
Abstract
Designing effective mRNA sequences for therapeutics remains a formidable challenge. Inspired by successes in protein design, language models (LMs) are now being applied to RNA, but progress is often impeded by the lack of comprehensive training data. Existing models are frequently limited to UTR or CDS regions, restricting their application for complete mRNA sequences. We introduce mRNABERT, a robust, all-in-one mRNA designer pre-trained on the largest available mRNA dataset. To enhance performance, we propose a dual tokenization scheme with a cross-modality contrastive learning framework to integrate semantic information from protein sequences. On a comprehensive benchmark, mRNABERT demonstrates state-of-the-art performance, outperforming previous models in the majority of tasks for 5’ UTR and CDS design, RNA-binding protein (RBP) site prediction, and full-length mRNA property prediction. It also surpasses large protein models in several related tasks. In conclusion, mRNABERT’s superior performance across these diverse tasks signifies a substantial leap forward in mRNA research and therapeutic development.
Similar content being viewed by others

Introduction
In recent years, mRNA therapeutics have emerged as a revolutionary technology with substantial potential in gene therapy1. A multitude of mRNA vaccines have been developed to combat a wide array of viruses2, including the Zika virus3, human immunodeficiency virus4, influenza virus5, cytomegalovirus6, respiratory syncytial virus7, varicella-zoster virus8, and rabies virus9. Notably, the rapid development and deployment of two COVID-19 mRNA vaccines stand as a testament to this potential, marking the advent of a novel biotechnological platform against SARS-CoV-2, other potential pathogens, and tumors10,11,12,13. mRNA vaccines have exhibited a remarkable ability to be customized for encoding specific antigens, aligning with the unique characteristics of a disease2. Unlike DNA vaccines, mRNA vaccines mitigate the risk of insertional mutagenesis within the host genome14 while simultaneously facilitating the modifiable expression of the targeted antigen15,16. From an industrial perspective, the remarkable efficiency of in vitro transcription reactions supports the rapid progress and large-scale production of mRNA vaccines via cell-free methodologies17,18,19, rendering them a cost-efficient solution. Consequently, the long-term prospects of mRNA therapeutics in disease prevention and treatment are becoming increasingly evident, heralding a new era of designer medicines20.
mRNA is a specific type of RNA molecule responsible for transporting genetic information from DNA to the protein synthesis machinery. Its core is composed of a coding sequence (CDS) that encodes proteins, flanked by 5′ and 3′ untranslated regions (UTRs) and stabilized by a 7-methylguanosine (m7G) 5′ cap and a 3′ poly(A) tail21 (Fig. 1A). The structural integrity and functional synergy of these regions ensure efficient translation and regulation of protein expression within cells. Although mRNA components exhibit high design flexibility, designing effective mRNA therapeutics remains complex22,23,24. A comprehensive understanding of how nucleotide sequences, nucleotide modifications, and RNA structures interplay to influence translation efficiency and mRNA stability is crucial for optimizing mRNA-based therapeutic protein production20.

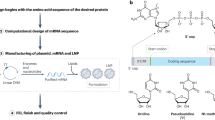
A The mRNABERT model is developed in two stages: In the first stage, pretraining is conducted on a set of 18 million mRNA sequences. This dataset is carefully curated and processed using the ORF finder tool from NCBI to identify different regions within the mRNA sequences. Subsequently, the sequences are tokenized using a custom tokenizer and fed into the model for the MLM task. The model architecture of mRNABERT includes 12 transformer blocks and incorporates advanced techniques like Flash Attention to enhance its overall performance. B In the second stage, a selected set of 500,000 CDS data and their corresponding amino acid sequences are processed by separate models. Embeddings from these models are projected into a shared dimensional space for a custom contrastive learning task to facilitate the full training of mRNABERT. C mRNABERT exhibits adaptability for various downstream tasks through the utilization of different strategies, illustrating its versatility. MLM Masked Language Model, pLM Protein Language Model.
Recently, the concept of using large language models (LLMs) that are pre-trained on the vast unlabeled text and fine-tuned for specific tasks25 has been extended to biological sequences, including proteins26,27,28, DNA29,30, and non-coding RNA31,32,33,34,35,36. This advancement offers a promising solution to the limitations of traditional experimental and computational methods in comprehensively exploring the vast sequence and structural space of mRNA37. mRNA molecules exhibit diverse mechanisms and interactions, requiring complex analysis techniques that frequently pose considerable challenges in experimental research. Given the similarities in nucleotide composition and sequence motifs between mRNA and other biological sequences, pre-trained models are well-positioned to significantly enhance mRNA research.
Although the interest in decoding biological sequences through language models has been steadily increasing, there remains a notable dearth of language models tailored specifically for learning the semantic representations of mRNA sequences. The practical application of existing mRNA language models faces three major challenges. Firstly, publicly available mRNA sequence data are relatively limited and exhibit significant variability in quality. This shortage undermines the creation of comprehensive mRNA libraries, which are vital for enhancing model performance during both pre-training and fine-tuning stages. Currently, RNA foundational models primarily rely on large-scale non-coding RNA datasets from resources like RNAcentral38 or Rfam39 for pre-training, excluding mRNA, a unique and distinct entity within the RNA domain. Secondly, existing efforts to decode mRNA sequences using machine learning techniques largely concentrate on developing specialized models for specific UTR40,41 or CDS42,43 regions, treating each mRNA region as an independent sample. These models are insufficient for addressing clinical mRNA design challenges since multiple components (5′UTR, CDS, 3′UTR) function synergistically during translation. Consequently, there is an urgent need for an integrated design approach that can effectively capture both local patterns within regions and global patterns spanning the entire mRNA sequence. Finally, the traditional approaches of enhancing model performance involve increasing either the model size26,27 or the volume of training data44, a process that is time-consuming and resource-intensive. In intricate biological processes such as protein engineering, splicing editing, and gene expression regulation, it is essential to incorporate not only mRNA sequence information but also the interactions among various biomolecules45. While some preliminary hypotheses suggest that integrating diverse biological data, including nucleotide and codon information43, amino acids, and coding sequences42, can boost mRNA model capabilities, a definitive path forward has yet to be established.
Existing RNA-related language models possess some glaring limitations due to the aforementioned issues. RNABERT33, RNAFM32, RNAErnie31, and ERNIE-RNA34, pre-trained on ncRNA from RNAcentral, have demonstrated utility in various ncRNA prediction tasks. RNA-MSM35, pre-trained on Rfam using an MSA-based BERT-style. RiNALMo36, pre-trained on 36 million ncRNA sequences sourced from a combination of databases such as RNAcentral, Rfam, and Nucleotide, represents advancements in both dataset size and model parameters. UNI-RNA46, pre-trained on a diverse range of RNA sequences from multiple sources encompassing both ncRNA and mRNA, remains inaccessible due to its lack of open-source availability. In the realm of mRNA-specific models, UTR-LM40, pre-trained on the 5′UTR sequences of multiple species, excels in tasks related to the 5′UTR, such as translation efficiency and mRNA expression levels. However, its performance on other mRNA downstream tasks remains to be evaluated, owing to its limited model size and pre-training dataset bias. CaLM42, trained on protein-coding DNA (cDNA), captures rich sequence information through codons, demonstrating outstanding performance in protein engineering tests. CodonBERT43, similarly trained on CDS sequences from multiple species using codons as inputs, is adept at various CDS prediction tasks. However, both models use a codon-based tokenizer, which can lead to improper segmentation and information confusion when encoding full-length mRNA if regions are not multiples of three. This leads to a loss of single-nucleotide resolution, complicating the extraction of valuable information for UTR-related tasks. 3UTRBERT41, pre-trained on human 3’UTR sequences using a k-mer tokenizer, outperforms other methods in specific 3’UTR tasks. However, its applicability to most mRNA tasks is constrained by its maximum input length of 512 (more details in Supplementary Information Part 1. Related Work). Furthermore, the current dense Transformer architecture faces high computational costs as input sequence length increases, due to its quadratic scaling with model width47. Therefore, it is necessary to adjust the model architecture and employ innovative strategies to enhance the capabilities of mRNA pre-training models.
In light of these observations, we developed mRNABERT, a robust language model pre-trained on a diverse and high-quality dataset of over 18 million non-redundant mRNA sequences (curated by the authors, with further details provided in the Methods section). To overcome the limitations of previous models, we incorporated several advanced techniques. Built on the well-established BERT architecture25, mRNABERT replaced traditional positional embeddings with Attention with Linear Biases (ALiBi)48 to handle long input sequences and integrated Flash Attention49 to improve computational efficiency. Furthermore, mRNABERT featured an innovative dual tokenization strategy, treating individual nucleotides as tokens for UTRs and codons for coding sequences (CDS). This unique tokenization approach not only aligns with the biological characteristics of mRNA but also lays a strong foundation for a wide range of downstream tasks (Fig. 1A). Additionally, we introduced a customized contrastive learning scheme to align mRNA and protein sequences in latent space (Fig. 1B), allowing mRNABERT to improve predictions of protein functions and mRNA-protein interactions. By effectively capturing the complex relationships between genetic sequences and protein sequences, this method enhances our understanding of biological processes and broadens the model’s range of applications.
Results
Overview of mRNABERT and benchmarks
To fully leverage the vast integrated dataset of full-length mRNA sequences, mRNABERT introduces a novel dual tokenization scheme for encoding the entire sequences. Tokenization, a critical initial step in language modeling, determines the types of semantic information the model can capture. RNA sequences are composed of four nucleotide bases, and traditional language models typically used character-based tokenizers that encode each nucleotide as an independent token to learn attention weights that may help a model understand nucleotide interactions within a sequence. However, when encoding full-length mRNA sequences, the maximum length constraints on the token input compromise the model’s representation capacity. Given the nature of mRNA codons, models based on CDS often treat each codon as an independent token, resulting in a complete loss of individual nucleotide information. Consequently, these models are only suitable for handling specific sequences. To address this, we employ an appropriate method to segment each region of the mRNA, refining local features at multiple granularities and integrating global features of the entire sequence. This approach yields a comprehensive embedding that is suitable for a wide range of downstream tasks.
Regarding the model architecture, mRNABERT is built on 12 bidirectional encoder blocks rooted in Transformers. To overcome the input length limitations of existing models, we introduced ALiBi, an alternative method for encoding positional information. By directly incorporating linear biases into the attention scores, ALiBi enhances the model’s ability to handle long sequences and improves overall performance. Additionally, we used IO-aware Flash attention to implement precise standard attention calculations in a more time- and memory-efficient way, thereby accelerating mRNABERT’s training process.
Considering the tight functional interplay between mRNA and protein sequences, we augmented our approach by incorporating contrastive learning to align codon and amino acid sequences following the masked language model (MLM) learning phase, a staple of BERT training. This step aimed to enrich the model’s understanding of the intricate biological landscape. Notably, mRNABERT’s performance exhibited marked improvement subsequent to the implementation of contrastive learning. Further details on the model architecture and training process are provided in the Methods section.
We conducted a comparative analysis of mRNABERT against the leading models in various tasks. For the eight 5′UTR ribosome load prediction tasks, the neural network baselines include Optimus50, FramePool51, and MTtrans52, as well as the state-of-the-art UTR-LM40. In the six CDS-related prediction tasks, mRNABERT was evaluated against methods such as Codon2vec53, TextCNN54, and the pre-trained CDS model, CodonBERT43. For the 3′UTR tasks, we focused on predicting 22 RBP binding sites using three baseline methods, including CNN-based iDeepE55 and DeepCLIP56, and the language model BERT-RBP57. In the m6A site prediction task, we collected data from nine different cell lines and compared mRNABERT against machine learning methods such as SRAMP58 and WHISTLE59. Both tasks also include the specially designed 3UTRBERT41. Furthermore, we evaluate the performance of all ncRNA models mentioned above on these tasks. Protein-related tasks include melting point and solubility prediction, as well as transcript abundance prediction across seven species. Here, mRNABERT was benchmarked against high-performing protein models, including ESM227, ProtTrans26, Ankh28, and the pre-trained codon language model CaLM42. Finally, we benchmarked all available RNA-related pre-trained models for eight full-length mRNA property prediction tasks. This comprehensive comparison demonstrates mRNABERT’s exceptional performance across all tasks.
Capturing multi-dimensional biological information of mRNA
To illustrate that mRNABERT can learn more biological knowledge from sequences than most baseline models, we performed an analysis of its embeddings, characterizing how it extracts functional and evolutionary knowledge from biological sequences.
The first aspect we investigated was the model’s vocabulary representation capabilities, focusing on its ability to discern the fundamental biological principles of the genetic code. Ideally, an mRNA model should identify similarities among synonymous codons, but this task is challenging due to the unannotated pre-training data and the model’s representation of biological sequences as tokens, which lack explicit information about nucleotides or codons. Additionally, we conducted ablation experiments to validate the effectiveness of contrastive learning. As illustrated in Fig. 2A, B, the mRNABERT model without contrastive learning exhibited disorganized clustering at the amino acid level. However, through the same t-SNE60 dimensionality reduction to mRNABERT’s vocabulary embeddings and projecting them onto a two-dimensional space, we observed that synonymous codons corresponding to the same amino acid type tended to cluster together (Fig. 2C). This clustering suggests that the model has successfully learned the genetic code from the extensive data it was trained on. Furthermore, by utilizing color to differentiate amino acids based on their distinct chemical properties (Fig. 2D), we found that the model effectively groups amino acids with similar properties, with ARI increasing from 0.166 to 0.498 and FMI from 0.325 to 0.596 (Methods). This clearly indicates that contrastive learning enables the model to capture additional semantic information about amino acids.

High-dimensional embeddings are projected into a two-dimensional space using t-SNE. Here, panels A and B depict the results of the mRNABERT model without contrastive learning, while the remaining four panels illustrate the results of the mRNABERT model. A–C The vocabulary embeddings from the model. Each point represents a codon or nucleotide, with colors corresponding to the amino acids of the codons. B–D Codons are then clustered based on amino acid properties. Codons encoding the same amino acid and those with similar biochemical properties tend to be spatially proximate. E Classification of different types of sequences, including lncRNA sequences that bear high similarity to mRNA and all regions of mRNA. F Species and sequence data were randomly sampled from the retained dataset, with each point representing a complete mRNA sequence. ARI Adjusted Rand Index, FMI Fowlkes-Mallows Index.
Next, we evaluated mRNABERT’s ability to classify various types of RNA data. As depicted in Fig. 2E, mRNABERT successfully discriminated between distinct mRNA regions, including the 5′UTR and 3′UTR sequences. Impressively, it also showed an ability to differentiate between long non-coding RNA (lncRNA) and mRNA sequences, despite not being explicitly trained on ncRNA data. This highlights mRNABERT’s capacity to encapsulate sufficient biological characteristics, enabling it not only to differentiate among various mRNA regions but also to distinguish mRNA from other RNA sequence types. By extracting profound semantic information from the entire mRNA sequence, it identifies sequence similarities that extend beyond mere length.
Subsequently, our analysis concentrated on the embeddings of sequences derived from six different species, carefully chosen to represent a broad range of biological classifications across diverse holdout datasets. The scatter plot depicted in Fig. 2F reveals a clear clustering of homologous sequences, with clear-cut boundaries delineating different species. This result highlights mRNABERT’s ability to recognize and retain evolutionary information embedded within biological sequences, emphasizing its robust capability to capture biological details at the sequence level.
Predicting ribosome load from 5′UTR sequences
Controlling translation efficiency hinges on the critical role of 5′UTR sequence. Ribosome load, defined as the number of ribosomes bound to an mRNA molecule at any given moment, stands as a pivotal marker of protein synthesis efficiency50. Therefore, accurately predicting ribosome load from 5′UTR sequences is paramount for optimizing mRNA sequence design to maximize protein expression, particularly when forging new sequences beyond existing 5′UTR templates.
To address this challenge, we leveraged a benchmark dataset sourced from previous studies that used massively parallel reporter assays (MPRA) to curate a library of 280,000 gene sequences with their respective ribosome loads50. Our approach involved fine-tuning the mRNABERT model to predict ribosome load from 5’UTR sequences (detailed in the “Method” section). Alongside mRNABERT, we benchmarked several machine-learning models tailored for this task, including Optimus50, FramePool51, and MTtrans52, as well as pre-trained language models such as UTR-LM40, RNABERT33, and RNA-FM32. The performance of mRNABERT was evaluated by comparing it against benchmark methods across eight synthetic libraries.
The results from our study, depicted in Fig. 3 and Supplementary Tables 4 and 5, highlight the exceptional performance of mRNABERT, which was comparable to the top-performing specialized model, UTR-LM. Notably, in the largest MPRA datasets (fixed-length random UTRs denoted as U1 and U2), our model achieved state-of-the-art results (Spearman R = 0.962 and 0.924). Across the remaining six datasets, our model led in three tasks (Ψ2, m1Ψ1, and mC-U1), effectively matching UTR-LM in the number of tasks with top performance (both achieving the best results in 4 of 8).

Predictive performance, evaluated by the Spearman correlation coefficient, of various models across eight different 5’UTR libraries. Each bar represents the correlation coefficient. The models are differentiated by color as indicated in the plot. The test set for each library comprised the top 20,000 reads with the highest expression levels from randomly selected 50-nucleotide sequences.
Evaluating mRNABERT on CDS prediction tasks
We collected multiple datasets to evaluate the performance of our model on CDS prediction tasks. These datasets include the mRFP Expression61, Fungal Expression62 and Escherichia coli Proteins63 datasets, comprising thousands of data points on protein expression in fungi and E. coli; the mRNA Stability64 and SARS-CoV-2 Vaccine Degradation65 datasets, containing mRNA stability data; and the Tc-Riboswitches66 dataset, highlighting tetracycline riboswitch dimer sequences. These datasets cover various downstream tasks related to mRNA translation, stability, and regulation, incorporating data ranging from newly published recombinant proteins to bio-computation for SARS-CoV-2 vaccine design (Supplementary Table 6 contains detailed information about the datasets).
After fine-tuning mRNABERT on these datasets, we compared its performance with several state-of-the-art CDS prediction methods, including TF-IDF67, TextCNN54, Codon2vec53, RNABERT33, RNA-FM32, and CodonBERT43. Our results indicate that mRNABERT outperformed or matched all other methods in all 6 CDS-related prediction tasks, demonstrating exceptional performance in the SARS-CoV-2 Vaccine Degradation dataset (Table 1).
Furthermore, our analysis revealed that codon-based models such as CodonBERT excel in protein expression tasks but exhibit subpar performance in stability-related tasks. This discrepancy may be attributed to the pivotal role codons play in protein expression61, whereas mRNA stability is closely tied to its secondary structure64. Notably, the performance of codon-based models declined in datasets where the local and global secondary structure patterns of RNA sequences are crucial68, such as the SARS-CoV-2 vaccine degradation and Tc-riboswitch datasets. In contrast, mRNABERT effectively integrates nucleotide and codon information, encoding the structurally relevant 5′UTR and 3′UTR regions. Consequently, it demonstrates superior performance in tasks where CodonBERT struggles, as it can learn co-evolutionary and structural characteristics from millions of mRNA sequences. This capability aids in designing highly expressive and stable mRNA sequences.
Detecting RBP binding sites from 3′UTR sequences
RNA-binding proteins (RBPs) specifically bind to RNA molecules, and this binding depends on both RNA sequences and spatial structure characteristics69. We downloaded and processed protein-RNA crosslinking sites for 22 RBPs70 and fine-tuned the mRNABERT to predict RBP binding sites using these experimentally determined data. When evaluating the predictive performance of our model, we benchmarked it against several computational methods, including neural network models iDeepE55, DeepCLIP56, RPI-Net71, GraphProt272, BERT-RBP57, all pre-trained RNA models such as RNABERT33 and RNAFM32, and the previously best model designed for 3’UTR tasks, 3UTRBERT41.
To assess the effectiveness of each model, we employed five-fold cross-validation and evaluated predictions using three metrics: accuracy (ACC), F1-score, and Matthews correlation coefficient (MCC) (The definition of the evaluation metrics is in Supplementary Table 7). Across all 22 RBPs, mRNABERT demonstrated superior performance with an average ACC of 0.786, F1-score of 0.751, and MCC of 0.501, comparable to the best specialized 3UTRBERT model with an average ACC of 0.785, F1-score of 0.751, and MCC of 0.503. Remarkably, mRNABERT outperformed other methods for 13 out of the 22 RBPs, exceeding 3UTRBERT’s performance for 9 RBPs. Except for 3UTRBERT, mRNABERT significantly outperformed all other models. The next best performance was achieved by iDeepE, with an ACC of 0.758, an F1 score of 0.565, and an MCC of 0.413, which were on average 20% lower than those of mRNABERT (Fig. 4A and Supplementary Table 8). It is worth noting that BERT-RBP lagged due to the lack of pre-training, while other deep learning methods underperformed due to insufficient model capacity. These comparative results suggest that mRNABERT is a highly effective method for accurately identifying RBP binding sites in the 3′UTR.

A The heatmap visually illustrates the prediction performance of different models for RBP-RNA interactions, with color intensity representing performance levels, where darker shades indicate better performance. B The bubble chart compares the performance of different models in predicting m6A modification sites. Each row represents a model, and data from different cell lines are indicated by bubbles of various colors. Larger bubbles positioned further to the right indicate better model performance. Each panel corresponds to a specific statistical metric.
Identifying m6A modification sites from 3′UTR sequences
N6-methyladenosine (m6A) is the most common covalent modification in cells, involved in numerous critical developmental processes and human diseases73. We downloaded real m6A modification sites from the m6A-Atlas database74 and enhanced the prediction capabilities of mRNABERT for potential m6A modification sites by fine-tuning (refer to Methods for detailed information).
We conducted a comparative analysis of mRNABERT’s predictive performance with various models found in the literature, such as the most effective model 3UTRBERT41, as well as different machine learning-based methods (SRAMP58, WHISTLE59, iMRM75) and deep learning-based methods (DeepM6ASeq76). The results displayed in Fig. 4B and Supplementary Table 9 indicated that mRNABERT achieved the second-best performance consistently across all nine cell lines, closely trailing the leading 3UTRBERT model while surpassing all other models. These findings demonstrate that mRNABERT possesses the ability to capture and utilize structural and functional information from the 3′UTR, exhibiting comparable performance to models extensively pre-trained exclusively on 3′UTR data.
Predicting splice sites and alternative polyadenylation
RNA splicing is a fundamental regulatory mechanism in eukaryotic gene expression, orchestrating the precise removal of non-coding intronic sequences from precursor mRNAs (pre-mRNAs) and the ligation of coding exons to generate mature transcripts77. This process critically depends on the accurate recognition of splice sites that demarcate exon-intron boundaries. At the 5′end of introns, donor sites initiate splicing, while acceptor sites at the 3′termini facilitate exon ligation.
Accurate identification of these splice sites constitutes a critical prerequisite for determining gene architecture and transcriptional isoforms. Computational approaches to this challenge are frequently framed as sequence-based binary classification tasks, where algorithmic models discriminate authentic splice signals from decoy sequences within pre-mRNA molecules. To this end, we utilized a widely adopted dataset of positive and negative splice site sequences78, which includes donor and acceptor site data from four distinct species. We fine-tuned the models using the same dataset and testing protocol to evaluate all RNA baseline models. mRNABERT exhibited the second-highest overall performance, outperformed solely by ERNIE-RNA and surpassing both RiNALMo and UNI-RNA (Supplementary Table 10).
Alternative polyadenylation (APA) is a widespread post-transcriptional regulatory mechanism that diversifies transcriptomes through selective 3′UTR processing79, thereby generating mRNA isoforms with distinct stability, localization, and protein-coding potential. This dynamic process fine-tunes gene expression networks and is indispensable for cellular differentiation, stress responses, and developmental patterning.
To systematically quantify APA dynamics, we integrated isoform-level predictions derived from the BEACON dataset80 into our analytical framework. Our approach specifically models the relative usage of proximal versus distal polyadenylation sites (PAS) within annotated 3′UTR regions, enabling precise resolution of APA-mediated regulatory outcomes. In this task, mRNABERT exhibited a significant advantage over all other RNA baseline models (Supplementary Table 11).
mRNABERT’s superior performance in these specific tasks provides compelling evidence for its profound understanding of post-transcriptional mRNA modifications, thereby significantly expanding its analytical capabilities within the broader landscape of mRNA research.
Applying mRNABERT to protein engineering tasks
We evaluated the performance of mRNABERT on protein-related tasks, noting that codon pLM models have previously shown superior results in certain amino acid sequence annotation tasks42. We assessed the performance of mRNABERT on several protein-related tasks, specifically predicting protein melting points and solubility. Additionally, we gathered and compiled transcriptome abundance data from seven organisms to evaluate the model’s effectiveness in key codon usage tasks. All datasets were mapped back to original codon sequences, with further details provided in the Methods.
We fed amino acid sequences into advanced pLMs such as ESM227, ProtTrans26, and the Ankh28 series, and mapped codon sequences into mRNA models, including CaLM42 and mRNABERT. We also tested mRNABERT without contrastive learning to better understand the impact of amino acid semantic integration. The resulting embeddings from these models were then utilized in the downstream task model, and performance was evaluated through the use of five-fold cross-validation (refer to Methods for detailed information).
Figure 5 and the supplementary file demonstrate that mRNABERT with contrastive learning exhibited significant improvement across all tasks. In melting point prediction (Fig. 5A), after contrastive learning, mRNABERT increased its R2 from 0.60 to 0.77, slightly below CaLM’s 0.78 but surpassing all other large-scale protein models (best ProtT5-XL with R2 = 0.73). In solubility prediction (Fig. 5B), mRNABERT achieved an R2 of 0.63, surpassing both its performance before contrastive learning and CaLM’s 0.61. Furthermore, mRNABERT’s performance in this task is comparable to most protein models, falling just behind larger-scale models like ProtT5-XL and Ankh-large (R2 = 0.66).

Predictions of protein melting point (A) and solubility (B). Each point represents a model, plotted by its R² value against the number of model parameters (log scale). Models are colored by their architectural family (e.g., mRNABERT, ESM, ProtT5). For each model, the central point is the mean R², and error bars represent the standard deviation (s.d.) across n = 5 folds of cross-validation. C Transcript abundance prediction across seven species. Results for each of the seven species are differentiated by color. Each box plot shows the distribution of R² values from n = 5 folds of cross-validation. The center line indicates the median, the box limits represent the upper and lower quartiles, and the whiskers extend to 1.5 times the interquartile range. Individual data points from each fold are overlaid as dots. In all panels, the cross-validation folds are considered computational replicates of the evaluation procedure. mRNABERT- refers to models that have not undergone contrastive learning.
Additionally, in the task of transcript abundance prediction across seven species, mRNABERT outperformed CaLM in five species(except E. coli and Haloferax hvolcanii). Remarkably, across multiple species, mRNABERT outperformed all other models. For instance, in predictions for Homo sapiens, mRNABERT achieved an R2 of 0.38, significantly higher than CaLM’s 0.35 and surpassing all other protein models (highest R2 = 0.36). In the predictions of Pichia pastoris and Saccharomyces cerevisiae, mRNABERT is also substantially superior to any other models, achieving respective best R2 of 0.56 and 0.53, while the highest performance among protein models is 0.53 and 0.52 (Fig. 5C).
The success of the CaLM model underscores the potential of codon-based pre-training to enhance the quality of protein models42. Furthermore, our mRNA model exhibited superior performance in certain protein-related tasks. Based on these results and ablation studies, the integration of amino acid information with encoding sequences emerges as a cost-effective approach to enhancing overall model performance substantially. This outcome highlights the potential of leveraging extensive biological data to enhance machine learning capabilities, thereby addressing model limitations and broadening its applicability.
Evaluating the applicability using full-length mRNA sequences
Redesigning complete mRNA sequences to maximize their stability and expression can significantly improve the overall performance of therapeutic mRNA23. However, designing such sequences faces challenges due to a limited understanding of how mRNA sequences and structures affect their expression and stability in solution and cells81,82,83. Therefore, accurately predicting the structural and functional properties of complete mRNA will aid in understanding mRNA design rules, greatly advancing mRNA vaccine development.
Rapidly synthesizing large quantities of full-length mRNA with different UTRs and CDS is challenging, making direct comparisons of their stability and expression capabilities through high-throughput experimental approaches impossible. To address this issue, we compiled a dataset of hundreds of reporter gene constructs that encompass a wide range of UTR and CDS mRNA sequences23, resulting in 233 usable mRNA sequences, with 112 distinct 5′ and/or 3′UTRs and 121 CDSs. The dataset included labels for four cell interior translation efficiencies and two stability-related properties that directly impact protein expression levels. To further explore the potential of mRNA models, we fine-tuned mRNABERT using the collected data and evaluated its performance in real-world mRNA tasks. Additionally, we assessed all currently available RNA baseline models, such as UTR-LM40 for 5′UTRs, codon-related CaLM42 and mRNA-FM, 3UTRBERT41 for 3′UTRs, and various RNA pre-trained models including RNABERT33, RNA-FM32, RNA-MSM35, ERNIE-RNA34, RNAErnie31 and RiNALMo.
The results in Fig. 6 and Supplementary Table 12 demonstrated that mRNABERT significantly outperformed other models across all tasks. Models pre-trained on ncRNA data struggled to generalize to full-length mRNA, and models excelling in specific mRNA region tasks performed poorly on complete mRNA tasks. This discrepancy likely arises because previous models used nucleotide-based tokenizers constrained by maximum input lengths, causing truncation and information loss for full-length mRNAs. Codon-based tokenizers often misinterpret non-triplet region segments, leading to information confusion. Our model adopted a dual-tokenizer approach for UTR and CDS regions and incorporates an advanced technique to enable a BERT architecture to extend input sequence lengths and improve practical application. Additionally, previous mRNA models were trained and evaluated on specific fragments, thereby limiting their efficacy on full-length mRNA tasks. In contrast, ncRNA models primarily focus on RNA structure prediction, which is often challenging to surpass mRNA-trained models.

A, B The bar charts in orange represent two stability-related prediction tasks. C–F The bar charts in blue represent four translation efficiency-related prediction tasks. The first four bars to the left of mRNABERT represent pre-trained models on 5′UTR, CDS, and 3′UTR regions, while the five bars to the right represent different ncRNA pre-trained models. G The average performance across the six tasks. The x-axis uses a logarithmic scale, with a break in the middle, to represent the number of model parameters. All models were fine-tuned using the same code, data, and parameter settings to ensure a fair comparison. Performance is compared using the R² metric, revealing that mRNABERT demonstrates a significant performance lead over most models of comparable size.
To rigorously evaluate mRNABERT’s predictive capabilities for ultra-long mRNA sequences, we conducted additional benchmark tasks focused on predicting the translation efficiency of full-length mRNA in mammalian cells84. The analysis leveraged a comprehensive dataset derived from thousands of ribosome profiling experiments paired with matched RNA-seq data across >140 human and mouse cell types. Notably, the human dataset (mean length: 4040 nt) contained 94.9% of sequences exceeding 1024 nt, with 82.2% surpassing 1022 tokens post-encoding. The mouse dataset (mean length: 3645 nt) demonstrated comparable proportions (94.6% and 80.8%, respectively). These sequence lengths substantially surpass the maximum input capacities of existing RNA models (typically limited to 1024 nt) and our training dataset (Supplementary Table 13). However, it is crucial to emphasize that the application of ALiBi enables mRNABERT to handle sequences longer than 1022 tokens. Sequences exceeding the model’s max_length parameter were systematically truncated to ensure computational feasibility.
To assess the generalization capabilities of mRNABERT on sequences exceeding the training length, we evaluated mRNABERT with maximum sequence lengths of 1022, 2044, and 3066 tokens. Our results reveal that mRNABERT consistently outperformed all existing RNA models, achieving a mean R² value of 0.66 across cell types (Table 2). This represents a substantial performance enhancement, ranging from 1.6 to 10.4-fold improvement over previous RNA models, which attained a maximum R² of 0.42 (range: 0.06–0.42). Moreover, the observed performance gains with increasing input length suggest that mRNABERT exhibits robustness and applicability to longer sequences. This finding underscores the benefits of our model design: dual tokenization facilitates the capture of comprehensive mRNA information, while the ALiBi mechanism enables generalization to extended sequence lengths. mRNABERT demonstrates applicability and a clear advantage in predicting the properties of longer mRNA sequences.
Overall, the success of mRNABERT in these challenging tasks fully illustrates the efficacy of our model design strategy and its tremendous potential in real-world application scenarios.
Discussion
In this study, we developed mRNABERT, a foundational model designed to analyze and predict mRNA properties. Pre-trained on our newly compiled repertoire of 18 million mRNA sequences from various species, mRNABERT lays the foundation to universally tackle all mRNA-related tasks with one model. Furthermore, it further integrates pLM-derived amino acid semantic information through contrastive learning. Similar to other unsupervised large language models, our goal was for mRNABERT to capture a wide range of information shaped by natural selection, thereby facilitating the design of mRNA sequences with high expression and stable structures. Our analyses demonstrate that mRNABERT indeed learns numerous insightful knowledge from various biological sequences.
We then evaluated mRNABERT’s performance on several supervised prediction tasks, including tests on UTRs, CDS, and protein-related datasets. Benchmark comparisons with state-of-the-art methods for each mRNA region indicate that mRNABERT generally outperforms or matches the best-specialized models for various mRNA tasks. Notably, mRNABERT surpassed all other models in every test in predicting properties of full-length mRNA, showcasing its broad applicability and practical potential.
Several key advantages contribute to mRNABERT’s success. Developing a powerful pre-training model relies on large-scale, high-quality data. We constructed a high-quality mRNA sequence database, which forms the foundation of our model’s success. Once trained on this extensive data, the model learns intrinsic structures and syntax, allowing it to adapt flexibly to specific subtasks with minimal fine-tuning. We identified the limitations of existing tokenization methods in computational and representational capacities and innovatively proposed a hybrid modeling approach suitable for full-length mRNA. Furthermore, we integrated various techniques, such as attention with linear biases (ALiBi) and Flash Attention, to enhance existing model structures and significantly boost overall performance through contrastive learning. Preliminary explorations were also made into enhancing mRNA model capabilities using multimodal data.
The ability of mRNABERT to accurately predict mRNA properties directly from sequences will assist researchers in exploring new mRNA mechanisms. Although the tasks focused on were inherently supervised, as a large language model, mRNABERT is also applicable for generative purposes. Specifically, we envision using this model to optimize various components of mRNA vaccine sequences based on the target protein (amino acid) sequences or to select and design specific mRNA sequences to achieve particular biological functions, such as genome editing. As future research progresses, we anticipate that optimizing mRNA design and sampling via mRNABERT will be of great significance for basic research, disease treatment, and the development of new therapies.
Despite the promising results achieved by mRNABERT, we acknowledge that it represents a foundational step, and we recognize several exciting avenues for future improvement. A key direction is the explicit integration of structural information. Future iterations could move beyond indirect inference from sequence towards the direct modeling of physical interactions by developing multi-modal architectures that incorporate predicted mRNA secondary structures or other biophysical properties. This would enhance the model’s mechanistic interpretability, particularly for tasks like RBP binding. Architecturally, to address the inherent computational constraints of Transformers on extremely long transcripts, exploring models with linear complexity is a promising path to improve scalability. Future work could investigate sparse attention mechanisms or emerging architectures, such as State Space Models, which have shown great potential for handling long-range dependencies more efficiently than traditional Transformers. Finally, future work can also build upon our large-scale dataset by refining data preprocessing strategies to enable more nuanced handling of complex genomic features. Pursuing these directions will be crucial for developing the next generation of more powerful and comprehensive mRNA language models.
In conclusion, mRNABERT stands as a pioneering pre-trained model for mRNA, bridging the gap between biological sciences and existing knowledge systems. Through techniques such as cross-modal feature alignment, mRNABERT helps us one step closer to mastering the complex rules governing the functionality of mRNA sequences.
Methods
Training datasets
Database construction and data collection
RNA molecules are classified into two categories: messenger RNA (mRNA) and non-coding RNA (ncRNA). As of January 2023, RNAcentral stands as the most extensive and comprehensive ncRNA database, integrating data from 56 expert databases and containing over 30 million sequences38. Current RNA models are predominantly pre-trained on RNAcentral data. However, there is a dedicated mRNA database specifically tailored for training large language models. To address this gap, we embarked on constructing an mRNA database by undertaking an extensive data collection process, similar to recent studies85.
For our dataset integration, we aggregated mRNA data from various sources, including the nt database from NCBI86, and transcriptome assembly data from MG-RAST87, GWH88,89, and MGnify90. From these diverse sources, we extracted all complete mature mRNA sequences (containing full CDS regions) to compile a dataset comprising approximately 36 million sequences.
Pre-processing the data
To ensure consistency and facilitate subsequent analysis, we mapped all collected sequences to the DNA alphabet. NCBI data (e.g., RefSeq or GenBank) constitutes over 70% of our dataset and serves as our primary, high-quality, and manually annotated data source. We directly used the provided CDS position annotations. For the remaining unannotated data where precise start positions could not be established, we sought to identify the most accurate and efficient open reading frame (ORF) prediction method. We benchmarked several widely used tools on a representative dataset and found that NCBI’s ORFfinder91, when combined with a 40% CDS length filter, demonstrated the best balance of high accuracy across diverse species and superior computational efficiency (Supplementary Table 1). Therefore, we adopted this method, defining each ORF from the start codon ATG to the nearest stop codon, with the longest continuous region designated as the coding sequence. Next, we conducted length and redundancy control measures. Sequences with predicted CDS lengths below 40% of the maximum possible were excluded, and those exhibiting high identity redundancy were removed. Furthermore, to balance computational efficiency with data quality, sequences exceeding 1022 nucleotides post-coding were excluded, despite the theoretical maximum length being 3066 nucleotides. Further details and discussions can be found in the Supplementary Information Part 2. Analysis of the Pre-training Dataset.
Subsequently, we meticulously curated a high-quality mRNA dataset consisting of approximately 18 million unique mRNA sequences. Each sequence was methodically classified based on its species of origin, enabling a more nuanced understanding of data provenance. Detailed information is provided in Supplementary Fig. 1 and Supplementary Tables 2 and 3. To rigorously evaluate model performance and generalizability, we randomly stratified the data and established an independent holdout dataset.
Model details
Sequence tokenization
Previous mRNA pretraining models typically employed two different encoding methods depending on the data and target region: models designed for UTRs encoded each nucleotide as a token40, while models for CDS used triplets of nucleotides (codons) as tokens42,43. In this study, we combined these approaches by applying different tokenization methods to UTRs and CDSs. This innovative method allowed us to capture hidden states and attention weights of each part and the full sequence. To standardize the sequence alphabet, we converted all uracil (U) bases to thymine (T) to align with RNA-to-cDNA sequencing protocols. The input sequence is a vector of T tokens, with each token being an integer representing a nucleotide, codon, or special character. The vocabulary includes 64 codons, 5 nucleotides (A, T, C, G, and the rare base N), and five special tokens: [MASK] for masking, [PAD] for padding, [UNK] for unknown codons, and [CLS] and [SEP] for denoting sequence boundaries. Notably, the model was not trained with any labels or prior information, emphasizing that it cannot differentiate whether token 6 represents nucleotide A or token 18 signifies the start codon ATG.
Model architecture
mRNABERT consists of 12 transformer layers with a hidden state dimension of 768. Configured similarly to DNABERT-230, mRNABERT replaces positional embeddings with Attention with Linear Biases (ALiBi) and integrates I/O-aware Flash attention to improve the accuracy of standard attention calculations while enhancing efficiency in terms of time and memory. After token embedding, mRNA sequences are then input into the Transformer.
where L represents the token sequence length.
Model training
We pre-trained the model using MLM loss with a masking ratio of 15%. Specifically, 80% of the tokens were replaced with the [MASK] token, 10% were randomly replaced with another token, and the remaining 10% were left unchanged. To balance training time and cost, we limited the sequence length to a maximum of 1022 tokens, which we found adequate for encompassing most mRNA transcripts. Each batch of sequences was padded to the maximum length.
For optimization purposes, we implemented the AdamW optimizer with a learning rate of 1 × 10−4 alongside default settings for other parameters. The learning rate underwent a linear increase from 0 to 1 × 10−4 within the initial 10,000 steps, followed by a linear decay to zero by 1,000,000 steps. To monitor training progress, we randomly reserved 1% of the training set as a validation set. The reported model was trained on NVIDIA A6000 GPUs, corresponding to 660,000 gradient steps over 10 epochs. We halted the training manually after observing no improvement in validation loss over 10,000 steps. The training loss curve is available for reference in Supplementary Fig. 3.
To incorporate more comprehensive sequence information and enable the model to learn multimodal molecular interactions, we further conducted contrastive learning after MLM. We selected 500,000 CDS data from the training set and input the translated amino acid sequences into a protein language model (pLM). To optimize computational efficiency without sacrificing performance, we utilized the half-precision version of the ProtT5-XL-UniRef50 model26 to generate amino acid or protein embeddings, thereby reducing GPU memory consumption. With the pLM weights kept frozen, we fed the sequences into their corresponding models and extracted embeddings from the final hidden layer. To encode the sequences as fixed-size vectors (768 dimensions for CDS and 1024 dimensions for amino acids), we averaged the obtained embeddings. To facilitate contrastive learning, a trainable linear layer was employed to project the 768- and 1024-dimensional vectors into 256 dimensions. Through the utilization of the OpenAI-CLIP library92, we trained the model with a contrastive loss function aimed at minimizing the distance between corresponding sequences while maximizing the distance between non-corresponding ones. The loss function used for this purpose is defined as follows93:
where C represents the CDS sequence embeddings and A represents the amino acid sequence embeddings, both projected to 256 dimensions. The final trained model is mRNABERT.
Model evaluation
We employed the t-SNE method60 to reduce the dimensionality of token and sequence embeddings to two dimensions for visualization. Initially, embeddings for all vocabulary words were extracted and clustered according to amino acid categories and properties. We used the adjusted_rand_score and fowlkes_mallows_score functions from the Scikit-learn library to calculate the ARI and FMI, respectively. These metrics are based on:
Subsequently, we randomly selected segments from 5′UTR, CDS, and 3′UTR from downstream datasets, and complete mRNA sequences were chosen from the validation set. Long non-coding RNA (lncRNA) data were obtained from the GENCODE database94. A total of 9287 entries were used for RNA classification. Finally, we selected six representative species from the validation dataset, covering mammals, insects, plants, bacteria, fungi, and viruses, using a total of 3452 complete mRNA sequence entries for species classification. All data were carefully curated to ensure less than 40% overlap in identity and exclusion from the training set.
Downstream task datasets
Ribosome load datasets of 5′ UTR sequences
The dataset is derived from an MPRA investigation carried out by Sample et al.50, consisting of random 50-nucleotide-long 5′ UTR sequences paired with their respective average ribosome load. To enhance data reliability, read counts are included. The dataset is divided into eight libraries, which are further classified into two groups. Six libraries contain a constant region that encodes the enhanced green fluorescent protein (eGFP), while the remaining two libraries have the coding sequences (CDS) of mCherry instead of eGFP. Within the eGFP group, there are two libraries with unmodified uridine (U) and libraries that have been modified with pseudouridine (Ψ) and 1-methylpseudouridine (m1Ψ). Details of dataset splitting and fine-tuning strategies can be found in the supplementary information.
Datasets related to CDS sequences
The mRFP dataset61, constructed through synonymous codon randomization, encompasses the complete coding sequence of red fluorescent protein (mRFP) and consists of 1459 gene variants in E. coli. It records the protein yield and investigates the correlation between codon usage and protein production. The Fungal dataset62 compiles information on protein-coding genes and tRNA genes from diverse fungal genomes across multiple species. The E. coli dataset63 contains experimental data on protein expression in E. coli, categorized into low, medium, and high expression levels with 2308, 2067, and 1973 sequences, respectively. The mRNA stability dataset64 provides insights into mRNA stability features from zebrafish, Xenopus laevis embryos, and mouse and human cells, aiming to elucidate the codon-dependent regulation of mRNA stability. The Tc-Riboswitches dataset66 features tetracycline (Tc) riboswitch dimer sequences positioned upstream of GFP, facilitating the evaluation of the switch factor to discern differential effects in the presence or absence of Tc. The SARS-CoV-2 Vaccine Degradation dataset65 comprises optimized mRNA sequences derived from structural features, stability, and translation efficiency. By employing a consistent data partitioning strategy, we refined the mRNABERT model and conducted a comparative analysis against existing literature. More details on these datasets are provided in the supplementary information.
RBP binding sites and human m6A modifications across nine cell lines
We collected and analyzed two datasets from Yang et al.41 The first dataset consolidates data from 31 CLIP (crosslinking immunoprecipitation) experiments, covering 19 RBPs. A unified data processing workflow and specific sequence window sizes were used to analyze these data, differentiating between positive datasets (RBP binding sites) and negative datasets (non-binding sites). Measures were taken to reduce redundancy and avoid interference between adjacent sites.
The second dataset includes human m6A modification data at single-nucleotide resolution across nine cell lines, resulting in 131,703 high-confidence m6A sites. By selecting non-m6A adenosines from the same transcripts’ 3′ UTR and removing duplicates, we generated a dataset with 79,021 m6A sites and 849,005 non-m6A sites, maintaining a 1:10 positive to negative ratio. To ensure accuracy and reliability, we employed 10-fold cross-validation and random down-sampling methods.
Splice sites and alternative polyadenylation datasets
The splice site dataset used was the GS_1 dataset95. This dataset maintains a balanced ratio of positive to negative samples, with negative samples consisting of exon, intron, or false positive sequences. The dataset was constructed by randomly selecting sequences from the exon and intron regions of the G3PO+ genomic sequences. It comprises error-free splice-site sequences derived from a diverse set of 148 eukaryotic organisms, including humans. Importantly, the test dataset incorporates sequences from four distinct species not represented in the training set. The APA dataset utilized was sourced from BEACON, which filtered 228,000 sequences from over 3 million APA reporter gene entries in Bogard’s96 dataset. This regression task aims to quantify and evaluate the relative proportion of proximal APA isoforms.
Protein engineering tasks datasets
To validate the quality of the embedding, several protein engineering task datasets constructed by Carlos et al.42 were utilized. These datasets encompass a series of melting temperatures reported in the FLIP97 study and solubility proxy data from solubility assays conducted by Sridharan et al.98 Amino acid sequences were mapped to nucleotide sequences using UniProt IDs, with exclusions made for sequences that could not be mapped or did not meet standards. Furthermore, transcriptome data, consisting of RNA sequences from seven model organisms, was used to estimate transcript abundance in assemblies through transcripts per million and to map these data to existing sequence databases.
Dataset of full-length mRNA sequences
The PERSIST-seq study conducted by Leppek et al.23 provides the full-length mRNA dataset, which systematically assesses the translation efficiency and stability of various mRNA sequences intracellularly and extracellularly. The mRNA ′library includes 233 different mRNA sequences with 112 unique 5′ and/or 3′ UTRs. Unlike the randomized short UTRs screened in Sample et al.‘s study50, this library uses full-length native UTRs to test mRNA expression, including sequences from cellular and viral genomes. By employing diverse algorithms and design methods22,99, the CDS sequences and structures of protein targets were diversified, resulting in 121 CDS variants. PERSIST-seq analyzed the polysome profiles of constructs in this mRNA library, calculating ribosome load and stability, aiding in a comprehensive understanding of the effects of different functional regions on mRNA properties. These datasets were used to fine-tune our mRNABERT model. To ensure fair performance comparison with benchmark models, we also fine-tuned other models using the multimolecule library100 from Hugging Face, despite some models not initially utilizing fine-tuning methods in their reports.
Polysome and ribosome profiling are established as direct methods for assessing translation rates101. Zheng et al.84 compiled and leveraged a comprehensive collection of 3819 ribosomal profiling datasets, distilling them into a transcriptome-wide atlas of translation efficiency (TE) measurements encompassing >140 human and mouse cell types. We utilized this resource to predict TEs in hundreds of cell types based on sequence-encoded mRNA features.
Detailed descriptions of benchmark models and methodologies for each downstream task are provided in the supplementary information.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The pre-training and downstream task datasets generated in this study have been deposited in the Zenodo database under accession https://doi.org/10.5281/zenodo.12516160102. Detailed statistics for downstream task datasets are provided in the supplementary information. Source data are provided with this paper.
Code availability
The code required to reproduce the findings of this study has been deposited in a permanent Zenodo archive under the https://doi.org/10.5281/zenodo.15112690103. The most up-to-date version of the code is available on GitHub at https://github.com/yyly6/mRNABERT. The pre-trained model is available on Hugging Face at https://huggingface.co/YYLY66/mRNABERT.
References
Rohner, E., Yang, R., Foo, K. S., Goedel, A. & Chien, K. R. Unlocking the promise of mRNA therapeutics. Nat. Biotechnol. 40, 1586–1600 (2022).
Zhang, G., Tang, T., Chen, Y., Huang, X. & Liang, T. mRNA vaccines in disease prevention and treatment. Signal Transduct. Target. Ther. 8, 365 (2023).
Pardi, N. et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 543, 248–251 (2017).
de Jong, W. et al. iHIVARNA phase IIa, a randomized, placebo-controlled, double-blinded trial to evaluate the safety and immunogenicity of iHIVARNA-01 in chronically HIV-infected patients under stable combined antiretroviral therapy. Trials 20, 1–10 (2019).
Lutz, J. et al. Unmodified mRNA in LNPs constitutes a competitive technology for prophylactic vaccines. Npj Vaccines. 2, 29 (2017).
John, S. et al. Multi-antigenic human cytomegalovirus mRNA vaccines that elicit potent humoral and cell-mediated immunity. Vaccine 36, 1689–1699 (2018).
Aliprantis, A. O. et al. A phase 1, randomized, placebo-controlled study to evaluate the safety and immunogenicity of an mRNA-based RSV prefusion F protein vaccine in healthy younger and older adults. Hum. Vaccines Immunother. 17, 1248–1261 (2021).
Monslow, M. A. et al. Immunogenicity generated by mRNA vaccine encoding VZV gE antigen is comparable to adjuvanted subunit vaccine and better than live attenuated vaccine in nonhuman primates. Vaccine 38, 5793–5802 (2020).
Alberer, M. et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 390, 1511–1520 (2017).
Baden, L. R. et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 384, 403–416 (2021).
Dong, Y. et al. Poly (glycoamidoamine) brushes formulated nanomaterials for systemic siRNA and mRNA delivery in vivo. Nano Lett. 16, 842–848 (2016).
Polack, F. P. et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 383, 2603–2615 (2020).
Barbier, A. J., Jiang, A. Y., Zhang, P., Wooster, R. & Anderson, D. G. The clinical progress of mRNA vaccines and immunotherapies. Nat. Biotechnol. 40, 840–854 (2022).
Guan, S. & Rosenecker, J. Nanotechnologies in delivery of mRNA therapeutics using nonviral vector-based delivery systems. Gene Ther. 24, 133–143 (2017).
Karik, O. K. et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 16, 1833–1840 (2008).
Thess, A. et al. Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol. Ther. 23, 1456–1464 (2015).
Pollard, A. J. & Bijker, E. M. A guide to vaccinology: from basic principles to new developments. Nat. Rev. Immunol. 21, 83–100 (2021).
Sahin, U. et al. mRNA-based therapeutics—developing a new class of drugs. Nat. Rev. Drug Discov. 13, 759–780 (2014).
Wang, Y. et al. mRNA vaccine: a potential therapeutic strategy. Mol. Cancer 20, 33 (2021).
Metkar, M., Pepin, C. S. & Moore, M. J. Tailor made: the art of therapeutic mRNA design. Nat. Rev. Drug Discov. 23, 67–83 (2024).
Chaudhary, N., Weissman, D. & Whitehead, K. A. mRNA vaccines for infectious diseases: principles, delivery and clinical translation. Nat. Rev. Drug Discov. 20, 817–838 (2021).
Zhang, H. et al. Algorithm for optimized mRNA design improves stability and immunogenicity. Nature 621, 396–403 (2023).
Leppek, K. et al. Combinatorial optimization of mRNA structure, stability, and translation for RNA-based therapeutics. Nat. Commun. 13, 1536 (2022).
Castillo-Hair, S. M. & Seelig, G. Machine learning for designing next-generation mRNA therapeutics. Acc. Chem. Res. 55, 24–34 (2021).
Devlin, J., Chang, M., Lee, K. & Toutanova, K. Bert: pre-training of deep bidirectional transformers for language understanding. Preprint at https://doi.org/10.48550/arXiv.1810.04805 (2018).
Elnaggar, A. et al. ProtTrans: Towards cracking the language of lifes code through self-supervised deep learning and high performance computing. IEEE Trans. Pattern Anal. Mach. Intell. 44, 7112–7127 (2020).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Elnaggar, A. et al. Ankh: optimized protein language model unlocks general-purpose modelling. Preprint at https://doi.org/10.48550/arXiv.2301.06568 (2023).
Ji, Y., Zhou, Z., Liu, H. & Davuluri, R. V. DNABERT: pre-trained bidirectional encoder representations from transformers model for DNA-language in genome. Bioinformatics 37, 2112–2120 (2021).
Zhou, Z. et al. Dnabert-2: Efficient foundation model and benchmark for multi-species genome. Preprint at https://doi.org/10.48550/arXiv.2306.15006(2023).
Wang, N. et al. Multi-purpose RNA language modelling with motif-aware pretraining and type-guided fine-tuning. Nat. Mach. Intell. 6, 548–557 (2024).
Chen, J. et al. Interpretable RNA foundation model from unannotated data for highly accurate RNA structure and function predictions. Preprint at https://doi.org/10.48550/arXiv.2204.00300 (2022).
Akiyama, M. & Sakakibara, Y. Informative RNA base embedding for RNA structural alignment and clustering by deep representation learning. Nar. Genom. Bioinform. 4, lqac012 (2022).
Yin, W. et al. ERNIE-RNA: An RNA language model with structure-enhanced representations. Preprint at bioRxiv https://doi.org/10.1101/2024.03.17.585376 (2024).
Zhang, Y. et al. Multiple sequence alignment-based RNA language model and its application to structural inference. Nucleic Acids Res. 52, e3 (2024).
Penić. R. J. et al. Rinalmo: general-purpose rna language models can generalize well on structure prediction tasks. Nat. Commun. 16, 5671 (2025).
Zhang, J., Fei, Y., Sun, L. & Zhang, Q. C. Advances and opportunities in RNA structure experimental determination and computational modeling. Nat. Methods 19, 1193–1207 (2022).
RNAcentral 2021: secondary structure integration, improved sequence search and new member databases. Nucleic. Acids. Res. 49, D212-D220 (2021).
Kalvari, I. et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 49, D192–D200 (2021).
Chu, Y. et al. A 5′ UTR language model for decoding untranslated regions of mRNA and function predictions. Nat. Mach. Intell. 6, 449–460 (2024).
Yang, Y. et al. Deciphering 3’UTR mediated gene regulation using interpretable deep representation learning. Adv. Sci. 11, 2407013 (2023).
Outeiral, C. & Deane, C. M. Codon language embeddings provide strong signals for use in protein engineering. Nat. Mach. Intell. 6, 170–179 (2024).
Li, S. et al. CodonBERT large language model for mRNA vaccines. Genome Res. 34, 1027–1035 (2024).
Ruffolo, J. A. & Gray, J. J. Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies. Biophys. J. 121, 155a–156a (2022).
Nguyen, E. et al. Sequence modeling and design from molecular to genome scale with Evo. Science 386, eado9336 (2024).
Wang, X. et al. UNI-RNA: universal pre-trained models revolutionize RNA research. Preprint at bioRxiv https://doi.org/10.1101/2023.07.11.548588 (2023).
Tay, Y. et al. Charformer: fast character transformers via gradient-based subword tokenization. Preprint at https://doi.org/10.48550/arXiv.2106.12672 (2021).
Press, O., Smith, N. A. & Lewis, M. Train short, test long: attention with linear biases enables input length extrapolation. Preprint at https://doi.org/10.48550/arXiv.2108.12409 (2021).
Dao, T. et al. Flashattention: Fast and memory-efficient exact attention with io-awareness. Adv. Neural Inf. Process. Syst. 35, 16344–16359 (2022).
Sample, P. J. et al. Human 5′ UTR design and variant effect prediction from a massively parallel translation assay. Nat. Biotechnol. 37, 803–809 (2019).
Karollus, A., Avsec, V. Z. I. & Gagneur, J. Predicting mean ribosome load for 5’UTR of any length using deep learning. Plos Comput. Biol. 17, e1008982 (2021).
Zheng, W. et al. Discovery of regulatory motifs in 5′ untranslated regions using interpretable multi-task learning models. Cell Syst. 14, 1103–1112 (2023).
Mikolov, T., Chen, K., Corrado, G. & Dean, J. Efficient estimation of word representations in vector space. Preprint at https://doi.org/10.48550/arXiv.1301.3781 (2013).
Kim, Y. Convolutional neural network for sentence classification. Preprint at https://doi.org/10.48550/arXiv.1408.5882 (2014).
Pan, X. & Shen, H. Predicting RNA-protein binding sites and motifs through combining local and global deep convolutional neural networks. Bioinformatics 34, 3427–3436 (2018).
Gr, O. et al. DeepCLIP: predicting the effect of mutations on protein-RNA binding with deep learning. Nucleic Acids Res. 48, 7099–7118 (2020).
Yamada, K. & Hamada, M. Prediction of RNA-protein interactions using a nucleotide language model. Bioinforma. Adv. 2, vbac023 (2022).
Zhou, Y., Zeng, P., Li, Y., Zhang, Z. & Cui, Q. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 44, e91 (2016).
Chen, K. et al. Whistle: a high-accuracy map of the human n 6-methyladenosine (m6a) epitranscriptome predicted using a machine learning approach. Nucleic Acids Res. 47, e41 (2019).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Nieuwkoop, T. et al. Revealing determinants of translation efficiency via whole-gene codon randomization and machine learning. Nucleic Acids Res. 51, 2363–2376 (2023).
Wint, R., Salamov, A. & Grigoriev, I. V. Kingdom-wide analysis of fungal protein-coding and tRNA genes reveals conserved patterns of adaptive evolution. Mol. Biol. Evol. 39, msab372 (2022).
Boël, G. et al. Codon influence on protein expression in E. coli correlates with mRNA levels. Nature 529, 358–363 (2016).
Medina-Mu, N. et al. Crosstalk between codon optimality and cis-regulatory elements dictates mRNA stability. Genome Biol. 22, 1–23 (2021).
Wayment-Steele, H. K. et al. Deep learning models for predicting RNA degradation via dual crowdsourcing. Nat. Mach. Intell. 4, 1174–1184 (2022).
Groher, A. et al. Tuning the performance of synthetic riboswitches using machine learning. Acs Synth. Biol. 8, 34–44 (2018).
Aizawa, A. An information-theoretic perspective of tf-idf measures. Inf. Process. Manag. 39, 45–65 (2003).
Groher, F. et al. Riboswitching with ciprofloxacin—development and characterization of a novel RNA regulator. Nucleic Acids Res. 46, 2121–2132 (2018).
Hentze, M. W., Castello, A., Schwarzl, T. & Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 19, 327–341 (2018).
Ma, H., Wen, H., Xue, Z., Li, G. & Zhang, Z. RNANetMotif: identifying sequence-structure RNA network motifs in RNA-protein binding sites. PLoS Comput. Biol. 18, e1010293 (2022).
Yan, Z., Hamilton, W. L. & Blanchette, M. Graph neural representational learning of RNA secondary structures for predicting RNA-protein interactions. Bioinformatics 36, i276–i284 (2020).
Uhl, M., Tran, V. D., Heyl, F. & Backofen, R. GraphProt2: a graph neural network-based method for predicting binding sites of RNA-binding proteins. Preprint at bioRxiv https://doi.org/10.1101/850024 (2021).
Dominissini, D. et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012).
Tang, Y. et al. m6A-Atlas: a comprehensive knowledgebase for unraveling the N 6-methyladenosine (m6A) epitranscriptome. Nucleic Acids Res. 49, D134–D143 (2021).
Liu, K. & Chen, W. iMRM: a platform for simultaneously identifying multiple kinds of RNA modifications. Bioinformatics 36, 3336–3342 (2020).
Zhang, Y. & Hamada, M. DeepM6ASeq: prediction and characterization of m6A-containing sequences using deep learning. Bmc Bioinforma. 19, 1–11 (2018).
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176, 535–548 (2019).
Chen, K. et al. Self-supervised learning on millions of pre-mRNA sequences improves sequence-based RNA splicing prediction. Biorxiv, 2021-2023 (2023).
Yuan, F., Hankey, W., Wagner, E. J., Li, W. & Wang, Q. Alternative polyadenylation of mRNA and its role in cancer. Genes \ Dis. 8, 61–72 (2021).
Ren, Y. et al. Beacon: benchmark for comprehensive RNA tasks and language models. Preprint at https://doi.org/10.48550/arXiv.2406.10391 (2024).
Wayment-Steele, H. K. et al. Theoretical basis for stabilizing messenger RNA through secondary structure design. Nucleic Acids Res. 49, 10604–10617 (2021).
Mauger, D. M. et al. mRNA structure regulates protein expression through changes in functional half-life. Proc. Natl. Acad. Sci. USA 116, 24075–24083 (2019).
Thess, A. et al. Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol. Ther. 23, 1456–64 (2015).
Zheng, D. et al. Predicting the translation efficiency of messenger RNA in mammalian cells. Nat. Biotechnol. https://doi.org/10.1038/s41587-025-02712-x (2025).
Chen, K., Litfin, T., Singh, J., Zhan, J. & Zhou, Y. MARS and RNAcmap3: the master database of all possible RNA sequences integrated with RNAcmap for RNA homology search. Genom. Proteom. Bioinform. 22, qzae018 (2024).
Sayers, E. W. et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 52, D33 (2024).
Wilke, A. et al. A RESTful API for accessing microbial community data for MG-RAST. PLoS Comput. Biol. 11, e1004008 (2015).
Chen, M. et al. Genome warehouse: a public repository housing genome-scale data. Genom. Proteom. Bioinforma. 19, 584–589 (2021).
Database Resources of the National Genomics Data Center. China National Center for Bioinformation in 2024. Nucleic Acids Res. 52, D18–D32 (2024).
Richardson, L. et al. MGnify: the microbiome sequence data analysis resource in 2023. Nucleic Acids Res. 51, D753–D759 (2023).
Rombel, I. T., Sykes, K. F., Rayner, S. & Johnston, S. A. ORF-FINDER: a vector for high-throughput gene identification. Gene 282, 33–41 (2002).
Shariatnia, M. M. OpenAI-CLIP. https://zenodo.org/record/6845731 (2021).
Hallee, L., Rafailidis, N. & Gleghorn, J. P. cdsBERT-Extending Protein Language Models with Codon Awareness. Preprint at bioRxiv https://doi.org/10.1101/2023.09.15.558027 (2023).
Frankish, A. et al. GENCODE: reference annotation for the human and mouse genomes in 2023. Nucleic Acids Res. 51, D942–D949 (2023).
Scalzitti, N., Jeannin-Girardon, A., Collet, P., Poch, O. & Thompson, J. D. A benchmark study of ab initio gene prediction methods in diverse eukaryotic organisms. Bmc Genomics. 21, 1–20 (2020).
Bogard, N., Linder, J., Rosenberg, A. B. & Seelig, G. A deep neural network for predicting and engineering alternative polyadenylation. Cell 178, 91–106 (2019).
Dallago, C. et al. FLIP: Benchmark tasks in fitness landscape inference for proteins. Preprint at bioRxiv https://doi.org/10.1101/2021.11.09.467890 (2021).
Sridharan, S. et al. Proteome-wide solubility and thermal stability profiling reveals distinct regulatory roles for ATP. Nat. Commun. 10, 1155 (2019).
Sharp, P. M. & Li, W. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281–1295 (1987).
Chen, Z. & Zhu, S. Y. MultiMolecule. https://doi.org/10.5281/zenodo.12638419 (2024).
Li, J. J., Bickel, P. J. & Biggin, M. D. System wide analyses have underestimated protein abundances and the importance of transcription in mammals. Peerj 2, e270 (2014).
Xiong, Y. Pre-trained Dataset of mRNABERT. Zendo, https://doi.org/10.5281/zenodo.12516160 (2024).
Xiong, Y. yyly6/mRNABERT: Version 1.0. Zendo, https://doi.org/10.5281/zenodo.15112690 (2025).
Acknowledgements
This work was financially supported by the National Key R&D Program of China (2024YFA1306400, 2024YFA1307501 to T.H.), the National Natural Science Foundation of China 999 (22373085 to C.Y.H.), and the Medical Interdisciplinary Innovation Program 2024, Zhejiang University School of Medicine.
Author information
Authors and Affiliations
Contributions
Y.X. and C.Y.H. designed and developed mRNABERT; Y.X. and A.W. performed the evaluation and wrote the code; Y.X., Y.K., and C.S. analyzed the data; Y.X. and C.Y.H. wrote the initial draft of the manuscript; and C.Y.H. and T.H. revised the manuscript and supervised the overall study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiong, Y., Wang, A., Kang, Y. et al. mRNABERT: advancing mRNA sequence design with a universal language model and comprehensive dataset. Nat Commun 16, 10371 (2025). https://doi.org/10.1038/s41467-025-65340-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65340-8
