
Abstract
Adenine base editors (ABEs) are powerful tools for gene therapy. However, efficient version of ABEs (e.g. ABE8e) always induce excessive bystander and off-target editing events and are large in size, hindering their potential in clinical disease treatment. Here, we develop a pre-trained Protein-Nucleic Acid Constrained Language Model to design ABE8e with high activity, reduced editing window and decreased size. By further engineering, the smallest ABE8e- PNLM-pcABE- with a 27% size reduction, exhibits high activity, precise 3-nt editing window, and reduced off-target events near background level in HEK293T cells. Compared to ABE8e, PNLM-pcABE has up to 133.5-fold precision improvement in pathogenic mutation correction. By PNLM-pcABE, the albino mouse model carrying desired base mutation is nearly 100% obtained via zygotes microinjection and the expression of PCSK9 substantially decreases in mice receiving in vivo delivery with lipid nanoparticle (LNP), indicating their great potential in gene therapy and disease modeling.
Similar content being viewed by others

Introduction
Fifty-eight percent of genetic diseases are caused by single-nucleotide variation and treatment of those heritable diseases requires safe, effective genome editing tools1. Traditional CRISPR/Cas9-mediated homologous recombination to repair pathogenic point mutations is inefficient1. The new generation of gene editing tools, particularly base editors such as ABEs, which can potentially target 47% of genetic diseases caused by single C•G-to-T•A base conversion, attract growing attention as promising tools for future genetic disease treatment1,2.
In recent years, research groups from the world have made series of improvements on ABEs to achieve high editing efficiency, desired editing windows, and reduced off-target effects. For example, direct evolution of TadA resulted in ABEs with high activity3,4,5; fusion of circularly permuted Cas9n and adenine deaminase shifted the editing window of ABE6; introduction of single-stranded DNA binding proteins into ABE expanded its editing window7; replacing SpCas9 in ABE with SaCas9 or SpCas9 variants expanded its target scope of PAM recognition6,8; engineering TadA reduced or eliminated ABE’s RNA off-target event9,10. In addition, recent studies reported ABE has inherent cytosine deaminase activities9,11. By evolving TadA-8e, ABE can be transformed into base editors with only adenine or cytosine deaminase activity, or both12,13,14,15,16. However, efficient ABEs usually have a wide editing window, potentially leading to increased bystander and off-target editing, which is the main safety concern of their application in genetic disease treatment. We previously developed ABE9 with 1-2nt editing window and no off-target editing through rational TadA-8e design. However, this didn’t shorten its size, making it difficult to deliver in vivo for gene therapy12.
AI-assisted discovery of functional enzymes has been applied in gene editing research. For example, structural prediction methods have been used to explore and develop novel deaminases17,18, and protein language models (PLMs) have shown promises in predicting amino acid mutation effects on enzyme properties19,20. Conventional gene editing-related enzyme evolution have relied on methods such as directed evolution and rational design, which are time-consuming and labor-intensive. In contrast, AI-assisted strategies could avoid meaningless mutations and reduce experimental workload. Although PLMs have been used in antibody engineering to induce affinity maturation for designing protein interactions21, most currently existing PLMs solely focus on the properties and the evolution of monomers, which could limit their application in more complexed interactive systems. Recent studies suggested that incorporating antigen information can induce antibody evolution more accurately22, therefore, an efficient PLM should consider not only the protein of design, but also the substrate it binds to. This theory is particularly applicable to the field of gene editing, as gene editing systems, such as CRISPR/Cas9, rely on the interaction between Cas proteins and nucleic acids to function. These proteins can be potentially fused with deaminases to form a new generation of gene editing tools-base editors that mainly includes adenine base editors (ABEs) and cytosine base editors (CBEs). Especially for ABEs, editing precision remains the crucial challenge for their clinical applications. There is an urgent need to develop more rigorous PLMs that integrate nucleic acid substrate specificity to enhance editing accuracy in therapeutic protein design.
Here, we developed a protein-nucleic acid constrained language model (PNLM) to design adenine deaminase variants for creating precise and compact ABEs. PNLM incorporates substrate information- nucleotide content, marking the first time to our knowledge that editing nucleotide position information is being incorporated into generative models to constrain the protein design. This approach enhances precision and enables the generation of protein sequences with insertions and deletions, facilitating the identification of smaller, functional proteins. There is an urgent need for an efficient ABE with high precision and compactness. Therefore, we selected classic ABE- ABE8e- as an example to generate a subset of TadA-8e variants using this model and experimentally validated their performance in HEK293T cells. Among all TadA-8e variants, the truncated 147–152 aa has a narrow (3nt) editing window while maintaining editing efficiency comparable to ABE8e. To further optimize the ABE, we combine truncated variants and linker deletion to obtain the smallest-size TadA-8e (45aa reduction, 27% size decrease excluding the SpCas9n), named PNLM-pcABE. It has high activity, a precise editing window (3nt), and reduced off-target events (including DNA or RNA off-targets) near background levels. We also demonstrate that PNLM-pcABE can precisely correct pathogenic point mutations with minimal bystander mutations.
Finally, we test PNLM-pcABE’s application in vivo. Via microinjecting PNLM-pcABE into mouse zygotes targeting the Tyrosinase gene, we achieve nearly 100% pups carrying the desired base mutation. In addition, by delivering PNLM-pcABE in mice with lipid nanoparticle (LNP), we precisely target the splice site of the Pcsk9 gene and substantially decrease its expression along with LDL-C level in mice. These two applications of PNLM-pcABE demonstrate that it has promising potential in both gene therapy and disease models.
Results
Establishment and characteristics of protein-nucleic acid constrained language model for adenosine deaminase generation
Generative protein language models are typically trained on diverse natural proteins whose sequences encode valuable information about evolutionary history and biological structure functionality, with sequence variances reflecting constraints and selection pressures23,24. With the growth of protein sequence databases, while the majority of which remain unlabeled in terms of structure or function, unsupervised training on large-scale sequences has become a practical approach and can be effectively applied for sequence generation.
More specifically, our focus is on ABEs, where the precision of base editor is governed by their distinct preferences for specific nucleic acid sequence contexts, reflecting intricate biophysical interactions that dictate their editing accuracy. To generate precise and efficient ABEs, we incorporated target information of ABE and developed a nucleic acid information-constrained protein generation model. We adopted a transfer learning approach by extracting embeddings from the pre-trained protein language model ESM-2 and aligned them with our model (Fig. 1a and Supplementary Fig. 1a). Correspondingly, we additionally extracted embeddings from nucleic acids and annotated editing positions of ABE in representation as constraints for fine-tuning (Fig. 1b). By doing so, we injected the editing target constraint information into the generative model and infer the generated sequences retain original adenine deaminase preferences.

a Transfer learning. Pre-trained protein language models leverage large-scale datasets of protein sequences to learn the relationships and patterns within the amino acid sequences, capturing the underlying grammar and structure of proteins. Generate embeddings on the collected tRNA-specific adenosine deaminase protein sequences by ESM-2 and align PNLM embeddings with them. b Pre-trained language models with expertise were fine-tuned on the collected TadA-8e-like protein sequences and their target ssDNA sequences. c During the autoregressive process, masks can be retained in the output to generate sequences with masks, allowing for the creation of truncated, mutated and inserted sequences.
Additionally, we made further modifications to address issues overlooked by existing protein language models. During protein translation, amino acid substitutions, deletions, duplications, and insertions offer diverse selection templates for evolution. We adopted a masked autoregressive approach to predict and generate each amino acid (Fig. 1c and Supplementary Fig. 1b). During the fine-tuning stage, we introduced a few mask tokens to replace original amino acid tokens, treating them as true labels and excluding them from loss calculations. This encourages the model to simulate the sequence deletions observed in natural evolution, enabling the generation of variable-length sequences that mimic natural variations.
Screening of precise and compact ABEs via PNLM
Using our constructed PNLM model, we explored using PNLM for further engineering TadA-8e proteins. We fine-tuned the PNLM with our curated TadA-8e variant dataset. We utilized the PNLM to generate 150 TadA-8e-like sequences, including 73 mutations, 39 insertions, and 38 truncated variants (Fig. 2a and Supplementary Data 1). During the fine-tuning process, we observed that incorporating nucleic acid and editing position information, compared to only using protein sequences, led to a reduction in the model’s fine-tuning loss (Fig. 2b). We used ESM-1v25 to predict the transformed log-likelihood of all generated sequences and PNLM has higher conservation degree of the catalytic domain (Fig. 2c). Existing machine learning scoring methods demonstrated diverse evaluation techniques. Using the sequence-based evaluation method, ESM-1v25 reported 21 of the generated sequences outperformed the wild-type (Supplementary Fig. 2c and Supplementary Data 1). We further combined ESM-1v25, MIF-ST26, Rosetta27 and AlphaFold228 to screen and characterize the generated truncated sequences(Fig. 2a and Supplementary Fig. 2).

a Schematic of applying PNLM to engineer ABE variants with precise against adenine. Following sequence generation using the PNLM model, truncated variants were selected based on protein sequence alignment. The candidate proteins were then screened using computational methods. Ultimately, the top 20 ranked sequences were chosen for experimental validation. b The validation loss during fine-tuning. Incorporating nucleic acid embeddings during fine-tuning improved the model’s performance by reducing the loss. c In the sequence-based evaluation methods ESM-1v, the 1 + log-likelihood estimation distribution of PNLM-generated sequences is compared to the 1 + log-likelihood estimation of ProGen2-generated sequences and ProtGPT2-generated sequences. Each method generated 50 sequences. The violin plots on the right represent probability density, with internal boxplots showing the median and interquartile range. The scatter plots on the left display the raw data points (n = 50 independent experiments). d The efficiency of A-to-G and C-to-A/T/G of the top 20 ABE8e truncated variants were examined at an endogenous genomic site (ABE site27) containing multiple adenosines and cytidines within the editing window in HEK293T cells, with ABE8e and ABE9 serving as controls. Data are mean ± s.d. (n = 3 independent experiments). e The efficiency of the combinations of truncated variants without XTEN linker, with ABE8e and ABE9 serving as controls, was examined at ABE site27 in HEK293T cells. Data are mean ± s.d. (n = 3 independent experiments). Source data are provided as a Source data file.
Based on the principles above, the top 20 ABE8e variants with size-truncated were selected for construction and a positive endogenous target containing multiple As and editable Cs (ABE site27) was used for testing their performance in HEK293T cells (Supplementary Fig. 3). High-throughput sequencing (HTS) showed that 3 out of 20 ABE8e variants- ABE8e Δ2–8, Δ158–167, and Δ147–152 had editing activity comparable to ABE8e by analyzing the most edited adenine at ABE site 27 (Fig. 2d). Notably, ABE8e Δ147–152 had reduced A-to-G bystander editing and narrowed major A-to-G editing window (A6) compared to ABE8e (A6-A9) (Fig. 2d). Notably, the C bystander editing (C5) activity decreased near background (Fig. 2d). Compared to ABE9, a high-precision version of ABE, the editing activity of ABE8e Δ147–152 was higher and the major editing window was similar (Fig. 2d). The remaining variants exhibited far lower or no activity (Fig. 2d). To further optimize the precision and size of ABE8e Δ147–152, we combined these ABE8e variants and deleted the linker between TadA-8e and Cas9n to narrow the editing window. The results show that ABE8e Δ2–8-NL also maintain comparable editing activity and its major editing window were not changed (Fig. 2e). ABE8e Δ2–8 + 147–152-NL and ABE8e 147–152-NL exhibited similar base editing activity, reduced bystander editing and narrowed major editing window compared to ABE8e (Fig. 2e). Moreover, ABE8e Δ2–8 + 147–152-NL and ABE8e 147–152-NL have high editing efficiency and consistent editing window compared to ABE9, indicating that 147–152aa truncated in TadA-8e may be the main factor for these two ABEs to maintain high efficiency and precision (Fig. 2e). Therefore, we selected ABE8e Δ2–8 + 147–152-NL with more compact structure (45aa reduced) for further investigation, and named it PNLM-pcABE.
Characterization of PNLM-pcABE
To further profile the characteristics of PNLM-pcABE, 21 endogenous targets (9 targets contain multiple As, 9 targets contain editable Cs, and 3 targets contain editable As and Cs) were tested in HEK293T cells with ABE8e and ABE9 as controls. HTS data showed the A-to-G editing efficiency of PNLM-pcABE was 43.8–78.6%, which were higher than that of ABE9 (6.2–84.0%) and slightly lower than that of ABE8e (59.9–92.2%) (Fig. 3a). The major editing window (efficiency >30%) of PNLM-pcABE (A5-A7) was parallel to ABE9 (A5-A6) and narrower than that of ABE8e (A3-A8) (Fig. 3a, b and Supplementary Fig. 4). Notably, at position A6 and A7, the A-to-G editing efficiency of PNLM-pcABE was significantly higher than that of ABE9 (Fig. 3a, b and Supplementary Fig. 4). On the contrary, PNLM-pcABE had lower editing efficiency at position A5 than that of ABE9 (Fig. 3b and Supplementary Fig. 4). We further analyzed the precision using the most edited A/the second-most edited A. Compared to ABE8e, we observed a 0.1–224-fold precision increase for ABE9 and a 0.2–126-fold precision increase for PNLM-pcABE (Fig. 3a, c). By further analyzing motif preference, we found that PNLM-pcABE had no obvious motif preference but had low efficiency at RA (R = A or G) motif, similarly to ABE8e and ABE9 (Fig. 3a and Supplementary Fig. 5). Moreover, like ABE9, the bystander Cs editing activity was nearly eliminated in PNLM-pcABE by analyzing 12 targets containing editable Cs (Fig. 3d). In addition, the indels of PNLM-pcABE were also significantly lower than that of ABE8e and ABE9 (Fig. 3e). These data suggested PNLM-pcABE was an efficient base editing tool with high precision and compactness in size.

a The A-to-G base editing efficiency of ABE8e, ABE9 and PNLM-pcABE weas examined at 12 endogenous genomic loci containing multiple As in HEK293T cells. Heatmap reflects averaged data from three biological replicates. b The average A-to-G base editing efficiency of ABE8e, ABE9 and PNLM-pcABE at 12 endogenous genomic loci containing multiple As in Fig. 3a. Data are mean ± s.d. (n = 3 independent experiments) and p values were determined by a two-sided paired Wilcoxon rank-sum test. (ABE9 vs PNLM-pcABE: p = 0.0002 at A5, p = 0.0044 at A6, p = 0.0005 at A7) c The normalized ratio of highest/sub-optimal A-to-G base editing efficiency for ABE8e and PNLM-pcABE at the 12 target sites in Fig. 3a. Data are mean ± s.d. (n = 3 independent experiments). d The C-to-D (T/G/A) editing efficiency of ABE8e, ABE9 and PNLM-pcABE was examined at 12 endogenous genomic loci containing one C or multiple Cs in HEK293T cells. Heatmap reflects averaged data from three biological replicates. e The indel frequency formation of ABE8e, ABE9 and PNLM-pcABE at 21 endogenous genomic loci in Fig. 3a, d. Data are mean ± s.d. (n = 3 independent experiments) and p values were determined by a two-sided paired Wilcoxon rank-sum test (ABE8e vs ABE9: p = 0.0078; ABE8e vs PNLM-pcABE: p <0.0001; ABE9 vs PNLM-pcABE: p = 0.0012). Source data are provided as a Source data file.
Off-target evaluation of PNLM-pcABE
Next, we performed DNA off-target assessment of PNLM-pcABE in three ways: sgRNA-dependent DNA off-target, sgRNA-independent DNA off-target and the whole-transcriptomic RNA off-target. First, for sgRNA-dependent DNA off-target, 63 off-targets in total were selected for evaluation-50 of which were in silico predicted off-target sites from PD-1-sg4, PCSK9-sg1, PCSK9-sgA, HAAVR-sg4, VEGFA-sg3 and TTR-sg6 using Cas-OFFinder29, and 13 were from previously known Cas9 off-target sites (HEK site 2 and HEK site 4) identified by GUIDE-seq or ChIP-seq30. Our results showed that 8 off-target sites were observed for ABE8e and none for PNLM-pcABE and ABE9 (Fig. 4a and Supplementary Fig. 6). Second, for sgRNA-independent DNA off-target, Modified R-loop assay31 was applied for evaluation. HTS data showed that PNLM-pcABE induced no sgRNA-independent DNA off-target, even a performance far superior than that of ABE8e (Fig. 4b and Supplementary Fig. 7). Third, to comprehensively assess the whole-transcriptomic RNA off-target, we co-transfected HEK293T cells with plasmids encoding ABE8e, ABE9, or PNLM-pcABE and on-target sgRNA (HEK site2) (Fig. 4c and Supplementary Fig. 8). Seventy-two hours after transfection and the total mRNA of the cells were harvested for RNA-Seq. the RNA-seq results show that ABE8e exhibited some RNA off-target events (Fig. 4c). However, PNLM-pcABE, similar to ABE9, induced minimal off-target events close to background level (Fig. 4c). In a summary, PNLM-pcABE was a highly efficient base editing tool with minimal off-target events.

a Cas9-dependent DNA on and off-target analysis at the indicated targets (HEK site2、PD-1-sg4、PCSK9-sg1 and HAAVR-sg4) of ABE8e, ABE9 and PNLM-pcABE in HEK293T cells. Data are mean ± s.d. (n = 3 independent experiments) and p values were determined by a two-tailed Student’s t-test (ABE8e vs PNLM-pcABE: for HEK site2, p < 0.0001 at GUIDE-seq-OT1, p = 0.0002 at CHIP-seq-OT1, p = 0.0002 at CHIP-seq-OT3, p <0.0001 at CHIP-seq-OT5; for PD-1-sg4, p = 0.0023 at OT5, p = 0.0021 at OT7, p = 0.4904 at OT10). b Cas9-independent DNA off-target analysis of the modified orthogonal R-loop by ABE8e, ABE9 and PNLM-pcABE. Data are mean ± s.d. (n = 3 independent experiments) and p values were determined by a two-tailed Student’s t-test (ABE8e vs PNLM-pcABE: p <0.0001 at R-loop1, p < 0.0001 at R-loop2, p = 0.0002 at R-loop3, p < 0.0001 at R-loop4, p = 0.0010 at R-loop5, p = 0.0019 at R-loop6). c RNA off-target editing activity by ABE8e, ABE9, PNLM-pcABE using RNA-seq, GFP is a negative control. Each biological replicate is listed on the bottom. Source data are provided as a Source data file.
Precise correction of pathogenic mutations using PNLM-pcABE
To further validate PNLM-pcABE’s potential for gene therapy with high precision and efficiency, stable HEK293T cell lines were generated with 2 pathogenic mutations (GALT c.413 C > T, variation ID: 25174, Transferase Deficiency Galactosemia32; OTC c.533 C > T, variation ID: 97237, Ornithine transcarbamylase deficiency33) adjacent to bystander mutations that are potentially deleterious in the ClinVar database. The results showed that PNLM-pcABE exhibited higher desired base editing efficiency than ABE9 at A6 in GALT (74.6%) and A6 in OTC (82.0%), though slightly lower than ABE8e at corresponding sites (Fig. 5a, b). However, ABE8e also induced deleterious no-desired base mutation with high efficiency, such as A3 in GALT and A3 in OTC, which bring the possibility of additional disease34, while PNLM-pcABE had far lower or no editing efficiency at these sites (Fig. 5a, b). We further compared the precision between ABE8e and PNLM-pcABE by analyzing the most edited adenines/the second-most edited adenines. The results showed PNLM-pcABE had 133.5- and 10.3-fold precision improvement than that of ABE8e, which has better performance than ABE9 at those two targets (Fig. 5a, c).

a Comparison of correction efficiencies for pathogenic mutations mediated by ABE8e and PNLM-pcABE in two stable HEK293T cell lines, including GALT c.413 C > T and OTC c.533 C > T. The heat map represents editing efficiency of A-to-G. The efficiency of ABE8e and PNLM-pcABE were determined by HTS. Heatmap reflects averaged data from three biological replicates. b The target sequences of the two gene pathogenicity locus in Fig. 5a. Above the sequences are the corresponding amino acids. The green sequence area represents the main editing window of ABE8e. The disease locus A is in bold red and labeled ▲. The potential pathogenicity locus is in bold blue and labeled×. The PAM sequence is in purple. Specific locus information and Variation IDs are listed. Below are alleles and frequencies of the pathogenic mutations corrected by ABE8e, ABE9 and PNLM-pcABE. The wild-type allele frequencies were omitted. c The normalized ratio of A-to-G base editing efficiency of ABE8e, ABE9 and PNLM-pcABE in the two previously mentioned stable HEK293T cell lines. Data are mean ± s.d. (n = 3 independent experiments) d The pie chart on the top left shows distribution ratio of pathogenetic point mutations that could potentially be corrected by base editors in the ClinVar database (accessed 16 July 2024). On the top right is a schematic diagram of pathogenic point mutations correctable by ABEs. Venn diagrams on the bottom show the pathogenic mutations that can be suitably corrected by ABE8e, ABE9 and PNLM-pcABE in NGG and NG PAM contexts without introducing bystander editing (ABE8e: Corrects pathogenic A in the 3–9 editing window without additional As; ABE9: Corrects pathogenic A at position 5, other As allowed in positions 3–9 except position 6; PNLM-pcABE: Corrects pathogenic A at position 6 or 7, other As allowed in positions 3–9 except position 5). Source data are provided as a Source data file.
Among the 47% pathogenic point mutations can be potentially corrected by ABEs in the ClinVar database. We further counted the pathogenic mutations targeted by ABE8e that could cause a risk of disease from bystander editing. In all pathogenic point mutations, 5413, 1841 and 3282 were suitable for correction without introducing risk mutations using ABE8e, ABE9, and PNLM-pcABE, respectively (Fig. 5d and Supplementary Data 4). When PAM was further expanded from NGG to NG, the number of precise disease-treatment events increased to 14,472 for ABE8e, 6033 for ABE9 and 10,354 for PNLM-pcABE, respectively (Fig. 5d and Supplementary Data 4). These data suggested that PNLM-pcABE has great potential in precise targeting specific base for future clinical gene therapy.
Generation of albinism mouse models with high precision using PNLM-pcABE
Accurate production of mouse disease models is essential for basic research and clinical treatment. The reported mouse disease models produced using efficient base editors often induce bystander editing in addition to targeted base editing, which potentially perturbs the analysis of the relationship between the disease phenotype and base mutations. We utilized PNLM-pcABE to target the Tyrosinase gene, disrupting the normal expression of the Tyr gene by destroying the splice site, leading to albinism12. Here, PNLM-pcABE mRNA and previously used sgRNA targeting the splicing acceptor site of the Tyrosinase gene were co-injected into mouse zygotes12 (Fig. 6a, b). The results showed that PNLM-pcABE efficiently and precisely induced desired base editing (A6) with minimal bystander editing (A9) in 12/13 born pups (Fig. 6c, f), similar to that of ABE9 but not for ABE8e12. In all F0 mice carrying single A-to-G mutation, the allele base editing efficiency of PNLM-pcABE was significantly higher than that of ABE8e, suggesting the high precision of PNLM-pcABE (Fig. 6f). These edited mice also exhibited albino phenotype in the eyes and fur color of the founders (Fig. 6d, e). Furthermore, the analysis of 9 off-targets from in silico prediction off-targets in all mice revealed that PNLM-pcABE produced no additional off-target editing (Fig. 6g). These data suggested that PNLM-pcABE is an efficient and precise base editing tool to be used for mouse or other mammalian embryo to generate desired disease models.

a Schematic diagram of mouse embryo injection. b The splice acceptor sequence of intron 3 of the mouse Tyr gene was targeted by PNLM-pcABE. The splice acceptor site “AG” is shown in red, and the neighboring adenine is in green. The sgRNA sequence is underlined, and the PAM sequence is in blue. c Genotyping of all F0 generation pups treated with ABE8e (n = 4) and PNLM-pcABE (n = 13). The alleles and frequencies were analyzed by CRISPResso2. The percentage values on the right represent the frequencies of the indicated mutant alleles. The frequency of the wild-type allele was omitted. d, e Phenotypes of F0 mice generated by microinjection of sgRNA and ABEs. In the left photo, the mice were 3 days old, showing the albino phenotype in eye. In the right photo, the mice were 14 days old, exhibiting the albino phenotype in their fur color. f Single A-to-G editing at the indicated Tyr site by ABE8e (n = 4) and PNLM-pcABE (n = 13) in F0 pups. Data are mean ± s.d. (n = 4 independent F0 mice for ABE8e and n = 13 independent F0 mice for PNLM-pcABE), and p values were determined by a two-sided unpaired Wilcoxon rank-sum test (p = 0.0017). g DNA off-target effects at the indicated Tyr site mediated by ABE8e (n = 4) and PNLM-pcABE (n = 13) in F0 pups. Data are mean ± s.d. (n = 4 independent F0 mice for ABE8e and n = 13 independent F0 mice for PNLM-pcABE). Source data are provided as a Source data file.
In vivo adenine base editing Pcsk9 in mice using PNLM-pcABE
Next, we tested PNLM-pcABE as a tool for in vivo gene therapy. We chose Pcsk9, a gene that has been extensively investigated in its relationship with hypercholesterolemia35, to verify PNLM-pcABE’s ability as a gene editing tool in vivo for treating hypercholesterolemia. After three weeks of delivery of ABE8e or PNLM-pcABE mRNA and a previous reported sgRNA targeting splice donor site of Pcsk9 packaged in LNP (Fig. 7a). We collected the genomic DNA for base editing efficiency measurement using HTS and blood samples for the quantification of PCSK9 and LDL-C level using ELISA in mice. The results showed that both ABE8e and PNLM-pcABE effectively induced base editing at the splicing site of Pcsk9. PNLM-pcABE precisely edited the desired base (A6) with minimal bystander editing (A4), whereas ABE8e induced both desired base mutation (A6) and bystander base mutation (A4) (Fig. 7b). By calculating the ratio of the targeted base editing efficiency (A6)/the bystander base editing efficiency (A4), the editing precision of PNLM-pcABE was 2.2-fold times than that of ABE8e (Fig. 7c). Furthermore, a substantial decrease in the expression levels of PCSK9 and LDL-C was observed in both ABE8e- and PNLM-pcABE- treated groups, although ABE8e had better performance (Fig. 7d, e). In conclusion, these data indicated that PNLM-pcABE provided an alternative platform for in vivo gene therapy of hypercholesterolemia and other genetic disorders by precisely targeting the desired base.

a Schematic diagram of delivering LNPs for in vivo editing of Pcsk9 in mice. The splice donor sequence of exon 1 of the mouse Pcsk9 gene was targeted by PNLM-pcABE. The blue sequence area represents the Exon1 of Pcsk9, and the yellow represents the Exon2. The splice donor site “GT” is shown in red, and the neighboring adenine is in green. Blood was collected on the 2nd day before injection and on the 7th, 14th, and 21st days after injection, respectively. b The A-to-G editing efficiency of Pcsk9 in mice 3-weeks after the delivery of LNPs packaged with ABE8e / PNLM-pcABE and sgRNA. Data are mean ± s.d. (n = 3 different mice) c The normalized ratio of A6 / A4 A-to-G base editing efficiency of ABE8e and PNLM-pcABE at the Pcsk9 site in Fig. 7b. Data are mean ± s.d. (n = 3 different mice). d, e The expression of PCSK9 and LDL-C in plasma of adult mice before and after the delivery of LNPs packaged with ABE8e/PNLM-pcABE and sgRNA. Data are mean ± s.d. (n = 3 different mice). Source data are provided as a Source data file.
Discussion
In this study, we introduce a protein-nucleic acid constrained Language Model, a pre-trained model to generate TadA-8e-like proteins. Additional analyses suggest that our model has learned domain-specific information, allowing it to generate functional proteins in a nucleic acid-constrained manner. However, due to the scarcity of structural on base editors and nucleic acid complexes, we find it challenging to construct multi-feature models. In the future, as deep learning methods mature, we can consider exploring more sophisticated network architectures and incorporating multi-feature information into language models, such as incorporating molecular surface fingerprints, interacting residues, and three-dimensional structural information to enhance their robustness and performance.
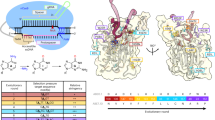
PNLM-pcABE, generated by a pre-trained Protein-Nucleic Acid constrained Language Model, exhibits high efficiency, a condensed editing window, and a shortened size. Truncation of 147–152 amino acids is the primary reason for PNLM-pcABE’s superior performance. The TadA-8e introduces 6 mutations at α53, splitting α5 into two separate helices that undergo a sharp 180° turn at P152. The R152P mutation has been shown to be critical for ABE8e deamination activity36. Through comparing the structure of PNLM-pcABE (AF3 predicted) and TadA-8e (PDB: 6VPC), deletion from D147 to P152 for PNLM-pcABE abolishes the sharp turn. Aligning the predicted structure of PNLM-pcABE to TadA-8e in 6VPC and compared the surface (electrostatic surface calculated in ChimeraX) of their terminal helices both with non-target stranded DNA (NTS) in 6VPC. The PNLM-pcABE complex appears to present a larger interface, which stables the U-turn conformation of NTS, facilitating for site-specific deamination. Thus, the deletion from D147 to P152 reform the C-terminal α-helix (α5), enabling additional interactions with NTS which may contribute to the reduction of bystander mutations (Supplementary Fig. 9). The 147–152 amino acids in TadA-8e can affect the editing activity and editing window of ABE8e, consistent with previous reports that the F148A in TadA narrowed ABE7.10’s editing window10 and Y149F mutation enhanced selectivity for adenine editing and reduced cytosine editing activity12.
Compared to ABE8e, PNLM-pcABE exhibited slightly decreased editing activity and a narrowed editing window from original 3–8 to 5–7. However, PNLM-pcABE’s editing window is similar to ABE9. ABE8e’s average editing efficiency at A5 was lower than ABE9, while ABE8e’s efficiencies at A6 and A7 were higher than ABE912, indicating PNLM-pcABE and ABE9 are complementary precision base editing tools. The 45aa reduction in PNLM-pcABE versus ABE8e and ABE9 suggests PNLM-pcABE or optimal TadA-8e truncations fused with SaCas9 or small-size nuclease are conducive to AAV packaging for in vivo delivery in gene therapy.
In summary, we developed the ABE-PNLM-pcABE via AI-assisted design. PNLM-pcABE is an elegant base editing tool with precision, high efficiency, and small size, holding great application prospects in gene therapy and beyond.
Methods
Training nucleic acid conditioned protein language model
A total 34,255 sequences were collected by searching for tRNA-specific adenosine deaminase in UniPortKB37 and applying filters on Enzyme Classification 3.5.4.33. Additionally, variants sequences of TadA-8e were obtained from published data3,4,5, resulting in a total of 27 sequences.
Such constrained language model was implemented by designing a Nucleic Acid conditioned language model. During pre-training, where only protein information was used to capture the general sequence patterns of tRNA-specific adenosine deaminases, the reverse of each sequence was added to the training set as a data augmentation strategy to enhance sequential learning. The PNLM aims to match the model output to the embeddings generated by ESM-238. Given a protein sequence, the overall loss function for pre-training is:
where \({x}_{i}\) denotes the \(i\)th amino acid of sequence, N is the length of the protein sequence, \({E}_{{xi}}\) is the representation of \({x}_{i}\) and \({\hat{E}}_{{xi}}\) is the prediction of representation. The model was optimized using Adam (β1 = 0.9, β2 = 0.999)39 with a learning rate of 1e–06. For the decoder, loss was computed as the cross-entropy between predicted logits and sequence labels. The best model checkpoint was selected according to validation loss.
In the fine-tuning process, single-stranded DNA editing data is used to achieve single-base resolution annotation of sequences and provide information about the enzyme’s substrate and editing sites. The embeddings of a batch of ssDNA sequences are represented as a tensor \({E}_{{s}{s}{D}{N}{A}}\in {\mathbb{R}}^{batch\times {L}_{{s}{s}{D}{N}{A}}}\), where batch corresponds to the batch size for the input and \({L}_{{s}{s}{D}{N}{A}}\) is the length of the nucleic acid sequence. The editing position is encoded by another encoder and represented as a tensor \({E}_{{p}{o}{s}{i}{t}{i}{o}{n}}\in {\mathbb{R}}^{batch\times {L}_{{s}{s}{D}{N}{A}}}\). The conditional embeddings were then concatenated with \({E}_{{s}{s}{D}{N}{A}}\) and \({E}_{{p}{o}{s}{i}{t}{i}{o}{n}}\), forming a unified tensor that serves as the input \({E}_{conditioned}\) for the language model. The loss function in the fine-tuning stage is:
where xi denotes the i-th animo acid of sequence. The model was allowed to produce mask tokens in place of amino acids and to continue predictions based on the masked context. Consecutive masks shorter than five tokens were treated as true labels, thereby encouraging truncated sequence generation during inference.
Sequence generation and screening
A total of 150 TadA-8e-like protein sequences were generated, with a sampling initiation temperature of T = 1 and top_p = 0.9. In this context, top_p refers to the cumulative probability used during the generation process when dynamically selecting the next token. After removing sequences containing insertions and mutations, 38 truncated proteins were retained to prioritize smaller candidates. These proteins were screened through a sequential sequence- and structure-based filtering pipeline. AlphaFold2 (ColabFold v1.5.537) was used to predict protein structures, and sequences with pLDDT <84 were excluded. ESM-IF was then applied to score the predicted structures according to log-likelihood. Rosetta energy evaluation was subsequently performed to assess structural stability and charge, retaining sequences with total scores within 100 units of the wild type and charge differences within 50 units. Finally, 20 candidates were selected for experimental validation.
Evaluation script
The scores derived from the ESM−1v25 models represent the mean of the logarithmic probabilities assigned to each amino acid at various positions within a sequence. MIF-ST scores are the average log-likelihood of query residues in predicted structures using alphafold2. Rosetta-based analyses were conducted using the Rosetta software suite, which is available through Rosetta Commons under a specific academic license (https://www.rosettacommons.org).
Plasmid construction
The plasmid DNA sequences employed in this research are listed in the Supplementary Data 5. The ABE8e (#138489) and lentiCRISPR v2 (#52961) plasmids were obtained from Addgene. To amplify the target fragment, polymerase chain reaction (PCR) was conducted using KOD-Plus-Neo DNA Polymerase (TOYOBO, Code: KOD-401). The plasmids in this study were generated using the ClonExpress MultiS One Step Cloning Kit (Vazyme), with ABE8e or lentiCRISPR v2 serving as the backbone vectors for molecular cloning. The construction of sgRNA expression plasmids was performed in accordance with the methodology previously described8. In brief, the oligonucleotides from Supplementary Data 2 were annealed at 95 °C for 5 min, then cooled to room temperature and ligated into BbsI-linearized vectors for sgRNA (Thermo Fisher Scientific).
Human cell culture and cell transfection
The HEK293T (ATCC; CRL-3216) cell lines were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco) supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco) and 1% penicillin-streptomycin (Gibco) antibiotic. All cell lines were maintained under standard conditions at 37 °C with 5% CO2 in the incubator. For both the on-target and off-target base editing experiments utilizing DNA, HEK293T cells were seeded into 24-well plates and transfected at approximately 80% confluency. Subsequently, 100 μl serum-free medium, comprising 3 μl of polyethyleneimine (PEI, Polysciences), 750 ng of the ABEs expression plasmid and 250 ng of the sgRNA expression plasmid (1 μg of plasmid DNA in total), was added to the cells. Three days following transfection, genomic DNA was extracted using the QuickExtract™ DNA Extraction Solution (QE09050, Epicenter), in accordance with the manufacturer’s instructions.
Stable cell line generation
To construct the HEK293T stable cell line, a 220-bp fragment containing disease-associated mutation flanked by ~100 bp was cloned into the lentiviral vector with puromycin-resistant gene expression maker. For lentiviral production, a total of 12 μg of the lentiviral specified transfer plasmid (Lenti-GALT-sg1 and Lenti-OTC-sg1), alongside 6 μg of pMD2.G (#12259) and 9 μg of psPAX2 (#12260) were co-transfected into HEK293T cells in a 10 cm dish at approximately 85% confluence. The virus-containing supernatant was harvested at 72 h post-transfection. The supernatant was subjected to centrifugation at 1699 × g for 10 min at 4 °C, with the objective of precipitating cell debris. Following this, the supernatant was filtered through a 0.45 µm low protein-binding membrane (Millipore). And then serially diluted to add into a 24-well plate, cultured with 5 × 104 HEK293T cells per well. After 24 h, transduced cells were replated in 2 μg/mL puromycin-containing medium for selection. Following 7 days of the puromycin selection, the pools cells were spread into a 96-well plate, and single clone cells were harvested and expand for cell transfection.
High-throughput DNA sequencing (HTS) and data analysis
On- and off-target genomic regions were amplified by PCR using primers detailed in Supplementary Data 2. HTS amplification libraries were prepared by PCR using KOD-Plus-Neo DNA Polymerase and, site-specific primers containing an adapter sequence (Forward 5′-ggagtgagtacggtgtgc-3′; Backward 5′-gagttggatgctggatgg-3′) at their 5′ ends. The resulting products underwent a second PCR using primers containing different barcode sequences. Subsequently, PCR products with different tags were pooled together for deep sequencing on Illumina HiSeq platform. The reference sequence between the positive direction primers was selected for sequencing analysis. The base editing or indel efficiencies were quantified using BE-Analyzer40 or CRISPResso241.
Enhanced orthogonal R-loop assay
In this study, the augmented orthogonal R-loop assay was employed for Cas9-independent DNA off-target analysis, substituting the dSaCas9-sgRNA plasmid with the nSaCas9-sgRNA plasmid at each R-loop site. In the transfection process, 100 μl serum-free medium, comprising 3 μl of polyethyleneimine (PEI, Polysciences), 375 ng nSaCas9-sgRNA plasmid, 375 ng base editor plasmid and 250 ng sgRNA plasmid (1 μg of plasmid DNA in total), was added to the cells. Following a three-day period following transfection, the cells were digested using 0.25% trypsin (Gibco) for sorting. Subsequently, genomic DNA was extracted with the utmost care using the QuickExtract™ DNA Extraction Solution (QE09050, Epicenter), in accordance with the manufacturer’s instructions.
Preparation of mRNAs and sgRNAs and microinjection in mice
Chemically modified sgRNAs with 2′-O-methyl and phosphorothioate modifications at the first three 5′ and 3′ terminal RNA residue was synthesized by GenScript (Nanjing, China) (Supplementary Data 5). mRNA preparation was performed as following. The T7 promoter was introduced into PNLM-pcABE template by PCR using the primers T7-mRNA-F/R (Supplementary Data 2). The base editor mRNA was transcribed in vitro using mMESSAGE mMACHINE T7 Kit (Invitrogen) and purified using a MEGAclear Kit (Invitrogen)30.
Six to eight weeks old C57BL/6 J and ICR mice, sourced from the Institutional Animal Care and Use Committees (IACUCs) at the Suzhou Institute of Systems Medicine and housed under a specific pathogen-free (SPF) condition in a controlled environment (12-h light/dark cycle, 20–22 °C, 40–60% humidity) with ad libitum access to food and water, were used as embryo donors and foster mothers, respectively. The methods of microinjection in mice are as follows: A 2 nl mixture of PNLM-pcABE mRNA (200 ng/μl) and sgRNA (100 ng/μl) was co-injected into one-cell stage wild-type embryos. About 20 days after transplantation, the mice were born, and genomic DNA from the tail of these born pups was isolated using the QuickExtract™ DNA Extraction Solution (QE09050, Epicenter) according to the manufacturer’s instructions.
LNP treatment and serum analysis
All mice cohorts were maintained at SPF facilities in Suzhou Institute of Systems Medicine and approved by Institutional Animal Care and Use Committee. Feeding conditions were as described above. A total 200 μl mix of LNP (2 mg/kg) packaged PNLM-pcABE mRNA and an sgRNA targeting the Pcsk9 at a 2:1 ratio (Starna Therapeutics Co., Ltd., Suzhou) was delivered to 6–8 weeks old C57BL/6 J mice intravenously via tail vein injection. A control group received an equivalent volume of normal saline. To track the serum levels of PCSK9 and low-density lipoprotein cholesterol (LDL-C), mice were fasted overnight for 12 h prior to blood collection via tail tip sampling. To minimize batch effects, serum samples from all the time points were collected and analyzed concurrently. Blood samples were allowed to clot at room temperature, after which serum was separated by centrifugation. PCSK9 levels were measured using an ELISA kit (Proteintech, #KE10050), while LDL-C levels were assessed using assay kits from Solarbio (#BC5335). All procedures were conducted in strict adherence to the manufacturers’ instructions. For terminal procedures, mice were euthanized using carbon dioxide inhalation. The median lobe of the liver was excised for DNA extraction to evaluate on-target editing efficiency.
Structure analysis based on Alphafold3
PNLM-pcABE and PNLM-pcABE-ssDNA binary structures were predicted using the AlphaFold3 web server and imported into ChimeraX (version 1.8), along with the structure of ABE8e (PDB: 6VPC). Upon inspection, the predicted binary complex did not position the ssDNA precisely within the ABE pocket. To resolve this, PNLM-pcABE was structurally aligned to TadA-8e using the “matchmaker” command, and the TadA-8e protein was hidden to visualize the composite structure of PNLM-pcABE bound to ssDNA. Electrostatic surface potentials were calculated using the “coulombic” command. The ssDNA chain from TadA-8e was colored in gray, and its base atoms were displayed to highlight potential interactions with the protein.
Statistics and reproducibility
All statistical analyses were performed on a minimum of three biologically independent experiments using a two-tailed Student’ s t-test, a two-sided paired Wilcoxon rank-sum test or a two-sided unpaired Wilcoxon rank-sum test via Prism software, version 10.6.1(GraphPad). A p-value of less than 0.05 was considered statistically significant, with the specific p-values indicated in the figure legend. No statistical method was used to predetermine sample size. No data were excluded from the analyses. The experiments were not randomized. The Investigators were not blinded to allocation during experiments and outcome assessment.
Ethical statement
The research strictly followed all applicable ethical standards and guidelines. All procedures involving mice were meticulously designed according to institutional and national standards and have received approval from the Institutional Animal Care and Use Committees (IACUCs) at the Suzhou Institute of Systems Medicine, Chinese Academy of Medical Sciences & Peking Union Medical College, Suzhou, China.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Targeted amplicon sequencing data and RNA-seq data used in this study have been deposited in the NCBI Sequence Read Archive Database under Accession Code PRJNA1291838. Plasmids encoding PNLM-pcABE are available from Addgene (#237618). The data generated in this study are provided in the Source Data file. Source data are provided with this paper.
Code availability
The code for generating sequences and scoring for TadA-8e variants is available under the Apache 2.0 license at our GitHub repository(https://github.com/yao-jiawei/PNLM). The code for calculating pathogenic point mutations suitable for PNLM-pcABE in the ClinVar database has been deposited in Code Ocean (https://codeocean.com/capsule/2242462/tree) without any access restrictions or usage limitations.
References
Rees, H. A. & Liu, D. R. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 19, 770–788 (2018).
Gaudelli, N. M. et al. Programmable base editing of A• T to G• C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017).
Richter, M. F. et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 38, 883–891 (2020).
Gaudelli, N. M. et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 38, 892–900 (2020).
Xiao, Y. L., Wu, Y. & Tang, W. An adenine base editor variant expands context compatibility. Nat. Biotechnol. 42, 1442–1453 (2024).
Huang, T. P. et al. Circularly permuted and PAM-modified Cas9 variants broaden the targeting scope of base editors. Nat. Biotechnol. 37, 626–631 (2019).
Xue, N. et al. Improving adenine and dual base editors through introduction of TadA-8e and Rad51DBD. Nat. Commun. 14, 1224 (2023).
Yang, L. et al. Increasing targeting scope of adenosine base editors in mouse and rat embryos through fusion of TadA deaminase with Cas9 variants. Protein Cell 9, 814–819 (2018).
Grunewald, J. et al. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat. Biotechnol. 37, 1041–1048 (2019).
Zhou, C. et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature 571, 275–278 (2019).
Kim, H. S., Jeong, Y. K., Hur, J. K., Kim, J. S. & Bae, S. Adenine base editors catalyze cytosine conversions in human cells. Nat. Biotechnol. 37, 1145–1148 (2019).
Chen, L. et al. Engineering a precise adenine base editor with minimal bystander editing. Nat. Chem. Biol. 19, 101–110 (2023).
Jeong, Y. K. et al. Adenine base editor engineering reduces editing of bystander cytosines. Nat. Biotechnol. 39, 1426–1433 (2021).
Neugebauer, M. E. et al. Evolution of an adenine base editor into a small, efficient cytosine base editor with low off-target activity. Nat. Biotechnol. 41, 673–685 (2023).
Chen, L. et al. Re-engineering the adenine deaminase TadA-8e for efficient and specific CRISPR-based cytosine base editing. Nat. Biotechnol. 41, 663–672 (2023).
Lam, D. K. et al. Improved cytosine base editors generated from TadA variants. Nat. Biotechnol. 41, 686–697 (2023).
Huang, J. et al. Discovery of deaminase functions by structure-based protein clustering. Cell 186, 3182–3195.e3114 (2023).
Xu, K. et al. Structure-guided discovery of highly efficient cytidine deaminases with sequence-context independence. Nat. Biomed. Eng. 9, 93–108 (2024).
Cheng, P. et al. Zero-shot prediction of mutation effects with multimodal deep representation learning guides protein engineering. Cell Res. 34, 630–647 (2024).
He, Y. et al. Protein language models-assisted optimization of a uracil-N-glycosylase variant enables programmable T-to-G and T-to-C base editing. Mol. Cell 84, 1257–1270 e1256 (2024).
Hie, B. L. et al. Efficient evolution of human antibodies from general protein language models. Nat. Biotechnol. 42, 275–283 (2024).
Shanker, V. R., Bruun, T. U. J., Hie, B. L. & Kim, P. S. Unsupervised evolution of protein and antibody complexes with a structure-informed language model. Science 385, 46–53 (2024).
Madani, A. et al. Large language models generate functional protein sequences across diverse families. Nat. Biotechnol. 41, 1099–1106 (2023).
Ferruz, N., Schmidt, S. & Hocker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nat. Commun. 13, 4348 (2022).
Meier, J. et al. Language models enable zero-shot prediction of the effects of mutations on protein function. Adv. Neural Inf. Process. Syst. 34, 29287–29303 (2021).
Yang, K. K., Zanichelli, N. & Yeh, H. Masked inverse folding with sequence transfer for protein representation learning. Protein Eng. Des. Sel. 36, gzad015 (2023).
Nivon, L. G., Moretti, R. & Baker, D. A Pareto-optimal refinement method for protein design scaffolds. PLoS ONE 8, e59004 (2013).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Bae, S., Park, J. & Kim, J. S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014).
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016).
Doman, J. L., Raguram, A., Newby, G. A. & Liu, D. R. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat. Biotechnol. 38, 620–628 (2020).
Yang, Y. P., Corley, N. & Garcia-Heras, J. Molecular analysis in newborns from Texas affected with galactosemia. Hum. Mutat. 19, 82–83 (2002).
Oppliger Leibundgut, E. O., Liechti-Gallati, S., Colombo, J. P. & Wermuth, B. Ornithine transcarbamylase deficiency: new sites with increased probability of mutation. Hum. Genet. 95, 191–196 (1995).
Yamaguchi, S., Brailey, L. L., Morizono, H., Bale, A. E. & Tuchman, M. Mutations and polymorphisms in the human ornithine transcarbamylase (OTC) gene. Hum. Mutat. 27, 626–632 (2006).
Fitzgerald, K. et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet 383, 60–68 (2014).
Li, J. et al. Structure-guided engineering of adenine base editor with minimized RNA off-targeting activity. Nat. Commun. 12, 2287 (2021).
UniProt Consortium, T. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 46, 2699 (2018).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Kingma, D. P. & Ba, J. Adam: A Method for Stochastic Optimization. In Proc. International Conference on Learning Representations (ICLR, 2014).
Hwang, G. H. et al. Web-based design and analysis tools for CRISPR base editing. BMC Bioinf 19, 542 (2018).
Clement, K. et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019).
Acknowledgements
We thank Professor Dali Li from East China Normal University for providing many base editors plasmids and some necessary experimental materials to accelerate the process of this project. We thank Jun-Jie Gogo Liu and Shuo Lin from the School of Life Sciences of Tsinghua University for their help in elucidating the working mechanism of PNLM-pcABE from a structural biology perspective. We thank Yuanqing He and Naiyun Ma from the Laboratory Animal Science Center of Suzhou Institute of Systems Medicine for their help in generating base-edited mice. This work was partially supported by grants from National Key R&D Program of China (No. 2022YFC3400200 to X.Z. and No. 2022YFA1103400 to X.Z.), the National Natural Science Foundation of China (No. 82522046 to X.Z., No. 82350003 to X.W. and No. 32201223 to X.Z.), the NCTIB Fund for R&D Platform for Cell and Gene Therapy, the CAMS Innovation Fund for Medical Sciences (No. 2022-I2M-1-024 to X.Z., No. 2022-I2M-2-004 to X.Z. and No. 2023-I2M-2-005 to X.Z.), the Suzhou Municipal Key Laboratory (No. SZS2022005), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (No. 2022-RC180-08 to X.Z.), the Ministry of Education, Singapore (MOE) (T1251RES2309 and T2EP20125-0039 to Y.Z.), and the Agency for Science, Technology and Research, Singapore (A*STAR) (H25J6a0034 to Y.Z.).
Author information
Authors and Affiliations
Contributions
X.Z. and X.W. designed and supervised the project; J.R. and Y.N.L. performed the experiments and data analysis for base editing in HEK293T cells and mouse embryos with the help of Z.Z.Z; J.R., and Q.C. performed the experiments and data analysis for base editing in vivo in mice; Q.C. performed the analysis for correcting pathogenic point mutations with base editors in the ClinVar database. J.Y., Y.L., and S.W. designed and completed Protein-Nucleic Acid constrained Language Model associated experiments with the crucial advice of X.W. and Y.Z.; X.Z., and X.W wrote the manuscript with the input from all the authors and X.G polished it. All the authors contributed to this manuscript.
Corresponding authors
Ethics declarations
Competing interests
X.Z., J.R., Y.N.L., Z.Z., and J.W. have submitted patent applications (application no. 202311487517.3, under review) based on the results reported in this study. This patent mainly relates to PNLM-pcABE in this paper. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Shun-Qing Liang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ren, J., Yao, J., Cao, Q. et al. Protein-nucleic acid language model-assisted design of precise and compact adenine base editor. Nat Commun 16, 11207 (2025). https://doi.org/10.1038/s41467-025-65311-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65311-z
