
Abstract
Diverse microorganisms can execute one or more steps in denitrification, during which nitrate or nitrite is successively reduced into nitric oxide, nitrous oxide, and ultimately dinitrogen. Many of the best-characterized denitrifiers are complete denitrifiers capable of executing all steps in the pathway, but their dominance in natural communities and what metabolic traits and environmental factors drive the global distribution of complete vs. partial denitrifiers are unclear. To address this, we conducted a comparative analysis of denitrification genes in 61,293 genomes, 3991 metagenomes, and 413 terrestrial and aquatic metatranscriptomes. We show that partial denitrifiers outnumber complete denitrifiers and the potential to initiate denitrification is more common than the potential to terminate it, particularly in nutrient rich environments. Our results further indicate that complete denitrifiers tend to be fast-growing organisms, favoring organic acid over sugar metabolism, and encoding the ability to oxidize and reduce a broader range of organic and inorganic compounds compared to partial denitrifiers. This suggests complete denitrifiers are metabolically flexible opportunists. Together, our results indicate an environmental footprint on the presence of denitrification genes which favors the genomic potential for partial over complete denitrification in most biomes and highlight that completion of the denitrification pathway is a community effort.
Similar content being viewed by others


Introduction
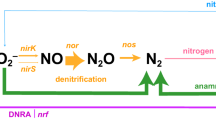
The potent greenhouse gas and ozone depleting agent nitrous oxide (N2O) is produced in several microbial N-cycle processes, of which denitrification plays a dual role in both producing and consuming N2O. In this facultative anaerobic microbial respiratory pathway, nitrate (NO3−) or nitrite (NO2−) are used as electron acceptors and successively reduced into nitric oxide (NO), N2O, and dinitrogen (N2) by a diverse range of predominantly bacterial species. As the genes encoding the enzymes involved in these reductive steps can be independently gained and lost1,2,3, they are found in different combinations among genomes4, such that denitrification can be executed by complete denitrifiers with genes encoding enzymes for all steps or, alternatively, split between multiple partial denitrifier community members. Recent studies suggest that genetically complete denitrifiers are scarce among genomes assembled from environmental data5,6,7,8,9,10. However, much of our understanding of the physiology and regulation of denitrification is based on studies of a very phylogenetically restricted set of complete denitrifiers11,12,13,14,15. Thus, there is pressing need for a broader look at the prevalence, diversity and ecology of organisms genetically capable of complete and partial denitrification, particularly considering their differential roles in serving as sources and sinks of N2O16.
Split pathways divide the protein production costs for a pathway between multiple cells and may enable higher ATP flux17,18, increasing the specific growth rate. Although this could be a fitness advantage under nitrite-rich conditions, ATP yields per molecule nitrate or nitrite would be lower for partial compared to complete denitrification. This is expected to lead to partial denitrification being favored when electron acceptor supply is not limiting, but complete denitrification being favored when the flux of electron donors such as carbon (C compounds is high relative to electron acceptor availability). Splitting the denitrification pathway also increases dependencies between community members, not only because the denitrification product of one organism serves as the electron acceptor for another19,20, but also because the regulation of specific reactions may rely on intermediates that those executing the reaction themselves cannot use21. Further, there is less buildup of toxic intermediate compounds, which may allow microorganisms only capable of carrying out the initial steps of denitrification avoid the cytotoxic effects of NO2−, particularly under low pH environments11,22. This jointly indicates that split pathways may open more niches, thereby supporting a greater diversity of denitrifiers20. If organisms encoding partial vs. complete denitrification fulfill different niches, this should also be reflected in additional traits, such as growth rate, abiotic optima, and the ability to metabolize different substrates and use electron acceptors beyond those in the denitrification pathway. For instance, if complete denitrification is indeed associated with C influx, complete denitrifiers would be expected to be capable of higher growth and broader substrate catabolism compared to partial denitrifiers. Addressing the linkages between the complete or partial denitrification and the capacity for other traits can help identify the broader roles and conditions that favor each denitrifier type.
Here we combined comparative genomics (Fig. 1a) with a global survey of denitrification gene ratios in environmental metagenomes and metatranscriptomes to evaluate the diversity of bacterial denitrifier types, their metabolic traits, and their distribution across engineered, environmental and host-associated biomes. We posited that environmental factors favoring or disfavoring execution of the different steps leave a genetic footprint in the form of presence and absence of genes encoding the enzymes responsible for them, as well as via differential associations between denitrification enzyme genes and redox traits. Furthermore, considering organisms encoding a complete denitrification pathway are able to use a broader range of nitrogenous terminal electron acceptors than those encoding fewer, we hypothesized that the former would have broader metabolic flexibility. We used a framework in which partial denitrifiers include genetically defined initiators, which encode genes involved in NO2− but not N2O reduction, and terminators, which encode genes for N2O but not NO2− reduction (Fig. 1b). We used NO2− reduction, the decisive step of denitrification23, as the first point of our analysis. We combined comparative genomics of 61,293 non-redundant bacterial isolate, metagenome assembled (MAGs), and single cell genomes with 3991 metagenomes derived from all major biomes and 413 terrestrial and aquatic metatranscriptomes to determine the frequency of a complete vs. partial denitrification pathway and the genetic potential to initiate vs. terminate denitrification at the genome and community level. Here we show evidence for dominance of partial over complete denitrifiers in genomes, metagenomes, and metatranscriptomes. We interpret our results in the context of resource availability and complementary genomic traits associated with the denitrifier types in our framework and propose that complete denitrifiers are metabolically flexible generalists.

a Overview of experimental approach for comparative genomics. Metagenome-assembled, single cell and isolate bacterial genomes covering environmental, host-associated, and engineered biomes were extracted from GTDB v.214.1 (Supplementary Data 2). Denitrification genes were annotated using a combination of HMM-based searches, phylogeny construction, and manual inspection of alignments. Denitrifier genomes were then searched for additional functional traits including maximum growth rate, transcription factors and potential to use various electron donors and acceptors. b Venn diagram of prevalence of denitrifier types. The number inside each circle denotes the percentage of denitrifier genomes with that genotype. Letters in italics denote whether genotype is considered an initiator (i), terminator (t) or is capable of both (init-term; it). Reactions completed by each enzyme and corresponding denitrifier type name are listed below the diagram, with the lines under each step defining the colors used in the Venn diagram. Genomes encoding nor without accompanying nir or nosZ genes are marked with an asterisk and classified as non-denitrifiers. c Stacked bar charts denoting relative proportion of genomes belonging to phyla within each denitrifier type, with non-denitrifiers defined as containing neither nir nor nosZ, or being archaeal nitrifiers or anammox bacteria (see “Methods”). The number of genomes corresponding to each denitrifier type is indicated above the corresponding bar. Phyla represented by fewer than 100 denitrifier genomes are aggregated under “other” and can be found in Supplementary Data 11. d Proportion of denitrifier genomes within dominant phyla encoding each denitrification enzyme gene as well as the complete denitrification pathway. Numbers to the right in (d) indicate the number of denitrifier genomes belonging to each phylum denoted in the graph. Source data are provided as a Source Data file.
Results
Partial denitrification dominates microbial genomes
We assessed the prevalence of partial and complete denitrification pathways among 61,293 bacterial genomes represented in GTDB release 214.1 (Fig. 1a). Genes for the denitrification enzymes NirK (nirK) and NirS (nirS) catalyzing NO2− reduction to NO, all proposed NO-reducing heme copper oxidases (i.e., Nor (nor)24), and NosZ (nosZI, nosZII) catalyzing N2O reduction were annotated using hidden Markov models trained on manually curated databases of these genes25. Genomes carrying nor but not nir or nosZ accounted for 5% of the total number of assemblies and were excluded in downstream analyses because Nor in nor-only genomes are likely involved in detoxification rather than respiration. With this conservative approach, 18% of the genomes (n = 11,126) were considered denitrifiers and included nir and/or nosZ with or without nor. Among these, 23% encoded genes for complete NO2− reduction to N2 (complete denitrifiers; Fig. 1b), with the complete denitrification trait more prevalent among isolates compared to MAGs (29% vs. 17%, respectively, Supplementary Table 1). However, there was considerable variation between phyla (Fig. 1c, Supplementary Fig. 1). Twenty-eight different bacterial phyla encoded the complete denitrification trait, with an overwhelming dominance of Pseudomonadota in our dataset (Fig. 1c). Among phyla represented by at least 100 denitrifier genomes, complete denitrifiers were most prevalent within Campylobacterota (38% of denitrifiers), Pseudomonadota (37%), and Myxococcota (26%, Fig. 1d).
Using our framework of initiators and terminators, we found that initiators encoding nir with or without nor but not nosZ were broadly more prevalent than terminators having nosZ with or without nor but not nir (49% vs. 24% of genomes with denitrification genes, respectively). This may represent the potentially divergent functions for NO2− versus N2O reduction as a means of growth or detoxification vs. maintenance26. Partial denitrifiers occurred in 68 bacterial phyla, with initiators being overrepresented within Actinomycetota (98% initiators, 1% terminators) and Nitrospirota (88%, 8%) (Fig. 1d). However, Nitrospirota accounted for just a small fraction of initiator genomes (2.7%), while the majority were Pseudomonadota (47%) and Actinomycetota (22%) (Fig. 1c). We found a higher proportion of terminators than initiators within Planctomycetota (76% terminators, 19% initiators) and Bacteroidota (60%, 18%) (Fig. 1d), with the latter phylum accounting for the majority of terminators in the dataset (53%; Fig. 1c). We also found genomes carrying nir and nosZ but not nor, suggesting a third category: the initiator-terminators. They accounted for just 4% of genomes with denitrification genes, including 13% of Gemmatimonadota, 13% of Chloroflexota, 9% of Bacteroidota and 5% of Verrucomicrobiota (Fig. 1c). It is possible that these organisms either depend on NO reduction by other organisms or produce N2O via pathways that do not depend on Nor27,28,29,30.
Among all genomes carrying nosZ (i.e., terminators, initiator-terminators, and complete denitrifiers), those encoding clade I nosZ were more likely to encode the complete denitrification trait compared to genomes encoding clade II (74% in clade I vs. 26% in clade II; Supplementary Fig. 2), as previously observed4. Within clade II, complete denitrification was most prevalent in Pseudomonadota and Aquificota, with 80% of Pseudomonadota and 94% of Aquificota encoding complete denitrification compared to an average of 20% in the remaining phyla. This suggests that the difference in clade I vs. clade II prevalence can be used as a proxy for complete vs. partial denitrifier prevalence within the N2O reducing community, but not if clade II is dominated by Pseudomonadota or Aquificota.
Inferred ecological preferences of complete vs. partial denitrifiers
We next assessed whether the genetic potential for complete denitrification or for initiating rather than terminating denitrification was associated with the predicted growth rate and resource use patterns among bacteria. Using the codon bias-based maximum growth rate prediction tool gRodon31, we categorized genomes into fast- (growth rate faster than 0.2 h−1) and slow-growing (growth rate lower than 0.2 h−1)31. Rapid growth was more common among complete denitrifiers (83%) compared to initiators (70%), terminators (65%) and initiator-terminators (62%; Fig. 2a upper panel) and for genomes encoding clade I (85%) compared to clade II NosZ (65%; Fig. 2a lower panel). Similarly, estimated median growth rates of complete denitrifiers (0.46 h−1) were greater than that of initiators (0.28 h−1), terminators (0.23 h−1) and initiator-terminators (0.20 h−1; overall Kruskall-Wallis χ2 = 401, Dunn P < 0.0001 in all cases). This faster potential growth rate was associated with a lower sugar to acid preference of complete denitrifiers compared to all three of the categories of partial denitrifiers, based on the total sum of genes involved in each pathway32 (Fig. 2b). This was accompanied by a higher density of transcription factors (Fig. 2c) and transporters (68 Mbp−1 in complete denitrifiers vs. 65 Mbp−1 in initiators, 40 Mbp−1 in terminators, and 42 Mbp−1 in initiator-terminators, P < 2 × 10−16). Furthermore, complete denitrifiers were inferred to grow on more of the 56 organic substrates examined (median of 20 corresponding to 4.75 Mbp−1), compared to initiators (13; 3.32 Mbp−1), terminators (11; 2.90 Mbp−1), and initiator-terminators (9; 2.55 Mbp−1; Fig. 2d). This was driven by complete denitrifiers using a broader range of organic and amino acids compared to partial denitrifiers (median of 15 vs. 6–7), rather than sugars (median of 2 vs. 2–3).

a Estimated maximum growth rate of organisms based on codon usage bias. Stacked bar charts show the maximum predicted growth rate for genomes separated by denitrifier type (upper panel) and NosZ clade (lower panel). Slow-growing taxa (maximum growth rate <0.2 h−1) are depicted to the left on the horizontal axis and fast-growing taxa to the right. Numbers to the right denote number of genomes used in maximum growth rate predictions and exclude genomes with fewer than 10 ribosomal proteins annotated. b Boxplots of sugar and organic acid preference of denitrifiers, based on KEGG annotation ratios32. c Genomic density of transcription factors inferred using DeepTFactor. d Boxplot of substrate use counts by denitrifier type and substrate category based on GapMind, with colors following (b, c). e Heatmap of terminal electron acceptors and donors, with intensity of color and number denoting proportion of genomes within denitrifier type encoding redox ability. Terminal oxidases are near-ubiquitous and are not included in figure or totals. In (b–d), box boundaries represent first and third quartiles, with midline denoting median. Whiskers denote the 1.5 IQR, and outliers are shown as individual points. Asterisks above boxes indicate a difference between complete denitrifiers and the partial denitrifier type and no comparisons between partial denitrifier types were made. Numbers to the right in (e) denote number of genomes used in (b–e). Elemental S elemental sulfur, Init-term initiator-terminator. Source data are provided as a Source Data file.
We subsequently examined the potential for partial and complete denitrifiers to use a range of inorganic compounds as electron donors and acceptors (Fig. 2e). While the ability to oxidize inorganic compounds such as hydrogen may be associated with an ability to tolerate energy-limited conditions33, the ability to reduce inorganic compounds may indicate that one type or another of denitrifiers is better able to tolerate lower redox conditions. Although most genomes lacked the genetic potential to oxidize inorganic substances or reduce non-nitrogenous terminal electron acceptors (median = 0) other than oxygen, complete denitrifiers had the genetic potential to both oxidize and reduce more compounds than partial denitrifiers (Kolmogorov-Smirnov statistic P < 0.0001 in all cases). Almost all (99%) genomes encoded the potential for aerobic respiration, with low affinity oxidases more prevalent in complete denitrifiers (87%) than initiators (83%; χ2 = 15.03, P = 0.0001), and similar in prevalence to terminators (85%; χ2 = 0.083, P = 0.7734) and initiator-terminators (86%; χ2 = 0.91, P = 0.3397). High affinity variants were more prevalent among complete denitrifiers (99%) vs. initiators (89%; χ2 = 228, P < 2.2 × 10−16), terminators (94%; χ2 = 84.63, P < 2.2 × 10−16) and initiator-terminators (88%; χ2 = 142.82, P < 2.2 × 10−16).
The inferred abiotic niche breadths of complete denitrifiers were not uniformly broader than that of partial denitrifiers. Complete denitrifiers had slightly broader inferred temperature ranges compared to initiators (26.84 vs. 25.12 °C; t = −24.583, P < 2 × 10−16), terminators (25.54 °C, t = −18.569, P < 2 × 10−16) and initiator-terminators (25.93 °C, t = −8.038, P = 1.02 × 10−15). pH ranges were narrower in complete denitrifiers compared to initiators (3.75 vs. 3.83; t = 8.136, P = 4.44 × 10−16) and broader compared to terminators (3.58; t = −6.756, P = 1.50 × 10−11). Salinity ranges were broader in complete denitrifiers than initiators (5.64 vs. 5.31; t = −3.775, P = 0.000162) but narrower than in initiator-terminators (5.92; t = −2.441, P = 0.014665).
We subsequently assessed the degree to which complete and partial denitrifiers were associated with high or low resource availability using CoverM34 to map reads from the TARA Oceans35 project to MAGs derived from the same data36. No subset of the metagenome data we collected or other databases we screened had a sufficient number of complete denitrifier MAGs and environmental data to allow for this analysis, highlighting the rarity of complete denitrifiers in global ecosystems. Nitrate was used as a proxy for denitrification electron acceptor availability and chlorophyll concentration as a proxy for electron donor supply because of its positive association with gross primary productivity37. The proportion of reads mapping to the collection of 170 MAGs in the dataset differed by sample and ranged from 5 to 30%. We calculated a standardized environmental response by correlating the proportion of reads mapping the denitrifier types among the MAGs (7 complete, 13 initiators, 14 terminators, 134 non-denitrifiers and 2 initiator-terminators) with chlorophyll and NO3− concentrations in different samples and standardizing this to the maximum observed relative abundance of each MAG. We found that terminators and complete denitrifiers tended to have a greater increase in relative abundance with increasing NO3− than non-denitrifiers, but there was no difference between complete denitrifiers and partial denitrifiers (Supplementary Fig. 3a). Mean standardized chlorophyll and chlorophyll: NO3− ratio responses were similar across all groups of organisms, independent of denitrifier type (Supplementary Fig. 3b, c). One possible reason for the apparent lack of correlation between denitrifier type and resource availability is that these bulk measurements likely do not represent the microhabitat that denitrifiers experience. For instance, pre-filtering of the samples depleted much of the particulate organic matter, where denitrification activity is concentrated38, and where modeling indicates that complete denitrifiers are favored20. Nonetheless, the results from this pelagic marine dataset do not indicate that measured resource availability affects the prevalence of complete or partial denitrifier genomes.
Prevalence of denitrification initiators and terminators varies across global biomes
Next, we assessed variation in denitrification initiator and terminator prevalence and complete and partial denitrifiers among terminators (i.e., N2O reducers) at the community level in a broad range of environments. We screened 3991 metagenomes derived from soil, aquatic, engineered and host-associated biomes for nirK, nirS, and nosZ clades I and II using an HMM-based search and phylogenetic placement approach39. The difference in the copy number of nosZ and nir genes normalized to Gbp sequenced in each metagenome (δnos-nir) was used as a proxy for the genetic potential for termination compared to initiation of denitrification, and nosZ clade I vs. clade II (δnosZI-nosZII) as a proxy for complete denitrifier dominance within the N2O reducing community. Delta values were chosen for their ability to account for differences in overall denitrifier prevalence between biomes, including cases where one or both of the denitrification genes being compared were not detected.
Across the majority of biomes, nir gene fragments were typically more abundant than nosZ, indicating a greater or similar potential to initiate than terminate denitrification within the denitrifier communities (Fig. 3a). Marine mats served as an exception, and nosZ prevalence exceeded that of nir by 60%. This would occur if there were higher rates of NO3− assimilation or low inputs of NO2− from nitrification40, as suggested by the overall low prevalence of nir and lack of ammonifier associated nirK clades observed previously in these samples25. nosZ Clade II was more prevalent than nosZ Clade I in nearly all biomes considered, (Fig. 3b). This indicates that partial denitrifiers generally dominate terminator communities because partial denitrification is more prevalent among Clade II nosZ. This is further supported by our observation that phyla depleted in complete denitrifiers, such as Bacteroidetes, Chloroflexota and Gemmatimonadota, dominated nosZ clade II across biomes (Fig. 3c, d). Therefore, we can conclude that the majority of organisms capable of terminating denitrification in global biomes do not also initiate this process. This is particularly the case in croplands, marshes, and activated sludge from municipal wastewater treatment plants, which were the biomes where δnosZI-nosZII is lowest for terrestrial, marine, and engineered environments (Fig. 3b). The dominance of nosZII coincides with the highest total nosZ and nir gene abundances (Supplementary Data 1), indicating conditions that are overall more favorable for denitrification also promote N2O reducers that are partial rather than complete denitrifiers. A notable exception to this pattern is sewage communities, which were dominated by clade I nosZ despite having high overall nir gene fragment prevalence, and seems to largely reflect the preponderance of Acidovorax in these samples41.

Boxplots showing difference in nos versus nir counts ((nosZ I + nosZ II) – (nirK + nirS)) (a); and clade I and clade II nosZ counts (b) per Gb sequenced. Biomes were compared using Benjamini-Hochberg FDR-corrected pairwise ranked comparisons following Kruskall-Wallis, and common letters to the right in each boxplot denotes biomes with similar median gene prevalences. Numbers to the right denote the number of metagenomes included for each biome. Box boundaries represent first and third quartiles, with midline denoting the median and whiskers the 1.5 IQR. Mean composition of clade I (c) and clade II nosZ reads (d), organized by biome and colored by phylum. Biomes represented by fewer than 20 metagenomes are excluded from the figure. The percentage of genomes having clade II nosZ and a complete denitrification pathway are indicated for each phylum in the legend and can be seen for both clades in Supplementary Fig. 2. Standard errors corresponding for each phylum are available in Supplementary Data 12. Host assoc. host-associated, Engineer. engineered. Source data are provided as a Source Data file.
We also assessed the correlation between the diversity of denitrifiers based on phylogenetic diversity of the dominant nitrite reductase gene, nirK and potential initiator-terminator or clade I and II differential abundance25. We found that nirK phylogenetic diversity was positively correlated with δnos-nir in soils (Spearman’s ρ = 0.46, P < 2.2 ×10−16) and marine samples (ρ = 0.19, P = 1.18 × 10−11), while nirK diversity was positively correlated with δnosZI-nosZII in soils (ρ = 0.29, P < 2.2 × 10−16) and uncorrelated in marine samples (ρ = 0.03, P = 0.35). These results indicate that more diverse denitrifier communities occur where capacities for initiating and terminating denitrification are relatively more balanced, such that complete denitrification at the community level is associated with greater niche partitioning.
Environmental drivers of nir vs. nosZ and nosZ clade prevalence were assessed using random forest modeling on the largest soil and marine datasets with complete metadata represented in our analysis42. We generated accumulated local effect plots to show the main effect of the target variable on predicted δnos-nir (Fig. 4) and δnosZI-nosZII (Fig. 5), while accounting for the other predictors. Models explained less than half of the variance in gene differences, except for δnosZI-nosZII in soils. Increases in soil organic carbon content up to ~1% positively affected δnos-nir, while the effect of soil NO3− content was always negative (Fig. 4a). nosZ was also predicted to become less prevalent compared to nir as available phosphorus increased, indicating that high nutrient conditions in soils favor NO2− reducers over N2O reducers. Within the marine dataset, predicted δnos-nir was lower at the highest concentrations of the sum of NO3− and NO2− compared to the lowest concentrations, and showed a dramatic shift in prediction from a decreasing to increasing relationship with temperature at 22 °C (Fig. 4b).

Abiotic predictors of the difference in nos and nir gene counts, calculated as (nosZ I + nosZ II) – (nirK + nirS) and normalized per Gb sequenced in soil (a) and marine metagenomes (b). Accumulated local effects plots show the differences in prediction of the δnos-nir (y-axis) compared to the mean prediction along the range of each predictor (x-axis), while accounting for potential correlations among predictor values. Values above zero indicate the model predicts higher than average dominance of nosZ over nir at a given value of the predictor variable. The analyses were performed on a subset of the soil (n = 298) and marine (n = 89) metagenomes for which relevant environmental metadata was available (see “Methods”). Root mean square error and variance explained were 37 and 27% in the soil (a) and 19 and 26% in marine (b) models, respectively. Vertical marks on the x axis in each panel denote data density. Five hundred trees were run, and mtry, nodesize, and sampsize parameters were 4, 8, and 238 for soil and 2, 3, and 71 for the marine models, respectively. Source data are provided as a Source Data file.

Abiotic predictors of the difference in nosZ clade I and II gene counts normalized per Gb sequenced in soil (a) and marine metagenomes (b). Accumulated local effects plots show the differences in prediction of the δnosZI-nosZII (y-axis) compared to the mean prediction along the range of each predictor (x-axis), while accounting for potential correlations among predictor values. Values above zero indicate the model predicts higher than average dominance of nosZ clade I over nosZ clade II at a given value of the predictor variable. The analyses were performed on a subset of the soil (n = 298) and marine (n = 89) metagenomes for which relevant environmental metadata was available (see “Methods”). Root mean squared error and variance explained were 41 and 58% in the soil (a) and 14 and 30% in marine (b) models, respectively. Vertical marks on the x axis in each panel denote data density. Five hundred trees were run, and mtry, nodesize, and sampsize parameters were 7, 8, and 238 for soil and 2, 10, and 62 for the marine models, respectively. Source data are provided as a Source Data file.
Available NO3−, phosphorus, potassium, and zinc had a negative effect on predicted δnosZI-nosZII (Fig. 5a), which suggest that partial N2O reducers increase with increasing nutrient levels. pH and clay content also had a negative effect on predicted δnosZI-nosZII, though only pH had a negative effect over the entirety of measured values. Increases in elevation at low altitude were associated with increased predicted δnosZI-nosZII, but additional increases in elevation were not associated with further increases. In the marine study, temperature and ammonium concentrations had opposing unimodal effects on predicted values over their observed ranges (Fig. 5b).
Expression of denitrification termination and initiation in the environment
We subsequently quantified the presence of nosZ and nir transcripts in 413 metatranscriptomes from soils and aquatic environments, distributed among five biomes, to assess whether the biome-level differences and associations between environmental variables and δnosZI-nosZII and δnos-nir gene abundance were also apparent in gene expression. nirK transcripts were strongly dominated by archaeal nitrifier reads (median 50%, range 0–100%), which we excluded in our analysis (see “Methods”). Consistent with the metagenome analysis, nir was more prevalent than nosZ and clade II nosZ was more prevalent than clade I nosZ (Fig. 6a). The soil studies either directly or indirectly manipulated carbon availability, and soils associated with higher carbon availability had higher δnos-nir expression than their paired lower-carbon samples (SMD 0.75, p = 0.015) but did not have higher δnosZI-nosZII (SMD 0.06, p = 0.73). Among aquatic studies, which were all observational, correlation coefficients were negative between δnos-nir and the NO3− + NO2− (mean −0.27, CI: −0.53 to 0.00) and positive between δnos-nir and chlorophyll concentrations (0.22, CI: 0.04, 0.41; Fig. 6b). δnos-nir was not correlated with dissolved oxygen or phosphate concentrations, or with bacterial production, another proxy for resource availability. δnosZI-nosZII was positively correlated with dissolved oxygen content, which would occur if the dominant organisms encoding clade I NosZ lowered oxygen concentrations around their N2O reductase better than those encoding clade II43.

a δnos-nir and δnosZI-nosZII normalized per Gbp mRNA sequenced across different biomes. Numbers to the right denote number of metatranscriptomes included in each category. b Mean and 95 % confidence intervals of spearman correlation coefficients between δnos-nir or δnosZI-nosZII and environmental factors in aquatic metatranscriptomes. Coloring follows gold for δnos-nir and red for δnosZI-nosZII as in (a). Circle size and numbers to the right denote the number of studies the effect size is calculated from. Soil samples with root exudate addition are categorized under rhizosphere in (a). Box boundaries represent first and third quartiles, with midline denoting median and whiskers the 1.5 IQR. Source data are provided as a Source Data file.
Discussion
By combining a comparative analysis of genomes with broad metagenomic and metatranscriptomic surveys, we provide fresh insight into the prevalence of the complete and partial denitrification trait among global microbial communities. In contrast to a previous comparative genomics study based on a small set of isolated microbes4 but consistent with more recent studies of MAGs from different environments5,9,44,45,46,47, we found that the genetic potential for complete denitrification is less common than that for partial denitrification, among both genome-sequenced microorganisms and environmental communities from all major biomes. Our results show denitrification is primarily a community trait based on division of labor, thereby relying cross feeding between microorganisms producing and consuming NO2−, NO and N2O to complete the pathway. Furthermore, other genes and genomic traits implicate complete denitrifiers as metabolically flexible generalists compared to any partial denitrifier type.
Various explanations exist for the dominance of partial denitrifiers. We show that partial denitrification enables greater overall niche partitioning, which was supported by narrower substrate ranges, lower capacity for oxidizing and reducing various inorganic electron donors and acceptors, and a lower genomic density of transcription factors and transporters among partial denitrifiers compared to complete denitrifiers. In the environment, higher nirK phylogenetic diversity, an indication of greater functional diversity among nitrite reducers25, was associated with an increasing balance between capacity to initiate and terminate denitrification at the community level. This would further support niche partitioning if the pathways were split among partial denitrifiers, which is likely considering that that we mainly observed negative δnos-nir values in the environment and the fact that MAGs are predominantly (84%) partial denitrifiers. Partial denitrifiers may also dominate communities due to the presence of a rate-efficiency tradeoff, i.e., that energy flux is slower through long pathways but potentially enables more complete usage of the terminal electron acceptor when it is limiting18. However, the dominance of partial denitrifiers among N2O reducers in the environment was not coherently explained by a rate-efficiency tradeoff. If this were the case, we would expect complete denitrification to decline as NO3− increases or as C:NO3− and C:NO2− ratios decrease, assuming denitrifiers are predominantly heterotrophs in the environment. This agrees with predictions based on modeling and observations in oxygen minimum zones in the oceans, proposing that the prevalence of a complete pathway increases as the limiting substrate shifts from C to N20. Accordingly, our random forest modeling inferred that the N2O reducing community in higher NO3− soils became more dominated by partial denitrifiers, i.e., those carrying nosZ clade II, and fertilized croplands had more negative δnosZI-nosZII than other soils in our cross-biome study. However, a similar reduction in δnosZI-nosZII was not associated with increasing NO3− + NO2− in aquatic metagenomes and metatranscriptomes or in the analysis mapping reads to marine MAGs. In addition, there was no relationship between increasing carbon content in soil or higher chlorophyll concentrations in aquatic samples and higher prevalence of complete denitrifiers in either metagenomes or metatranscriptomes. This is consistent with a recent qualitative analysis of ~1600 MAGs44, which concluded that a rate-efficiency tradeoff cannot explain observed partial denitrifier dominance. Based on our findings of broader substrate ranges and greater genomic allocation to substrate uptake and transcriptional regulation, we instead propose that complete denitrifiers are adapted to flexibly take advantage of resources varying in time and space, while partial denitrifiers are restricted to slower and therefore less variable growth rates. This may explain the over-representation of complete denitrifiers among model denitrifiers and isolates that grow readily under standard resource-rich lab conditions. Under carbon-rich conditions supporting rapid growth in the environment, complete denitrifiers may benefit from using available NO3−/NO2− more efficiently20, while under rapidly fluctuating conditions many have the flexibility to use whichever electron donor and terminal electron acceptors are most readily available, including oxygen via high affinity terminal oxidases. This indicates that partial denitrifiers dominate because they have cheaper metabolisms to run, that do not require such complex regulatory mechanisms. This is consistent with the overall dominance of slow-growing organisms in natural environments31,48,49.
The community level genetic balance between initiation and termination of denitrification indicates a higher capacity for N2O production than reduction in nearly all biomes. The variations between biomes and among the specific ecosystems within biome categories were readily explained by environmental variables. We found that high NO3− levels were associated with lower δnos-nir in both soils and aquatic systems, indicating greater imbalance between the two genes and a decrease in the relative importance of N2O reducers. In soils, the same pattern was observed for phosphorous. High concentrations of NO3− may also allow denitrifiers to outcompete ammonifiers, which both theoretical50,51 and empirical evidence indicate prevail under high C:NO3− 52,53, including in the soil metagenomes used in this study42. Overall, this shows that nutrient rich environments promote denitrifier communities dominated by initiators and similar patterns were observed at the transcriptional level in aquatic environments (no data for soils). The observed imbalance in the denitrifier community could explain why nutrient loaded environments like agricultural soils are major sources of N2O. However, complete denitrifiers have been observed to express only enzymes involved in denitrification initiation under high NO3− and/or NO2− 54,55, which also lead to accumulation of N2O. Furthermore, high fertilization rates commonly favor nitrifier-denitrification56, and hydroxylamine from nitrification and high NO2− concentrations can both induce N2O production from chemodenitrification56,57. Finally, N2O may also be produced during the detoxification of NO from host immune responses58,59. Therefore, there are a wealth of pathways that are distinct from denitrification and can reduce NO2− and/or produce N2O, and it is plausible that inter-pathway competition for NO2− favors the presence of initiators ready to consume NO2− in denitrification rather than letting it enter other pathways. Terminators could conceivably take advantage of the accumulated N2O from all these possible sources, in particular obligate terminators (i.e., predominantly nosZ clade II) which use N2O as terminal electron acceptor and thereby serve as important N2O sinks16,60. This reasoning agrees with the observed low δnosZI-nosZII in metagenomes from agricultural soils and wastewater treatment plants. We conclude that high levels of NO3− allow both initiation and termination specialists to prevail, but with a dominance of initiators which altogether increase the risk of N2O emissions.
Fixed carbon availability was associated with reduced dominance of nir over nosZ in aquatic transcriptomes and soil metagenomes, in particular in soils with less than 1% SOC. The soil metatranscriptomes also indicated increased carbon availability could help NosZ overcome a low competitiveness for electrons61,62,63. However, the chemical composition of the available carbon is expected to affect the balance between initiation and termination of denitrification and complete vs. partial denitrifiers. Organic acids have short catabolic pathways and fewer opportunities for enzyme bottlenecks to slow the flow of electrons into the electron transport chain compared to sugars and should favor complete denitrifiers able to use an array of terminal electron acceptors, in agreement with our comparative genome analysis. At the community level however, organisms metabolize and co-metabolize64 substrates from among tens of thousands of molecularly-distinct potential electron donors of varying bioavailability65,66, leading to conflicting effects of sugar and organic acids addition on N2O to N2 production from soils67,68,69. High dissolved oxygen concentrations were also positively associated with dominance of clade I over clade II nosZ I expression but not gene abundance in aquatic samples. There is no clear consensus on clade I being less sensitive to high oxygen than clade II NosZ70,71, and oxygen niche of N2O reducers may be a function of organism oxygen consumption43 or even indirectly their competition for NO2− with nitrite oxidizing bacteria20. Thus, while the mechanism for oxygen preference cannot be inferred from our data, our results nonetheless indicate that terminators and complete denitrifiers make distinct contributions to N2O reduction as a function of environmental conditions.
Our results indicate that the prevalence of denitrification functional types within and between environments should be considered in light of not only the potential organism level advantages to having a particular denitrification genotype under given environmental conditions, but also through biotic interactions. Denitrification does not occur in isolation, and its steps may occur in response to its intermediates being produced by other pathways. Further, spatiotemporal variation in electron donor and acceptor ratios can enable coexistence of organisms completing different steps of denitrification. Future work addressing phenotypic plasticity of denitrifiers under realistic environmental conditions is necessary to resolve this uncertainty and disentangle biotic and abiotic drivers of denitrifier community assembly. This will be particularly enhanced by improved generation of high-quality assemblies of representative environmental genomes, and by the ability to sample microbial communities on the scale at which they interact with one another and the environment.
Methods
Denitrification enzyme databases
Alignments and phylogenies for NirK and NirS were obtained from Pold et al.25. The Nor reference database was derived from Murali et al.24 and consisted of 67 Nor sequences (cNOR, qNOR, bNOR, eNOR, gNOR and sNOR, and nNOR) and 865 other heme copper oxidases. A hidden Markov model for NosZ was generated downloading all bacteria and archaea genomes from NCBI (7 October 2021) and using hmmsearch v.3.2.172 to identify NosZ using an HMM built from the alignment from in Graf et al.73. DNA sequences were translated to amino acids before being dereplicated at 100% identity with CD-HIT (v 4.674). After initial maximum likelihood (ML) trees reconstruction with FastTree v. 2.1.1175, removal of sequences derived from genomes either identified as contaminated in the GenBank metadata or as <80% complete or >5% contaminated by BUSCO v. 5.3.176, we manually checked the alignment for conserved residues, aligned poorly-conserved regions of the sequence, trimmed the alignment to its conserved core in Arb v.7.077, and built a phylogeny using IQ-TREE v. 2.1.3 with the best model identified by modelfinder (LG + R10)78. Nitrate reductases, catalyzing the first step in the denitrification pathway, were excluded from the analyses because they are found in a wide variety of microorganisms and their activity often occurs in isolation from the characteristic gas-producing denitrification process itself23,79.
Genome annotation
We used the representative set of genomes and associated genome quality metadata from GTDB v214.1 for our analysis (Supplementary Data 280). Genomes less than 80% complete, more than 5% contaminated, or which failed taxonomic consistency of contigs based on GUNC v.1.0.681 were excluded from our analysis, resulting in 61,293 genomes in our final dataset. We included both isolate (49% of assemblies) and culture-independent genome assemblies (metagenome assembled and single cell genomes; 51%) in our analysis to balance genome quality with inclusion of genetic diversity representative of environmental samples. We identified genes for NirK, NirS, Nor, and NosZ in each genome using an HMMsearch72 against the corresponding amino acid databases as described above. After aligning potential proteins to the reference, we imported them into ARB v.7.077 and used a combination of phylogeny building with FastTreeMP v.2.1.1175 and manual curation to exclude reads lacking conserved ligand binding domains. Nor and NosZ were also classified according to protein class (i.e., bNOR, cNOR, eNOR, gNOR, nNOR, qNOR and sNOR for Nor and halophiles, clade I and clade II for NosZ). We then classified each genome into non-denitrifiers (lacking nir and nosZ), complete denitrifiers (nir, nor, and nosZ), and partial denitrifiers (lacking nir or nosZ), with partial denitrifiers further divided into initiators (nir with or without nor), terminators (nosZ with or without nor) and initiator-terminators (nir and nosZ; Fig. 1b). Anammox genomes carrying nir (n = 47) were classified as non-denitrifiers because their nitrite reductase is not thought to be involved in denitrification82. Similarly, genomes encoding just nor were denoted as non-denitrifiers (n = 3064) as a conservative measure since they are likely involved in detoxification rather than respiration. Since we only used bacterial genomes, our analysis includes ammonia oxidizing bacteria potentially capable of nitrifier-denitrification, but not ammonia oxidizing archaea for which a respiratory function for nitrite reductase has not yet been established83.
A range of tools were used to identify and validate genes associated with potential electron donors and acceptors used by microorganisms. Genes for proteins involved in arsenite oxidation (AioBA; validated with alignment from Quemeneur et al.84), anammox (Hzs, Hdh) and iron redox were identified in genomes using MagicLamp v.1.0 with final annotations curated to match minimum subunit composition proposed for each enzyme85. We used AmoA as a marker for ammonia oxidation in bacteria using an HMM built from the alignment of AmoA, HmoA and PmoA from Diamond et al.5. Respiratory reductive dehalogenases were identified using the RDaseDB alignment (v.202086) combined with a literature search to exclude catabolic enzymes; we note that the TAT motif proposed to be indicative of respiratory enzymes was absent from the majority of sequences and could not be used. Sulfur redox genes were identified using HMSS2 and categorized into oxidation or reduction-associated enzymes following Tanabe and Dahl87. Sulfide: quinone reductase (SQR) enzymes were excluded from our analysis, both because their physiological role is variable, and because we found the HMMs were unspecific. Hydrogenases were identified using MagicLamp v.1.0, refined using the residues in Greening et al.33, and verified as to their clade and probable physiological function using the HydDB web server88. Clades 1a–d, 1 g, h and j and clade 2 were considered involved in hydrogen oxidation, and clades 4a, b, c, and h were considered involved in reduction. ArrA and ArxA involved in arsenate reduction and arsenite oxidation, respectively, were identified using an HMM built from sequences from Wells et al.89, and validated using the residues from Ospino et al.90. Genomic potential for aerobic respiration was identified by screening the HMM-based HCO family search used to annotate Nor for HCO classes A, B and C, and completing a second HMM-based search using the cytochrome bd oxidase family reference alignment from Murali et al.91. The proteins were categorized into high (HCO classes B and C and cytochrome bd oxidase) and low affinity oxidases (HCO class A)92. All redox gene annotations are reported in Supplementary Data 3.
Sugar-acid preference of organisms was inferred using the sum of KEGG orthologues identified by enrichM (v0.6.3, default settings) that were annotated to sugar vs. acid metabolism32, noting that the majority of the genomes fall outside the originally-calibrated range of GC contents (Supplementary Data 4). GapMind carbon93 (March 22nd, 2021 database version) was run to infer ability to grow on 62 compounds. We used custom pathway completeness cutoff scores for each pathway based on annotations for organisms for which growth had been reported on that substrate in BacDive (Supplementary Data 5). Our positive controls dataset did not have data for 6 substrates, leaving 56 for analysis. Transporters were annotated using DIAMOND (2.1.6.16094) against the TCDB database (download date: 16th May 2023)95, with hits showing at least 40% identity to the reference and 70% coverage of both reference and query sequence, with no more than 10% difference in alignment length kept for analysis96. Transcription factors were annotated using DeepTFactor97 (v. 2020-11-09) with default settings (Supplementary Data 6). We used GenomeSPOT v.1.0.1 to infer temperature, pH, and salinity ranges98, and used the difference between the maximum and minimum values for each genome in our analysis (Supplementary Data 7).
Unless otherwise noted, all subsequent analyses were completed in R v.4.199. The maximum growth rate of each taxon was estimated using gRodon231, which used codon usage bias of ribosomal proteins annotated in the genomes using hmmearch against the ribosomal proteins extracted from Uniprot PFAM v. 35.0 with profile-specific cutoffs (Supplementary Data 8100). Differences in growth rates between denitrifier types were assessed using Kruskall-Wallis tests (stats::kruskal.test) followed by Dunn tests with Benjamini-Hochberg correction for multiple testing (FSA::dunnTest), where all maximum growth rates less than 0.2 doublings h−1 were given the same rank. This cutoff was needed because codon use bias saturates with growth rates slower than this31.
To determine whether complete and partial denitrifiers differ in transcription factor, transporter, carbohydrate-active enzyme, or catabolized substrate count or sugar acid preference, we fit linear models, logging or taking the exponent of variables where needed. However, we analyzed GapMind data with a generalized linear mixed model with a Tweedie distribution in glmmTMB101 (v.1.1.10) due to the poor fit of OLS and high proportion (10-37%) of assemblies where no substrates within a chemical class were predicted to be used. We subsequently ascertained that the model residuals were not phylogenetically clustered using phytools::phylosig (v.2.3.0102) and an organism tree derived from the GTDB reference tree80. This tree was rerooted in Fusobacteriota103 and forced to be ultrametric using the castor::date_tree_red function104 (v.1.8.2). Blomberg’s K105 of model residuals was less than one in all instances (P < 0.05), so we did not include phylogenetic relatedness as a random effect in our analysis. We evaluated differences between complete and partial denitrifier genome content using post-hoc multiple comparisons with the sandwich::vcovHC function (v.3.1.1106) to account for heteroskedasticity. Due to the large number of zeroes, we assessed the hypothesis that partial denitrifiers encode the potential to oxidize and reduce a smaller number of inorganic compounds than complete denitrifiers by comparing the cumulative density distributions using a one-sided Kolmogorov-Smirnov test using stats::ks.test.
Metagenome and metadata collection
To assess the dominance of complete vs. partial denitrifier communities across biomes, we selected metagenomes with at least 100,000 reads ≥150 nt, primarily from larger sampling campaigns where uniform metadata were available (Supplementary Data 9). The majority of soil metagenomes came from the Australian Microbiome Project/BASE107, the National Ecological Observatory Network108, Topsoils Microbiome Project109, Long-Term Soil Productivity experiment110, and the Stordalen Mire111. The majority of aquatic metagenomes came from the Australian Microbiome Project112, Linnaeus Microbial Observatory113, the Amazon Continuum Project114 and BATS, GEOTRACE and HOTS115. Engineered metagenomes included those from drinking water116 sewage41, and wastewater treatment plants117. The host-associated metagenomes dataset consists primarily of plant-associated sequences, such as from leafy greens118, beans119, citrus rhizosphere120, switchgrass121 and Arabidopsis118, though we also included a handful of sequences from invertebrates such as sponges and gutless marine worms. For unpublished metagenomes where contact details were provided, we emailed authors to request permission to use them.
We completed a similar search for metatranscriptomes, focusing on soil (including rhizosphere) and marine studies where carbon and/or nutrient availability had either been manipulated or logic or metadata indicated they differed between samples (Supplemental Data S10). These metatranscriptomes were identified based on searching the American Society for Microbiology journals website for metatranscriptomes and google databases for the terms “soil” or “marine” and “carbon” or “nutrient” and “metatranscript*”.
Where multiple sequencing runs or files existed for the same sample, we combined the fastq files prior to searching. We used only the forward read in the case of paired end reads, and only the first 150 nt of any reads longer than that. In total, our dataset comprised 3991 metagenomes, with 1489 from aquatic habitats, 1642 from soil, 658 host-associated, and 202 from engineered habitats. Our final set of metatranscriptomes overlaps minimally with our metagenome dataset and contains 413 metatranscriptomes (64 rhizosphere or soil with root exudates added, 92 other soil, 185 primarily coastal and estuarine marine, 24 lake sediment, and 48 river metatranscriptomes).
Biomes were assigned to metagenomes based on The Nature Conservancy Terrestrial Ecoregions for terrestrial samples122 and latitude for marine samples (polar: latitude > 60°; westerlies: latitude 30–60°; trades: latitude 0–30°). Terrestrial biome assignment required use of the packages sp v.1.4-5123, rgdal v.1.523124, and rgeos v0.5-5125. Soils under cultivation were excluded from the biome-based approach and instead categorized as croplands.
Dominance of complete vs. partial denitrifier communities based on proxy gene searches across biomes
We selected mRNA reads from metranscriptomes using SortMeRNA v.4.3.7 with the smr_v4.3_default_db database and settings “--other --fastx --num-alignments 1 --no-best”. We used GraftM v.0.13.1 to identify denitrification genes (nirK, nirS and nosZ) in the metagenomes and metatranscriptomes, as previously described25. Briefly, GraftM uses a two-step process in which a HMM search identifies candidate reads, followed by phylogenetic placement on a reference tree using pplacer (v.1.1.19126;). We used the accumulate function in gappa v.0.8.0127 to find the position on the tree where at least 95% of the mass for each read descended from, and excluded all reads with any mass placed in the outgroup. We further excluded sequences placed in the non-denitrifying anammox NirS clade 1 h (median 0% of nirS reads in metagenomes and 0% in metatranscriptomes), archaeal nitrifier NirK clades 2 and 4 (1.4% of nirK reads in metagenomes and 50% in metatranscriptomes), as well as the eukaryotic NirK clades 1b and 1e (0% and 0%)25. Our analysis also excluded the reads placed in the recently reported clade III nosZ128, which forms the outgroup in our reference tree. This metagenome and metatranscriptome search depended on: hmmer (v.3.2.172;), OrfM (v.0.7.1129;), bbtools (v.38.90; https://sourceforge.net/projects/bbmap/), lbzip2 (v.2.5; https:// lbzip2.org/), and fxtract (v.2.3; https://github.com/ctSkennerton/fxtract).
We validated these search and placement and post-processing parameters by generating 150 nt fragments of full-length sequences that were picked up by the NirK, NirS and NosZ HMMs, which in addition to the target proteins also included homologous multicopper oxidase proteins from Cyanobacteriota and Thermoprotetoa, NirN and NirF, and various members of clade III NosZ, respectively. Sensitivity is the fraction of total ingroup fragments searched which were placed in the ingroup, and specificity is one minus the fraction of outgroup fragments incorrectly placed in the ingroup (or, in the case of nosZ, in a clade other than the target clade). Sensitivity was 76% for nirK, 93% for nirS, 91% for clade I nosZ and 94% for clade II nosZ. Specificity was 97% for nirK, 100% for nirS, 100% for clade I nosZ and 100% for clade II nosZ. These specificity scores do not preclude the possibility of reads from other related proteins not included in these outgroups from being placed within the phylogeny.
We compared gene prevalence across biomes using the absolute difference in counts of clade I and II nosZ or nir and nosZ genes standardized by the number of Gbp sequenced (or Gbp mRNA sequenced for metatranscriptomes) to account for differences in sequencing depth. For nir vs. nosZ abundance we used Eq. 1 such that positive values denote nosZ is more abundant than nir:
For nosZ, we used Eq. 2 such that positive values denote clade I is more abundant than clade II:
This method of calculating gene prevalence enables the abundance of both genes to be considered concurrently. This is particularly relevant for the 14% of metagenomes and 26% of metatranscriptomes where nosZI was not detected but nosZII was found in 1–13,041 (1–10,926 in metatranscriptomes) copies, and the 0.8% of metagenomes where nosZ was not found but 3-28 copies of nir were identified.
Environmental correlates with biome gene counts and denitrifier MAGs
We used the TARA Oceans35 MAGs present in the OceanDNA MAG database to test the hypothesis that complete denitrifiers are more abundant under conditions with high electron donor to acceptor ratios. These MAGs were chosen because the database is sufficiently large that multiple MAGs from each denitrifier type category were present, and where multiple conspecific MAGs were present, those with inconsistent denitrification gene repertoires could be excluded. After quality-controlling the raw paired-end reads with cutadapt (-m 100 -q 20 –max-n 0 –trim-n), we mapped them to all MAGs using CoverM (v. 0.6.134; -m relative abundance –min-read-aligned-percent 0.75 –min-read-percent-identity 0.95 –min-covered-fraction 0) then determined the total relative abundance of each MAG-OTU in each sample (95% ANI). Only the reads for the 3 µm filter fraction were used in this analysis to allow for bigger cells or cell aggregates than using the 0.22 µm fraction. We subsequently determined the standardized relative abundance by dividing the observed relative abundance within each sample to the maximum relative abundance observed across all samples. We then fit a linear model predicting the standardized relative abundance of each MAG OTU based on the combination of NO3− and chlorophyll concentrations (i.e., NO3− after accounting for chlorophyll, chlorophyll after accounting for NO3−, and the logarithm of chlorophyll: NO3−). The mean slope and standard error for each MAG OTU were extracted and used to calculate the weighted mean response and 95% confidence intervals for each denitrifier type. Differences in mean response to NO3−, chlorophyll and their ratios between denitrifier types were established based on non-overlapping 95% confidence intervals.
We evaluated the relationship between differences in normalized gene counts and resource availability using random forests. This analysis was restricted to metagenomes from the BASE and Australian marine projects107,112 as examples of studies with a large number of metagenomes with uniformly-collected metadata for variables associated with denitrification, including NO3−/NO2− levels, pH, carbon, and micronutrients. A pre-selection of variables for inclusion in the model was completed to exclude collinear variables (Spearman’s ρ > 0.7 or variance inflation factor > 4); in the collinear groups, the variable hypothesized to be most strongly and directly explicable of gene ratio was retained (Supplementary Fig. 3; Supplementary Tables 2 and 3). We subsequently ran VSURF v. 1.1.0130 100 times to identify the best predictors for each ratio and biome, keeping only those variables retained in at least 95 of the 100 iterations. We then used the randomForest package v. 4.7.1–1 to model the relationship between gene ratios and the final set of retained variables. A grid search was used to find the combination of tuning parameters yielding lowest out-of-bag root-mean-square error. Accumulated local effects plots were generated to visualize results using iml v. 0.11131.
Finally, we verified whether high carbon and/or low nutrient availability favored the transcription of nosZ over nir or clade I nosZ over clade II nosZ by calculating δnos− nir and δnosZI-nosZII in the metatranscriptomes, respectively. We calculated Hedges G in a meta-analysis of effect sizes for soil studies (meta::metacont v 8.2.1), taking the lower carbon soil as the reference and carbon-enriched soil as the treatment (e.g., bulk soil or unamended soil compared to rhizosphere, glucose, glycine, or root exudate amended soils). Aquatic studies were observational, so we fit a Spearman correlation coefficient between δnos-nir or δnosZI-nosZII and environmental variables (NO3− + NO2−, phosphate, bacterial production and chlorophyll A concentrations as proxies of resource availability, and oxygen). Where communities were captured and analyzed on two different filter sizes from the same sample, we determined the correlation separately for the two size fractions but counted them as a single study in our overall effect size calculations. We used psychmeta::ma_r (v2.7.0) to calculate overall correlations in the aquatic data.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The annotation data generated in this study are provided in the Supplementary Information. All genomes, metagenomes, and metatranscriptomes used are publicly available in NCBI or other sources under the identifiers denoted in Supplementary Data 2, 9, and 10. Hidden Markov Models, and reference databases used for denitrification gene searches are available in FigShare (https://doi.org/10.6084/m9.figshare.23913078 for NirK and NirS and https://doi.org/10.6084/m9.figshare.30122335 for NosZ). Source data are provided with this paper.
Code availability
Scripts used to generate figures and annotate genomes are available in FigShare under (https://doi.org/10.6084/m9.figshare.30122335).
References
Alvarez, L., Bricio, C., Gómez, M. J. & Berenguer, J. Lateral transfer of the denitrification pathway genes among Thermus thermophilus strains. Appl. Environ. Microbiol. 77, 1352–1358 (2011).
Rees, E., Siddiqui, R. A., Köster, F., Schneider, B. & Friedrich, B. Structural gene (nirS) for the cytochrome cd1 nitrite reductase of Alcaligenes eutrophus H16. Appl. Environ. Microbiol. 63, 800–802 (1997).
Schwintner, C., Sabaty, M., Berna, B., Cahors, S. & Richaud, P. Plasmid content and localization of the genes encoding the denitrification enzymes in two strains of Rhodobacter sphaeroides. FEMS Microbiol. Lett. 165, 313–321 (1998).
Graf, D. R. H., Jones, C. M. & Hallin, S. Intergenomic comparisons highlight modularity of the denitrification pathway and underpin the importance of community structure for N2O emissions. PLoS ONE 9, e114118 (2014).
Diamond, S. et al. Mediterranean grassland soil C–N compound turnover is dependent on rainfall and depth, and is mediated by genomically divergent microorganisms. Nat. Microbiol. 4, 1356–1367 (2019).
Orellana, L. H., Chee-Sanford, J. C., Sanford, R. A., Löffler, F. E. & Konstantinidis, K. T. Year-round shotgun metagenomes reveal stable microbial communities in agricultural soils and novel ammonia oxidizers responding to fertilization. Appl. Environ. Microbiol. 84, e01646–17 (2018).
Dove, N. C., Torn, M. S., Hart, S. C. & Taş, N. Metabolic capabilities mute positive response to direct and indirect impacts of warming throughout the soil profile. Nat. Commun. 12, 1–13 (2021).
Karthikeyan, S. et al. Metagenomic characterization of soil microbial communities in the Luquillo experimental forest (Puerto Rico) and implications for nitrogen cycling. Appl. Environ. Microbiol. 87, e00546–21 (2021).
Pessi, I. S. et al. In-depth characterization of denitrifier communities across different soil ecosystems in the tundra. Environ. Microbiome 17, 30 (2022).
Mosley, O. E. et al. Nitrogen cycling and microbial cooperation in the terrestrial subsurface. ISME J. 16, 2561–2573 (2022).
Lilja, E. E. & Johnson, D. R. Segregating metabolic processes into different microbial cells accelerates the consumption of inhibitory substrates. ISME J. 10, 1568–1578 (2016).
Lilja, E. E. & Johnson, D. R. Substrate cross-feeding affects the speed and trajectory of molecular evolution within a synthetic microbial assemblage. BMC Evol. Biol. 19, 1–11 (2019).
Dolinšek, J., Ramoneda, J. & Johnson, D. R. Initial community composition determines the long-term dynamics of a microbial cross-feeding interaction by modulating niche availability. ISME Commun. 2, 1–10 (2022).
Vitale, A. et al. Mapping of the Denitrification Pathway in Burkholderia thailandensis by Genome-Wide Mutant Profiling. J. Bacteriol. 202, e00304–e00320 (2020).
Zumft, W. G. & Kroneck, P. M. H. Respiratory transformation of nitrous oxide (N2O) to dinitrogen by bacteria and archaea. in Advances in Microbial Physiology Vol. 52 (ed. Poole, R. K.) 107–227 (Academic Press, 2006).
Jones, C. M. et al. Recently identified microbial guild mediates soil N2O sink capacity. Nat. Clim. Chang. 4, 801–805 (2014).
Heinrich, R., Schuster, S. & Holzhütter, H.-G. Mathematical analysis of enzymic reaction systems using optimization principles. EJB Rev. 1991, 167–187 (1991).
Kreft, J.-U., Griffin, B. M. & González-Cabaleiro, R. Evolutionary causes and consequences of metabolic division of labour: why anaerobes do and aerobes don’t. Curr. Opin. Biotechnol. 62, 80–87 (2020).
Wang, Y., Gao, H. & Wells, F. G. Integrated omics analyses reveal differential gene expression and potential for cooperation between denitrifying polyphosphate and glycogen accumulating organisms. Environ. Microbiol. 23, 3274–3293 (2021).
Sun, X. et al. Ecological dynamics explain modular denitrification in the ocean. Proc. Natl. Acad. Sci. 121, e2417421121 (2024).
LaSarre, B., Morlen, R., Neumann, G. C., Harwood, C. S. & McKinlay, J. B. Nitrous oxide reduction by two partial denitrifying bacteria requires denitrification intermediates that cannot be respired. Appl. Environ. Microbiol. 90, e01741–23 (2023).
Crocker, K. et al. Environmentally dependent interactions shape patterns in gene content across natural microbiomes. Nat. Microbiol 9, 2022–2037 (2024).
Mahne, I. & Tiedje, J. M. Criteria and methodology for identifying respiratory denitrifiers. Appl. Environ. Microbiol. 61, 1110–1115 (1995).
Murali, R. et al. Diversity and evolution of nitric oxide reduction in bacteria and archaea. Proc. Natl. Acad. Sci. 121, e2316422121 (2024).
Pold, G. et al. Phylogenetics and environmental distribution of nitric oxide-forming nitrite reductases reveal their distinct functional and ecological roles. ISME Commun. 4, ycae020 (2024).
Park, D., Kim, H. & Yoon, S. Nitrous oxide reduction by an obligate aerobic bacterium, Gemmatimonas aurantiaca strain T-27. Appl. Environ. Microbiol. 83, e00502–e00517 (2017).
Costa, C. et al. Regulation of the hexaheme nitrite/nitric oxide reductase of Desulfovibrio desulfuricans, Wolinella succinogenes and Escherichia coli. FEBS Lett. 276, 67–70 (1990).
Kim, S. O., Orii, Y., Lloyd, D., Hughes, M. N. & Poole, R. K. Anoxic function for the Escherichia coli flavohaemoglobin (Hmp): reversible binding of nitric oxide and reduction to nitrous oxide. FEBS Lett. 445, 389–394 (1999).
Xu, Z. et al. Unprecedented N2O production by nitrate-ammonifying Geobacteraceae with distinctive N2O isotopocule signatures. mBio 15, e02540–24 (2024).
Simon, J. & Klotz, M. G. Diversity and evolution of bioenergetic systems involved in microbial nitrogen compound transformations. Biochim. Biophys. Acta 1827, 114–135 (2013).
Weissman, J. L., Hou, S. & Fuhrman, J. A. Estimating maximal microbial growth rates from cultures, metagenomes, and single cells via codon usage patterns. Proc. Natl. Acad. Sci. 118, e2016810118 (2021).
Gralka, M., Pollak, S. & Cordero, O. X. Genome content predicts the carbon catabolic preferences of heterotrophic bacteria. Nat. Microbiol. 8, 1799–1808 (2023).
Greening, C. et al. Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilised energy source for microbial growth and survival. ISME J. 10, 761–777 (2016).
Aroney, S. T. N. et al. CoverM: read alignment statistics for metagenomics. Bioinformatics 41, btaf147 (2025).
Sunagawa, S. et al. Structure and function of the global ocean microbiome. Science 348, 1261359 (2015).
Nishimura, Y. & Yoshizawa, S. The OceanDNA MAG catalog contains over 50,000 prokaryotic genomes originated from various marine environments. Sci. Data 9, 305 (2022).
Morin, A., Lamoureux, W. & Busnarda, J. Empirical models predicting primary productivity from chlorophyll a and water temperature for stream periphyton and lake and ocean phytoplankton. J. North Am. Benthol. Soc. 18, 299–307 (1999).
Wan, X. S. et al. Particle-associated denitrification is the primary source of N2O in oxic coastal waters. Nat. Commun. 14, 8280 (2023).
Boyd, J. A., Woodcroft, B. J. & Tyson, G. W. GraftM: a tool for scalable, phylogenetically informed classification of genes within metagenomes. Nucleic Acids Res. 46, e59 (2018).
Coban, O., Rasigraf, O., de Jong, A. E. E., Spott, O. & Bebout, B. M. Quantifying potential N turnover rates in hypersaline microbial mats by using 15N tracer techniques. Appl. Environ. Microbiol. 87, e03118–e03120 (2021).
Hendriksen, R. S. et al. Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nat. Commun. 10, 1–12 (2019).
Saghaï, A., Pold, G., Jones, C. M. & Hallin, S. Phyloecology of nitrate ammonifiers and their importance relative to denitrifiers in global terrestrial biomes. Nat. Commun. 14, 8249 (2023).
Wang, Z., Vishwanathan, N., Kowaliczko, S. & Ishii, S. Clarifying microbial nitrous oxide reduction under aerobic conditions: tolerant, intolerant, and sensitive. Microbiol. Spectr. 11, e04709–e04722 (2023).
Roothans, N., van Loosdrecht, M. C. M. & Laureni, M. Metabolic labour division trade-offs in denitrifying microbiomes. ISME J. 19, wraf020 (2025).
Hester, E. R., Jetten, M. S., Welte, C. U. & Lücker, S. Metabolic overlap in environmentally diverse microbial communities. Front. Genet. 10, 989 (2019).
Valk, L. C. et al. Exploring the microbial influence on seasonal nitrous oxide concentration in a full-scale wastewater treatment plant using metagenome assembled genomes. Water Res. 219, 118563 (2022).
Zhang, I. H. et al. Partitioning of the denitrification pathway and other nitrite metabolisms within global oxygen deficient zones. ISME Commun. 3, 1–14 (2023).
Stone, B. W. G. et al. Life history strategies among soil bacteria—dichotomy for few, continuum for many. ISME J. 17, 611–619 (2023).
Caro, T. A., McFarlin, J., Jech, S., Fierer, N. & Kopf, S. Hydrogen stable isotope probing of lipids demonstrates slow rates of microbial growth in soil. Proc. Natl. Acad. Sci. 120, e2211625120 (2023).
Tiedje, J. M., Sexstone, A. J., Myrold, D. D. & Robinson, J. A. Denitrification: ecological niches, competition and survival. Antonie Leeuwenhoek 48, 569–583 (1983).
Jia, M., Winkler, M. K. H. & Volcke, E. I. P. Elucidating the competition between heterotrophic denitrification and DNRA using the resource-ratio theory. Environ. Sci. Technol. 54, 13953–13962 (2020).
Stremińska, M. A., Felgate, H., Rowley, G., Richardson, D. J. & Baggs, E. M. Nitrous oxide production in soil isolates of nitrate-ammonifying bacteria. Environ. Microbiol. Rep. 4, 66–71 (2012).
Yoon, S., Cruz-García, C., Sanford, R., Ritalahti, K. M. & Löffler, F. E. Denitrification versus respiratory ammonification: environmental controls of two competing dissimilatory NO3−/NO2− reduction pathways in Shewanella loihicastrain PV-4. ISME J. 9, 1093–1104 (2015).
Saleh-Lakha, S. et al. Effect of nitrate and acetylene on nirS, cnorB, and nosZ expression and denitrification activity in Pseudomonas mandelii. Appl. Environ. Microbiol. 75, 5082–5087 (2009).
Thomas, K. L., Lloyd, D. & Boddy, L. Effects of oxygen, pH and nitrate concentration on denitrification by Pseudomonas species. FEMS Microbiol. Lett. 118, 181–186 (1994).
Wrage-Mönnig, N. et al. The role of nitrifier denitrification in the production of nitrous oxide revisited. Soil Biol. Biochem. 123, A3–A16 (2018).
Venterea, R. T. Nitrite-driven nitrous oxide production under aerobic soil conditions: kinetics and biochemical controls. Glob. Chang. Biol. 13, 1798–1809 (2007).
Poole, R. K. Flavohaemoglobin: the pre-eminent nitric oxide–detoxifying machine of microorganisms. F1000Res 9, F1000 Faculty Rev–7 (2020).
Wonderen, J. H., van, Burlat, B., Richardson, D. J., Cheesman, M. R. & Butt, J. N. The nitric oxide reductase activity of cytochrome c nitrite reductase from Escherichia coli. J. Biol. Chem. 283, 9587–9594 (2008).
Onley, J. R., Ahsan, S., Sanford, R. A. & Löffler, F. E. Denitrification by Anaeromyxobacter dehalogenans, a Common Soil Bacterium Lacking the Nitrite Reductase Genes nirS and nirK. Appl. Environ. Microbiol. 84, e01985–17 (2018).
Wei, Q. et al. Molecular mechanisms through which different carbon sources affect denitrification by Thauera linaloolentis: Electron generation, transfer, and competition. Environ. Int. 170, 107598 (2022).
Pan, Y., Ni, B.-J., Bond, P. L., Ye, L. & Yuan, Z. Electron competition among nitrogen oxides reduction during methanol-utilizing denitrification in wastewater treatment. Water Res. 47, 3273–3281 (2013).
Schalk-Otte, S., Seviour, R. J., Kuenen, J. G. & Jetten, M. S. M. Nitrous oxide (N2O) production by Alcaligenes faecalis during feast and famine regimes. Water Res. 34, 2080–2088 (2000).
McGill, S. L. et al. Pseudomonas aeruginosa reverse diauxie is a multidimensional, optimized, resource utilization strategy. Sci. Rep. 11, 1457 (2021).
Dufour, L. J. P. et al. Potential energetic return on investment positively correlated with overall soil microbial activity. Soil Biol. Biochem. 173, 108800 (2022).
LaRowe, D. E. & Van Cappellen, P. Degradation of natural organic matter: a thermodynamic analysis. Geochim. Cosmochim. Acta 75, 2030–2042 (2011).
Morley, N. & Baggs, E. M. Carbon and oxygen controls on N2O and N2 production during nitrate reduction. Soil Biol. Biochem. 42, 1864–1871 (2010).
Curtright, A. J. & Tiemann, L. K. Chemical identity of carbon substrates drives differences in denitrification and N2O reduction within agricultural soils. Soil Biol. Biochem. 184, 109078 (2023).
Henry, S. et al. Disentangling the rhizosphere effect on nitrate reducers and denitrifiers: insight into the role of root exudates. Environ. Microbiol. 10, 3082–3092 (2008).
Sennett, L. B. et al. Determining how oxygen legacy affects trajectories of soil denitrifier community dynamics and N2O emissions. Nat. Commun. 15, 7298 (2024).
Bertagnolli, A. D., Konstantinidis, K. T. & Stewart, F. J. Non-denitrifier nitrous oxide reductases dominate marine biomes. Environ. Microbiol. Rep. 12, 681–692 (2020).
Eddy, S. R. Accelerated Profile HMM Searches. PLOS Comput. Biol. 7, e1002195 (2011).
Graf, D. R., Jones, C. M. & Hallin, S. Intergenomic comparisons highlight modularity of the denitrification pathway and underpin the importance of community structure for N 2 O emissions. PLoS ONE 9, e114118 (2014).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Ludwig, W. et al. ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371 (2004).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Sun, Y., De Vos, P. & Heylen, K. Nitrous oxide emission by the non-denitrifying, nitrate ammonifier Bacillus licheniformis. BMC Genomics 17, 68 (2016).
Parks, D. H. et al. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 38, 1079–1086 (2020).
Orakov, A. et al. GUNC: detection of chimerism and contamination in prokaryotic genomes. Genome Biol. 22, 178 (2021).
van de Vossenberg, J. et al. The metagenome of the marine anammox bacterium ‘Candidatus Scalindua profunda’ illustrates the versatility of this globally important nitrogen cycle bacterium. Environ. Microbiol. 15, 1275–1289 (2013).
Stein, L. Y. Insights into the physiology of ammonia-oxidizing microorganisms. Curr. Opin. Chem. Biol. 49, 9–15 (2019).
Quéméneur, M. et al. Diversity surveys and evolutionary relationships of aoxB genes in aerobic arsenite-oxidizing bacteria. Appl. Environ. Microbiol. 74, 4567–4573 (2008).
Garber, A. I. et al. FeGenie: a comprehensive tool for the identification of iron genes and iron gene neighborhoods in genome and metagenome assemblies. Front. Microbiol. 11, 499513 (2020).
Hug, L. A. et al. Overview of organohalide-respiring bacteria and a proposal for a classification system for reductive dehalogenases. Philos. Trans. R. Soc. B Biol. Sci. 368, 20120322 (2013).
Tanabe, T. S. & Dahl, C. HMS-S-S: A tool for the identification of sulphur metabolism-related genes and analysis of operon structures in genome and metagenome assemblies. Mol. Ecol. Resour. 22, 2758–2774 (2022).
Søndergaard, D., Pedersen, C. N. S. & Greening, C. HydDB: a web tool for hydrogenase classification and analysis. Sci. Rep. 6, 34212 (2016).
Wells, M., Kim, M., Akob, D. M., Basu, P. & Stolz, J. F. Impact of the dimethyl sulfoxide reductase superfamily on the evolution of biogeochemical cycles. Microbiol. Spectr. 11, e04145–22 (2023).
Ospino, M. C., Kojima, H. & Fukui, M. Arsenite oxidation by a newly isolated Betaproteobacterium possessing arx genes and diversity of the arx gene cluster in bacterial genomes. Front. Microbiol. 10, 1210 (2019).
Murali, R., Gennis, R. B. & Hemp, J. Evolution of the cytochrome bd oxygen reductase superfamily and the function of CydAA’ in Archaea. ISME J. 15, 3534–3548 (2021).
Morris, R. L. & Schmidt, T. M. Shallow breathing: bacterial life at low O2. Nat. Rev. Microbiol. 11, 205–212 (2013).
Price, M. N., Deutschbauer, A. M. & Arkin, A. P. Filling gaps in bacterial catabolic pathways with computation and high-throughput genetics. PLOS Genet. 18, e1010156 (2022).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Saier, M. H. et al. The Transporter Classification Database (TCDB): 2021 update. Nucleic Acids Res. 49, D461–D467 (2020).
Alballa, M. & Butler, G. TooT-T: discrimination of transport proteins from non-transport proteins. BMC Bioinform. 21, 25 (2020).
Kim, G. B., Gao, Y., Palsson, B. O. & Lee, S. Y. DeepTFactor: a deep learning-based tool for the prediction of transcription factors. Proc. Natl. Acad. Sci. 118, e2021171118 (2021).
Barnum, T. P. et al. Predicting microbial growth conditions from amino acid composition. 2024.03.22.586313 Preprint at https://doi.org/10.1101/2024.03.22.586313 (2024).
R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing https://www.r-project.org/ (2023).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419 (2021).
Brooks, M. et al. glmmTMB: generalized linear mixed models using template model builder. https://cran.r-project.org/web/packages/glmmTMB/index.html (2025).
Revell, L. J. phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223 (2012).
Coleman, G. A. et al. A rooted phylogeny resolves early bacterial evolution. Science 372, eabe0511 (2021).
Louca, S. castor: Efficient Phylogenetics on Large Trees. https://cran.r-project.org/web/packages/castor/index.html (2024).
Blomberg, S. P., Garland, T. Jr & Ives, A. R. Testing for phylogenetic signal in comparative data: behavioral traits are more labile. Evolution 57, 717–745 (2003).
Zeileis, A., Köll, S. & Graham, N. Various versatile variances: an object-oriented implementation of clustered covariances in R. J. Stat. Softw. 95, 1–36 (2020).
Bissett, A. et al. Introducing BASE: the Biomes of Australian Soil Environments soil microbial diversity database. Gigascience 5, s13742–016 (2016).
National Ecological Observatory Network (NEON). Soil microbe metagenome sequences. NEON https://doi.org/10.48443/fzzj-g053 (2021).
Bahram, M. et al. Structure and function of the global topsoil microbiome. Nature 560, 233–237 (2018).
Wilhelm, R. C. et al. A metagenomic survey of forest soil microbial communities more than a decade after timber harvesting. Sci. Data 4, 1–8 (2017).
Woodcroft, B. J. et al. Genome-centric view of carbon processing in thawing permafrost. Nature 560, 49–54 (2018).
Brown, M. V. et al. Systematic, continental scale temporal monitoring of marine pelagic microbiota by the Australian Marine Microbial Biodiversity Initiative. Sci. Data 5, 1–10 (2018).
Hugerth, L. W. et al. Metagenome-assembled genomes uncover a global brackish microbiome. Genome Biol. 16, 1–18 (2015).
Satinsky, B. M. et al. The Amazon continuum dataset: quantitative metagenomic and metatranscriptomic inventories of the Amazon River plume, June 2010. Microbiome 2, 1–7 (2014).
Biller, S. J. et al. Marine microbial metagenomes sampled across space and time. Sci. Data 5, 1–7 (2018).
Hu, W. et al. Metagenomics unravels differential microbiome composition and metabolic potential in rapid sand filters purifying surface water versus groundwater. Environ. Sci. Technol. 54, 5197–5206 (2020).
Singleton, C. M. et al. Connecting structure to function with the recovery of over 1000 high-quality metagenome-assembled genomes from activated sludge using long-read sequencing. Nat. Commun. 12, 1–13 (2021).
Bandla, A., Pavagadhi, S., Sridhar Sudarshan, A., Poh, M. C. H. & Swarup, S. 910 metagenome-assembled genomes from the phytobiomes of three urban-farmed leafy Asian greens. Sci. Data 7, 1–7 (2020).
Mendes, L. W., Raaijmakers, J. M., de Hollander, M., Mendes, R. & Tsai, S. M. Influence of resistance breeding in common bean on rhizosphere microbiome composition and function. ISME J. 12, 212–224 (2018).
Xu, J. et al. The structure and function of the global citrus rhizosphere microbiome. Nat. Commun. 9, 1–10 (2018).
Howe, A. et al. Seasonal activities of the phyllosphere microbiome of perennial crops. Nat. Commun. 14, 1039 (2023).
Olson, D. M. & Dinerstein, E. The global 200: priority ecoregions for global conservation. Ann. Mo. Bot. Gard. 89, 199–224 (2002).
Bivand, R. S., Pebesma, E. & Gomez-Rubio, V. Applied Spatial Data Analysis with R 2nd edn (Springer, New York, 2013).
Bivand, R. et al. rgdal: bindings for the ‘geospatial’ data abstraction library. https://CRAN.R-project.org/package=rgdal (2022).
Bivand, R. & Rundel, C. Rgeos: Interface to Geometry Engine—Open Source (‘GEOS’). https://CRAN.R-project.org/package=rgeos (2020).
Matsen, F. A., Kodner, R. B. & Armbrust, E. V. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinform 11, 538 (2010).
Czech, L., Barbera, P. & Stamatakis, A. Genesis and Gappa: processing, analyzing and visualizing phylogenetic (placement) data. Bioinformatics 36, 3263–3265 (2020).
He, G. et al. A novel bacterial protein family that catalyses nitrous oxide reduction. Nature 1–9. https://doi.org/10.1038/s41586-025-09401-4 (2025).
Woodcroft, B. J., Boyd, J. A. & Tyson, G. W. OrfM: a fast open reading frame predictor for metagenomic data. Bioinformatics 32, 2702–2703 (2016).
Genuer, R., Poggi, J.-M. & Tuleau-Malot, C. VSURF: variable selection using random forests. https://cran.r-project.org/web/packages/VSURF/index.html (2022).
Molnar, C., Bischl, B. & Casalicchio, G. iml: an R package for interpretable machine learning. J. Open Source Softw. 3, 786 (2018).
Acknowledgements
This work was supported by the Swedish University of Agricultural Sciences Senior Career Grant 2019-2025 to S.H. and the Swedish Research Council Grant Agreements No. 2016-03551 to S.H. and 2023-03627 to G.P. Computing resources were provided by the Department of Forest Mycology and Plant Pathology, Swedish National Infrastructure for Computing project SNIC 2022/22-1119 and NAISS 2023/22-1278, funded by the Swedish Research Council through Grant Agreement No. 2022-06725, and the Swedish National Infrastructure for Computing (SNIC) at the PDC Center for High Performance Computing, KTH Royal Institute of Technology, partially funded by the Swedish Research Council through grant agreement no. 2016-07213 (NAISS 2024/22-133).
Funding
Open access funding provided by Swedish University of Agricultural Sciences.
Author information
Authors and Affiliations
Contributions
Conceptualization: S.H. Data curation: G.P., A.S. and C.M.J. Formal analysis: G.P. Funding acquisition: S.H. and G.P. Investigation: G.P. Methodology: G.P., A.S., C.M.J., and S.H. Project administration: G.P. and S.H. Resources: S.H. Software: C.M.J., G.P., and A.S. Visualization: G.P., A.S. Writing—original draft preparation: G.P., A.S., and S.H. Writing—review and editing: G.P., A.S., C.M.J., and S.H.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Lisa Stein, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pold, G., Saghaï, A., Jones, C.M. et al. Denitrification is a community trait with partial pathways dominating across microbial genomes and biomes. Nat Commun 16, 9495 (2025). https://doi.org/10.1038/s41467-025-65319-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65319-5
