
Abstract
Eco-friendly electrocatalytic production of alkaline H2O2 via the two-electron oxygen reduction reaction offers a promising alternative to the conventional anthraquinone process, which is energy-intensive and poses explosion risks. However, the industrial adoption of this approach is hindered by sluggish proton-feeding kinetics of heterogeneous electrocatalysts, which restricts the achievable current densities. Herein, we develop Ni-based metal-organic frameworks with a bio-inspired B-coordination (B-NiBDC). The B-NiBDC shows Faradaic efficiency of ~90% for H2O2 electrosynthesis at 1000 mA cm−2, obtaining ~7.49 wt.% H2O2 alkaline solution for bleaching under urea oxidation-assisted energy-saving system. This has led to a significant increase in pulp brightness by 25-39 % (International Organization for Standardization, ISO). We propose that B-O-Ni motifs of B-NiBDC induce the formation of electron-deficient Ni active sites. Experiments and theoretical calculations elucidate that the electron-deficient Ni has accelerated water dissociation kinetics for proton-feeding to produce *OOH intermediate with suitable adsorption energy, boosting overall H2O2 electrosynthesis activity.
Similar content being viewed by others

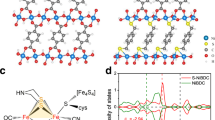

Introduction
Hydrogen peroxide (H2O2), an essential versatile green oxidant, has been used in a wide range of industrial processes with increasing demand, including wastewater treatment, semiconductor cleaning, paper/pulp bleaching, chemical synthesis, pharmaceutical disinfection, to name a few1,2,3. H2O2 is utilized extensively in the alkaline bleaching industry, accounting for nearly half of its total consumption4. Green on-site continuous H2O2 electrosynthesis, via a two-electron oxygen reduction reaction (2e− ORR), can efficiently prevent the high energy consumption and contamination of the traditional anthraquinone process. This promising alternative approach eliminates potential explosion risk associated with high concentration H2O2 transportation5,6. Notably, the development of selective heterogeneous electrocatalysts is imperative for its industrial application7.
Although numerous efforts have been devoted to searching for highly efficient heterogeneous electrocatalysts for 2e− ORR in recent years, it is still difficult to precisely control their fine structures and to elucidate the corresponding structure-activity relationship8,9. Among all explored heterogeneous electrocatalysts, carbon-rich Ni-based metal-organic frameworks (MOFs) with controllable structures, featured by the well-coordinated tunable electronic structure of metal centers and functionalized organic ligands, have been studied as model catalysts for efficient 2e− ORR10,11. Owing to the low activity and selectivity of MOFs-based electrocatalysts, the large-scale electrosynthesis of H2O2 has been limited, which requires industrial production with high selectivity at a high current density (>500 mA cm−2)12.
Electrochemical 2e− ORR in alkaline involves the following elementary reactions: (i) adsorption of O2 and subsequent proton-coupled electron transfer (PCET) reaction from H2O to generate the critical *OOH (electrochemical hydrogenation step): * + O2 + H2O + e−→ *OOH + OH−; and (ii) desorption of *OOH with second PCET reaction to produce H2O2 (desorption step): *OOH + H2O + e− → H2O2 + OH− + *. It has been observed that the dominant factor for limiting the intrinsic activity and selectivity of 2e− ORR is the slow proton-feeding kinetics in the step (i) for *OOH formation13,14. The appropriate disruption of the perfectly repetitive ordered coordination structure in MOFs can be used to optimize the local micro-environment favorable for modulating the electronic structure to improve the water dissociation ability for enhancing the proton-feeding kinetics15,16.
Cytochrome c oxidase (CcO), a typical metalloenzyme in natural biological systems for catalyzing oxygen reduction in respiration, consists of a Fe heme site of the cytochrome subunit and a Cu site cofactor of the critical second-sphere that synergistically serves to efficiently facilitate the PCET reaction17,18,19. Inspired by the nature, we take an enzyme-mimicking strategy, beyond the direct first shell coordination regulation, to adjust the secondary coordination sphere of active metal nodes within the MOFs-based matrix for promoting the rate-determining PCET process, leading to an enhanced H2O2 electrosynthesis production for paper-bleaching industry20,21.
Herein, we report a catalyst based on B-doped Ni-based MOF (B-NiBDC) synthesized through an organic linker modulation method, and regulated the coordination sphere of the center Ni sites as inspired by biological systems. The developed B-NiBDC exhibited a great 2e− ORR performance at an industrial-level current density of 1000 mA cm−2 with a stable Faradaic efficiency (FE) of ~90% for H2O2 electrosynthesis. Also, a high concentration of ~7.49 wt.% H2O2 alkaline solution was produced in an energy-saving electrolyzer, which was directly applied to bleaching without any treatment and raised pulp brightness by 25-39 % (International Organization for Standardization, ISO). Spectroscopic analysis results suggested that B-dopants in the second coordination shell likely modulate Ni active sites toward electron-deficient states via localized B-O-Ni motifs, correlating with enhanced 2e− ORR activity. In-situ attenuated total reflectance infrared absorption spectroscopy and theoretical calculations demonstrated that the electron-deficient Ni in the B-NiBDC could reduce the water dissociation barrier and accelerate the proton-feeding PCET process (Fig. 1a), realizing the H2O2 electrosynthesis with high activity and selectivity.

a Efficient 2e- ORR pathway by proton-feeding and suitable *OOH adsorption. Constructed models of (b) NiBDC and (c) B-NiBDC structures. d Free energy diagrams of water dissociation for NiBDC and B-NiBDC structures. Inset: schematic illustration of water dissociation on B-NiBDC model. Color legend for atoms: gray, C; white, H; green, Ni; red, O; orange, B.
Results
Theory predictions for proton-feeding
We utilized density functional theory (DFT) to evaluate the effect of the second shell B-site on the H2O2 electrosynthesis activity (Supplementary Data 1). Figure 1b-c display two constructed models for NiBDC-based MOF ([Ni3(OH)2(1,4-BDC)2(H2O)4]·2H2O, CCDC 638866)22,23. NiBDC typically adopts a two-dimensional layered structure, formed by parallel chains of edge-sharing NiO6 octahedra (-O-Ni-O- linkages), cross-linked via BDC organic linkers. Partial substitution of BDC linkers with 4-carboxyphenylboronic acid (CPBA) in NiBDC transforms the native C-O-Ni coordination into distinct B-O-Ni motifs within the B-NiBDC framework. This ligand exchange disrupts the invariant local configuration and alters second-shell coordinating atoms. As the source of proton-feeding for 2e− ORR, the pivotal water dissociation step determines the kinetics of the overall reaction24,25,26. The Gibbs free energy (ΔG) of water dissociation for two models reveals that B-NiBDC exhibits the lower ΔG value of 0.46 eV than that of NiBDC (2.31 eV), which indicates that the energy barrier of water dissociation for pristine NiBDC is decreased after B-doping (Fig. 1d). These results disclose that the B-NiBDC model with B-O-Ni motifs can efficiently promote the proton-feeding in electrochemical hydrogenation, thus accelerating the electrosynthesis kinetics of H2O2.
Synthesis and structural characterization
Inspired by the above-predicted results, catalyst B-NiBDC was synthesized by dissolving NiCl2·6H2O, BDC, and CPBA in N, N-dimethylformamide (DMF)/ethanol/water, followed by triethylamine (TEA) addition to deprotonate ligands and initiate coordination polymerization under ambient stirring (Fig. S1)27,28. Two control samples were also synthesized without B-doping and with entirely CPBA ligand-coordinated Ni-based polymers, denoted as NiBDC and Ni-CPBA, respectively (Figs. S2, S3). After systematically optimizing the relationship between the additive ratio in the linker precursor of CPBA and the morphology, structure, and electrochemical properties, the boron-containing ligand with an optimal molar percentage of 20% was determined (Figs. S4–S6). The B content in the B-NiBDC was determined by inductively coupled plasma-optical emission spectrometry (ICP-OES) to be 0.84 wt.% (Table S1).
The nanosheet morphology of the as-prepared B-NiBDC with a thickness of ~11 nm is observed on field-emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM) (Fig. 2a, b and Fig. S7). A high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image and EDX elemental mapping images display a homogeneous distribution of C, O, Ni, and B in the B-NiBDC nanosheets (Fig. 2c). X-ray diffraction (XRD) patterns and Raman spectra show that the NiBDC-based MOF crystal structure for the B-NiBDC remains unchanged after doping with boron-containing CPBA linkers (Fig. 2d and Figs. S8–S10).

a, b FESEM and TEM images of B-NiBDC. c HAADF-STEM image and EDX elemental mapping images of B-NiBDC. d XRD patterns of NiBDC and B-NiBDC. e Ni K-edge XANES spectra of NiBDC, B-NiBDC, Ni foil, and NiO. WT-EXAFS spectra of f NiO, g NiBDC, and h B-NiBDC. i Ni K-edge k2-weighted EXAFS spectra of NiBDC, B-NiBDC, Ni foil, and NiO. j B K-edge XANES spectrum of B-NiBDC.
To investigate the influence of the B dopant on the electronic and local coordination structures, the measurements of X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) were performed. The high-resolution Ni 2p1/2 and Ni 2p3/2 peaks of B-NiBDC exhibit a shift of ~0.2 eV towards higher binding energies compared to that of NiBDC (Fig. S11). This shift remains lower than Ni-CPBA. The B 1 s peaks for B-NiBDC display two peaks located at 191.2 and 192.1 eV, referring to the B-O and B-C bonds in CPBA ligand (Figs. S12–S15). Under the mediation of B-O-Ni motifs arising from the introduction of electron-deficient B, the electrons of center Ni atoms are transferred out, forming a higher-valent Ni site29,30. The result is further supported by the XAS test. X-ray absorption near-edge structure (XANES) spectra of Ni K-edge show that the absorption edge of the B-NiBDC is located between Ni foil and NiO (Fig. 2e). Further analysis reveals the Ni oxidation state for B-NiBDC is +1.5 (Fig. S16), which is higher than that of NiBDC ( + 1.4), in agreement with the XPS observations31. The highest intensity of B-NiBDC in wavelet transform (WT) is situated at 4.1 Å−1, originating from the light O atoms coordinated in the first shell (Fig. 2f–h and Fig. S17). The coordination environments of Ni atoms are further analyzed through Fourier-transformed extended X-ray absorption fine structure (EXAFS) spectra (Fig. 2i). A prominent peak located at 1.58 Å for B-NiBDC can be assigned to the Ni-O bond in the first coordination shell. Further EXAFS fitting analyses demonstrate that the introduction of B dopants does not affect the first shell octahedral coordination environment in the framework structure (Figs. S18–S20 and Table S2)32,33. The B K-edge XANES spectrum indicates a sharp peak located at 193.9 eV and a broad absorption band at 203.0 eV, assigned to the transitions from B 1 s to the unoccupied B 2p π* and σ* states, respectively, of the trigonal [BO2C] unit in CPBA linker (Fig. 2j)34,35. The 11B solid-state nuclear magnetic resonance (NMR) spectra also confirm the tri-coordinated boron sites in B-NiBDC (Fig. S21). Fine structure analysis suggests that the bio-inspired secondary-sphere B-coordination strategy facilitates the generation of electron-deficient Ni sites36.
H2O2 electrosynthesis performances
The H2O2 electrosynthesis performance of B-NiBDC was evaluated via 2e− ORR in O2-saturated 0.1 M KOH (Fig. S22), using a rotating ring-disk electrode (RRDE) with optimized catalyst loading ( ~ 0.24 mg cm−2). The collection efficiency of RRDE was calculated as 0.423 (Fig. S23). To study the role of B dopants on the 2e− ORR activity, a control sample of NiBDC was evaluated. The cyclic voltammetry (CV) curve for B-NiBDC showed an obvious oxygen reduction peak in O2-saturated 0.1 M KOH, but no peak was observed in Ar-saturated solution (Fig. S24). B-NiBDC exhibited larger disk and ring current densities (Fig. 3a), indicating its better 2e− ORR activity than NiBDC. B-NiBDC could reach the disk current density of 1.0 mA cm−2 at 0.64 V vs. RHE, larger than that of NiBDC (0.5 V vs. RHE). The calculated H2O2 selectivity of B-NiBDC was >95% in a wide potential range from 0 to 0.56 V vs. RHE (Fig. 3b), higher than that of NiBDC (67-71%). The corresponding electron transfer number results showed that the value of B-NiBDC was less than 2.2, which is more suitable for the 2e− pathway compared to NiBDC (2.6) (Fig. 3b and Fig. S25). The favorable 2e− ORR kinetics for B-NiBDC was further verified by a smaller Tafel slope of 71 mV dec−1, lower than 123 mV dec−1 for NiBDC (Fig. 3c)37. A smaller charge transfer resistance for B-NiBDC than NiBDC further confirmed its fast reaction kinetics (Fig. S26). Based on the double-layer capacitance (Cdl), the electrochemically active surface area (ECSA) for B-NiBDC (1.4 mF cm−2) was higher than that of NiBDC (1.1 mF cm−2) (Figs. S27–S29).

a Polarization curves (solid lines) at a rotation of 1600 rpm for NiBDC and B-NiBDC, and simultaneous H2O2 detection currents on the ring electrode (dashed lines) at 1.2 V vs. RHE in O2-saturated 0.1 M KOH electrolyte (solution resistance = 55.8 ± 3.2 Ω). b Calculated H2O2 selectivity (%) and electron transfer number (n). c Tafel slopes of NiBDC and B-NiBDC. d Illustration of gas diffusion electrode (GDE) in a three-phase flow cell set-up for H2O2 production. e Polarization curves and f corresponding H2O2 FEs of NiBDC and B-NiBDC in a flow cell using 1.0 M KOH as electrolyte (solution resistance = 2.6 ± 0.4 Ω). Error bars correspond to the standard deviation of the three measurements. g Chronopotentiometric curves at 1000 mA cm-2 for B-NiBDC and the corresponding H2O2 FEs.
Inspired by the H2O2 electrosynthesis performance, we further tested the B-NiBDC in a flow cell to enhance oxygen diffusion mass transfer capability (Fig. 3d and Fig. S30). As evidenced by the polarization curves, B-NiBDC achieved the current densities of 500 and 1000 mA cm−2 at potentials of 0.34 and 0.18 V vs. RHE, respectively, which are much higher than those of NiBDC (0.19 and -0.08 V vs. RHE) (Fig. 3e and Fig. S31). The H2O2 FEs at different current densities were further assessed by the cerium sulfate titration method (Fig. S32). The H2O2 FEs for B-NiBDC maintained over 90% when reaching a 1000 mA cm−2, which outperformed the NiBDC ( ~ 75%), demonstrating high performance of industrial-level H2O2 electrosynthesis for B-NiBDC (Fig. 3f). Compared to the reported performance of 2e− ORR to H2O2 for other electrocatalysts, B-NiBDC displays a competitive activity, especially at the industrial current density of >500 mA cm−2 (Table S3)38,39. The stability of B-NiBDC was tested in RRDE and flow cell set-up (Fig. 3g and Fig. S33). The results showed good stability for B-NiBDC, which is stable over 24 hours at 1000 mA cm−2, while maintaining >85% H2O2 FE and achieving a high production rate of 65.4 mol gcatalyst−1 h−1 (Fig. 3g). Moreover, the 2e− ORR post-characterizations revealed that the primary structure and nanosheet morphology of the B-NiBDC remained unchanged, serving as the active component (Figs. S34–S41 and Table S2)40.
Insight on promoted H2O2 electrosynthesis activity
To explore the 2e− ORR pathway, a deep H2O2 electroreduction reaction (H2O2RR) was conducted in Ar-saturated 0.1 M KOH with 10 mM H2O2. In Fig. 4a, b, B-NiBDC had virtually no H2O2RR activity (0.12 mA cm−2 at 0.2 V vs. RHE), whereas a significant reduction current was observed for NiBDC (0.85 mA cm−2 at 0.2 V vs. RHE), which in turn confirms the inherently high 2e− ORR selectivity of B-NiBDC41,42. This result also reflects that the Ni sites regulated by B-O-Ni motifs in the B-NiBDC can efficiently inhibit its further reduction of the produced H2O2 to OH−.

Polarization curves of a B-NiBDC and b NiBDC in Ar-saturated 0.1 M KOH with 10 mM H2O2 at 1600 rpm (solution resistance = 52.3 ± 2.6 Ω). Color legend for atoms: gray, C; white, H; green, Ni; red, O; orange, B. c, d In-situ ATR-SEIRAS spectra of B-NiBDC and NiBDC with the potentials of 1.0 to 0.2 V vs. RHE in O2-saturated 0.1 M KOH. e Polarization curves of B-NiBDC and NiBDC in O2-saturated 0.1 M KOH in H2O and D2O at 1600 rpm (solution resistance = 54.5 ± 2.2 Ω). f, g Corresponding H/D KIE values.
To further probe the promotional role of B species in the H2O2 electrosynthesis process, in-situ attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) measurements were performed. As shown in Fig. 4c, a significant absorption band at 1257 cm−1 began to emerge at 0.7 V vs. RHE and gradually strengthened as the applied potential decreased to 0.2 V vs. RHE on B-NiBDC, which can be attributed to the formation of the surface-adsorbed O-O stretching mode of OOH (*OOH) intermediates43. In contrast, the weaker *OOH absorption band in the same tested potential range was identified for NiBDC, signifying that the doped B species of B-O-Ni motifs reinforce the reaction kinetics to boost the production of the crucial *OOH (Fig. 4d).
As previously shown in Fig. 3c, the Tafel slope of NiBDC (123 mV dec−1) is close to 120 mV dec−1 which corresponds to the first-step rate-determining PCET step (O2 + H2O + e− + * → *OOH + OH−) in the elementary reaction of 2e− ORR for H2O2 production. In contrast, the significantly reduced Tafel slope of the B-NiBDC (71 mV dec−1) proves that the electrochemical hydrogenation step was no longer the rate-determining step after introducing B44,45. Accordingly, we examined the kinetic isotope effects (KIE) of H/D during 2e− ORR to ascertain the specific contributing factors (Fig. 4e–g and Fig. S42). The average KIE values for B-NiBDC were around ~1.2, which is close to the value of the secondary KIE, accompanied by the almost unchanged Tafel slope, suggesting no significant primary isotope effect occurred in B-NiBDC. In contrast, NiBDC showed a pronounced kinetic isotope effect (KIE > 1.0). These observations demonstrate that the rate-determining step of the 2e− ORR is no longer associated with O-H bond cleavage in water molecules after the B-O-Ni motifs formed46,47, owing to the efficient proton-feeding caused by accelerated water dissociation kinetics.
Theoretical calculation of H2O2 electrosynthesis mechanism
DFT calculations were used to elucidate the mechanism of bio-inspired B-coordinated regulation for enhancing the activity (Supplementary Data 1). Considering that the key *OOH of optimal 2e− ORR catalyst should not be adsorbed too strongly or too weakly, we constructed models for different boron-containing ligand substitution ratios and calculated the Gibbs free energies with the potential influence of solvent effect considered (Fig. S43, S44)48. The formation energy remains negative with increasing doping of boron-containing monomers, demonstrating the relative stability of the structural models (Table S4). Figure 5a illustrates a volcano plot for 2e− ORR. The excessively weak ΔG*OOH of pristine NiBDC is enhanced as the boron-containing ligand content increases. The density of states (DOS) calculations find that the Ni site d-band center also decreases relevantly with the increasing B-doping (Fig. 5b, c)49. 25.0% B-NiBDC with B-O-Ni motifs has more favorable ΔG*OOH values (4.15 and 3.45 eV at U = 0 and 0.7 V) as compared to NiBDC (4.57 and 3.87 eV at U = 0 and 0.7 V) (Fig. 5d, e). The introduction of the optimal boron-ligand (25.0%) achieves the most suitable *OOH adsorption, which matches well with the experimental result that the optimal 2e− ORR performance of B-NiBDC is at 20% doping. The free energy diagrams for 2e− and 4e− ORR pathways demonstrate high 2e− catalytic selectivity for the B-NiBDC (Fig. S45), which is experimentally validated by the improved activity of B-O-Ni motif-regulated sites in 2e− ORR (Figs. S46–S49). Notably, DFT methods have certain limitations due to the difficulty in simulating complex conditions of real catalytic environments.

a 2e- ORR volcano plot between ∆G*OOH and UL for NiBDC and B-NiBDC models. b PDOS of d-states for Ni site in NiBDC and B-NiBDC models. c Calculated *OOH adsorption energies and d-band center of Ni site in NiBDC and B-NiBDC models. Free energy diagrams of 2e- ORR for NiBDC and B-NiBDC at d U = 0 V and e U = 0.7 V.f Charge density differences of NiBDC and B-NiBDC. Color legend for atoms: gray, C; white, H; green, Ni; red, O; orange, B. The yellow color indicates charge accumulation and the cyan color indicates charge depletion. DOS for (g) NiBDC and (h) B-NiBDC.
To reveal the effect of the introduced B on the electronic structure of center Ni sites, the analysis of charge density difference was performed for B-NiBDC and NiBDC models. The B-atom mediated Ni site transfers out more electrons to form an electron-deficient Ni center (Fig. 5f), which is well supported by the results of the Bader charge analysis. Specifically, the number of electron loss of the Ni site increases from +1.23 in NiBDC to +1.26 in B-NiBDC, in accordance with the experimental characterizations (Fig. 2e, Figs. S11, and S16).
As shown in Fig. 5g, h, pristine NiBDC has distinct semiconductor features due to the lack of electron distribution near the Fermi level (EF). After B-doping, the total DOS of B-NiBDC is elevated around the EF, demonstrating that the modulation of B improves the electrical conductivity of NiBDC, which can be well supported by experimentally measured results (~ 2.5 × 10−11 S cm−1 for B-NiBDC vs. ~ 5.4 × 10−12 for NiBDC). Based on the above results, one can conclude that the electron-deficient Ni sites accelerate the water dissociation kinetics to intensify the electrochemical hydrogenation step of O2 to *OOH, and optimize the adsorption energy of the *OOH simultaneously, resulting in a significant increase of 2e− ORR activity and selectivity.
Energy-saving H2O2 electrosynthesis for sustainable bleaching
Intrigued by the enhanced H2O2 electrosynthesis activity, we further assembled an energy-efficient two-electrode urea oxidation reaction (UOR)//2e− ORR flow-cell electrolyzer by replacing the anodic oxygen evolution reaction (OER) with UOR, which has a much lower thermodynamic equilibrium potential of 0.37 V vs. RHE and is promising for urea-containing wastewater treatment (Fig. 6a)50. Ni-based material was prepared as a representative catalyst for efficient UOR (Fig. S50)51. The UOR//2e− ORR energy-saving system required a cell voltage of only 1.62 V to achieve a current density of 500 mA cm−2, significantly lower than the other reported OER//2e− ORR system (Fig. 6b, c and Fig. S51). Considering that a certain concentration of H2O2 alkaline solution can be used directly in the paper/pulp bleaching without further purification, different concentrations of NaOH alkaline electrolytes were used for the preparation of H2O2 bleaching solution (Fig. S52). Typically, H2O2 solution with a concentration of ~7.49 wt.% can be produced within 1 h at 500 mA cm−2 in 6 wt.% NaOH (Fig. 6d). The prepared H2O2 alkaline solution can effectively bleach the different types of pulp (wood, bamboo, straw, and recycled pulp), markedly increasing the brightness by 25–39 % ISO (Fig. 6e, f). This energy-efficient, sustainable H2O2 on-demand electrosynthesis system excludes the transportation, dilution, and alkalization of highly concentrated H2O2 from the conventional anthraquinone process before utilization for paper/pulp bleaching.

a Schematic diagram of UOR//2e- ORR two-electrode cell. b Polarization curves and c corresponding cell voltages at different current densities in 1.0 M KOH and 1.0 M KOH + 0.33 M urea without iR compensation. Error bars correspond to the standard deviation of the three measurements. d Chronopotentiometric curve at 500 mA cm-2 for UOR//2e- ORR in 6 wt.% NaOH solution without iR compensation. Inset: accumulated H2O2 concentrations. Error bars correspond to the standard deviation of the three measurements. e Digital photographs and f corresponding brightness of different types of pulp before and after bleaching treatment by the obtained alkaline H2O2 solution. Error bars correspond to the standard deviation of the three measurements.
Discussion
In summary, we have developed a B-NiBDC catalyst with the elaborately well-coordinated B-O-Ni motifs for H2O2 electrosynthesis. B-NiBDC can produce H2O2 with an FE of ~90% at 1000 mA cm−2 through the 2e− ORR process. In an energy-saving H2O2 electrosynthesis system based on UOR//2e− ORR flow-cell electrolyzer, B-NiBDC yielded ~7.49 wt.% H2O2 alkaline solution for direct use in bleaching, realizing a distinct increase in the brightness of the bleached pulp by 25–39 % ISO. Structural characterizations verified that the sphere coordination contributed to the formation of electron-deficient Ni sites through the B-O-Ni unit bonding micro-environment. Preliminary studies revealed that this strategy of modulating electron-deficient metal sites through the B-O-M motifs of the second shell layer to enhance 2e− ORR performance is universal and can be extended to CoBDC or FeBDC-based catalysts (Fig. S53–S56). Experimental measurements and theoretical calculations identified that the modulated electron-deficient Ni acted as accelerated water dissociation centers for rapid proton-feeding with more suitable adsorption energies for *OOH, thus promoting the alkaline 2e− ORR process. This work provides a bio-inspired modification method to rationalize the design of high-performance MOFs-based catalysts, which can potentially be applied to a broader range of industrial-level electrochemical applications, such as water splitting, carbon dioxide reduction, and nitrate reduction reaction.
Methods
Materials
1,4-dicarboxybenzene acid (BDC, ≥99.0%) and 4-carboxyphenylboronic acid (CPBA, ≥97.0%) were purchased from Macklin Biochemical Co., Ltd; Nickel (Ⅱ) chloride hexahydrate (NiCl2·6H2O, ≥98.0%), nickel (Ⅱ) nitrate hexahydrate (Ni(NO3)2·6H2O, ≥98.0%), triethylamine (TEA, ≥99.0%), potassium hydroxide (KOH, ≥85.0%), urea ( ≥ 99.0%), ammonium fluoride (NH4F, ≥96.0%), N, N-dimethylformamide (DMF, ≥99.5%) and ethanol absolute ( ≥ 99.7%) were bought from Sinopharm Chemical Reagent Co., Ltd. The water used was deionized (DI) water. All the chemicals were used as obtained without further purification.
Synthesis of B-NiBDC
178 mg of NiCl2·6H2O, 99.7 mg of BDC, and 25 mg of CPBA (the molar ratio of CPBA in total ligand precursors is 20%) were dissolved in a mixed solvent of water, ethanol, and DMF (volume ratio = 2:2:32 mL), and stirred for 20 minutes to form a homogeneous solution. Then, 0.8 mL TEA was quickly injected into the solution and was stirred for 24 h. Finally, the B-NiBDC product was obtained through centrifugation, washed with DMF and ethanol (3 ~ 5 times), and dried in a vacuum.
Synthesis of NiBDC
A total of 178 mg of NiCl2·6H2O and 125 mg of BDC were dissolved in a mixed solvent of water, ethanol, and DMF (volume ratio = 2:2:32 mL), and stirred for 20 minutes to form a homogeneous solution. Then, 0.8 mL TEA was quickly injected into the solution and was stirred for 24 hours. Finally, the NiBDC product was obtained through centrifugation, washed with DMF and ethanol (3 ~ 5 times), and dried in a vacuum.
Synthesis of 10% B-NiBDC
178 mg of NiCl2·6H2O, 112 mg of BDC, and 12.4 mg of CPBA (the molar ratio of CPBA in total ligands is 10%) were dissolved in a mixed solvent of water, ethanol, and DMF (volume ratio = 2:2:32 mL), and stirred for 20 minutes to form a homogeneous solution. Then, 0.8 mL TEA was quickly injected into the solution and was stirred for 24 hours. Finally, the 10% B-NiBDC product was obtained through centrifugation, washed with DMF and ethanol (3 ~ 5 times), and dried in a vacuum.
Synthesis of 30% B-NiBDC
178 mg of NiCl2·6H2O, 87 mg of BDC, and 37.2 mg of CPBA (the molar ratio of CPBA in total ligands is 30%) were dissolved in a mixed solvent of water, ethanol, and DMF (volume ratio = 2:2:32 mL), and stirred for 20 minutes to form a homogeneous solution. Then, 0.8 mL TEA was quickly injected into the solution and was stirred for 24 h. Finally, the 30% B-NiBDC product was obtained through centrifugation, washed with DMF and ethanol (3 ~ 5 times), and dried in a vacuum.
Synthesis of 40% B-NiBDC
178 mg of NiCl2·6H2O, 75 mg of BDC, and 50 mg of CPBA (the molar ratio of CPBA in total ligands is 40%) were dissolved in a mixed solvent of water, ethanol, and DMF (volume ratio = 2:2:32 mL), and stirred for 20 minutes to form a homogeneous solution. Then, 0.8 mL TEA was quickly injected into the solution and was stirred for 24 hours. Finally, the 40% B-NiBDC product was obtained through centrifugation, washed with DMF and ethanol (3 ~ 5 times), and dried in a vacuum.
Synthesis of Ni-CPBA
178 mg of NiCl2·6H2O and 124.5 mg of CPBA were dissolved in a mixed solvent of water, ethanol, and DMF (volume ratio = 2:2:32 mL), and stirred for 20 min to form a homogeneous solution. Then, 0.8 mL TEA was quickly injected into the solution and was stirred for 24 h. Finally, the Ni-CPBA product was obtained through centrifugation, washed with DMF and ethanol (3 ~ 5 times), and dried in a vacuum.
Synthesis of Ni-C
930 mg of Ni(NO3)2·6H2O and 498 mg of BDC were dissolved in a mixed solvent of water, ethanol, and DMF (volume ratio = 2.5:2.5:30 mL) and stirred for 20 min to form a homogeneous solution. The green transparent solution was transferred into a Teflon-lined autoclave with a piece of Ni foam and kept at 120 oC for 12 h in an oven, followed by naturally cooling down to room temperature. After being washed with DMF, ethanol, and DI water, Ni-MOF precursor was dried in a vacuum. The precursor Ni-MOF was then placed in a tubular furnace and annealed at 450 °C for 2 h with a heating rate of 2 °C min−1 under Ar atmosphere.
Synthesis of B-CoBDC
A total of 178 mg of CoCl2·6H2O, 99.7 mg of BDC, and 25 mg of CPBA were dissolved in a mixed solvent of water, ethanol, and DMF (volume ratio = 2:2:32 mL), and stirred for 20 minutes to form a homogeneous solution. Then, 0.8 mL TEA was quickly injected into the solution and was stirred for 24 h. Finally, the B-CoBDC product was obtained through centrifugation, washed with DMF and ethanol (3 ~ 5 times), and dried in a vacuum.
Synthesis of CoBDC
178 mg of CoCl2·6H2O and 125 mg of BDC were dissolved in a mixed solvent of water, ethanol, and DMF (volume ratio = 2:2:32 mL), and stirred for 20 min to form a homogeneous solution. Then, 0.8 mL TEA was quickly injected into the solution and was stirred for 24 h. Finally, the CoBDC product was obtained through centrifugation, washed with DMF and ethanol (3 ~ 5 times), and dried in a vacuum.
Synthesis of B-FeBDC
A total of 203 mg of FeCl3·6H2O, 99.7 mg of BDC, and 25 mg of CPBA were dissolved in a mixed solvent of water, ethanol, and DMF (volume ratio = 2:2:32 mL), and stirred for 20 minutes to form a homogeneous solution. Then, 0.8 mL TEA was quickly injected into the solution and was stirred for 24 h. Finally, the B-FeBDC product was obtained through centrifugation, washed with DMF and ethanol (3 ~ 5 times), and dried in a vacuum.
Synthesis of FeBDC
203 mg of FeCl3·6H2O and 125 mg of BDC were dissolved in a mixed solvent of water, ethanol, and DMF (volume ratio = 2:2:32 mL), and stirred for 20 min to form a homogeneous solution. Then, 0.8 mL TEA was quickly injected into the solution and was stirred for 24 h. Finally, the FeBDC product was obtained through centrifugation, washed with DMF and ethanol (3 ~ 5 times), and dried in a vacuum.
Synthesis of Ni(OH)2
473 mg of Ni(NO3)2·6H2O, 451 mg of urea, and 148 mg of NH4F were dissolved in 25 mL water. Then the obtained solution was transferred into a Teflon-lined autoclave and was heated at 120 °C for 8 h in an oven, which was then cooled down to room temperature naturally. The resulting material was rinsed with water several times, and then dried in a vacuum.
Characterization
The morphologies of samples were observed by FESEM (Hitachi SU-8010) with EDX (Oxford, X-max80), TEM (HT7700), and HRTEM (Tecnai G2 F20 S-TWIN). The crystal structures of the samples were measured by XRD (ZETIUM DY powder XRD unit) using Cu Kα radiation at 4.0 KW. The chemical environments of samples were measured by XPS (Escalab 250Xi) with Al Kα radiation. The contents of nickel and boron in the samples were analyzed by ICP-OES (Agilent 720ES). Raman spectra of samples were measured with a LabRAM HR Evolution at 532 nm. The N2 adsorption-desorption curves of the samples were tested by BET (Micromeritics/ 3FLEX-3MP). Thermogravimetric analysis (TGA) was tested by TA SDT650 at N2 atmosphere. The 11B solid-state NMR spectra were measured by Bruker AVANCE III 400WB. The XAS measurements of the samples were performed at the BL17C station in Taiwan Synchrotron Radiation Research Center. Electrical conductivity was measured by a high-resistance meter (ST2643). Pulp brightness was measured by WSB-3C.
Electrochemical measurements
The electrochemical workstation (CHI 760E) with a current amplifier was used to conduct all electrochemical experiments in a three-electrode configuration with a rotating ring disk electrode (RRDE) system (RRDE-3A, ALS Co., Ltd, Japan) and a custom-made flow cell. To prepare the ink, 2.5 mg catalyst and 1.5 mg carbon black (CB, Vulcan XC-72R) were dispersed in 225 μL isopropanol and 25 μL of 0.5 wt.% Nafion solution with ultrasonication for 1 hour. CB was added to enhance the electrical conductivity of MOF-based catalysts (Figs. S57–S59). The well-dispersed 3 μL ink was drop-cast onto a disk electrode (4 mm) with a Pt ring. The catalyst loading was ~0.24 mg cm−2, and the same amount was loaded on the gas diffusion layer (SGL 29BC) in the flow cell test. A Hg/HgO electrode and a graphite rod were used as the reference and counter electrode, respectively. The potential was converted to a reversible hydrogen electrode (RHE) via a Nernst equation (ERHE = EHg/HgO + 0.059 × pH + 0.098). The scan rate of linear sweep voltammetry (LSV) was set to 10 mV s−1 within the potential range from 1.065 and -0.035 V vs. RHE in 0.1 M KOH electrolyte (pH = 12.96 ± 0.06) to investigate the 2e− ORR activities of the catalysts at 1600 rpm. The preparation of the electrolyte solution involves dissolving KOH in deionized water and should be used immediately after preparation without storage. The applied potential on the Pt ring of the electrode was set at 1.2 V vs. RHE. An electrochemical impedance spectroscopy (EIS) test was measured at -0.7 V vs. RHE with a frequency range from 105 to 0.01 Hz. The system solution resistance was obtained from the EIS Nyquist plots. The polarization curves were calibrated with 80% iR compensation unless specifically mentioned. Cyclic voltammetry cycles (CVs) with scan rates from 20 to 100 mV s−1 were applied to analyze the electrochemically active surface area (ECSA). The 5000 CV cycles within the potential range from 1.065 and -0.035 V vs. RHE were conducted for the accelerated degradation test. The H2O2 selectivity and electron transfer number (n) were obtained from the equations:
where Ir is the ring current, Id is the disk current, and N is the collection efficiency of 0.423 by the calibration from the reduction of K3Fe(CN)6.
For flow cell tests, the Fumasep FAA-3-PK-130 membrane (130 μm, 2.5 × 2.5 cm², pretreated by soaking in a 1.0 M KOH solution) and 1.0 M KOH solution (pH = 13.87 ± 0.06) were used as the anion exchange membrane and electrolyte, respectively. The gas diffusion layer (1 cm2) loaded with catalyst was used as the working electrode. Hg/HgO electrode and nickel foam (1 cm2) were used as the reference and counter electrode. The flow rate of the cathode and anode electrolyte was controlled by a peristaltic pump at 8 mL min−1, and the flow rate of O2 was controlled at 20 mL min−1 by a gas flow meter.
Product analysis
The concentration of H2O2 was quantified by the reaction of cerium sulfate Ce(SO4)2 with H2O2: 2Ce4+ + H2O2 → 2Ce3+ + 2H+ + O2. The concentration of yellow Ce4+ before and after the reaction can be obtained from the standard curve with the absorbance at 317 nm by ultraviolet-visible spectroscopy to quantify the concentration of produced H2O2. The H2O2 Faradaic efficiency (FE) was obtained when a certain amount of charges (Q) was accumulated:
where C is the H2O2 concentration (mol L−1), V is the volume of electrolyte (L), F is the Faraday constant (96484 C mol-1), and Q is the total amount of charge transferred.
In-situ ATR-SEIRAS experiments
The in-situ ATR-SEIRAS tests were conducted on an Invenio-R spectrometer, which was configured with a mercury-cadmium-telluride (MCT) detector maintained at liquid nitrogen temperature. The three-electrode system comprised a Pt counter electrode and an Ag/AgCl reference electrode, with the catalysts-loaded Au-coated ZnSe prism functioning as the working electrode. Spectral data are reported in absorbance units defined by Abs = -log(R/R0), with R and R0 denoting the sample and background reflectance, respectively16.
In-situ Raman experiments
The in-situ Raman tests were measured at 532 nm in a three-electrode system, employing the catalysts as the working electrode, with Pt and Ag/AgCl electrode as the counter and reference electrodes, respectively16.
Calculation method
For first-principles calculations, the spin-polarized DFT calculations were performed using Vienna Ab Initio Simulation Package (VASP). The ion-electron interactions were described by the projector-augmented wave (PAW) pseudopotentials. The electron-electron interactions were described by the Perdew-Burke-Ernzerhof (PBE) functional with generalized gradient approximation (GGA). The plane-wave basis was set to 450 eV, and the Brillouin-zones were sampled with a grid of 1 × 1 × 1 to sample reciprocal space for geometry optimization and 2 × 2 × 1 k-points for electronic structure calculation. For the geometry optimizations, all atoms were relaxed to a self-consistent field tolerance level of 1.0 × 10−5 eV, and the residual force was smaller than 0.05 eV/Å. The DFT-D3 method was used to describe the van der Waals interactions. The climbing image nudged-elastic band (CI-NEB) method was employed to determine the transition states. The residual force was less than 0.08 eV/Å in the transition states calculation.
The (010) surface of NiBDC was used as the pristine Ni-MOF. The models of B-NiBDC were constructed by replacing the carbon atom with boron atom in the NiBDC model. NiBDC and B-NiBDC models were expanded to contain eight organic ligands. Therefore, doping each B atom increased the doping rate by 12.5%. A vacuum space of at least 15 Å in the direction perpendicular was applied to eliminate the interactions between the adjacent cells of slab models.
The Gibbs free energy (G) was calculated using the computational hydrogen electrode (CHE) method proposed by Nørskov et al52, where the free energy of the electron-proton pair (H+ + e−) can be referenced to the chemical potential of gaseous H2 at equilibrium (0 V vs. SHE). The free energy can be calculated as follows:
where E is the total energy obtained by DFT calculation, ZPE is the zero-point energy correction, T is the temperature (298.15 K), S is the entropy change, and U is the applied potential. For each system, its ZPE can be calculated by summing vibrational frequencies overall normal modes ν (ZPE = 1/2Σħν). Specifically, the vibrational frequencies were calculated by fixing the catalyst substrate and releasing the adsorbed molecules, followed by data processing using the VASPKIT program to obtain the ZPE and S53. The Bader charge analysis was performed using the code from the Henkelman group54.
The 2e− ORR competes with 4e− pathway, leading to H2O2 formation. The 2e− ORR is split into two consecutive reaction steps:
where * denotes the catalyst substrate and *OOH denotes the intermediate adsorbed on the catalyst surface.
DFT + U was taken into account for 3d-states Ni by applying an effective Hubbard-parameter Ueff = U − J within the Dudarev approach55. For the NiBDC surfaces, a value Ueff = 5 eV was applied, which was approached by previously published articles on systems containing Ni elements56,57,58,59. For reaction thermodynamics, the explicit solvent model was considered with the water overlayer on catalyst surfaces60.
Data availability
The data that support the findings of this study are available within the main text and supplementary information. Source data are provided with this paper.
References
Lewis, R. J. et al. Highly efficient catalytic production of oximes from ketones using in situ-generated H2O2. Science 376, 615–620 (2022).
Xu, J. et al. Organic wastewater treatment by a single-atom catalyst and electrolytically produced H2O2. Nat. Sustain. 4, 233–241 (2021).
Jung, E., Shin, H., Hooch Antink, W., Sung, Y.-E. & Hyeon, T. Recent advances in electrochemical oxygen reduction to H2O2: Catalyst and cell design. ACS Energy Lett. 5, 1881–1892 (2020).
Yang, S. et al. Toward the decentralized electrochemical production of H2O2: A focus on the catalysis. ACS Catal. 8, 4064–4081 (2018).
Perry, S. C. et al. Electrochemical synthesis of hydrogen peroxide from water and oxygen. Nat. Rev. Chem. 3, 442–458 (2019).
Xia, C., Xia, Y., Zhu, P., Fan, L. & Wang, H. Direct electrosynthesis of pure aqueous H2O2 solutions up to 20% by weight using a solid electrolyte. Science 366, 226–231 (2019).
Lu, Z. et al. High-efficiency oxygen reduction to hydrogen peroxide catalysed by oxidized carbon materials. Nat. Catal. 1, 156–162 (2018).
Xia, Y. et al. Highly active and selective oxygen reduction to H2O2 on boron-doped carbon for high production rates. Nat. Commun. 12, 4225 (2021).
Wu, Q. et al. Unveiling the dynamic active site of defective carbon-based electrocatalysts for hydrogen peroxide production. Nat. Commun. 14, 6275 (2023).
Wang, M. et al. An efficient interfacial synthesis of two-dimensional metal-organic framework nanosheets for electrochemical hydrogen peroxide production. Angew. Chem. Int. Ed. 60, 11190–11195 (2021).
Wang, M. et al. Partially pyrolyzed binary metal-organic framework nanosheets for efficient electrochemical hydrogen peroxide synthesis. Angew. Chem. Int. Ed. 59, 14373–14377 (2020).
Yang, X. et al. Tuning two-electron oxygen-reduction pathways for H2O2 electrosynthesis via engineering atomically dispersed single metal site catalysts. Adv. Mater. 34, 2107954 (2022).
Li, Y. et al. Single-atom iron catalyst with biomimetic active center to accelerate proton spillover for medical-level electrosynthesis of H2O2 disinfectant. Angew. Chem. Int. Ed. 62, e202306491 (2023).
Chen, G. et al. Promoted oxygen reduction kinetics on nitrogen-doped hierarchically porous carbon by engineering proton-feeding centers. Energy Environ. Sci. 13, 2849–2855 (2020).
Cheng, F. et al. In situ identification of the electrocatalytic water oxidation behavior of a nickel-based metal-organic framework nanoarray. Mater. Horiz. 8, 556–564 (2021).
Cheng, F. et al. Accelerated water activation and stabilized metal-organic framework via constructing triangular active-regions for ampere-level current density hydrogen production. Nat. Commun. 13, 6486 (2022).
Ishigami, I. et al. Structural insights into functional properties of the oxidized form of cytochrome c oxidase. Nat. Commun. 14, 5752 (2023).
Xie, L. et al. Enzyme-inspired iron porphyrins for improved electrocatalytic oxygen reduction and evolution reactions. Angew. Chem. Int. Ed. 60, 7576–7581 (2021).
Le, J. M. & Bren, K. L. Engineered enzymes and bioinspired catalysts for energy conversion. ACS Energy Lett. 4, 2168–2180 (2019).
Zheng, W. et al. Accelerating industrial-level CO2 electroreduction kinetics on isolated zinc centers via sulfur-boosted bicarbonate dissociation. Energy Environ. Sci. 16, 1007–1015 (2023).
Zhang, Z., Liu, W., Zhang, Y., Bai, J. & Liu, J. Bioinspired atomic manganese site accelerates oxo-dehydrogenation of N-heterocycles over a conjugated tri-s-triazine framework. ACS Catal. 11, 313–322 (2021).
Li, F. L. et al. Large-scale, bottom-up synthesis of binary metal-organic framework nanosheets for efficient water oxidation. Angew. Chem. Int. Ed. 58, 7051–7056 (2019).
Xu, X. et al. Leveraging metal nodes in metal-organic frameworks for advanced anodic hydrazine oxidation assisted seawater splitting. ACS Nano 17, 10906–10917 (2023).
Cheng, F. et al. Boosting alkaline hydrogen evolution and Zn-H2O cell induced by interfacial electron transfer. Nano Energy 71, 104621 (2020).
Wang, K. et al. Kinetically accelerating elementary steps via bridged Ru-H state for the hydrogen-evolution in anion-exchange membrane electrolyzer. Adv. Funct. Mater. 33, 2212321 (2023).
Chen, J. et al. Promoting CO2 electroreduction kinetics on atomically dispersed monovalent ZnI sites by rationally engineering proton-feeding centers. Angew. Chem. Int. Ed. 61, e202111683 (2022).
Ge, K. et al. Facile synthesis of two-dimensional iron/cobalt metal-organic framework for efficient oxygen evolution electrocatalysis. Angew. Chem. Int. Ed. 60, 12097–12102 (2021).
He, F. et al. Metal-organic frameworks with assembled bifunctional microreactor for charge modulation and strain generation toward enhanced oxygen electrocatalysis. ACS Nano 16, 9523–9534 (2022).
Li, Y. et al. Boron containing metal-organic framework for highly selective photocatalytic production of H2O2 by promoting two-electron o2 reduction. Mater. Horiz. 8, 2842–2850 (2021).
Deng, L. et al. Unveiling coordination transformation for dynamically enhanced hydrogen evolution catalysis. Energy Environ. Sci. 16, 5220–5230 (2023).
Zhao, S. et al. Structural transformation of highly active metal-organic framework electrocatalysts during the oxygen evolution reaction. Nat. Energy 5, 881–890 (2020).
Zhao, S. et al. Ultrathin metal-organic framework nanosheets for electrocatalytic oxygen evolution. Nat. Energy 1, 16184 (2016).
He, F. et al. Spin-state modulation on metal-organic frameworks for electrocatalytic oxygen evolution. Adv. Mater. 35, 2304022 (2023).
Min, Y. et al. Integrating single-cobalt-site and electric field of boron nitride in dechlorination electrocatalysts by bioinspired design. Nat. Commun. 12, 303 (2021).
Hemraj-Benny, T. et al. Investigating the structure of boron nitride nanotubes by near-edge X-ray absorption fine structure (NEXAFS) spectroscopy. Phys. Chem. Chem. Phys. 7, 1103–1106 (2005).
Wang, L. et al. Theory-guided design of electron-deficient ruthenium cluster for ampere-level current density electrochemical hydrogen evolution. Nano Energy 115, 108694 (2023).
Zhang, F. et al. High-efficiency electrosynthesis of hydrogen peroxide from oxygen reduction enabled by a tungsten single atom catalyst with unique terdentate N1O2 coordination. Adv. Funct. Mater. 32, 2110224 (2022).
Yan, M. et al. Sb2S3-templated synthesis of sulfur-doped Sb-N-C with hierarchical architecture and high metal loading for H2O2 electrosynthesis. Nat. Commun. 14, 368 (2023).
Zhang, X. et al. Electrochemical oxygen reduction to hydrogen peroxide at practical rates in strong acidic media. Nat. Commun. 13, 2880 (2022).
Zhang, L. et al. Self-reconstructed metal-organic framework heterojunction for switchable oxygen evolution reaction. Angew. Chem. Int. Ed. 61, e202214794 (2022).
Zhou, Z. et al. Cation-vacancy-enriched nickel phosphide for efficient electrosynthesis of hydrogen peroxides. Adv. Mater. 34, 2106541 (2022).
Tang, C. et al. Tailoring acidic oxygen reduction selectivity on single-atom catalysts via modification of first and second coordination spheres. J. Am. Chem. Soc. 143, 7819–7827 (2021).
Cao, P. et al. Metal single-site catalyst design for electrocatalytic production of hydrogen peroxide at industrial-relevant currents. Nat. Commun. 14, 172 (2023).
Holewinski, A. & Linic, S. Elementary mechanisms in electrocatalysis: Revisiting the orr tafel slope. J. Electrochem. Soc. 159, H864 (2012).
Cheng, F. et al. Carbon-confinement modulated interfacial water structure for accelerated dissociation kinetics towards hydrogen evolution. Chem. Eng. J. 520, 166181 (2025).
Yang, Y. et al. Inverse kinetic isotope effects in the oxygen reduction reaction at platinum single crystals. Nat. Chem. 15, 271–277 (2023).
Yu, L. et al. Disclosing the natures of carbon edges with gradient nanocarbons for electrochemical hydrogen peroxide production. Matter 5, 1909–1923 (2022).
Wang, Y. et al. Charge-polarized selenium vacancy in nickel diselenide enabling efficient and stable electrocatalytic conversion of oxygen to hydrogen peroxide. Adv. Sci. 10, 2205347 (2023).
Lee, B.-H. et al. Supramolecular tuning of supported metal phthalocyanine catalysts for hydrogen peroxide electrosynthesis. Nat. Catal. 6, 234–243 (2023).
Geng, S.-K. et al. Nickel ferrocyanide as a high-performance urea oxidation electrocatalyst. Nat. Energy 6, 904–912 (2021).
Li, J. et al. Deciphering and suppressing over-oxidized nitrogen in nickel-catalyzed urea electrolysis. Angew. Chem. Int. Ed. 60, 26656–26662 (2021).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886–17892 (2004).
Wang, V., Xu, N., Liu, J.-C., Tang, G. & Geng, W.-T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 267, 108033 (2021).
Tang, W., Sanville, E. & Henkelman, G. A grid-based bader analysis algorithm without lattice bias. J. Phys.: Condens. Matter 21, 084204 (2009).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 57, 1505–1509 (1998).
Cococcioni, M. & de Gironcoli, S. Linear response approach to the calculation of the effective interaction parameters in the LDA+U method. Phys. Rev. B 71, 035105 (2005).
Bengone, O., Alouani, M., Blöchl, P. & Hugel, J. Implementation of the projector augmented-wave LDA+U method: Application to the electronic structure of NiO. Phys. Rev. B 62, 16392–16401 (2000).
Füngerlings, A. et al. Crystal-facet-dependent surface transformation dictates the oxygen evolution reaction activity in lanthanum nickelate. Nat. Commun. 14, 8284 (2023).
Tamerd, M. A. et al. Magnetoelectric coupling at the NiFe2O4/PZT (001) interface: A density functional theory investigation. Superlattices Microstruct. 139, 106401 (2020).
Skúlason, E. et al. Density functional theory calculations for the hydrogen evolution reaction in an electrochemical double layer on the Pt(111) electrode. Phys. Chem. Chem. Phys. 9, 3241–3250 (2007).
Acknowledgements
We thank for the support from the National Key Research and Development Program of China (2022YFB4002100 to Y.H.), the National Natural Science Foundation of China (22278364, 22425805, 22211530045, 22178308, U22A20432, 22238008, 22578393 to Y.H. and 22408283 to F.C.), the development project of Zhejiang Province’s “Jianbing” and “Lingyan” (2023C01226 to Y.H.), the Fundamental Research Funds for the Central Universities (226-2024-00060 to Y.H.), Key Technology Breakthrough Program of Ningbo “Science and Innovation Yongjiang 2035” (2024H024 to Y.H.), the Open Project of Key Laboratory of Green Chemical Engineering Process of Ministry of Education (GCX2024001 to F.C.), the Australian Research Council for financial support (ARC, CE230100032 to L.D.). We thank Yi Huang and Dashuai Wang for their valuable contributions to this work.
Author information
Authors and Affiliations
Contributions
F.C. and Y.H. conceived the idea. F.C. performed experiments. Y.L. performed DFT calculations. Z.Z. performed in situ ATR-SEIRAS spectra. C.-L.D. performed XAS characterizations. X.Y., W.Z., B.Y., Z.L., Q.Z., and L.L. provided helpful suggestions. F.C., Y.H., and L.D. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cheng, F., Liu, Y., Zhao, Z. et al. Enhanced proton-feeding kinetics of metal-organic framework toward industrial-level H2O2 electrosynthesis for sustainable bleaching. Nat Commun 16, 10183 (2025). https://doi.org/10.1038/s41467-025-65276-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65276-z
