. Besides, we could also leverage data from rapidly expanding cell atlases to enhance our foundation model. This integration across different modalities and cell types would enable a more comprehensive analysis of sequence regulatory activities51. Third, we showed the pilot applications of OmniReg-GPT model on functional sequence generation with the improvement of CODA MinGap activities. Further experimental validation is needed to prove the generative abilities. Finally, while our model’s architecture reduces the computational cost of training on long sequences, we have not performed a post-training stage for context window extension. Consequently, the full potential of its RoPE positional embeddings for extrapolation to sequences far beyond the 20 kb pre-training length has not yet been explored.
Overall, OmniReg-GPT provides a valuable pretraining framework for genomics, paving the way for the next generation of foundation models trained on multi-scale sequence data. We demonstrate its applications in pinpointing critical regulatory components. Meanwhile, OmniReg-GPT stands out as the first genomic-sequence foundation model to be applied across several domains, including single-cell omics, spatial genomic interactions and enhancer generation. These advanced predictive abilities, coupled with its potential for hypothesis generation, offer substantial support for experimental design and hold the promise of accelerating scientific discovery.
Methods
OmniReg-GPT architecture
OmniReg-GPT maintains a decoder-only transformer architecture. First, we employed a BPE (Byte-Pair Encoding) tokenizer inherited from Gena, which iteratively merges the most frequent pairs of characters or character sequences to create a vocabulary of subword units, enabling efficient tokenization of genome sequences. This tokenizer segments genome sequences into meaningful tokens. Subsequently, an embedding layer maps these discrete tokens into continuous vector representations in a high-dimensional space. The token embeddings are then processed through a transformer layer stack that comprises 12 blocks with local attention and 2 blocks with global attention, with a model width of 1024 dimensions. Each block contains a multi-head self-attention module for token mixing and a feed-forward network consisting of two-layer perceptron for channel mixing. To understand the role of global attention, we consider the self-attention mechanism applied at each layer of the OmniReg-GPT. Let hl denote the sequence representation at layer l. The vanilla attention can be expressed through the following operations:
where Wq, Wk, and Wv are the weight matrices for the query, key and value, respectively. The standard attention function itself is computed as follows:
where dk represents the dimensionality of the key vectors.
The computational complexity of global attention can be broken into three main components:
-
1.
Computing \(Q{K}^{T}\)(Forward Pass): For a batch size of bs, sequence length L, and hidden dimension d, the matrix multiplication and addition requires \(2\times {bs}\times L\times L\times d\) FLOPs.
-
2.
Computing Attention Output (Forward Pass): The multiplication of attention weights with values requires another \(2\times {bs}\times L\times L\times d\).
-
3.
Backward Pass: The backward pass requires computing gradients through both matrix multiplications, which typically costs twice the FLOPs of the forward pass, resulting in \(8\times {bs}\times L\times L\times d\) FLOPs.
Therefore, the total computational complexity for both forward and backward passes is O(bsL2d), with the exact FLOPs being 12bsL2d. This quadratic dependency on sequence length L becomes a significant computational bottleneck for long sequences.
In the local sliding window attention mechanism, each query vector only attends to a subset of key vectors with a specified window size. This approach significantly reduces the computational complexity compared to the standard global attention mechanism, where each query attends to all key vectors across the entire sequence.
Formally, the attention mechanism for a query at position I in the sequence involves the following operations:
where Wq, Wk, and Wv are the weight matrices for the query, key and value, respectively. Each query Q interacts with keys Kj and values Vj within its local window, reducing the number of comparisons from the entire sequence length L to the window size w.
The attention scores are the computed as:
This localized attention computation effectively constrains the operation to a much smaller subset of the input. Specifically, by partitioning the input sequences of Length L into non overlapping windows with size w, the effective sequence length for attention computation is reduced from L to w. Meanwhile, the batch dimension is transformed from bs to bs*L/w, where each window is processed independently.
Thereby reducing computational complexity:
where L/w represents the number of windows across the sequence, and each window involves w2 × d operations. This formulation provides an efficient way to compute attention with reduced computational burden. Moreover, as the number of layers increases in the OmniReg-GPT model, the receptive field for each query increases to cover a progressively wider context without a corresponding increase in computational complexity per layer. This expansion allows each query to integrate information from distant areas.
Within the global blocks of the OmniReg-GPT model, we employ the classic attention mechanism, which considers the entire sequence to calculate attention scores. This global approach ensures comprehensive context integration. To enhance computational efficiency across all attention functions in our model, we utilize FlashAttention19, an optimized algorithm designed to accelerate the calculation of attention mechanisms. In the final layer of our model, we compute a single vector representation by taking the mean of all token embeddings, which is then used for downstream applications.
Training procedure
For pretraining, we utilized the Human T2T v2 genome assembly, which was downloaded from the NCBI database (Accession number GCF_009914755.1). Following the methodology established by Gena-LM16, we segmented the sequence into ‘sentences’, each comprising 20,000 base pairs (bp). We then processed these sentences using Gena’s tokenizer, which is configured with a dictionary size of 32,000. In total, this process generated approximately 5 billion tokens as training corpus. We employed an autoregressive form of a language model as the loss function, aiming to predict each subsequent token based on the previous tokens:
For the training process, we used the AdamW optimizer with a weight decay setting of 0.001 and a batch size of 32. The training was carried out over 3 epochs, incorporating 5000 warmup steps for linear scaling of the learning rate, which was set to a maximum of 0.0001. In total, the model was trained on four NVIDIA GeForce RTX 3090 GPUs, totaling 60 h of training time.
Evaluation expriments
Our evaluation explicitly compared OmniReg-GPT with existing models trained with different window sizes to demonstrate the advantages of our large-window approach. We list the baseline model configurations: DNABERT-2: 112 M parameters, trained on 512 bp; GENA: 330 M parameters, trained on 1 kb; GENA-BigBird: 108 M parameters, trained on 1 kb; Nucleotide-Human: 500 M parameters, trained on 6 kb; Nucleotide-V2-Multispecies: 500 M parameters, trained on 6 kb; HyenaDNA−1kb: 436 K parameters, trained on 1 kb; HyenaDNA-32kb: 3.3 M parameters, trained on 32 kb. For comparing performance on tasks requiring long sequences, we focused on models whose architectures support extrapolation beyond their training sequence length. Specifically, we evaluated: DNABERT2 (utilizing Alibi position embedding), Gena-bigbird (employing sparse attention), Nucleotide-V2-Multispecies (leveraging relative position embeddings like RoPE), and HyenaDNA-32k (trained for long contexts). The evaluation dataset splits, task types, loss functions and baseline model for each task are summarized in Supplementary Table 7.
benchmark of genomic foundation models
To evaluate the performance of the OmniReg-GPT model in predicting short-sequence genetic elements and epigenomic data, we compared it against other foundation models using a variety of benchmarks. We utilized the Nucleotide Transformer benchmark17, the Bend benchmark22, and the chromatin-profiling dataset from DeepSEA23.
Firstly, we downloaded consistent datasets from the HuggingFace for the revised Nucleotide Transformer benchmark, which comprises ENCODE Histone ChIP-seq data for 10 histone marks in the K562 human cell line (1000 bp), human enhancer elements from ENCODE’s SCREEN database (400 bp), and promoter sequences (300 bp) from the Eukaryotic Promoter Database. We used the Matthews Correlation Coefficient (MCC) as the metric of comparison. The baseline models included DNABERT-215 with 112 million parameters, HyenaDNA-1kb21 with 436 thousand parameters, Hyena-32kb with3.3 M parameters, Nucleotide Transformer HumanRef17 with 500 million parameters, Nucleotide Transformer Human 1000 G17with 500 million and 2.5 billion parameters and Nucleotide Transformer Multispecies v217 with 500 million parameters. We trained and tested OmniReg-GPT on the dataset, whereas for the remaining models, we utilized the MCC values already provided in the Nucleotide Transformer17. To systematically evaluate the impact of sequence length on model performance, we extended the sequences for the Promoter_No_TATA and Enhancer binary classification tasks to three different window sizes (1 kb, 2 kb, and 4 kb). We employed a linear probe training approach with consistent hyperparameters (learning rate = 5e-3, batch size = 32, 5 epochs) across all experiments to compare performance across the extended window sizes. Additionally, we evaluated the classification ability for distal enhancers by combining distal enhancer-positive samples from the enhancer dataset with all negative samples and calculating F1, recall, and precision metrics.
We further extended our evaluation by incorporating the histone modification and CpG methylation datasets from the Bend benchmark22, both of which contained coordinates mapped to the human genome. For histone modification dataset, each sequence was 512 bp and encompasses 18 labels. The test set comprised chromosomes 1, 8, and 9 (120,567 samples), the validation set included chromosomes 2 and 4 (70,801 samples), and the remaining chromosomes were used for training (420,713 samples). Similarly, the CpG methylation dataset also featured sequences of 512 bp, but with a total of 7 labels. The test set included chromosomes 4, 13, 19, and 21 (106,227 samples), the validation set included chromosomes 5, 9, and 22 (109,717 samples), and the remaining chromosomes were used for training (743,095 samples). We compared OmniReg-GPT with DNABERT-215, Gena16, and Nucleotide Transformer Multispecies v217 mentioned above. During the training process, we uniformly froze the parameters of the foundation models and trained only a single MLP layer for classification. We employed cross-entropy loss to optimize both classification tasks.
Regarding the DeepSEA dataset23, we downloaded the original DeepSEA dataset and converted it into nucleotide sequences. Each sequence is 1000 bp and the dataset includes a comprehensive set of labels: 690 transcription factor (TF) binding profiles for 160 different TFs, 125 DNase I hypersensitive sites (DHS) profiles, and 104 histone-mark profiles. Chromosomes 8 and 9 were excluded from training to serve as the test set for chromatin feature prediction. We then fine-tuned the OmniReg-GPT and DNABERT-215 models on this dataset by Cross-entropy loss and calculated the AUROC (Area Under the Receiver Operating Characteristic curve) for each label.
For the regulatory variants benchmark, we obtained the eQTLs and pathogenic ClinVar datasets with sequence length of 10 kb. Variants from the fine-mapped eQTL set were assigned positive labels if their posterior inclusion probability (PIP), as determined by SuSiE52, exceeded 0.9. Conversely, variants from the matched negative set were labeled as negative if their PIP was below 0.01. To explore the impact of sequence context, we constructed two additional datasets by extracting sequences of 2k and 6k base pairs centered on each variant. Chromosomes 9 and 10 were excluded from the training set and reserved for testing purposes.
In the pathogenic variant task, the positive class comprised pathogenic SNPs identified in ClinVar53, while the negative class consisted of common SNPs with a minor allele frequency (MAF) > 5% from gnomAD54.
For the NT and DeepSEA benchmark, we perform full parameters tuning and uniformly set the learning rate to 2e-5 and the batch size to 16. Additionally, we employed a learning rate scheduling strategy that included a warm-up phase of 1 epoch with cosine decay.
For Bend benchmark and variants benchmark, we kept the backbone frozen and trained only a simple linear MLP layer on top. The training protocol was standardized across all experiments, with the following hyperparameters: a learning rate of 5e-3 and a batch size of 32 for both binary classification task. Additionally, we employed a learning rate scheduling strategy that included a warm-up phase of 1 epoch with cosine decay.
Gene expression prediction of cell-type agnostic and single-cell RNA
For the cell-type-agnostic mRNA level regression, we downloaded protein-coding mRNA sequences along with their corresponding median expression values for human and mouse from Xpresso. The human median expression values are derived from 56 tissues and cell lines from Roadmap Epigenomics Consortium, while the mouse values are derived from 254 curated RNA-seq datasets from the ENCODE project. The median expression level was calculated across all cell types for each gene. We applied a log transformation to address the right-skewed distribution of the expression data. Specifically, we used the formula \(\hat{y}={\log }_{10}(y+0.1)\,\), where y represents the expression value. Unlike the original setup, which used the optimized region from 7 kb upstream to 3.5 kb downstream of the transcription start site (TSS), we opted to retain complete sequences with a length of 20 kb to preserve the full genomic context, enhancing the potential for capturing regulatory elements influencing gene expression. We adopted the pre-established train-test split from the Xpresso dataset, maintaining a 9:1 ratio. All models were configured with fixed pre-training parameters to ensure a consistent basis for performance comparison. We augmented each model with a single regression layer to predict gene expression level, where the input to the regression layer is the mean-pooled embedding of the token representations. This approach allowed us to directly assess the impact of the underlying architecture of each model on the gene expression prediction tasks. The performance of each model was quantified using the coefficient of determinations (R2) as the metric.
For the analysis of single-cell gene expression, we downloaded genome sequences, including a window spanning 6.5 kb upstream to 6.5 kb downstream of the TSS, along with expression labels for human, mouse, and Drosophila from the referenced sources. For human and mouse, all genes on chromosome 8 were held out for testing, while genes on other chromosomes were used for training and validation. For Drosophila dataset, we randomly split left-out 1000 genes for testing, 1000 genes for validation and the remaining genes for training. We applied the same models and training methodologies used in the cell-type agnostic RNA prediction. We treated the scRNA-seq prediction task as a multi-label classification problem. As in Nvwa, the expression label was binarized into labels (expressed=1, unexpressed=0) using species-specific cutoffs based on the typical number of expressed protein-coding genes per cell. We evaluated the prediction performance using the AUROC scores. To visualize the learned representations, we projected the trainable classifier weights into a t-SNE embedding. These weights correspond to the classification head parameters that map from the 1024-dimensional sequence representation space to single cell predictions, where each cell is characterized by a 1024-dimensional weight vector. To quantitatively assess the clustering similarity of the predicted cell atlas with the original cell atlas, we used Adjusted Mutual Information (AMI).
For both tasks, we kept the backbone frozen and trained only a simple linear MLP layer mapping hidden dimension to multi-label shape. We adopted a learning rate of 5e-3 and a batch size of 32 for all models, utilizing a cosine decay learning rate scheduler with a 1-epoch warm-up phase. The Mean Squared Error (MSE) Loss was used to train cell-type agnostic mRNA level regression task and the Cross Entropy Loss was used to train single-cell RNA classification task.
scATAC-seq prediction
We downloaded the data for Buenrostro 2018 from scBasset32 and adopted their data processing method to generate sequences and labels for fine-tuning OmniReg-GPT. The data was randomly split into training and test sets in a 9:1 ratio. For each peak, we predicted the accessibility for each cell type using Cross-Entropy Loss to optimize the classification task. We trained a single MLP classification layer using a learning rate of 5e-3 and a batch size of 32, utilizing a cosine decay learning rate scheduler with a 1-epoch warm-up phase. After training, we utilized the classifier weights that map from the 1024-dimensional sequence representation space to single cell predictions as cell embeddings and constructed a k-nearest neighbor (kNN) graph for clustering, which identified 10 distinct clusters for the Buenrostro 2018 dataset. For visualization, t-SNE embeddings were independently computed from the classifier weights. we compared the clustering outcome to the ground-truth cell type labels using ARI, AMI and homogeneity as implemented in sklearn.metrics. For evaluation of cell embeddings, we compared OmniReg-GPT to scBasset32, snapATAC33, ArchR34, chromVAR35 with motifs or k-mer features. To estimate transcription factor (TF) activity at single-cell resolution, we applied motif-insertion perturbation with Omnireg-GPT. Using the motif catalog and background sequences adopted by scBasset, we drew an instance from each TF’s PWm and embedded it at the midpoint of every background sequence. OmniReg-GPT produced cell-wise accessibility predictions for both motif-inserted and background sequences. Motif influence was defined as the change in predicted accessibility between the motif-inserted and background sequences. For each cell, we averaged this effect across the 1000 backgrounds to obtain a raw estimate of TF activity. The per-cell TF scores were subsequently standardized (z-scored) to yield the final activity values.
We implemented In Silico Mutagenesis (ISM) to evaluate the impact of nucleotide substitutions within specific sequences using the OmniReg-GPT model. For each nucleotide position, we generated three variants by replacing the reference nucleotide with each possible alternative. We then calculated the cell-specific changes in accessibility by comparing predictions for each mutated sequence against the reference sequence. To quantify these effects, we normalized the importance scores for the quartet of nucleotides at each position, ensuring their cumulative sum was zero. The importance of the original nucleotide was then determined by its normalized ISM score. In the β-globin enhancer ISM analysis, for each motif, we assigned a score of the dot product between the PWM and ISM.
3D chromatin contact prediction
We downloaded the processed Hi-C matrix (IMR90), DNA reference sequences (GRCh38), CTCF ChIP-seq (IMR-90) and ATAC-seq (IMR-90) from C.origami42. For training data generation, we utilized 2-Mb sliding windows with a step size of 40 kb across the genome, ensuring comprehensive coverage for model training. For genome-wide predictions, we maintained the same 2-Mb window size but implemented a step size of 262,144 bp (equivalent to 1/8 of the 2-Mb window) to enhance resolution and computational efficiency. The output target was the Hi-C matrix at the corresponding region. The Hi-C matrix was originally called at 10-kb resolution and downscaled to 8192-bp to match the model output resolution. Chromosomes 10 and 15 were used as the validation set and the test set, respectively. The rest of the chromosomes were used as the training set. To handle the 2 Mb input regions, we directly fed the entire 2 Mb sequences into the pretrained OmniReg-GPT to generate their corresponding embeddings. Following a similar model architecture as C.Origami, our setup included a 1D convolutional encoder, a transformer module, and a task-specific 2D convolutional decoder. We made a slight modification to the encoder’s convolutional module by adjusting the hidden size to ensure that the 1280-dimensional sequence representation was effectively reduced. The output of the decoder was characterized by a matrix of 256 by 256 channels. We employed Mean Squared Error (MSE) to quantify the discrepancies between the predicted output and the experimental Hi-C map. In alignment with C.Origami, we utilized the original study’s hyperparameters and codebase for training two distinct models: one that incorporated both sequence and genomic features as inputs, and another that relied solely on the sequence data. Stratified intensity at distance i was calculated by aggregating the line that is parallel to the Hi-C diagonal with offset of i. Stratified correlation was calculated as Pearson’s r between the shifted diagonal line of prediction and ground truth.
Enhancer and promoter compatibility prediction
We collected the data and plasmid sequences in the referenced study47. We positioned the promoter and enhancer fragments into their respective locations in the plasmid47. Subsequently, we conducted forward passes with the processed plasmid sequences using OmniReg-GPT and Enformer. For OmniReg-GPT, the log-likelihood obtained from these passes was used as a zero-shot score, while for Enformer, we utilized its CAGE predictions. Both outputs served as oracles for STARR-seq. To impute the intrinsic strengths of the promoter and enhancer sequences, we employed a Gamma model:
where P is the intrinsic promoter activity and E is the intrinsic enhancer activity.
This approach allowed us to derive the predicted strengths of promoters and enhancers independently and in combination.
We then compared these predictions to the experimentally derived STARR-seq activity levels, which were modeled using a Poisson regression model:
Here, RNA represents the measured RNA count, DNA is the plasmid input, and P and E are the promoter and enhancer indicators, respectively. By fitting both the Gamma and Poisson models, we were able to directly compare the predicted enhancer and promoter strengths from OmniReg-GPT and Enformer to the experimentally validated STARR-seq data. Finally, we performed a linear correlation analysis between the predicted enhancer and promoter strengths from OmniReg-GPT and Enformer and the experimentally derived STARR-seq activity levels.
Enhancer generation
We obtained the library of natural and synthetic sequences tested by experiments from the Supplemental materials of the reference study43. For three different cell types, we selected the top 2000 sequences from three algorithm-generated sequence sources and DHS-natural sequence source. The selection was based on the MinGap score, which was defined as the activity prediction in the target cell type minus the maximum off-target cell type activity prediction. The CODA score model was used to predict the activity of 200 bp generated sequences across three cell types (K562, HepG2, and SK-N-SH), representing the strength of the enhancers in each cell type. For each cell type, we employed a two-round sequence generation strategy using OmniReg-GPT. In the first round, we used the first 50% of each sequence length as the prompt for the generation process. For each prompt, we generated 20 new sequences. At each step, the model computed logits for the next potential token, applied a top-k filtering to focus on the most probable outcomes and then sampled the next token using Gumbel sampling with a specified temperature parameter to add controlled randomness. The entire process was performed without updating the model weights, ensuring that it was used purely for inference. In the first round, we obtained a total of 40,000 generated sequences from each of the four sources. We then employed the score model in CODA to perform in silico activity prediction and selected the top 10% of sequences. In the second round, we randomly sampled 3000 sequences from each source’s generated pool, using 75% of their length as prompts for further generation, and subsequently selected the top 10% of the resulting sequences. During the two-round generation process, we used the PairwiseAligner in local mode from the BioPython package to align the sequences generated from each source with the top sequences from CODA. To assess transcription factor (TF) motif enrichment, we analyzed original sequences (n = 2000 per cell line) and sequences generated over two rounds (n = 4000 per round per cell line for K562, HepG2, SK-N-SH). Motifs were identified using STREME48 and annotated against the JASPAR CORE (2022)49 database using TOMTOM (default parameters, retaining motifs with E-value < 1). To further investigate the functional importance of individual nucleotides and identify cell-type-specific TFs, we performed in silico mutagenesis (ISM) on all sequences. Using a zero-shot methodology, we generated embeddings for single-point mutated 200 bp sequences and compared them to the original sequence embeddings to calculate normalized contribution scores for each nucleotide position. Functional sequence motifs were extracted using TF-MoDISco Lite50 (10,000 seqlets, 200 bp window) and matched to known human TF-binding motifs in the JASPAR CORE (2022) database via TOMTOM.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data used in this study are publicly available and the usages are illustrated in the Methods. The dataset used for downstream tasks can be downloaded from the following links: Nucleotide Transformer benchmark (https://github.com/instadeepai/nucleotide-transformer); Bend benchmark (https://github.com/frederikkemarin/BEND); DeepSEA dataset (http://deepsea.princeton.edu/media/code/deepsea_train_bundle.v0.9.tar.gz); Xpresso dataset (https://github.com/vagarwal87/Xpresso); Nvwa dataset (https://bis.zju.edu.cn/nvwa/dataset.html); Buenrostro2018 dataset (https://github.com/pinellolab/scATAC-benchmarking/blob/master/Real_Data/Buenrostro_2018/input/combined.sorted.merged.bed); C.Origami dataset (https://github.com/tanjimin/C.Origami); Promoter and enhancer compatability dataset (https://zenodo.org/records/7076228); CODA library from the supplementary materials of its paper (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10441439/#) Source data are provided with this paper.
Code availability
The code used to pretrain and fine-gune the OmniReg-GPT model is publicly available and has been deposited in OmniReg-GPT at github (https://github.com/wawpaopao/OmniReg-GPT) and at the Zenodo (https://doi.org/10.5281/zenodo.16933275)55.
References
Kim, S. & Wysocka, J. Deciphering the multi-scale, quantitative cis-regulatory code. Mol. cell 83, 373–392 (2023).
Zeitlinger, J. Seven myths of how transcription factors read the cis-regulatory code. Curr. Opin. Syst. Biol. 23, 22–31 (2020).
Preissl, S., Gaulton, K. J. & Ren, B. Characterizing cis-regulatory elements using single-cell epigenomics. Nat. Rev. Genet. 24, 21–43 (2023).
Moore, J. E. et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020).
Meuleman, W. et al. Index and biological spectrum of human DNase I hypersensitive sites. Nature 584, 244–251 (2020).
Roadmap Epigenomics Consortium. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Eraslan, G., Avsec, Ž., Gagneur, J. & Theis, F. J. Deep learning: new computational modelling techniques for genomics. Nat. Rev. Genet 20, 389–403 (2019).
Novakovsky, G., Dexter, N., Libbrecht, M. W., Wasserman, W. W. & Mostafavi, S. Obtaining genetics insights from deep learning via explainable artificial intelligence. Nat. Rev. Genet. 24, 125–137 (2023).
Vaishnav, E. D. et al. The evolution, evolvability and engineering of gene regulatory DNA. Nature 603, 455–463 (2022).
de Almeida, B. P., Reiter, F., Pagani, M. & Stark, A. DeepSTARR predicts enhancer activity from DNA sequence and enables the de novo design of synthetic enhancers. Nat. Genet. 54, 613–624 (2022).
Chen, K. M., Wong, A. K., Troyanskaya, O. G. & Zhou, J. A sequence-based global map of regulatory activity for deciphering human genetics. Nat. Genet. 54, 940–949 (2022).
Brown, T. et al. Language models are few-shot learners. Adv. neural Inf. Process. Syst. 33, 1877–1901 (2020).
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. BERT: pre-training of deep bidirectional transformers for language understanding. In Proc. 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies (eds Burstein, J. et al.) 4171–4186 (Association for Computational Linguistics, 2018).
Luo, X. et al. A Closer Look At Few-shot Classification Again. In International Conference on Machine Learning 23103–23123 (PMLR, 2023).
Zhou, Z., Ji, Y., Li, W., Dutta, P., Davuluri, R. V. & Liu, H. DNABERT-2: efficient foundation model and benchmark for multi-species genomes. In International Conference on Learning Representations (ICLR, 2024).
Fishman, V. et al. GENA-LM: a family of open-source foundational DNA language models for long sequences. Nucleic Acids Res. 53, gkae1310 (2025).
Dalla-Torre, H. et al. Nucleotide Transformer: building and evaluating robust foundation models for human genomics. Nat. Methods. 22, 287–297 (2025).
Gankin, D. et al. Species-aware DNA language modeling. bioRxiv https://doi.org/10.1101/2023.01.26.525670 (2023).
Dao, T., Fu, D., Ermon, S., Rudra, A. & Ré, C. Flashattention: fast and memory-efficient exact attention with IO-awareness. Adv. Neural Inf. Process. Syst. 35, 16344–16359 (2022).
Su, J. et al. Roformer: enhanced transformer with rotary position embedding. Neurocomputing 568, 127063 (2024).
Nguyen, E. et al. HyenaDNA: long-range genomic sequence modeling at single nucleotide resolution. In Proc. 37th International Conference on Neural Information Processing Systems. (eds Oh, A. et al.) 43177–43201 (Curran Associates, Inc., 2023).
Marin, F. I. et al. BEND: benchmarking DNA Language Models on biologically meaningful tasks. Preprint at https://doi.org/10.48550/arXiv.2311.12570 (2024).
Zhou, J. & Troyanskaya, O. G. Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods 12, 931–934 (2015).
Kellis, M. et al. Defining functional DNA elements in the human genome. Proc. Natl Acad. Sci. 111, 6131–6138 (2014).
Agarwal, V. & Shendure, J. Predicting mRNA abundance directly from genomic sequence using deep convolutional neural networks. Cell Rep. 31, 107663 (2020).
Li, J. et al. Deep learning of cross-species single-cell landscapes identifies conserved regulatory programs underlying cell types. Nat. Genet 54, 1711–1720 (2022).
Buenrostro, J. D. et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015).
Yao, Z. et al. A transcriptomic and epigenomic cell atlas of the mouse primary motor cortex. Nature 598, 103–110 (2021).
Klemm, S. L., Shipony, Z. & Greenleaf, W. J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 20, 207–220 (2019).
Thurman, R. E. et al. The accessible chromatin landscape of the human genome. Nature 489, 75–82 (2012).
Buenrostro, J. D. et al. Integrated single-cell analysis maps the continuous regulatory landscape of human hematopoietic differentiation. Cell 173, 1535–1548.e16 (2018).
Yuan, H. & Kelley, D. R. scBasset: sequence-based modeling of single-cell ATAC-seq using convolutional neural networks. Nat. Methods 19, 1088–1096 (2022).
Fang, R. et al. Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nat. Commun. 12, 1337 (2021).
Granja, J. M. et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411 (2021).
Schep, A. N., Wu, B., Buenrostro, J. D. & Greenleaf, W. J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017).
Pierce, S. E., Granja, J. M. & Greenleaf, W. J. High-throughput single-cell chromatin accessibility CRISPR screens enable unbiased identification of regulatory networks in cancer. Nat. Commun. 12, 2969 (2021).
Tuan, D., Solomon, W., Li, Q. & London, I. M. The ‘beta-like-globin’ gene domain in human erythroid cells. Proc. Natl Acad. Sci. 82, 6384–6388 (1985).
Tallack, M. R. et al. A global role for KLF1 in erythropoiesis revealed by ChIP-seq in primary erythroid cells. Genome Res 20, 1052–1063 (2010).
Hafner, A. & Boettiger, A. The spatial organization of transcriptional control. Nat. Rev. Genet 24, 53–68 (2023).
Rowley, M. J. & Corces, V. G. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 19, 789–800 (2018).
Jerkovic, I. & Cavalli, G. Understanding 3D genome organization by multidisciplinary methods. Nat. Rev. Mol. Cell Biol. 22, 511–528 (2021).
Tan, J. et al. Cell-type-specific prediction of 3D chromatin organization enables high-throughput in silico genetic screening. Nat. Biotechnol. 41, 1140–1150 (2023).
Gosai, S. J. et al. Machine-guided design of cell-type-targeting cis-regulatory elements. Nature 634, 1211–1220 (2024).
de Boer, C. G. & Taipale, J. Hold out the genome: a roadmap to solving the cis-regulatory code. Nature 625, 41–50 (2024).
de Almeida, B. P. et al. Targeted design of synthetic enhancers for selected tissues in the Drosophila embryo. Nature 626, 207–211 (2024).
Bergman, D. T. et al. Compatibility rules of human enhancer and promoter sequences. Nature 607, 176–184 (2022).
Karollus, A., Mauermeier, T. & Gagneur, J. Current sequence-based models capture gene expression determinants in promoters but mostly ignore distal enhancers. Genome Biol. 24, 56 (2023).
Bailey, T. L., Johnson, J., Grant, C. E. & Noble, W. S. The MEME Suite. Nucleic Acids Res. 43, W39–W49 (2015).
Castro-Mondragon, J. A. et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 50, D165–D173 (2022).
Shrikumar, A. et al. Technical note on transcription factor motif discovery from importance scores (TF-MoDISco) version 0.5.6.5. Preprint at arxiv.org/abs/1811.00416 (2018).
Gschwind, A. R. et al. An encyclopedia of enhancer-gene regulatory interactions in the human genome. bioRxiv https://doi.org/10.1101/2023.11.09.563812 (2023).
Wang, G., Sarkar, A., Carbonetto, P. & Stephens, M. A simple new approach to variable selection in regression, with application to genetic fine-mapping. J. R. Stat. Soc. B 82, 1273–1300 (2020).
Landrum, M. J. et al. Clinvar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067 (2018).
Chen, S. et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 625, 92–100 (2024).
Wang, A. et al. OmniReg-GPT: a high-efficiency foundation model for comprehensive genomic sequence understanding. Zenodo https://doi.org/10.5281/zenodo.16933275 (2025).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. U24A201401 (J.Z.), No. 32500539 (J.L.)).
Author information
Authors and Affiliations
Contributions
J.Z. and J.L. conceived and designed the research study. A.W. developed the method and trained the OmniReg-GPT model. A.W., J.L. and H.D. conducted benchmarking experiments and model performance evaluation. A.W., J.L., H.D., B.X., Q.Y., Y.X., and J.F. performed the analysis. A.W., J.L., H.D., B.X. and J.Z. wrote and revised the manuscript. J.Z. supervised the project. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Chong Wu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, A., Li, J., Dong, H. et al. Omnireg-gpt: a high-efficiency foundation model for comprehensive genomic sequence understanding. Nat Commun 16, 10139 (2025). https://doi.org/10.1038/s41467-025-65066-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65066-7
