
Abstract
Genomic coupling theory predicts that progress towards speciation involves a transition from the dominant effects of selection on individual barrier loci to the aggregate effects of direct and indirect selection across loci that collectively produce stronger barriers to gene flow through genetic associations. However, our ability to test this prediction and to understand the factors that lead to the buildup and maintenance of these associations has been limited by a lack of methods to estimate variation in coupling across the genome. Here we develop approaches to quantify coupling using window-based estimates of Barton’s coupling coefficient and apply these to a dataset of 118 genomes from a rattlesnake hybrid zone. Our results provide empirical evidence for genomic coupling that is consistent with the predicted relationships of coupling with recombination, linkage, and inferences of selection. Applying these approaches, we find evidence for coupling within and among chromosomes, and highlight the roles of coupling in complex barrier effects, including the Large-Z effect, cytonuclear incompatibilities, and incompatibilities related to venom resistance. Together, our findings demonstrate the mechanism by which coupling is predicted to lead to speciation, and highlight how genome-wide quantification of coupling presents a promising framework for understanding progress towards speciation and the processes that underlie this progress.
Similar content being viewed by others

Introduction
A fundamental goal of biology is to understand the mechanisms that promote reproductive isolation (RI) and drive the generation and maintenance of species1,2,3,4,5. A persistent question in speciation research relates to the number of factors (i.e., traits and their underlying genes) that act as barriers to gene flow6,7, and the processes by which these barriers arise and strengthen to complete RI8,9. As lineages diverge in allopatry, RI is predicted to increase through the buildup of barrier effects and their underlying genetic basis (i.e., barrier loci)10,11. Following secondary contact, theory predicts that the combined effects of selection across loci can produce a strong cumulative barrier to gene flow causing the barrier effects and underlying loci to remain coupled or statistically associated despite admixture12. Here, loci with even weak effects on hybrid fitness may still contribute to strong RI through coupling with other barrier loci13. This view of coupling, based on multilocus-cline theory12, predicts a transition from a genic to a genomic phase of speciation as allopatric divergence between lineage pairs becomes greater. During the genic phase, direct selection acts on individual barrier loci, such that barrier effects are largely localized to distinct genomic regions. As speciation proceeds towards the genomic phase, the aggregate effect of both direct and indirect selection among barrier loci lead to genome-wide patterns of RI12,14. Based on the interaction of selection and recombination, coupling theory predicts this transition will produce strong genome-wide barrier effects and a non-linear buildup of linkage disequilibrium (LD) with greater divergence in allopatry between lineage pairs. This genic-to-genomic (or weak to strong coupling12) transition represents a critical step known as genome-wide congealing, predicted to result in speciation at its final phase12,15,16. However, a lack of frameworks for estimating genome-wide variation in coupling has limited our ability to test these predictions and to identify factors that contribute to the buildup of RI in natural systems16.
Speciation research has long been motivated to place lineages along a “speciation continuum”17 and to capture the relationship between RI at the organismal versus genomic level17,18, although practically meeting these goals has remained challenging19,20. Recently, coupling theory has experienced renewed interest because it provides a generalizable framework to quantify progress towards speciation by measuring the aggregation of direct and indirect barrier effects, while also inherently integrating evolutionary processes and genome architecture6,7,15,21,22,23. Coupling theory also complements various paradigms of speciation involving the accumulation and association of reproductive barriers, such as Large-X/Large-Z effects24, Bateson–Dobzhansky–Muller incompatibilities25,26, cytonuclear incompatibilities27, and snowball effects11,28,29, making it broadly applicable to any phenomena which manifest in stronger aggregate reproductive barriers through the buildup of direct and indirect selective effects.
Despite its utility to empirical speciation research16, evidence supporting key tenets of coupling theory, including evidence for genome-wide variation in the buildup of associations, and for interactive effects of intra- and interchromosomal coupling, have been primarily theoretical or from simulation-based studies6,7. Simulations have provided evidence that coupling can complete RI in speciation scenarios with15,16,21,30 or without13,22 gene flow and suggest that even weak per-locus selection across many individual barrier loci can rapidly lead to genome-wide barrier effects. Some empirical evidence of coupling has also emerged. For example, studies have revealed remarkably high numbers of loci in LD and under selection in natural hybrids, patterns that are generally consistent with predictions of coupling31,32,33,34,35,36. Additionally, geographic cline analyses applied to empirical data have shown evidence for stepped clines31,37,38, an expected outcome of coupling12. Other empirical studies have even provided evidence for coupling among multiple barrier loci that lead to stronger aggregate barriers to introgression23,35,39,40. However, empirical evidence for how coupling varies on genome-wide scales, and how this manifests in aggregate barrier effects, is lacking.
While coupling is viewed—in this study—as the accumulation of genome-wide LD in hybrids, impacting both neutral and selected loci (following Perspective 27), the observation of LD alone does not necessarily translate to direct evidence for the degree of coupling6. This is because some degree of LD (proportional to differences in allele frequencies between populations) is expected to result from admixture even in the absence of selection6,41, and measures of LD do not account for levels of genomic admixture (i.e., “hybrid index”) among individuals. Barton’s coupling coefficient12, which is defined by the ratio of the mean selection to mean recombination among loci, provides a parametric approach for quantifying the impact of indirect selection that can predict coupling. Accordingly, a dominant role of indirect selection is indicated when the coefficient exceeds 112. However, estimating this metric directly from empirical data is impractical due to the difficulty of identifying causal mutations and measuring their fitness effects. Because coupling theory stems from Barton’s cline theory12, coupling coefficients can be empirically quantified by estimating the coincidence of genomic cline parameters (slopes and intercepts) among selected and neutral loci12,22, which inherently accounts for hybrid index. With greater progress towards coupling, theory predicts a transition from independent to “coupled” behavior among barrier loci, as each barrier locus shifts towards experiencing the total selection rather than the locus-specific selection. With this progression, neutral loci experience increasingly strong indirect selection and LD with barrier loci leading to the coincidence of clines for barrier loci and neutral loci (Fig. 1A). Because coupling depends on both selection and recombination, some regions of the genome might become coupled earlier in the speciation process than others, and strongly coupled regions of the genome will converge on similar cline slopes and intercepts (Fig. 1A). Thus, quantification of genomic cline coincidence in hybrids can be used to empirically estimate the extent of coupling6,12,22,23.

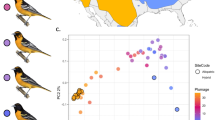
A Theoretical predictions of the progression of coupling as divergence in allopatry increases. Representation of the genomic region for which coupling is illustrated is shown with barrier loci in red and neutral loci in grey. Due to increased selection (represented by the transparency, bolder less-transparent colored bars along the genomic locus indicate stronger selection), LD among barrier loci increases as does the effect on surrounding neutral sites that are impacted by indirect selection with the progression of coupling (assumed to represent progress towards speciation). Below, cline center and cline slope of genomic regions converge more as coupling becomes stronger; color scales on the right represent cline center (top) and cline slope (bottom). Respective examples of Bayesian genomic clines are shown below each divergence point that reflect increasing coincidence of cline parameters for loci in the genomic region. While it is expected that cline slope will increase with increased progress of coupling, cline center may react in various ways (i.e., bias towards either parental species, or be neutral); the example shown is a scenario that leads to bias towards one parental species. B ADMIXTURE plot displaying ancestry coefficient of all sampled individuals. C Genomic cline plot of 1000 variants in different hybrid zone systems with the rattlesnake hybrid zone studied here shown in dark grey, and other systems analyzed in Firneno et al.22 in light grey; the number at the top left shows the genome-wide coupling coefficient for each hybrid system.
To empirically test predictions of coupling theory and gain insight into the causes and consequences of coupling, new methods are needed to quantify coupling in nature. Here we go beyond prior advances by quantifying cline variance to estimate variation in the extent of coupling across the genome. To do so, we develop methods to quantify variation in coupling using window-based estimates of coupling coefficients derived from genomic cline variances. We use these approaches to test key tenets of theory by applying them to data from a hybrid zone between two rattlesnake species (Crotalus scutulatus and Crotalus viridis)42,43. We leverage recent advances that apply Bayesian Genomic Cline approaches (BGC)44,45 to genome-scale data46, which we use in a novel way to estimate cline variation and infer genome-wide variation in coupling. We first confirm the validity of our inferences of coupling by comparing our coupling estimates to inferences of recombination, LD, and genomic cline parameters, which each have predicted relationships with coupling. We then apply our approach to address the overarching goals of empirically testing genome-scale predictions of coupling theory and demonstrating evidence for the impact of coupling in specific multilocus contexts related to other broad concepts of speciation and the biology of the empirical system. Because rattlesnakes have highly heteromorphic Z and W sex chromosomes47, we test for evidence for coupling contributing to Large-Z effects48,49. We also investigate coupling in multilocus effects related to intergenomic cytonuclear incompatibilities, based on evidence that these contribute to RI in related species50. Finally, we test for evidence of coupling related to self-resistance to venom, based on evidence that parental species exhibit major differences in venom composition, including highly neurotoxic Phospholipase A2 (PLA2) venom components present in one parental species42,51 that are absent in the other. Together, our results provide genome-wide empirical evidence for key tenets of coupling theory that illustrate the importance of the buildup of associations within and among chromosomes in the strengthening of RI. Our approach and empirical findings further highlight the potential of genomic coupling to be applied as a quantitative framework for comparing the buildup of RI and progress towards speciation.
Results
Variant dataset, population structure, differentiation, and demography
Whole genome resequencing data was collected for 118 individuals, including 31 parental C. viridis, 24 parental C. scutulatus, and 63 putative hybrids at an average of ~23-fold genome coverage per sample (Supplementary Table S1). Read mapping percentages are similar for both parental species (Supplementary Table S1), indicating that no significant biases exist due to mapping to the reference genome of C. viridis47 (Supplementary Fig. S1). Data were phased (average switch error rate =4.7%), and after filtering our gVCF contains 2.75 million variants (average SNP quality score =21,086.4; Supplementary Fig. S2), representing 0.2% of the 1.3 gigabase reference genome47. Based on analysis of a reduced version of this dataset (thinned to 1 random SNP per 10 kb), and a best-fit model of K = 2 populations (Supplementary Fig. S3), Admixture52 ancestry inferences are consistent with two distinct species and a clinal hybrid zone between them (Fig. 1B; Supplementary Fig. S4). PCA analyses of this thinned dataset are consistent with Admixture inferences, with hybrid individuals clustering between parental species individuals (PC1 = 68.6% and PC2 = 4.5% variation explained; Supplementary Fig. S5A). We also find a significant positive relationship between Admixture ancestry coefficients and PC1 scores (p < 10−15, R2 = 0.998; Supplementary Fig. S5B), indicating that ancestry coefficients are predictive of genetic variation among parentals and hybrids (Supplementary Fig. S5B). Additionally, mitochondrial haplotypes tend to correspond with major parent ancestry, except for a few hybrid individuals (Supplementary Fig. S6). Individuals with >99.999% Admixture ancestry coefficients were assigned as parentals (n = 24 C. scutulatus; n = 31 C. viridis) and the remaining individuals as hybrids (n = 63). Genome-wide genetic differentiation is moderately high between the two parental species (mean Fixation Index FST = 0.40, standard deviation σ = 0.34), with 4.4% of variants being fixed for different alleles (FST = 1) and 21% of variants above FST = 0.8 (Supplementary Fig. S7). Inferences of demography in each parental lineage using SMC++53 suggest that present-day effective population sizes are similar in both species, with the population-scaled mutation rate being slightly higher in C. scutulatus (4.16 × 10−5) versus C. viridis (3.14 × 10−5; Supplementary Fig. S8).
Genome-wide Bayesian genomic cline inferences
BGCs model the probability of ancestry for an allele as a function of hybrid index and are defined by two parameters: cline center and cline slope. Cline center indicates the hybrid index value at which the transition in ancestry is steepest, while cline slope reflects how rapidly this probability changes around the center. Our bgc-hm46 results indicate substantial variation in patterns of introgression across the genome (Figs. 1C, and 2A). We identify 548,631 variants with credible deviations from null patterns of introgression (i.e., exceeding the 95% CI of genome-wide average introgression) for cline center, cline slope, or both. Among these, 409,074 variants show excess ancestry (biased cline centers) in hybrids, with 211,145 showing excess C. viridis-biased ancestry, and 197,929 with excess C. scutulatus ancestry. A total of 186,560 variants show credible deviations from null expectations for cline slope, 64,201 with a credibly steep cline and 122,359 with a flatter cline slope. The Z chromosome is significantly enriched (chi-squared test p < 10−15) for both excess ancestry and extreme cline slopes (20.4% and 12.9% percent of all Z variants, respectively), compared to macro- and microchromosomes (14.2% and 13.8% for excess ancestry variants, respectively; 6.0% and 6.3% for cline slope outliers). While the average chromosome-wide cline center and cline slope values for autosomes are close to null expectations (macrochromosomes=0.503 and microchromosomes=0.507 for cline center; 1.01 and 0.967 for cline slope, respectively; Supplementary Fig. S9), the Z chromosome shows a significant chromosome-wide average biased ancestry for C. viridis with a mean cline center of 0.469 ± 1.94 × 10−4, and overall steeper clines (1.127 ± 3.4 × 10−4) (Wilcoxon signed-rank test p < 10−15 for both comparisons).

A Plot of genome-wide mean genomic cline parameters for 100 kb windows, with the mean cline center displayed on the outer track (top color scale: cline center biased towards C. viridis are shown downward and green, C. scutulatus upward and purple) and mean cline slope on the inner track (bottom color scale: flat clines are shown downward and blue, steep clines upward and red). Centromeric regions are indicated with black bars between tracks. B Empirical chromosome-wide distribution of the coupling coefficient computed over 100 kb windows for our rattlesnake hybrid zone dataset (top to bottom, n = 3118, 2306, 1799, 1033, 874, 696, 615, 226, 200, 168, 168, 138, 138, 124, 110, 68, 54, 1118; boxplots show the median (center line), interquartile range (box), and whiskers extending to the furthest point within 1.5×IQR; outliers not shown). Dotted lines indicate the highest and lowest median values for autosomes, while the dashed line indicates the median value of the Z.
Hybrid zone simulations and estimation of coupling
Because coupling predicts decreased variance in genomic cline parameters, we first quantified variation in cline slope (σv) and center (σc) separately22 for adjacent non-overlapping 100 kb genomic windows. Comparing the distribution of per-window cline variance across chromosomes, we observe very similar patterns for both σc and σv (Supplementary Fig. S10), with the average of both cline variance estimates being lower overall for macrochromosomes (mean of σc: μσc = 0.42, mean of σv: μσv = 0.16) compared to microchromosomes (μσc = 0.49 and μσv = 0.18). The Z chromosome has the lowest mean per-window variance for both cline parameters (μσc = 0.36 and μσv = 0.14) followed by ma6 for per-window cline center variance (μσc = 0.38) and ma4 for per-window cline slope variance (μσv = 0.15; Supplementary Fig. S10).
Next, to integrate cline slope and center variation into a single metric, we used the simulation-derived estimator of ‘coupling coefficient’ described in Firneno et al.22. This approach uses linear equation to estimate the coupling coefficient (θ12) as a function of variation in cline slope (σv) and cline center (σc). While both the coefficients of cline slope and cline center variation are negative, cline slope variation plays a larger role in the estimation of the coupling coefficient in the specific simulations from Firneno et al.22, with a weight ~15 times greater than that of cline center variation (see detailed equation in “Methods”). To evaluate the utility of using this equation with our data set, we conducted two sets of simulations: one with the parameters described in Firneno et al. and another with larger population sizes per deme. As expected, simulations using Firneno et al.’s parameters produce a linear equation for estimating the coupling coefficient based on cline variance with coefficients closely matching those previously reported (Supplementary Table S2). By contrast, simulations with modified parameters yield an equation where the contribution of cline center variation and the interaction coefficient are comparatively high. Additionally, we conducted a PCA of empirical 100 kb window σc and σv estimates (Supplementary Fig. S11A) to compare with estimates obtained from linear equations. We find strong correlation between the PC1 values and the coupling coefficient estimates obtained with both linear equations (p < 10−15, R2 = 0.84; Supplementary Fig. S11B, C), justifying the use of linear equations to capture overall cline variance and to approximate the coupling coefficient. Since both simulation-based equations fit empirical PC1 values similarly well, we opted to use the equation from Firneno et al., which has the advantage of being directly comparable to multiple prior studies22,23. Finally, to directly compare our empirical dataset to a prior study that used randomly sampled sets of loci to roughly approximate a coupling coefficient across multiple systems, we calculated a genome-wide coupling coefficient as per Firneno et al.22 based on cline variance in 1000 randomly sampled genome-wide SNPs. Using this approach, we obtain a coupling coefficient of 0.84 for our rattlesnake species (Fig. 1C), which is relatively high when compared with other hybrid zones analyzed using this framework22. This estimate of the coupling coefficient is fundamentally distinct from those estimated from 100 kb windows because it is based on sampling a subset of predominantly unlinked genome-wide variants, rather than capturing variation across genomic windows. This value ranks as the fifth highest among 26 hybrid zones examined by Firneno et al. (a subset of which are shown in Fig. 1C), suggesting that the rattlesnakes studied here are at a relatively advanced stage of the speciation continuum when compared to other systems.
Relationships between recombination and coupling
By estimating the coupling coefficient across non-overlapping 100 kb windows of entire chromosomes, we find evidence for substantial variation in coupling coefficients among and within chromosomes. Overall, window-based estimates of coupling coefficients are highest on the Z chromosome (μ = 1.29, σ = 0.26), and relatively higher on average for macrochromosomes (μ = 1.12, σ = 0.23) compared to microchromosomes (μ = 1.00, σ = 0.24) (Fig. 2B; Supplementary Fig. S12). We also observe substantial variation in the coupling coefficient among regions within chromosomes (Fig. 3A, Supplementary Fig. S13), notably with several regions displaying peaks of elevated coupling coefficient values, which also corresponds to substantial dips in per-window cline center and slope variance (Fig. 3B).

A Coupling coefficient computed over 100 kb windows for three autosomes and the Z chromosome, with a black line indicating smoothed values. B Cline slope variance (σv; tope panels) and cline center variance (σc; bottom panels) over chromosomes, with a black line indicating smoothed values. Hybrid recombination deficit (C) and mean cline slope (D) for 100 kb windows. Neutral values for hybrid recombination deficit (0) and cline slope (1) are represented by a dashed line. A–D Dark grey bars indicate centromeric regions, grey backgrounds highlight region around centromeres with elevated coupling, and orange backgrounds highlight hotspots of coupling in areas distal from the centromeres. E Genome wide scatterplot of hybrid recombination rate versus minimum parental recombination rate; colors indicate the coupling coefficient values (with coupling coefficient value colors following panel (A). Scatterplot of coupling coefficient versus hybrid recombination deficit (F) and versus cline slope (G). E, F regression lines are shown in dark grey, with R2 and p-value (obtained from a two-sided t-test st; p < 10−15 in all three tests) indicated in the top left corner.
Because coupling represents the tension between recombination and selection, we expect strong aggregate effects of selection to result in elevated coupling that should also manifest in excess intrachromosomal LD and the reduced observation of recombination events (hybrid recombination deficit, hereafter) in hybrids. To detect evidence of selection leading to relatively increased hybrid LD, and to test if our coupling coefficient captured these effects, we compared inferences of parental and hybrid recombination rates alongside the coupling coefficient (Fig. 3A, C). As expected, the minimum parental recombination rate overall is positively correlated with hybrid recombination rate (p < 10−15, R2 = 0.55; Fig. 3E), with per-window cline center variance (p < 10−15, R2 = 0.19; Supplementary Fig. S14A), cline slope variance (p < 10−15, R2 = 0.26; Supplementary Fig. S14B), and negatively correlated with the coupling coefficient (p < 10−15, R2 = 0.29; Supplementary Fig. S14C). While we do observe strong correlations between estimates of coupling coefficients and parental species recombination, we also identify many genomic regions with high recombination deficits in hybrids (Fig. 3C). Indeed, genome-wide, we find that the hybrid recombination deficit is positively correlated with the coupling coefficient (100 kb windows; p < 10−15, R2 = 0.04; Fig. 3F). Congruent with our recombination inferences, we also find that, for 100 kb windows, the frequency of genomic ancestry block breaks (corresponding to interspecies recombination events) is positively correlated with cline variance (p < 10−15, R2 = 0.10 for both center and slope variance; Supplementary Fig. S15A, B) and negatively correlated with coupling coefficients (p < 10−15, R2 = 0.12, Supplementary Fig. S15C). These data together are consistent with predictions that coupling manifests as reduced recombination in hybrids and in the coincidence of cline parameters across adjacent genomic windows, and that our coupling coefficient effectively captures these effects, as intended.
By estimating variation in coupling across the genome, we find that the Z chromosome harbors the largest contiguous regions of elevated coupling (along with reduced cline variation) and higher recombination deficit in hybrids compared to autosomes (Fig. 3A–C). We also find that regions with elevated estimates of coupling coefficients tend to overlap with centromeric regions of chromosomes, particularly for macrochromosomes and the Z (Fig. 3A, Supplementary Fig. S13). Many near-centromeric regions with elevated coupling coefficients are also associated with high hybrid recombination deficit (Fig. 3A–C). However, we also identify many regions distant from the centromeres with high coupling coefficient and high hybrid recombination deficit (Fig. 3A–C, Supplementary Fig. S13), suggesting the existence of multiple distinct hotspots of elevated coupling distributed across the genome independent of centromeric regions.
Cline theory predicts that strong selection on hybrid genotypes manifests as steep clines, and that coupling leads to the aggregation of selective effects across barrier loci into a single stronger barrier effect that affects both selected barrier and linked neutral loci6,12,22. Consistent with these predictions, we find that coupling coefficients and cline slopes over 100 kb windows are positively correlated genome-wide (p < 10−15; R2 = 0.22; Fig. 3D, G), while the relationship between coupling coefficients and per-window mean cline center is weak (Supplementary Fig. S16). We note that the relationship between coupling coefficient values and cline slope is not an automatic outcome of coupling estimation because it is inferred from cline variance and not cline slopes or centers. Because genomic clines represent deviations relative to an average (as captured by hybrid index), the mean cline slope on a log scale across all loci is 0. Thus, the genome-wide variance is not correlated with the mean slope because the mean slope should not itself vary, though such correlations are possible for individual windows. These comparisons also highlight the extreme density and magnitude of steep clines on regions of ma6 and the Z chromosome that correlate with high coupling (Fig. 3A, D, Supplementary Fig. S13). Thus, the correspondence between high coupling coefficients and steep clines support the inference of an enhanced role of selection (relative to recombination) underlying coupling in hybrids.
Coincidence of cline parameters in coupling hotspots
In addition to the aggregation of barrier effects leading to the higher recombination deficit in hybrids, theory predicts that coupling should manifest as the coincidence of cline parameters among non-adjacent loci6,15,21. Consistent with these predictions, on most chromosomes we observe an excess of variants (i.e., hotspots) that exhibit coincident cline parameters (Supplementary Table S3; Supplementary Fig. S17). In many cases, these hotspots consist of many loci located in near-centromeric regions, along with additional distant loci on the same chromosome, all of which show coincidence in cline parameters (Supplementary Fig. S17). For example, ma7 and mi1 have hotspots that deviate from expectations of genome-wide ancestry (Fig. 4A, E), each of which include loci distributed across the chromosome (Fig. 4B, F) that are further associated with elevated coupling coefficients (Fig. 4C, G) and excess intrachromosomal LD per 100 kb windows in hybrids (Fig. 4D, H), which is the difference in the per-window mean intrachromosomal LD between hybrids and the parental species (see detailed explanation in “Methods”).

A Density plot of cline slope and center for all ancestry informative variants on macrochromosome 7 (ma7); colors represent the relative density of variants (number of neighboring points) with similar cline parameter values. White circles highlight ‘hotspots’ of highly frequent convergent cline parameters (see Supplementary Table S3). B Density of variants belonging to the hotspots in panel A plotted along ma7 with arrows from Panel A indicating the distribution of SNPs from each hotspot. Points representing variants for panels A, B were downsampled 10-to-1 for visualization purposes; Supplementary Fig. S17 shows non-downsampled versions. C Coupling coefficient across the ma7, with smoothed values across the chromosome indicated by the black line. D Heatmap showing excess intrachromosomal hybrid LD (difference between the minimum R2 of parental species and the R2 of hybrids) across ma7. E–H Density plot of ancestry informative variants, density of variants, respective coupling coefficient, and excess intrachromosomal hybrid LD across microchromosome 1 (mi1), following the same format as panels A–D.
Some of the most extreme signatures of coupling and coupling hotspots occur on the Z. We identified high frequencies of variants with similar cline parameters that involve loci primarily located in one of three regions of the Z (Z1–Z3 on Fig. 5A–C). These three regions are all associated with C. viridis-biased steep clines and with elevated coupling coefficients (Fig. 5A–D). Furthermore, these three regions are outstanding in the degree that they demonstrate coincident clines across nearby and distant loci, consistent with theoretical predictions of strong coupling. Also consistent with predictions of strong coupling, the density of observed recombination events between parental species (i.e., breaks in inferred ancestry blocks) in hybrids is greatly reduced along the ancient stratum (e.g., Z1), even in parts of the chromosome distant from the centromere (Fig. 5E). This inferred hybrid recombination deficit is also consistent with large contiguous ancestry blocks observed on the ancient stratum (Fig. 5E; Supplementary Fig. S13). For example, within Z1 we identified two ~6 Mb regions where no recombinant haplotypes are observed in hybrids (Fig. 5E).

A Density plot of cline slope and center for all ancestry informative variants on the Z chromosome showing hotspots of cline convergence (indicated with white circles); colors represent the density of convergent cline values (number of neighboring points). Points representing variants were downsampled 10-to-1 for visualization purposes; Supplementary Fig. S17 shows non-downsampled version. B Density plots showing the distribution of variants belonging to three hotspots from panel A along the Z chromosome. Arrows from Panel A to B indicate the distribution of SNPs from each hotspot. The green, blue and red rectangles show regions enriched for variants with convergent cline parameters corresponding to hotspots in panel A, a grey rectangle indicates the PAR, and the recent stratum is shown in pink. C Coupling coefficients for 100 kb windows across the Z chromosome, with a black line indicating the smoothed coupling coefficient values. D Cline center across the Z chromosome. E Number of heterospecific recombination events per 100 kb window across the Z chromosome. F Genome-wide intrachromosomal coupling with C. viridis Z-linked interchromosomal LD. The outer track shows mean cline slope over 100 kb windows, and the inner track displays the coupling coefficient over 100 kb windows and a smoothed coupling coefficient line in black. Interchromosomal conspecific LD between the Z and autosomes (above threshold of R2 > 0.7) is shown with green lines at the center of the circle. G Distribution of cline center (left panel) and cline slope (center panel) for variants in C. viridis Z linked conspecific LD and high-FST variants not in LD (for both panels: n = 24,260 for variants in LD and n = 385,351 for uncorrelated variants), along with the distribution of coupling coefficients (right panel) for windows containing at least one variant in C. viridis Z linked conspecific LD compared to windows containing highly differentiated variants without variants in LD (n = 470 and n = 9237, respectively). Boxplots show the median (center line), interquartile range (box), and whiskers extending to the furthest point within 1.5×IQR (outliers not shown) and p-values were obtained from a Wilcoxon test (***p < 0.001).
Buildup of coupled associations within and among chromosomes
The buildup and maintenance of associations through direct and indirect selection within and among chromosomes is predicted to be the underlying mechanism that leads to genomic congealing and the transition from weak to strong coupling in the final stages of speciation6,12,15,16,54. To test for interchromosomal associations within the hybrid population, we estimated conspecific LD by calculating LD between interchromosomal variants with FST values > 0.9 between parental species (referred to as high-FST variants hereafter) that also showed significant excess ancestry (i.e., variants where the 95% CI of the estimated cline center does not contain the neutral value) based on bgc-hm, and considered in LD all pairs of high-FST variants with a coefficient of determination above 0.7 (see details and justification for threshold in Supplementary Fig. S18).
We find that 97% of conspecific C. viridis interchromosomal LD involved the Z chromosome, with more than 800,000 pairs of high-FST variants (23,693 distinct variants) in strong LD (R2 > 0.7; Supplementary Fig. S18) compared to <3000 for autosomes-only pairs of LD variants (Fig. 5F; Supplementary Fig. S19). C. viridis-biased interchromosomal LD variants involving the Z chromosome are associated with steep clines (p < 10−15; Fig. 5G), and 100 kb windows containing these variants are enriched for lower variance for both cline slope and center (p < 10−15; Supplementary Fig. S20) and elevated coupling coefficient compared to windows containing high-FST variants without LD (p < 10−15; Fig. 5G). The highest concentration of variants in C. viridis conspecific LD is located on the non-recombining ancient stratum of the Z chromosome on Z1 (Fig. 5F; Supplementary Fig. S21). This region shows intermediate levels of coupling coefficient values and extremely strong evidence of interchromosomal associations, including loci in LD with distinct regions of nearly all autosomes. Regions Z2 and Z3 also show an abundance of interchromosomal LD links and are associated with extremely high coupling coefficient values. Considering only the subset of C. viridis conspecific LD variants on autosomes (n = 806), we identify complex networks of interchromosomal LD that include clustered hubs of LD on ma1, mi7 and mi8 (Supplementary Fig. S19). In contrast to other examples, while C. viridis interchromosomal LD loci-containing windows are enriched for steep clines, they are not enriched for elevated coupling coefficient (Supplementary Fig. S19B–D) or for enriched categories of genes.
To further explore interactions between interchromosomal LD and coupling, we examined C. scutulatus conspecific interchromosomal associations (Fig. 6), and identified 1200 high-FST variants with C. scutulatus conspecific LD distributed across nearly all chromosomes (Fig. 6A). These 1200 variants are significantly enriched for steep cline slopes (p < 10−15, Fig. 6B) compared to non-correlated high-FST variants, and 100 kb windows containing at least one variant in C. scutulatus conspecific LD are also associated with elevated coupling coefficients (p < 10−15, Fig. 6B) and lower cline variance (p < 10−15, Supplementary Fig. S22) compared to other windows containing high-FST variants. Among these regions, ma6 and ma7 contain the largest contiguous regions with the highest number of conspecific interchromosomal LD variants for C. scutulatus, and stand out as hubs of interchromosomal LD (Fig. 6A). These hubs on ma6 and ma7 contain multiple contiguous regions that show interchromosomal LD with other chromosomes, and are also in LD with one another (Fig. 6A). To test if these interchromosomal interactions may be driven by functional gene interactions (i.e., functional interchromosomal multilocus effects), we tested functional enrichment of the 324 genes within 100 kb windows of variants in C. scutulatus LD. We find significant enrichment for multiple functional categories, including 22 genes related to ion channel activity, which have STRING-predicted interactions with one another and with other genes in LD (FDR = 0.0033; Fig. 6C; Supplementary Fig. S23).

A Genome-wide coupling and interchromosomal LD for C. scutulatus conspecific highly differentiated variants. The outer track shows the mean cline slope over 100 kb windows, and the inner track displays the coupling coefficient over 100 kb windows, as well as a smoothed coupling coefficient line in black. Interchromosomal conspecific C. scutulatus LD is shown at the center of the circle. B Distribution of cline center (left panel) and cline slope (center panel) for variants in C. scutulatus conspecific LD compared to highly differentiated variants that are not in LD LD (for both panels: n = 1234 for variants in LD and n = 385,351 for uncorrelated variants; Wilcoxon test; ***p < 0.001). The right panel shows the distribution of coupling coefficients in windows containing at least one variant (n = 227) in C. scutulatus LD compared to windows containing at least one high-FST variants with none in LD (n = 9477; Wilcoxon test; ***p < 0.001). Boxplots show the median (center line), interquartile range (box), and whiskers extending to the furthest point within 1.5×IQR (outliers not shown). C Gene enrichment analysis of genes within 100 kb from a variant in C. scutulatus LD; enlarged, colored dots represent genes that are part of up to four gene sets functionally related to ion channels. Distribution of coupling coefficients for 100 kb windows and interchromosomal LD (R2) with PLA2 crotoxin venom haplotype in regions of ma2 (D) and ma7 (E) that spans genes related to GABA receptors. Thick black line represents smoothed coupling coefficient values and the grey ribbon shows the 95% CI. Genes associated with GABA receptors or ion channel activity in these regions are labeled at the top of each plot. F Illustration of the mechanism by which venom PLA2 neurotoxin (crotoxin; labeled C on the figure) is thought to inhibit GABA receptor signaling through its binding to these receptors.
A key phenotypic difference between C. viridis and C. scutulatus is the presence of a PLA2 neurotoxin (crotoxin) in the latter, which is known to target ion channels55,56,57 (Fig. 6F). To test if the enrichment for ion channel genes in the C. scutulatus conspecific LD network is related to the presence of this neurotoxin, we computed LD between haplotypes (with and without this neurotoxin) at the PLA2 gene cluster locus (on mi7) and all variants genome-wide. We find that 9 of 22 genes were located within 100 kb of a variant in LD with the PLA2 haplotype of hybrids (R2 > 0.65; Supplementary Fig. S24). These nine genes include four GABA receptor genes (GABRA1, GABRA2, GABRA4, GABRB1) that are located within contiguous regions showing a significant increase in interchromosomal coupling coefficient values and LD with PLA2 haplotypes compared to the rest of the chromosome (Wilcoxon signed-rank test, p < 0.05, Fig. 6D, E, Supplementary Fig. S25). Two other genes on ma1 outside of the GABA receptor clusters (TRPM8, UNC80) also occur in regions of elevated PLA2 haplotype LD and coupling (Supplementary Fig. S25).
Inter-genomic coupling and cytonuclear incompatibilities
Because cytonuclear interactions present among the most common and well-understood hybrid incompatibilities, we investigated nuclear-mitochondrial genome associations by first identifying high-FST nuclear variants in LD with the mitochondrial haplotype (mtDNA) in hybrids. Across the nuclear genome we identified 8278 of mtDNA-LD variants (R2 > 0.75; Supplementary Fig. S26), which were particularly abundant and dense on ma6 (Fig. 7A), corresponding to a region of the genome with among the highest coupling coefficient values. Variants in LD with the mitochondria are significantly enriched for steep clines and C. scutulatus excess ancestry (p < 10−15; Fig. 7B). Further, 100 kb windows containing at least one high-FST variant in LD with the mtDNA are significantly enriched for higher coupling coefficient values (p < 10−15; Fig. 7B) and lower cline slope and center variance (p < 10−15; Supplementary Fig. S27). Analysis of the 534 genes in 100 kb windows surrounding mitochondrial-LD variants using STRING revealed a subset of 378 genes that comprise a large interaction network; tests for functional enrichment of the full set of 534 genes identified a significant enrichment of genes (360) related to organelle function (FDR = 0.0029; Supplementary Fig. S28). Also among these were multiple nuclear-encoded genes (NDUFB5, NDUFA6, NDUFAF1) that function in mitochondrial complex I.

A Genome-wide coupling and LD with the mitochondrial genome. The outer track shows the mean cline slope over 100 kb windows, and the inner track displays the coupling coefficient over 100 kb windows, as well as a smoothed coupling coefficient line in black. Nuclear-mitochondrial LD is shown at the center of the circle. B Distribution of cline center (left) and cline slope (right) for variants in LD with the mtDNA and highly differentiated variants that are not in LD (for both panels: n = 8502 for variants in LD and n = 402996 for uncorrelated variants; Wilcoxon test; ***p < 0.001). The last panel on the right shows the distribution of coupling coefficients in windows containing at least one variant in LD with the mtDNA (n = 415) compared to windows containing at least one high-FST variants but not in LD (n = 9294; Wilcoxon test; ***p < 0.001). Boxplots show the median (center line), interquartile range (box), and whiskers extending to the furthest point within 1.5×IQR (outliers not shown).
Discussion
Despite renewed interest in applying coupling theory to empirical systems to quantify associations among barriers and their relevance to speciation6,7,12,16,21,22,23, a lack of approaches for estimating variation in coupling across the genome has limited its application and testing in nature16. Here, we develop new approaches that bridge this gap and apply them to investigate predictions of coupling, and its causes and consequences, in a rattlesnake hybrid zone. Our findings provide empirical evidence for the aggregation of barrier effects at genome-wide scales, providing confirmation of key predictions of coupling theory and its role in the strengthening of RI. Our results demonstrate that distinct chromosomes and genomic regions span a continuum of coupling within a single empirical system, with multiple regions and even chromosomes exceeding a coupling coefficient of 1, indicating a dominant role of aggregate and indirect effects on these loci consistent with predictions of coupling for a system in the mid-to-late stages of speciation15,22. These examples also illustrate how the quantification of coupling can provide insight into how (and even why) genome-wide aggregate barrier effects arise. Our evaluation of Large-Z effects, cyto-nuclear incompatibilities, and other multilocus effects in this system provides further evidence that coupling of barrier effects strengthens RI in each of these scenarios. Together, these findings provide among the most compelling genome-wide evidence in support of coupling theory, including the identification of genome-wide networks of intra- and interchromosomal coupling that act as barrier to gene flow. Importantly, such genome-wide coupled interactions are the mechanism that coupling theory predicts may eventually lead to speciation via genome congealing15,16,30.
To address our goals of testing predictions of coupling at genome-wide scales and understanding its causes and consequences, we first developed approaches for empirically estimating coupling that leverage cline variation across genomic windows. Coupling effectively represents a genome-wide tension between selection and recombination (s/r)12, and our approach estimates the coupling coefficient based on inferences of cline variation across the genome. We observe expected relationships between coupling and inferences related to recombination, linkage, and selection, suggesting that this estimator of the coupling coefficient reasonably captures the effects of coupling. In particular, our inferences of coupling correspond with observations of relatively reduced recombination (and inferences of excess intrachromosomal LD) in hybrids compared to parentals consistent with expectations of coupling. Importantly, we also identify elevated coupling in specific regions not associated with low recombination in parentals but associated with a high hybrid recombination deficit (e.g., ma6), consistent with expectations that coupling is maintained and strengthened by selection. Steep genomic clines, indicative of strong selection, are also enriched in regions of elevated coupling. Finally, non-adjacent regions within chromosomes with elevated coupling also show excess hybrid LD and cline coincidence (e.g., hotspots; Fig. 4).
Our analyses identified the Z chromosome as the single most coupled chromosome (mean coupling coefficient =1.29) in our rattlesnake system, which led us to further investigate the relevance of this pattern in the context of key paradigms in speciation. In particular, a well-known “rule” that commonly applies in species formation is the Large-X (or Large-Z) effect5,24,58,59, which predicts that sex chromosomes play a disproportionally large role in RI as lineages diverge24,59,60. Considering the importance of recombination to coupling, the lower recombination rates of sex chromosomes predicts they are particularly prone to intrachromosomal coupling22. While prior studies have found evidence that sex chromosomes show reduced introgression and exceptionally steep and center-biased clines (relative to the genome-wide average) in other ZW systems39,61, the relationship between coupling and Large-Z (or Large-X) effects has not been investigated. We estimate high coupling (>1) across the entire ancient stratum of the Z chromosome, with large tracks showing no evidence of interspecies recombinants. This suggests the Z is strongly dominated by selection and indirect effects that lead to nearly the entire chromosome acting as a single aggregate coupled barrier locus. Also consistent with selection driving strong coupling, multiple large regions of the Z are associated with steep cline slopes, highly biased ancestry, and high recombination deficit in hybrids. The Z also contains multiple hotspots of loci with coincident cline parameters associated with elevated coupling. Further, we find evidence for extremely high levels of interchromosomal LD between the Z and autosomes (99% of all interchromosomal C. viridis conspecific LD connections), indicating strong and numerous multilocus effects between Z and autosomal regions that are also enriched for elevated coupling. These findings are strongly suggestive of Z-autosome incompatibilities, consistent with multiple hypotheses for the Large-Z effect (e.g., Dominance62 and Dosage Theory33). Taken together, these results provide evidence for a significant role of both intra- and interchromosomal coupling that leads to strong aggregate genome-wide effects underlying the Large-Z effect. They also provide evidence for the potential for sex chromosomes to act as primary hubs of coupling capable of pulling the entire genome towards congealing and speciation.
While the field of speciation has long appreciated the importance of epistatic interactions and multilocus incompatibilities11,26,62,63,64,65, the importance of coupling in the aggregation and strengthening of multilocus effects remains untested except for a few studies that analyzed small sets of loci21,23,39. Among multilocus effects, cytonuclear incompatibilities are the most well-documented27,66,67, and have been implicated in prior studies to drive elevated LD between the mtDNA and specific nuclear genomic regions67. Here, we also find evidence for cytonuclear multilocus effects, including nuclear regions that show strong LD with the mtDNA in hybrids and that are enriched for steep clines, biased ancestry, and genes associated with organelle function, including multiple nuclear-encoded mitochondrial complex I genes. In this case, the buildup of associations is presumably driven by direct selection on multiple nuclear loci to be maintained with the corresponding mitochondrial haplotype, which itself represents coupling based on our definition here because it led to the coincidence of clines and the buildup of LD. Our results further show that nuclear loci in LD with the mtDNA in hybrids are enriched for elevated intrachromosomal coupling, and include some of the most highly coupled regions of the genome (e.g., ma6; Fig. 7A). Together these findings provide evidence that multilocus cytonuclear incompatibilities may manifest in strong coupled barrier effects, which result from the aggregation of indirect and direct effects, that can even span the mitochondrial and nuclear genomes. The mtDNA and W chromosomes are co-inherited in ZW systems, and while we did not explicitly include the W chromosome in our analysis, the mtDNA-nuclear interaction network identified inherently includes the W. While our enrichment analyses suggest that these mtDNA-associated effects are largely driven by cytonuclear interactions, it is likely that additional barrier effects associated with the W chromosome contribute to this coupled network. These results further suggest that both sex chromosomes are associated with remarkably strong yet distinct coupled barrier effects.
Coupling theory is fundamentally based on the hypothesis that associations between barrier traits, and their underlying loci, may build up and manifest in stronger cumulative barrier effects6,7,12. Indeed, the few empirical examples that have identified coupled barrier traits have provided compelling prior evidence for coupling theory and its potential role in nature35,39,40,68,69. However, while the presence of coupled barriers has been demonstrated, the ability to quantitatively measure and compare their genome-wide effects on RI has remained limited. A major phenotypic difference between hybridizing parental species studied here is their venom composition; C. scutulatus venom contains a presynaptic neurotoxic PLA2 (Crotoxin; also known as Mojavetoxin), which is absent in C. viridis42,51. Crotoxin targets neuronal synapses and neuromuscular junctions through its activity on ion channels55,56,57. C. scutulatus conspecific interchromosomal LD in hybrids identifies these regions as being enriched for steep clines and coupling, and for containing genes related to ion channels. Further analyses of genes specifically in LD with PLA2 neurotoxin haplotypes showed that multiple ion channel genes encoded on the same chromosomes, including the known Crotoxin target GABA receptor subunits70,71,72,73, are located in regions with elevated coupling. This suggests coupling of venom toxins and their ion channel targets is maintained by selection due to removal of incompatible allele combinations related to self-resistance to venom. This example provides evidence for associations related to key adaptive barrier traits involved in well-known protein-protein interactions being further strengthened by coupled aggregate barrier effects.
It is reasonable to question what specific role and added value integrating coupling into studying the process of speciation brings to the field. A persistent motivation in speciation research is to develop approaches that enable objective and quantitative comparisons across diverse systems, which has additional potential to illuminate paradigms of speciation17,18,74. Stankowski and Ravinet17 made a strong argument to define a speciation continuum as a continuum of RI, while others have argued for a multivariate view that can capture variation in distinct “pathways” lineages can take towards speciation75,76. Westram et al.18 made significant contributions to the characterization of RI and proposed objective methods for quantifying it between populations18, which are well-suited to a two-deme model. Westram et al.18 also discuss the potential to leverage geographic clines to estimate Barton’s “barrier strength”14 (which can be used to estimate effective migration) that could be applied to more continuous hybrid zones. However, Westram et al.18 also identify key challenges to implementing this later approach, including the difficulty in estimating total migration in such models, and the application to hybrid zones that do not adhere to the assumptions of geographic cline models. Thus, while the Westram et al.18 approach for a two deme model does enable objective estimation of RI in a comparable way across systems, approaches that can objectively compare RI or progress towards speciation in objective ways for more continuous or mosaic hybrid zones remain largely unavailable.
Among options for an objective and quantitative framework for comparing progress towards speciation, genomic coupling is attractive because it describes the buildup and maintenance of associations to be described on a scale that is objectively comparable across systems, and has biological meaning directly related to recombination and selection (e.g., Barton’s coupling coefficient)12. A key innovation of the approach developed here is that it makes the estimation of this fundamental process that is critical for speciation—variation in the buildup of barrier effects and the strengthening of RI genome-wide—now feasible and applicable to diverse empirical systems. While distinct from estimating RI directly (e.g., Westram et al.18), estimation of this buildup of barrier effects inherently reflects the strength of RI, because RI is directly related to the buildup of barriers that impede gene flow18. Our approach enables the estimation of the full genomic landscape of coupling in continuous hybrid zones by computing the variation in cline slope and center to infer a coupling coefficient6,41. This method can be applied to small genomic windows, allowing for comprehensive analysis of whole-genome data, and enables the inference of the degree of aggregation of barrier effects and the roles of indirect selection that is directly quantitatively comparable across systems. While comparison and interpretation of interchromosomal LD across systems is challenging, the coupling coefficient and cline variance (e.g., variance in cline slope, cline center, or both) of regions involved in interchromosomal LD can be compared in a quantitative way between systems to compare aggregate multilocus barrier effects.
The quantitative estimate of the coupling coefficient from cline variance applied here, and elsewhere22,23, is dependent on a simulation-derived linear equation, and may require re-calibration with different simulation conditions for systems with distinct population structure, demography or evolutionary histories22. In the case of our empirical system, the Firneno et al. equation22 fits our observed cline variance particularly well, although future studies are required to understand the degree to which this equation should be refined with new simulations to fit a particular empirical system, or is robust to such variation across systems. While the importance of refining this equation to estimate the coupling coefficient in a way that is directly comparable across taxa remains an ongoing question for future work, it is important to appreciate that cline variance itself represents an objective and informative measure of coupling. As we refine our understand of how to accurately infer a quantitative coupling coefficient that fits diverse empirical systems (and maintains its intended quantitative relationship to selection versus recombination)6, we suggest that cline variance should be reported alongside estimates of coupling coefficients in future studies.
Taken together, our study provides genome-scale evidence supporting key predictions of coupling theory and its relevance to existing paradigms of speciation, and new approaches developed enable future work to test other key predictions. Our findings illustrate genome-wide variation in the buildup and maintenance of coupled barriers to gene flow, which at later stages of the speciation process may lead to complete RI through the cohesion of genome-wide barriers. While our results do not demonstrate genome congealing per se, they do provide compelling empirical evidence for the buildup of associations within and between chromosomes that remain coupled following secondary contact. Such genome-wide aggregation of barrier effects is the underlying mechanism that theory predicts can lead to complete RI and speciation through genome congealing6,7,15,16,30, but which has otherwise only been demonstrated by simulation studies15,30. Our focus on a single empirical system here does, however, limit our ability to test other important predictions of coupling theory, including those related to how reproductive barriers build up and become associated across the genome at distinct stages of lineage divergence and speciation. Future applications of coupling to compare different hybrid zones, with reproductive barriers that range from weak to nearly full RI, are critical next steps for testing remaining tenets of coupling theory and for expanding our understanding of how the aggregation of barrier effects varies across a continuum of progress towards speciation.
Methods
Sample collection, genome resequencing and variant calling
Collection of specimens was conducted under permits issued for New Mexico (New Mexico Department of Game and Fish: 3605, 3418), Arizona (State of Arizona Game and Fish Department: #SP620063), California (California Department of Fish and Wildlife: SC200770003), and Texas (Texas Parks and Wildlife Department: SPR-0604-391, SPR-0814-159). All animal collection and sampling were carried out in accordance with approved IACUC protocols (APF# 22-07-008C from San Diego State University Institutional Animal Care and Use Committee, and 2303D-SM-S-26 from University of Northern Colorado Institutional Animal Care and Use Committee). We sampled a total of 118 individuals from across a hybrid zone previously identified by Zancolli et al.43, and geographically expanded our sampling further east and west to integrate the full extent of the hybrid zone and adjacent parental lineage populations. DNA was extracted from either snap frozen tissue or tissue in lysis buffer stored at −80 °C, using standard phenol-chloroform-isoamyl extraction. Whole-genome sequencing libraries were generated from purified DNA using Illumina’s Nextera Flex Library Prep kit and sequenced on multiple Illumina NovaSeq 6000 S4 flow cell lanes using 150-bp paired-end reads, targeting 10x–30x whole genome coverage per sample.
Sequencing libraries were demultiplexed using the FastQ Generation application on Illumina’s BaseSpace Sequence Hub (basespace.illumina.com). We quality trimmed raw reads using Trimmomatic v0.3677 using the following flags: LEADING:20, TRAILING:20, MINLEN:32 AVGQUAL:30. Trimmed and quality filtered reads were mapped to the C. viridis reference genome47 using BWA v0.7.10 ‘mem’ option78 with default settings. We used SAMtools version v.1.278 to quantify overall mapping and read quality statistics from each sample using the’-flagstat’ option.
We called variants using GATK v.4.0.8.1 with the best practices workflow79 by first generating individual gVCFs using “HaplotypeCaller” and specifying the ‘-ERC GVCF’ option. We then combined all individual gVCFs into one cohort using the “GenomicsDBImport” and called population variants using the “GenotypeGVCFs” tool. We hard filtered variant calls using the recommended parameter thresholds from GATK’s best practices workflow (QD < 2, QUAL < 30.0, SOR > 3.0, FS > 60.0, MQ < 40.0, MQRankSum < −12.5, ReadPosRankSum < −8.0) with the ‘VariantFiltration’ tool. We kept only non-singleton, biallelic variants located on known chromosomes that passed these criteria, and masked variants located within repeat regions (identified with RepeatMasker80). To avoid potential downstream impacts of structural variants (i.e., misassembly errors, paralogs, etc.), we used the program CNVseq81 to identify copy number variants (CNVs) shared across parental populations. This approach uses a coverage-based method that identifies CNVs by highlighting genomic regions that show significant deviations in coverage between individual pairwise comparisons. We randomly sampled individuals from both known parental allopatric populations and extracted alignments across all chromosomes and ran CNVseq using the following parameters: --log2 0.6 --p 0.001 --bigger-window 1 --window-size 2500 –minimum-windows 2 for each chromosome and masked shared CNVs across populations. We then used VCFtools v0.1.1782 to exclude any variant located within regions identified as CNV between parental species, any sites that were heterozygous for known female individuals and variants that had a minor allele frequency of greater than 0.05 (--maf 0.05). To ensure that sites with no coverage are correctly called as missing, we used BCFtools83 +setGT function with the following flags: -- -t q. -n./. -i ‘FMT/DP = = 0’. Finally, we filtered sites with extremely high read depths as missing data to avoid impacts of potential paralogous mappings by excluding sites that had coverage estimates above the 97.5th quantile.
We phased our final variant dataset using SHAPEIT484 by parsing our VCF by chromosome and then specifying the following parameters: --seed 123456 --mcmc-iterations 6b,1p,2b,1p,2b,8 m. Switch error rates were estimated by computing a second phased VCF with a different seed (726381), and comparing both runs with vcftools --diff-switch-error function.
Population structure and hybrid index analysis
We used the program ADMIXTURE v1.3.052 to estimate overall population structure and infer ancestry coefficients for a series of K values ranging from 1 to 10. We first thinned our SNP dataset by 10 kb using VCFtools (--thin 10000) to avoid the impacts of linkage on individual ancestry assignments. We used the program PLINK v1.9085 to convert our VCF in STRUCTURE format for input, ran each K value for 10 iterations and chose the best K model using the cross-validation method implemented in ADMIXTURE. To visualize the ordination and genetic variance across all individuals, we performed a principal component analysis (PCA) with PLINK (--pca). From both analyses we identified putative hybrid individuals as those who had ancestry coefficients <99.999% for either parental lineage and grouped spatially within our PCA. We further validated hybrid classification by visualizing the relationship between our ADMIXTURE ancestry coefficients and values from the first principal component loading (PC1; Supplementary Fig. S5).
Local ancestry inferences was inferred for all putative hybrids using Loter86. Individuals with ancestry coefficients >99.999% of either parental lineage were used as ancestral population. We then computed the number of observed hybrid recombination within our dataset by summing the number of ancestry haploblock start/end within 100 kb windows across the genome.
Mitochondrial genome assembly, annotation, and analysis
We used our WGS data to assemble mitochondrial genome sequences from each individual, and to identify the parental species mitochondrial haplotype of each individual. We assembled the mitochondrial genomes for each individual using the program MITObim v.1.9.187. This program uses the assembly tool MIRA v4.0.188 which we used to perform reference-guided assemblies in MITObim using a previously-assembled mitochondrial genome for C. viridis47 as a reference. To format raw reads for input in MITObim we used a script (https://github.com/dib-lab/khmer/blob/master/scripts/interleave-reads.py) that interleave paired reads and then we performed a maximum of 10 iterations of baiting and mapping for each sample by mapping paired reads that were derived from the mitochondrial genome using default parameters. We annotated mitochondrial genomes using the MITOs webserver89. We aligned mitochondrial protein-coding genes for all individuals using MAFFT V7.49090. Using these aligned sequences, we estimated phylogenetic relationships among mitochondrial haplotypes using MrBayes v3.2.191. One Crotalus atrox and one Crotalus ruber were used as outgroups. MRBAYES analyses were conducted using the default substitution model with inverse gamma rate of variation across sites, and run in two parallel runs for 108 generations each, with sampling every 2000 generations. We used Tracer v1.792 to confirm MCMC convergence and mixing, and used TreeAnnotator v2.6.793 to infer a consensus phylogram by combining sampling from both runs (excluding the first 10% as burnin). These analyses were used to assign each individual’s mitochondrial haplotype to one of the two parental species.
Bayesian genomic cline analysis
Site-specific genomic cline measures in a Bayesian framework were estimated using bgc-hm46 to identify genomic regions that show deviations from null expectations based on genome-average patterns of admixture and introgression. Deviations from null expectations are credible when the 95% credible interval does not encompass the null value of cline slope (=1) and cline center (=0.5). The bgc-hm method estimates the specific allelic posterior probability of inheritance from each parental population (Φ) by using the overall hybrid index (h) for each hybrid individual that represents the degree of inheritance from each parental population and the two genomic cline parameters, c & v, (genomic cline center and cline slope, respectively). This is done using a hierarchical model (as in Gompert and Buerkle94) where the variance in cline centers and slopes are estimated as model parameters22,46. We fit the bgc-hm model in three steps in R using Hamiltonian Monte Carlo (HMC) via rstan95 interface with Stan. First, Bayesian estimates of hybrid indexes were obtained for each of the 63 hybrids based on 1000 randomly selected, ancestry informative SNPs (i.e., here, SNPs with a parental allele frequency difference >0.3). For this analysis, we ran four HMC chains, each comprising a 1000 iteration warmup and 1000 post-warmup samples (HMC is often much more efficient at sampling the posterior than standard Markov chain Monte Carlo approaches and thus requires for fewer iterations95). Second, to speed up the estimation of inferring clines from all genome-wide variants, we conducted an initial cline estimation using the 1000 randomly sampled variants and the inferred hybrid indexes to approximate the genome-wide standard deviation of cline parameters. For this approximation, normal priors with means of 0 and standard deviations of 1 were used for σc and σv (see Gompert et al.46 for additional details of the model structure). This analysis involved running four HMC chains of 2000 total iterations each. Finally, we estimated genomic cline center and slope parameters all of the 2,565,420 SNPs, using parameter estimates from our 1000 SNP estimation of the cline variance (cline slope and cline center variance) and hybrid indices for this full SNP analysis. This approach allowed us to significantly increase the computational efficiency of estimation of clines for all genomic SNPs by starting full analyses with informed priors. Cline parameters for each SNP were estimated using four HMC chains with a 1000 iteration warmup and 1000 post-warmup samples. We imposed a sum-to-zero constraint on the cline parameters, specifically on logit(c) and log(v), as suggested by refs. 46,94. The bgc-hm analyses were done in R version 4.1.2 with rstan version 2.21.795.
High frequency cline parameters, which we refer to as “hotspots”, were then identified by plotting BGC parameters for variants with ggplot296 and the package ggpointdensity97, with the “adjust” argument set to 0.02. Hotspots were then identified by visual inspection of these density plots, and variants associated with these hotspots were isolated based on a density threshold and cline parameters (Supplementary Table S3).
Hybrid zone simulations
To evaluate methods for quantifying coupling in hybrid zones, we conducted simulations following the approach of Firneno et al.22, using dfuse98. First, we replicated the simulations from Firneno et al.22 with identical parameters to confirm that our results matched their derived equation. Simulations were run for a range of coupling coefficients (θ = 0.05, 0.1, 0.3, 0.5, 0.7, 0.9, 1.0, 1.1 and 1.5) and the number of loci under selection (L = 2, 10, 100, 200, 500, and 1000), all involving 110 demes arranged in a one-dimensional stepping-stone model, 50 individuals per deme, and a migration rate of either 0.1 or 0.2 between adjacent demes22. Next, we conducted simulations with modified parameters to explore conditions that may better represent our empirical hybrid zone. Specifically, we (i) increased the population size from 50 to 250 individuals per deme, (ii) explored a broader range of parameters by adding θ = 1.3 and θ = 1.7 to the set of simulated coupling coefficient values to improve resolution near higher coupling transitions, and (iii) expanded the range of the number of loci under selection to include 5, 20 and 50. All simulations and subsequent analyses were performed using scripts and code available on the GitHub repository (https://github.com/zgompert/ClineCoupling). For simulations replicating Firneno et al.’s parameters, linear equations estimating coupling were derived using runs that included 10 or more hybrids. For simulations with modified parameters, we adjusted the threshold to include runs with 50 or more hybrids, accounting for the fivefold increase in population size in these simulations.
Estimation of the coupling coefficient
To quantify coupling for non-overlapping 100 kb windows across the genomes of hybrids, we estimated the standard deviation of the logit-transformed cline (\({\sigma }_{c}\)) center and log-transformed cline (\({\sigma }_{v}\)) slope following Firneno et al.22 for sites with an allele frequency difference > 0.3. A coupling coefficient was then estimated using the linear equation from Firneno et al.22: \(\theta=-0.511-0.0167\times {\sigma }_{c}-0.24\times {\sigma }_{v}+0.013\times {\sigma }_{c}\times {\sigma }_{v}\) and the equation we obtained with the modified simulations: \(\theta=-0.508-0.08\times {\sigma }_{c}-0.13\times {\sigma }_{v}+0.19\times {\sigma }_{c}\times {\sigma }_{v}\). To assess the fit of empirical cline variance (including empirical variance in cline slope and variation in cline center) to various simulation-derived equations, we summarized variation in clines via PCA, and assessed the correlation between PC1 values and coupling coefficients obtained with the linear equations (Supplementary Fig. S11). Since both equations showed equally-strong correlations with PC1 (R2 = 0.84), we used the coupling coefficient values obtained from the original Firneno et al. equation for all subsequent analysis.
Analysis of LD in hybrids
To infer interchromosomal Admixture Linkage Disequilibrium (LD) across the genome, we computed inter-chromosomal R2 LD values using VCFtools (--interchrom-hap-r2) using only putative hybrid individuals, and keeping variants with significant excess ancestry as determined by bgc-hm and an FST value between parental species above 0.9. Pairs of variants with excess ancestry for the same species and an R2 above 0.7 (threshold based on visual inspection on the distribution of R2 across all comparisons; Supplementary Fig. S18), were considered to be in conspecific LD within hybrids.
To generate heatmaps of Intrachromosomal LD, LD within chromosomes was computed using VCFtools (--hap-r2) for hybrids and both parental species using a 1 kb thinned datasets (--thin 1000). We summarized LD using the mean R2 of pairs of variants between 100 kb windows. We combined the parental intrachromosomal LD maps by selecting the maximum R2 between both LD maps for each pair of 100 kb windows. This combined map was then subtracted from the hybrid intrachromosomal LD map to obtain the hybrid excess intrachromosomal LD.
To infer LD between nuclear variants and the mitochondrial haplotype, we added the mitochondrial genotype as a scaffold containing one variant (either homozygous for A for C. viridis or G for C. scutulatus) to the VCF. We then computed interchromosomal LD (--interchrom-geno-r2) between that variant representing the mitochondrial haplotype and highly differentiated nuclear variants (FST between parental species above or equal to 0.9). After visual inspection of the distribution of R2 (Supplementary Fig. S26), nuclear genome variants with an R2 value above 0.75 with this mitochondrial genotype were considered in LD with the mitochondrial genome.
Due to low differentiation between parental species around venom genes and the absence of the crotoxin sequence from C. scutulatus in the C. viridis genome reference, we identified LD between variants and haplotypes at the PLA2 locus using a method similar to the mitochondrial haplotype LD mentioned above. The haplotypes at the PLA2 locus were determined using local ancestry inferences from Loter, and encoded it as a variant on mi7 in the VCF. We then computed interchromosomal LD between this variant and all variants genome-wide using VCFtools and, after visual inspection of the R2 distribution (Supplementary Fig. S24), considered variant pairs with an R2 value above 0.65 to be in LD.
Recombination analysis in parentals
Recombination maps were constructed using pyrho v0.1.799,100. To provide pyrho with the demography of our samples, we used SMC++ v1.15.453 to infer population size histories of C. scutulatus, C. viridis, and hybrids. We generated VCFs for each macrochromosome, microchromosome, and the Z chromosome, only retaining biallelic SNPs. While running SMC++, we used a chunk size of 50,000 base pairs and set the time range of demographic inference to 1000 to 200,000 generations. We assumed a generation mutation rate of 2.4 × 10−9 following the protocols of Schield et al.101 on Crotalus recombination rates. Then, pyrho was run using N number of samples per lineage x 1.25 and a per-generation mutation rate of 8.4 × 10−9 to generate a lookup table using the approx. flag. As our data were phased, we used a ploidy level of 1 and while simulating data and performing parameter optimization. Mean recombination rate maps were then created for 10 kb, 100 kb, and 1000 kb windows for C. viridis, C. scutulatus, and hybrids. To combine both parental lineage recombination maps, we used the minimum recombination rate of either species for every genomic windows in order to be conservative, as we are focused on detecting regions where hybrids have a lower recombination rate than parentals. Then, to compute the excess of linkage in hybrids, we log2-transformed recombination rates and subtracted the combined parental recombination map from the hybrid one.
Gene ontology, enrichment, and network analysis
To determine functionality of genes in LD, we used two gene sets: (1) genes within 100 kb of all variants that are in conspecific LD for C. scutulatus, (2) genes within 100 kb of any variant in LD with the mitochondrial haplotype. To identify gene enrichment and gene interactions within our gene sets, we used the STRING database102 and the stringDB package in Cytoscape103, applying an FDR < 0.05 for all enrichment tests. Interaction networks were visualized in Cytoscape and filtered for the GO Biological Process, GO Cellular Component, GO Molecular Function, KEGG Pathways, UniProt, and Reactome Pathways databases. Networks were visualized using a confidence cutoff of 0.40 and singletons hidden.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All raw sequencing data are publicly available on NCBI under the BioProject PRJNA1137891. Intermediate data files are available on Dryad [https://doi.org/10.5061/dryad.kh18932]. Source data are provided as a Source Data file. Source data are provided with this paper.
Code availability
All scripts for analyses are available on Github [github.com/yzfranci/GenomicCoupling].
References
Mayr, E. Systematics and the Origin of Species, from the Viewpoint of a Zoologist (Harvard University Press, 1942).
Mayr, E. Animal Species and Evolution (Belknap Press of Harvard University Press, Cambridge, MA, 1963).
Darwin, C. On the Origins of Species by Means of Natural Selection (London: Murray, 1859).
Barton, N. H. & Hewitt, G. M. Analysis of hybrid zones. Annu. Rev. Ecol. Syst. 16, 113–148 (1985).
Coyne, J. A. & Orr, H. A. Speciation (Sinauer Associates, Sunderland, MA, 2004).
Butlin, R. K. & Smadja, C. M. Coupling, reinforcement, and speciation. Am. Natur. 191, 155–172 (2018).
Dopman, E. B., Shaw, K. L., Servedio, M. R., Butlin, R. K. & Smadja, C. M. Coupling of barriers to gene exchange: causes and consequences. Cold Spring Harb. Perspect. Biol. a041432, https://doi.org/10.1101/cshperspect.a041432 (2024)
Liu, X., Glémin, S. & Karrenberg, S. Evolution of putative barrier loci at an intermediate stage of speciation with gene flow in campions (Silene). Mol. Ecol. 29, 3511–3525 (2020).
Elmer, K. R. Barrier loci and progress towards evolutionary generalities. J. Evolut. Biol. 30, 1491–1493 (2017).
Kulmuni, J., Butlin, R. K., Lucek, K., Savolainen, V. & Westram, A. M. Towards the completion of speciation: the evolution of reproductive isolation beyond the first barriers. Philos. Trans. R. Soc. B 375, 20190528 (2020).
Satokangas, I., Martin, S. H., Helanterä, H., Saramäki, J. & Kulmuni, J. Multi-locus interactions and the build-up of reproductive isolation. Philos. Trans. R. Soc. B 375, 20190543 (2020).
Barton, N. H. Multilocus clines. Evolution 37, 454–471 (1983).
Bierne, N., Welch, J., Loire, E., Bonhomme, F. & David, P. The coupling hypothesis: why genome scans may fail to map local adaptation genes. Mol. Ecol. 20, 2044–2072 (2011).
Barton, N. & Bengtsson, B. O. The barrier to genetic exchange between hybridising populations. Heredity 57, 357–376 (1986).
Feder, J. L. et al. Genome-wide congealing and rapid transitions across the speciation continuum during speciation with gene flow. J. Heredity 105, 810–820 (2014).
Nosil, P., Feder, J. L., Flaxman, S. M. & Gompert, Z. Tipping points in the dynamics of speciation. Nat. Ecol. Evol. 1, 1–8 (2017).
Stankowski, S. & Ravinet, M. Defining the speciation continuum. Evolution 75, 1256–1273 (2021).
Westram, A. M., Stankowski, S., Surendranadh, P. & Barton, N. What is reproductive isolation?. J. Evolut. Biol. 35, 1143–1164 (2022).
Mallet, J. & Mullen, S. P. Reproductive isolation is a heuristic, not a measure: a commentary on Westram et al., 2022. J. Evolut. Biol. 35, 1175–1182 (2022).
Stuckert, A. M. M. & Matute, D. R. Using neutral loci to quantify reproductive isolation and speciation: a commentary on Westram et al., 2022. J. Evolut. Biol. 35, 1169–1174 (2022).
Schilling, M. P. et al. Transitions from single- to multi-locus processes during speciation with gene flow. Genes 9, 274 (2018).
Firneno, T. J. et al. Quantitative analyses of coupling in hybrid zones. Cold Spring Harb. Perspect Biol. a041434, https://doi.org/10.1101/cshperspect.a041434 (2023).
Le Moan, A. et al. Coupling of twelve putative chromosomal inversions maintains a strong barrier to gene flow between snail ecotypes. Evol. Lett. qrae014, https://doi.org/10.1093/evlett/qrae014 (2024).
Coyne, J. A. & Orr, H. A. Two rules of speciation. in Speciation and Its Consequences 180–207 (Sinauer Associates, Sunderland, MA, 1989).
Orr, H. A. & Turelli, M. The evolution of postzygotic isolation: accumulating Dobzhansky-Muller incompatibilities. Evolution 55, 1085–1094 (2001).
Welch, J. J. Accumulating Dobzhansky-Muller Incompatibilities: reconciling theory and data. Evolution 58, 1145–1156 (2004).
Burton, R. S. The role of mitonuclear incompatibilities in allopatric speciation. Cell. Mol. Life Sci. 79, 103 (2022).
Orr, H. A. The population genetics of speciation: the evolution of hybrid incompatibilities. Genetics 139, 1805–1813 (1995).
Moyle, L. C. & Nakazato, T. Hybrid Incompatibility “Snowballs” Between Solanum Species. Science 329, 1521–1523 (2010).
Flaxman, S. M., Feder, J. L. & Nosil, P. Genetic hitchhiking and the dynamic buildup of genomic divergence during speciation with gene flow. Evolution 67, 2577–2591 (2013).
Szymura, J. M. & Barton, N. H. Genetic analysis of a hybrid zone between the fire-bellied toads, Bombina bombina and B. variegata, near Cracow in southern Poland. Evolution 40, 1141–1159 (1986).
Payseur, B. A. & Hoekstra, H. E. Signatures of reproductive isolation in patterns of single nucleotide diversity across inbred strains of mice. Genetics 171, 1905–1916 (2005).
Masly, J. P. & Presgraves, D. C. High-resolution genome-wide dissection of the two rules of speciation in Drosophila. PLoS Biol. 5, e243 (2007).
Schumer, M. et al. High-resolution mapping reveals hundreds of genetic incompatibilities in hybridizing fish species. eLife 3, e02535 (2014).
Hench, K., Vargas, M., Höppner, M. P., McMillan, W. O. & Puebla, O. Inter-chromosomal coupling between vision and pigmentation genes during genomic divergence. Nat. Ecol. Evol. 3, 657–667 (2019).
Nikolakis, Z. L. et al. Evidence that genomic incompatibilities and other multilocus processes impact hybrid fitness in a rattlesnake hybrid zone. Evolution 76, 2513–2530 (2022).
Szymura, J. M. & Barton, N. H. The genetic structure of the hybrid zone between the fire-bellied toads Bombina bombina and B. variegata: comparisons between transects and between loci. Evolution 45, 237–261 (1991).
Gay, L., Crochet, P.-A., Bell, D. A. & Lenormand, T. Comparing clines on molecular and phenotypic traits in hybrid zones: a window on tension zone models. Evolution 62, 2789–2806 (2008).
Walsh, J. et al. A complex genomic architecture underlies reproductive isolation in a North American oriole hybrid zone. Commun. Biol. 6, 1–11 (2023).
Schield, D. R. et al. Sexual selection promotes reproductive isolation in barn swallows. Science 386, eadj8766 (2024).
Barton, N. H. & De Cara, M. A. R. The evolution of strong reproductive isolation. Evolution 63, 1171–1190 (2009).
Glenn, J. L. & Straight, R. C. Venom characteristics as an indicator of hybridization between Crotalus viridis viridis and Crotalus scutulatus scutulatus in New Mexico. Toxicon 28, 857–862 (1990).
Zancolli, G. et al. Is hybridization a source of adaptive venom variation in rattlesnakes? A test, using a Crotalus scutulatus × viridis hybrid zone in southwestern New Mexico. Toxins 8, 188 (2016).
Gompert, Z. & Buerkle, C. A. Bayesian estimation of genomic clines. Mol. Ecol. 20, 2111–2127 (2011).
Gompert, Z. & Buerkle, C. A. bgc: software for Bayesian estimation of genomic clines. Mol. Ecol. Resour. 12, 1168–1176 (2012).
Gompert, Z., DeRaad, D. A. & Buerkle, C. A. A next generation of hierarchical bayesian analyses of hybrid zones enables model-based quantification of variation in introgression in R. Ecol. Evol. 14, e70548 (2024).
Schield, D. R. et al. The origins and evolution of chromosomes, dosage compensation, and mechanisms underlying venom regulation in snakes. Genome Res. 29, 590–601 (2019).
Presgraves, D. C. Evaluating genomic signatures of “the large X-effect” during complex speciation. Mol. Ecol. 27, 3822–3830 (2018).
Nelson, E. D., Cong, Q. & Grishin, N. V. Influence of the large-Z effect during contact between butterfly sister species. Ecol. Evol. 11, 11615–11626 (2021).
Schield, D. R. et al. Insight into the roles of selection in speciation from genomic patterns of divergence and introgression in secondary contact in venomous rattlesnakes. Ecol. Evol. 7, 3951–3966 (2017).
Mackessy, S. P. Evolutionary trends in venom composition in the Western Rattlesnakes (Crotalus viridis sensu lato): Toxicity vs. tenderizers. Toxicon 55, 1463–1474 (2010).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Terhorst, J., Kamm, J. A. & Song, Y. S. Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat. Genet. 49, 303–309 (2017).
Flaxman, S. M., Wacholder, A. C., Feder, J. L. & Nosil, P. Theoretical models of the influence of genomic architecture on the dynamics of speciation. Mol. Ecol. 23, 4074–4088 (2014).
Georgieva, D., Arni, R. K. & Betzel, C. Structure-function relationship in heterodimeric neurotoxin PLA2s from Viperidae snakes inhabiting Europe, South America, and Asia. In Venom Genomics and Proteomics (Gopalakrishnakone, P. & Calvete, J. J. Eds.), 269–289 (Springer Netherlands, Dordrecht, 2016). https://doi.org/10.1007/978-94-007-6416-3_16.
Ho, C. L. & Lee, C. Y. Presynaptic actions of Mojave toxin isolated from Mojave rattlesnake (Crotalus scutulatus) venom. Toxicon 19, 889–892 (1981).
Valdes, J. J., Wolff, V. L., Menking, D. E., Thompson, R. G. & Chambers, J. P. Mojave Toxin: A Selective Ca(++) Channel Antagonist. U.S. Army Chemical Research, Development & Engineering Center (CRDEC), (Aberdeen Proving Ground, MD, USA. 1988).
Coyne, J. A. Genetics and speciation. Nature 355, 511–515 (1992).
Coyne, J. A. ‘Two rules of speciation’ revisited. Mol. Ecol. 27, 3749–3752 (2018).
Presgraves, D. C. Sex chromosomes and speciation in Drosophila. Trends Genet. 24, 336–343 (2008).
Caeiro-Dias, G., Brelsford, A., Meneses-Ribeiro, M., Crochet, P.-A. & Pinho, C. Hybridization in late stages of speciation: strong but incomplete genome-wide reproductive isolation and ‘large Z-effect’ in a moving hybrid zone. Mol. Ecol. 32, 4362–4380 (2023).
Turelli, M. & Orr, H. A. Dominance, epistasis and the genetics of postzygotic isolation. Genetics 154, 1663–1679 (2000).
Dobzhansky, T. Studies on hybrid sterility. II. Localization of sterility factors in Drosophila Pseudoobscura hybrids. Genetics 21, 113–135 (1936).
Muller, H. J. Isolating mechanisms, evolution, and temperature. Biol. Symp. 6, 71–125 (1942).
Moran, B. M. et al. A lethal mitonuclear incompatibility in complex I of natural hybrids. Nature https://doi.org/10.1038/s41586-023-06895-8 (2024).
McFarlane, S. E., Sirkiä, P. M., Ålund, M. & Qvarnström, A. Hybrid dysfunction expressed as elevated metabolic rate in male Ficedula flycatchers. PLoS ONE 11, e0161547 (2016).
Wang, S. et al. Signatures of mitonuclear coevolution in a warbler species complex. Nat. Commun. 12, 4279 (2021).
Xu, M. & Shaw, K. L. Genetic coupling of signal and preference facilitates sexual isolation during rapid speciation. Proc. R. Soc. B: Biol. Sci. 286, 20191607 (2019).
Kunerth, H. D. et al. Consequences of coupled barriers to gene flow for the build-up of genomic differentiation. Evolution 76, 985–1002 (2022).
Cecchini, A. L., Soares, A. M., Giglio, J. R., Amara, S. & Arantes, E. C. Inhibition of L-glutamate and GABA synaptosome uptake by crotoxin, the major neurotoxin from Crotalus durissus terrificus venom. J. Venom. Anim. Toxins incl. Trop. 10, 260–279 (2004).
Hannan, S., Mortensen, M. & Smart, T. G. Snake neurotoxin α-bungarotoxin is an antagonist at native GABA(A) receptors. Neuropharmacology 93, 28–40 (2015).
Kudryavtsev, D. S. et al. Neurotoxins from snake venoms and α-Conotoxin ImI inhibit functionally active ionotropic γ-aminobutyric acid (GABA) receptors. J. Biol. Chem. 290, 22747–22758 (2015).
Rosso, J.-P. et al. MmTX1 and MmTX2 from coral snake venom potently modulate GABAA receptor activity. Proc. Natl Acad. Sci. USA 112, E891–E900 (2015).
Sobel, J. M. & Chen, G. F. Unification of methods for estimating the strength of reproductive isolation. Evolution 68, 1511–1522 (2014).
Bolnick, D. I. et al. A multivariate view of the speciation continuum. Evolution 77, 318–328 (2023).
Johannesson, K. et al. Diverse pathways to speciation revealed by marine snails. Trends Genet. 40, 337–351 (2024).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Xie, C. & Tammi, M. T. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinform. 10, 80 (2009).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Delaneau, O., Zagury, J.-F., Robinson, M. R., Marchini, J. L. & Dermitzakis, E. T. Accurate, scalable and integrative haplotype estimation. Nat. Commun. 10, 5436 (2019).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Dias-Alves, T., Mairal, J. & Blum, M. G. B. Loter: a software package to infer local ancestry for a wide range of species. Mol. Biol. Evol. 35, 2318–2326 (2018).
Hahn, C., Bachmann, L. & Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res. 41, e129–e129 (2013).
Chevreux, B. MIRA: An Automated Genome and EST Assembler. (Ph.D. Thesis, University of Heidelberg, Heidelberg, Germany, 2007).
Bernt, M. et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319 (2013).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Bouckaert, R. et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10, e1003537 (2014).
Gompert, Z. & Buerkle, C. A. A Hierarchical Bayesian model for next-generation population genomics. Genetics 187, 903–917 (2011).
Stan Development Team. RStan: the R Tnterfance to Stan (2022).
Villanueva, R. A. M. & Chen, Z. J. ggplot2: elegant graphics for data analysis (2nd ed.). Meas. Interdiscip. Res. Perspect. 17, 160–167 (2019).
Kremer, L., P. M. & Anders, S. ggpointdensity (2019).
Lindtke, D. & Buerkle, C. A. The genetic architecture of hybrid incompatibilities and their effect on barriers to introgression in secondary contact. Evolution 69, 1987–2004 (2015).
Kamm, J. A., Spence, J. P., Chan, J. & Song, Y. S. Two-locus likelihoods under variable population size and fine-scale recombination rate estimation. Genetics 203, 1381–1399 (2016).
Spence, J. P. & Song, Y. S. Inference and analysis of population-specific fine-scale recombination maps across 26 diverse human populations. Sci. Adv. 5, eaaw9206 (2019).
Schield, D. R. et al. The roles of balancing selection and recombination in the evolution of rattlesnake venom. Nat. Ecol. Evol. 6, 1367–1380 (2022).
Szklarczyk, D. et al. The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646 (2023).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504 (2003).
Acknowledgements
Support for this work was provided by NSF Grants DEB-1655571 (to TAC, SPM, and JMM), (IOS-2307044) to TAC, SPM, and RHA, and DBI-2208959 (to JMB). We thank the California Academy of Sciences for providing several specimens used in this study.
Author information
Authors and Affiliations
Contributions
Y.Z.F., J.M.B., R.H.A., Z.G., and T.A.C. conceived of and designed the study, conducted analyses and wrote the manuscript. Y.Z.F., J.M.B., H.D.G., D.R.S., S.S.G., D.W.M., J.M.M., T.J., R.W.C., and S.P.M. collected samples and data. D.R.S., S.N.S., S.S.G., and J.P.D. analyzed data. All authors participated in editing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Francioli, Y.Z., Bernstein, J.M., Adams, R.H. et al. Estimation of genome-wide coupling in rattlesnake hybrids provides insight into the process of speciation and its progress. Nat Commun 16, 10242 (2025). https://doi.org/10.1038/s41467-025-65457-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65457-w
