
Abstract
In the pursuit of sustainable development, the world faces two key challenges: effective waste management and improved energy efficiency. In this context, it is necessary to propose a chlorine-centered sustainable solution, as the related industries consume large amounts of electricity and precious metals while generating significant quantities of plastic waste. Here, we report a chloride electrocatalyst of titanium nitride-supported ruthenium single-atoms (Ru1@TiN) that exhibits high efficiency for chlorine evolution reaction of NaCl in multi-conditions, with a nearly 100% selectivity in a wide range of pH values, potentially saving some of the electricity for the chlor-alkali industry. Moreover, Ru1@TiN can decompose polyvinyl chloride waste into chlorine and other valuable chemicals, providing an approach to plastic upcycling and green chlorine production. A coordination-derived activation mechanism on TiN support is uncovered and a dynamic polyatomic active site is proposed to explain the achieved catalytic performances. This work will benefit developing sustainable chlorine chemical industries.
Similar content being viewed by others


Introduction
Chlorine-related chemical processes and plastic products play a crucial role in modern society; however, they are associated with significant energy consumption and environmental pollution. Upgrading the chlorine chemical industry is essential for achieving sustainability. Traditionally, chlorine is produced through the electrolysis of sodium chloride, which is the largest industrial electrolysis and provides basic chemicals, such as hydrogen and caustic soda, thereby known as the chlor-alkali process1,2,3,4. While chlor-alkali is an energy-intensive industry, consuming nearly 1% of global electrical power (approximately 150 TW·h·y-1)5,6. Its energy consumption is primarily determined by the chlorine evolution reaction (CER) occurring at the anode7,8. The state-of-the-art anode is Ti mesh supported RuO2-IrO2-TiO2 solid solution, named dimensionally stable anode (DSA), which has high catalytic efficiency and long lifespan9,10,11. However, DSA requires a large amount of precious metal, accounting for about 20% of production costs, and the overvoltage of DSA is still high, responsible for the high energy consumption12,13.
Chlorinated plastics, such as polyvinyl chloride (PVC), are indispensable in industrial and agricultural applications, as well as in everyday life. However, the disposal of chlorinated plastics poses significant environmental challenges due to their resistance to decomposition. Consequently, these wastes are often incinerated or landfilled, releasing hazardous substances and causing severe environmental pollution (Fig. 1a)14,15. Alternatively, PVC waste can be mechanically or chemically recycled into valuable monomers and polymers16,17. With growing concerns about environmental sustainability and public health, there is increasing interest in developing sustainable methods for chlorine production through the recycling of chlorinated plastics18,19. Whether the goal is to reduce the energy consumption and costs associated with the traditional chlor-alkali process or to harness waste PVC for chlorine production, the critical component remains an efficient electrolysis catalyst for chlorides.

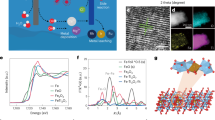
a The schematic diagram of this work and the proposed chloride cycle in this work. b HAADF-STEM image of the atomically dispersed Ru1 on TiN as highlighted by circles. c k2-weighted FT-EXAFS of indicated materials and spectrum fitting based on the inserted RuNTi4 structure with one adsorbed O2. The gold, blue, gray and red balls refer to Ru, Ti, N and O atoms, respectively. d Ru 3p XPS spectra of Ru1@TiN and Ru1/TiN (the intermediate before the heat treatment). Source data for Fig. 1 are provided as a Source Data file.
Recent advances have shown that single-atom catalysts (SACs, especially Pt-based SACs) achieve remarkable performance under low current densities (e.g., 5 mV@ 1 mA·cm-2) and exhibit superior kinetic characteristics in laboratory settings20,21. Yet, these catalysts often face significant degradation under industrial conditions, including high current densities and prolonged operational stability challenges. It leads to reduced efficiency and impractical scalability for large-scale electrochemical chlor-alkali processes. The reported current density is normally lower than 200 mA cm-2, and the stability test is usually less than 200 h. This discrepancy underscores the ongoing need for catalyst design innovations to enhance robustness in real-world environments. Here, we report a highly efficient, selective, and stable chloride electrolysis catalyst, namely Ru1@TiN SACs, i.e., titanium nitride nanoparticle (NP) supported ruthenium single-atoms. Ru1@TiN requires a small overpotential and has nearly 100% selectivity in acidic and neutral media for CER, and thereby can potentially save some of the electricity for the chlor-alkali industry. The noble metal efficiency of Ru1@TiN is hundreds of times higher than DSA. Operando characterization and theoretical calculations reveal that the high catalytic efficiency of Ru1@TiN is associated with the formation of a dynamic polyatomic active site due to the activation of four substrate Ti atoms on the support by one Ru atom. Moreover, Ru1@TiN also exhibits high dechlorination capability for PVC, enabling both chloride oxidation and reduction reactions. A new chlorine production pathway is thereby proposed with PVC wastes as the raw material (Fig. 1a). Overall, the efficient Ru1@TiN is expected to save electricity, reduce costs for the traditional chlor-alkali industry, and enable the chlorine upcycling from PVC wastes. These advancements will help the development of sustainable chlorine chemical industry.
Results and discussions
Ru1@TiN was synthesized via an impregnation method followed by a low-temperature annealing, which is easy to scale up for mass production (see Methods and Supplementary Fig. 1). Compared with commonly used carbon substrates, TiN has the advantages of much higher corrosion resistance and electrical conductivity, which allow Ru1@TiN to survive in the harsh CER environment and sustain the high CER current density. Ru1@TiN contains 1.23 wt% ruthenium as determined by inductively coupled plasma-mass spectrometry. The uniformly and atomically dispersed Ru atoms on the TiN substrate are confirmed by the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), X-ray diffraction (XRD), selected area electron diffraction (SAED) and elemental mapping in Fig. 1b, Supplementary Figs. 2 and 3. The valence state of Ru1 should be higher than +3 by comparing with Ru0 and Ru3+ in the X-ray absorption near-edge structure (XANES) spectra (Supplementary Fig. 4). Ru K-edge Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) spectrum of Ru1@TiN (Fig. 1c) shows a main peak of Ru-N(O) at 1.5 Å and a nearby small peak at 2.3 Å in R space. The wavelet transform (WT) comparison of Ru1@TiN and references excludes the presence of Ru-Ru bonds (Supplementary Fig. 5). According to the FT-EXAFS fitting results using the inserted structural model, the Ru atom is predicted to coordinate with one N atom of TiN substrate and adsorb an oxygen molecule on the other side. The small peak at 2.3 Å may be associated with the weak interaction between Ru1 and adjacent Ti atoms of the substrate (see Table S1 for fitting parameters)22. Atomic coordinates of the optimized computational models for fitting are provided in Supplementary Data 1. It should be noted that following heat treatment under an Ar/H₂ atmosphere, the Ru species supported on TiN is reduced. However, subsequent X-ray absorption spectroscopy (XAS) characterization is performed in air, leading to O2 adsorption, which was also supported by the soft XAS (Supplementary Fig. 6). During the electrocatalysis process, the adsorbed oxygen dissolves into the electrolyte. Upon application of a positive anodic potential, oxygen-coordinated unsaturated sites (denoted as the Oc) form and serve as the potential active sites for the CER, which will be further explained in the latter section. X-ray photoelectron spectroscopy (XPS) (Fig. 1d, Supplementary Fig. 7) shows that the Ru 3p peak of Ru1@TiN has a chemical shift of ~1 eV towards high binding energy compared with Ru1/TiN (an intermediate of Ru1@TiN before the heat treatment, Supplementary Fig. 8). In other words, the heat treatment enhances the electronic metal–support interaction between Ru1 and TiN substrate, which is crucial to the high catalytic activity and stability of Ru1@TiN as shown later23,24.
The energy saving by Ru1@TiN was first evaluated through a parallel comparison with an industrial DSA (denoted as DSA-1, Supplementary Fig. 9). It should be noted that DSA-1 is the best commercial catalyst we can purchase, but may not be the best in the industry. Since the corrosion-resistant Ti plate has significant catalytic activities toward both CER and OER, Ru1@TiN was loaded on a piece of carbon paper for an interference-free evaluation of the catalyst activity25. The CER test was performed in a 5 M NaCl solution in a three-electrode system26. At an industrial operation temperature of 90 °C, the overpotential for Ru1@TiN to reach 1000 mA·cm-2 is 77 mV, lower than that of DSA-1 (Fig. 2a). Moreover, Ru1@TiN has a super high mass activity of 32265 A·gRu−1 at the potential of 1.4 V vs. NHE, much higher than 192 A·gRu−1 of RuO2 and 1020 A·gRu+Ir−1 of DSA-1 (Fig. 2b). Therefore, Ru1@TiN can significantly reduce the catalyst cost. The charge transfer resistance (Rct) tests indicate that the Rct of Ru1@TiN decreases rapidly with the increase of CER potential, and approaches zero at the potential above 1.5 V (Fig. 2c, Supplementary Fig. 10). It means that a small overpotential is sufficient for the massive charge transfer on Ru1@TiN. The small Rct under working condition (above 1.4 V) is attributed to the high conductivity of TiN and the strong electronic interaction between Ru1 and TiN, which will be further proved in the following part of theoretical analysis where another contribution of dynamic polyatomic active sites will be proposed. To compare with literature reports, the activity of Ru1@TiN is also measured under room temperature (all denoted as 25 °C) as shown in Supplementary Figs. 11 and 12, respectively (Table S5). It can be indicated from Table S5 that there are also some SACs that exhibit high efficiency under low current density (<100 mA·cm-2)20,21. And Ru1@TiN performs well under a wide range of temperatures and current densities due to the specific support effect of TiN, which will be further explained later.

a Polarization curves and b mass activities of Ru1@TiN, RuO2 and DSA-1 in 5 M NaCl (pH = 2) at 90 °C for product of Cl2. The scan rate is 10 mV·s-1. The resistance of the 5 M NaCl (pH = 2) is 3.2 ± 0.2 Ω. The data is not iR corrected. c Charge transfer resistance (Rct) of the indicated catalysts at different potentials. d Polarization curves and e selectivity of Ru1@TiN and DSA-1 in 5 M NaCl (pH = 7) at 90 °C for product of NaClO. The scan rate is 10 mV·s-1. The resistance of the 5 M NaCl (pH = 7) is 5.1 ± 0.2 Ω. The 48 h tests of Ru1@TiN in f acidic media and g neutral media. Data are mean ± s.d. of at least four independent experiments. h A long-term stability test of Ru1@TiN under varying current densities and temperatures in 5 M NaCl (pH = 2) in the H-cell. i The atomic image of Ru1@TiN after the stability test showing no Ru1 aggregation. Source data for Fig. 2 are provided as a Source Data file.
Besides the activity advantages, Ru1@TiN also has superior selectivity. It exhibits a negligible OER current in both acidic (pH = 2 ± 0.1) and neutral media (pH = 7 ± 0.1) in the whole voltage range (Supplementary Fig. 13). When the electrolyte changes from acidic (pH = 2) to neutral media (pH = 7), the CER product will change from Cl2, H2 and NaOH to H2 and NaClO. The chloride ion oxidation process (denoted as COP) under neutral media is crucial for NaClO production. As the pH value increases, the side reaction of OER also becomes more and more significant, leading to a relatively low selectivity. The NaClO selectivity of DSA-1 is only ~87%. Ru1@TiN exhibits a much higher selectivity of ~97% at 90 °C (Fig. 2d and e) and 99% at room temperature (Supplementary Fig. 14), which means considerable energy saving at neutral media too. The selectivity of Ru1@TiN is high for both Cl2 and NaClO production, and stable over the whole testing period of 48 h (Fig. 2f, g).
Moreover, Ru1@TiN shows stable performance through a 22-day test with varying current densities (10 and 1000 mA·cm-2) and temperature (30, 50, and 90 °C) as shown in Fig. 2h, Supplementary Figs. 15–18. The HAADF-STEM image in Fig. 2i and EXAFS spectra in Supplementary Fig. 19 demonstrate the high stability of atomic Ru1 and its coordination structure after the long-term test. As a reference, the intermediate Ru1/TiN exhibits a fast performance decay (Supplementary Fig. 20). Therefore, the excellent stability of Ru1@TiN is associated with the strong electronic interaction between Ru1 and TiN. Moreover, via the two-electrode tests, the electricity consumption of the system in the chlor-alkali industry could be calculated and potentially cut down the energy consumption compared to DSA-1 (Supplementary Fig. 21).
Beyond being an anode catalyst, Ru1@TiN has also been applied to the cathode to drive the dechlorination reaction27,28. Fig. 3a shows the schematic of electrolysis of PVC waste with Ru1@TiN as the catalyst for both anode and cathode (Table S2-3). The system is carried out in an undivided two-electrode system, where the organic electrolyte (NBu4BF4 in DMF) is applied to increase the mass efficiency. Carbon substrates are chosen for both sides to avoid the interference of metal components. In this process, the dechlorination of PVC occurs on the cathode, and then Cl- migrates to the anode side, where the desired chloride species can be produced depending on the pH value of electrolyte, such as Cl2 (in acidic media), NaClO (neutral media), and ethylene oxide (EO, acidic media). Like the chlor-alkali process, Ru1@TiN also exhibits nearly 100% selectivity towards the Cl2 and NaClO (Fig. 3b, c). When it comes to EO, the selectivity decreases to ~90% (Fig. 3d), which may be due to the overoxidation of ethylene. On the aspect of activity, Ru1@TiN shows the higher rate than DSA-1 again for PVC dechlorination (Fig. 3e). Furthermore, to make PVC dechlorination completely green, a solar-driven (solar cell: 19.5×15.8 cm) electrochemical apparatus was constructed, as shown in Fig. 3f. A 10 × 10 cm electrode was used in this system. Ru1@TiN shows high and stable activity (over 90% selectivity) toward five targeted products, including Cl2, NaClO, EO, propylene oxide, and chlorobenzene (Fig. 3g). Therefore, we may achieve sustainable and green recycling of chlorine with Ru1@TiN catalyst.

a The schematic diagram for the PVC electrolysis based on Ru1@TiN as both anode and cathode catalysts. b A 24-hour test in acidic media for Cl2 product. The PVC is dissolved in the DMF and H2SO4 is applied to adjust pH value. Data are mean ± s.d. of at least four independent experiments. c A 24 h test in neutral media for NaClO product. Data are mean ± s.d. of at least four independent experiments. d A 24 h test in neutral media for EO product. Data are mean ± s.d. of at least four independent experiments. e Efficiency comparison of Ru1@TiN and DSA-1 for PVC dechlorination, assisted by ICP-MS. Data are mean ± s.d. of at least four independent experiments. f Solar-driven apparatus for green and large-scale PVC electrolysis. g The 72 h production of Cl2, NaClO, EO, propylene oxide, and chlorobenzene by the apparatus of f. Source data for Fig. 3 are provided as a Source Data file.
To explore the electrolysis mechanism of chloride on Ru1@TiN, we conducted operando XAS and Raman spectroscopy characterizations (Supplementary Figs. 22 and 23). The signals were collected through cell voltages of 0, 1.3 (onset potential), 1.6 V (normal working condition), 2.0 V (high current density condition), and back to 0 V. During CER, the Ru1 would steadily adsorb one oxygen atom (O) due to the oxidation of water, forming Ru1Oc in which the adsorbed O acts as one active site to capture Cl–29,30,31,32,33,34,35. As cell voltage increases from 0 to 2.0 V, the stretching force of Cl to O elongates the Ru-N/O bonds, which is reflected by the shifts of WT maximum (k = 4.3 Å-1) and FT-EXAFS main peak to the higher R values (Fig. 4a, Supplementary Figs. 24 and 25). Meanwhile, new local maxima assigned to Ru1-Cl (k = 5.3 Å-1) and Ru1-Ti (k = 7.6 Å-1) appear in WT plots36, and the intensity of the corresponding FT-EXAFS peak is obviously enlarged. In addition, operando Raman spectra (Fig. 4b, Supplementary Fig. 26) show an additional Ti-Cl peak at 380 nm for Ru1@TiN under the CER state with reference to the blank TiN support37,38. From these operando data, it is concluded that Ru1 may activate the surrounding Ti atoms that adsorb Cl- ions at high CER voltage. A model of the polyatomic active site (Ru1Oc-Ti4) with the multiple adsorbed Cl- ions is thus obtained as illustrated in Fig. 4c.

a Operando WT of Ru1@TiN at Ru K-edge and b operando Raman spectra of Ru1@TiN at the indicated voltages during CER in 5 M NaCl (pH = 2) at room temperature. The local maxima at k = 4.3, 5.3 and 7.6 Å-1 are assigned to Ru-N/O, Ru-Cl and Ru-Ti, respectively. c Schematic diagram of adsorption of Clˉ ions on the dynamic polyatomic active site of Ru1Oc-Ti4. The Ti atoms on the first shell around Ru are the activated Ti (a-Ti). Charge difference density of Ru1@TiN with the adsorption of a Cl atom on d Ru1Oc and on e one a-Ti atom. The isosurface value is 0.01 e/Å3. The blue and red isosurfaces refer to the positive and negative charge, respectively. f ΔGmax of CER as marked by the color of star on each atom around the active site. The x and y axes have the same scale. g The free energy of CER and OER on Ru1@TiN. Source data for Fig. 4 are provided as a Source Data file.
The above dynamic active-site structure is further proved by DFT calculations. (Supplementary Figs. 27–36) The atomic coordinates of the optimized computational models used for calculations are provided in Supplementary Data 1. The charge density difference in Fig. 4d, e shows two paths for electron transfer: 1. the transfer along Cl-O-Ru1 path if Cl is adsorbed on Ru1Oc; 2. the transfer from Cl to the activated Ti (a-Ti) and Ru1 when Cl is adsorbed on a-Ti. The calculated free energy change (ΔGmax) of CER in Fig. 4f indicates that Ru1Oc and a-Ti have similar single-spot activity (ΔGmax = 0.745 and 0.752 eV), but the second-shell Ti has negligible activity (ΔGmax = 1.732 eV). Combined with density of states (DOS), electronic localization function (ELF), crystal orbital hamilton population (COHP), electrostatic potential (ESP) and Bader charge, results in Supplementary Figs. 34-36 and Table S4, it is concluded that only the four Ti atoms in the first shell around Ru1 may be activated, i.e., a-Ti. The electron near the α-Ti and Ru sites is in a low localization state, as demonstrated by high ELF values (>0.5, Supplementary Fig. 34). Additionally, the COHP results show distinct positive peaks below the Fermi level (Supplementary Fig. 35), indicating a strong interaction. Since the single-spot activity of Ru1Oc and a-Ti in Ru1@TiN is obviously higher than that of RuO2 NP (ΔGmax = 0.811 eV)39,40,41,42, the integration of five such single spots around one Ru atom generates a super active polyatomic active site of Ru1Oc-Ti4, which can be the main reason for the high CER catalytic efficiency of Ru1@TiN. It is also supported by the ESP analysis that there are districts with similar ESP value (Supplementary Fig. 36), which means that the Ru1 and a-Ti site may exhibit similar properties for the reactant adsorption.
Compared with the ΔGmax of 1.88 eV for OER, the lower ΔGmax of CER on Ru1Oc-Ti4 suggest that CER is more likely to occur on Ru1@TiN (Fig. 4g), consistent with the observed ~100% CER selectivity. In addition, the adsorption energies of Cl2 on a-Ti and Ru1Oc are calculated to be -0.24 and -0.36 eV respectively, implying the easy desorption of product gas from Ru1@TiN, consistent with the observed large contact angle on the Ru1@TiN powder and electrode (Supplementary Fig. 37). Moreover, the proper adsorption (negative value of ΔGmax) and desorption (positive value of ΔGmax) of the Ru1Oc-Ti4 site endow Ru1@TiN bifunctional properties on both electrodes, leading to excellent efficiency in the dechlorination process.
In summary, a Ru1@TiN chloride electrolysis catalyst has been designed with high activity, ~100% selectivity, and excellent stability towards the dechlorination of NaCl and PVC. Ru single atoms activate the substrate Ti atoms, forming Ru1Oc-Ti4 dynamic polyatomic active sites that facilitate the electrolysis. This study establishes a novel electrode material platform with distinct advantages for industrial chlorine production. The specifically designed TiN support unlocks robust operation at high current densities, coupled with demonstrated stability meeting industrial demands. Importantly, the material exhibits dual functionality for core chlor-alkali processes and PVC treatment, thereby contributing to the sustainable development of society.
Methods
Chemicals
Titanium nitride (TiN, 99.9%), ruthenium trichloride (RuCl3, 99.9%), hexamine ruthenium chloride (Ru(NH3)6Cl3, 99.9%), ruthenium dioxide (RuO2, 99.9%), sodium borohydride (NaBH4, 99.9%), and sodium chloride (NaCl, 99.9%), were purchased from Macklin Chemical Reagent Corp. (China). Ethanol (C2H5OH, 99.9%), hydrochloric acid (HCl, 35.5 wt%) and sulfuric acid (H2SO4, 98%) were purchased from Beijing Chemical Corp. (China). Carbon black (BP-2000, 99.9%) was provided by CABOT. The industrial DSA is Ti mesh supported catalysts RuO2-IrO2-TiO2 with a molar ratio of Ru: Ir: Ti = 7: 1: 2 and a total metal loading of 1.43 mg·cm-2.
Synthesis of a single-atom catalyst of Ru1@TiN
The support TiN powder was pretreated in 1 M H2SO4 at 40 °C for 6 h. 20 mg of pretreated TiN was added into a three-port flask (50 mL) containing DI water (13.5 mL) and ethanol (1.5 mL, DI water:ethanol = 9:1) and then dispersed by ultrasonication for 30 min to form a uniform suspension. 4 mg of RuCl3 was dissolved in 1 mL of DI water, followed by the addition of 0.05 mL of 0.1 M HCl. Subsequently, the solution was added dropwise into the TiN suspension. Each drop has a volume of 5 ~ 10 μL. The time interval of two consecutive drops was about 30 s. A total 500 μL of RuCl3 solution was added into the suspension. The mixture solution was then stirred for 9 h at 50 °C. After cooling to room temperature (all denoted as 25 °C), the solid was thoroughly washed and collected by a filtration method, and then dried in a vacuum oven at 25 °C overnight. After a mild heat treatment at 310 °C under Ar/H2 (5%) atmosphere for 86 min, Ru-grafted TiN (Ru1@TiN) catalyst was obtained. The heating rate was 5 °C min-1. Immediately after heating, we opened the furnace and quickly cooled it using a fan. The cooling rate was about 20 °C min-1.
Synthesis of nanoparticle catalyst of Ru NP@TiN
TiN supported Ru nanoparticles (Ru@TiN) was prepared by wet reduction method. Typically, 20 mg of pretreated (as above) support (TiN) was added into a three-port flask (50 mL) containing DI water (13.5 mL) and ethanol (1.5 mL) and then dispersed by ultrasonication for 30 min to form a uniform suspension. 4 mg of RuCl3 was dissolved in 1 mL DI water, and then 0.05 mL of 0.1 M HCl was added into the solution. A total 500 μL of RuCl3 solution was added dropwise into the TiN suspension. The time interval of two consecutive drops was about 30 s. The volume of each drop was about 5 ~ 10 μL. Then, the mixture was stirred for 9 h at 50 °C. After cooling to room temperature, 0.5 g of NaBH4 was added into the suspension and stirred for 2 h at 60 °C. The solid was thoroughly washed and collected by a filtration method, and then dried in a vacuum oven at 25 °C overnight.
Synthesis of carbon supported single-atom catalyst of Ru1@C
Typically, 20 mg of pretreated (as above) carbon black support was added into a three-port flask (50 mL) containing DI water (13.5 mL) and ethanol (1.5 mL) and then dispersed by ultrasonication for 30 min to form a uniform suspension. 4 mg of RuCl3 was dissolved in 1 mL DI water, and then 0.05 mL of 0.1 M HCl was added into the solution. A total 500 μL of RuCl3 solution was added dropwise into the carbon black suspension. The time interval of two consecutive drops was about 30 s. The volume of each drop was about 5 ~ 10 μL. Then, the suspension was stirred for 9 h at 50 °C. After cooling to room temperature, the solid was thoroughly washed and collected by a filtration method, and then dried in a vacuum oven at 25 °C overnight. After a heat treatment at 200 °C under Ar/H2 (5%) atmosphere for 1 h, Ru single-atoms grafted C (Ru1@C) catalyst was obtained.
Characterization
The morphology and chemical composition of the as-prepared samples were characterized by scanning electron microscopy (SEM, JSM7500), transmission electron microscopy (TEM, Model JEM-2100F, JEOL) and scanning transmission electron microscopy (STEM, JEM-ARM200F, JEOL) equipped with energy dispersive spectrum (EDS-mapping). X-ray diffraction (XRD) measurements were conducted with a scanning step of 0.1°/s in the 2θ range of 35–150° (D/MAX2200pc). X-ray photoelectron spectroscopy (XPS) measurements were performed on an ESCALABMK II X-ray photoelectron spectrometer using Mg as the exciting source. The content of Ru in catalyst was measured by inductively coupled plasma–mass spectrometry (ICP-MS) on an Optima-7000DV system. X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) measurements were recorded in solid-state detector (SSD) mode at beamline BL14W1 station at Shanghai Synchrotron Radiation Facility (SSRF) and 1W1B station at Beijing Synchrotron Radiation Facility (BSRF). The energy resolution of the X-ray is 2×10-4@10 keV, Si(111). The Photon flux is >5 × 1012phs/s @10 keV Si(111) focused, 200 mA. The focus spot size at sample point is 0.3 × 0.3 mm2 (H × V, focused). The irradiation time is 21 min per curve. The operando X-ray absorption spectroscopy (XAS) test was carried out on a homemade cell using a three-electrode configuration with a graphite rod as the counter electrode and an Ag/AgCl (in saturated KCl electrolyte) electrode as the reference electrode. The homemade cell is made of polymethyl methacrylate. The Ru1@TiN was directly subjected on the carbon paper and was infused in 5 M NaCl solution for XANES and EXAFS measurements at room temperature. All the operando tests were carried out after a constant voltage test for 20 min. Data was analyzed using the Athena and Artemis software in the Demeter package43.
General statement for the electrochemical measurements
All electrochemical performance tests were carried out on an electrochemical workstation (CHI 760E, CH Instrument) using a three-electrode configuration with a graphite rod as the counter electrode and an Ag/AgCl (in saturated KCl electrolyte) electrode as the reference electrode. The reference electrode was calibrated versus Normal Hydrogen Electrode (NHE), ENHE = ESCE + 0.197 V. The reference electrode can also be calibrated versus the Reversible Hydrogen Electrode (RHE), ERHE = ENHE + 0.05917×pH (V). The catalyst support was a Ti-mesh that was polished with a sand paper (2000 mesh) to remove the oxide surface first. Then it was washed with 10 M NaOH at 40 °C for 2 h, followed by 5 M H2C2O4 (99.9%, Macklin) at 50 °C for 1 h. The above treatment formed a rough surface on the Ti-mesh, which was then used to support Ru1@TiN. The stability test of Ru1@TiN was conducted in constant voltage or constant current modes. The purchased industrial DSA (named as DSA-1) consists of RuO2-TiO2 and a total metal loading of 3 mg·cm-2, of which some other metal species will be added to stabilize the DSA.
The detection of the concentration of effective chlorine (ClO-) and ClO3 -
To reach the accurate concentration of ClO-, 50 μl of electrolyte is collected and dissolved into 3 mL of DI water. Then the sample is detected by the UV-Vis spectrophotometry. A blank solution containing only DI water (pH = 7) is also prepared as reference or background. The main peak arising from 292 nm is attributed to the ClO-. A quartz cuvette is rinsed multiple times with the blank solution, then fill it with the blank and record the baseline absorbance. After the standardization process, the absorbance of the sample at 292 nm is recorded. Repeat this rinsing and measurement process for all standard solutions and the unknown sample(s), ensuring consistent measurement conditions. Then plot the absorbance values of the standards against their known concentrations to generate a calibration curve, and the concentration of the ClO- is obtained. The ClO3- is extracted from the electrolyte with a similar process above and detected by the ion chromatography.
The preparation of anode
We mixed the catalyst (5 mg) with iso-propyl alcohol (IPA), Nafion® solution (5%) and deionized (DI) water (volume ratio = 19:5:11) and stirred for 2 h to get the catalyst ink (500 μl). The ink was then dropped onto a piece of carbon paper (gas diffusion layer, GDL) or a pre-treated Ti-mesh, followed by drying at 20 °C in a vacuum oven. A following ambient heat treatment is needed at 90 °C under Ar atmosphere for 1 h. The catalyst loading is detected to be 5.5\(\pm\)0.3 mg/cm2 on average, with the electrode area of 1 cm*1 cm.
The preparation of membrane electrode assembly (MEA) and H-type cell
The cathode catalyst ink was prepared by dispersing a commercial Pt/C (20 wt%) catalyst in a solution of DI water, n-propanol, and 5 wt% Nafion to make a ∼2 mg·mL-1 catalyst ink. The cathode gas diffusion electrode (GDE) was prepared by spray-coating the catalyst ink onto a 2.5 × 2.5 cm piece of a carbon-paper using an ultrasonic spray coating system to achieve a platinum loading of 0.5 mg cm−2. The electrode was dried on a heated metal plate at a temperature of 60 °C for 10 min. A thin layer of Nafion ionomer was then deposited on top of the catalyst layer by ultrasonic spray to help the adhesion of the GDE to the membrane during the hot-pressing and to reduce the electrode/membrane contact resistance. The electrolyte membrane was a piece of Nafion 211 (4 × 4 cm) that was washed in 5 M NaOH at 60 °C for 2 h before being applied in the equipment. During the stability test, the GDL is replaced by a metal mesh. The cathode GDE was hot-pressed onto one side of a Nafion N211 membrane at 110 °C for 5 min at 3.7 MPa. The anode preparation is shown as the above section. It should be noted that some chlor-alkali industry apply Pt mesh as the cathode, which is also efficient for the oxygen depolarized cathode system in this study.
For the assembly of H-type electrolytic cell, the distance between the anode and cathodes is about 8 cm.
The calculation of equilibrium voltage
In NHE scale, the equilibrium potential of CER (ECER) is dependent on temperature, concentration of Cl−, and partial pressure of Cl2 on electrolyte. ECER was derived from the Nernst equation:
where R, T, F and a represent the universal gas constant, the temperature, the Faraday constant, and the chemical activity, respectively. The value of a(Cl2) was assumed to be 0.01 for the partial pressure of evolving Cl2 under Ar purging. a(Cl−) was determined by experimental conditions (i.e., a(Cl−) = 5.0 for 5.0 M NaCl).
Product yielding rate and Faradaic efficiency (FE)
Before the test, we performed a 2 h constant voltage test to obtain a Cl2-satutured electrolyte. And the electrochemical cell is sealed during this period. After the reaction beginning, the product of Cl2 is led into NaOH (solution A). The after-treated gas goes into the GC, and the by-product is detected. After that, solution A was mixed with DI water. Then FeO was added to the diluted solution to reduce the ClO-. The Cl- is quantitatively detected by ion chromatography. The FE of CER was calculated using the following equation:
where [Cl2] is the measured Cl2 concentration by colorimetric method; V is the volume of the cathodic reaction electrolyte; t is the time; F is the Faraday constant, and Q is the total charge of applied electricity.
The above is an example of measuring the amount of Cl2, while other products are detected by GC with a similar operation.
In-situ Raman measurements
In-situ Raman spectra were collected by a Horiba spectrometer (LabRAM HR Evolution). The catalyst inks are prepared by mixing 5 mg of electrocatalysts, 0.95 mL of ethanol, and 50 μL of Nafion solution (5 wt%). Then, 10 μL of ink solution is dropped onto the central area of an Au film (confined by an O-ring with Φ = 8 mm) that is deposited on a glassy carbon. The glassy carbon electrode was assembled in a spectroelectrochemical cell with a piece of Pt wire as the counter electrode, an Ag/AgCl electrode as the reference electrode, and 5 M NaCl (pH = 2) and 0.5 M NaCl (pH = 7) solutions as electrolytes. A CHI 760E electrochemistry workstation (Shanghai CH Instruments, Inc.) was used for potential control from OCV to 1.6 V vs. NHE. The laser wavelength was 633 nm, and the exposure time was 5 s. For each sample, five Raman spectra at random positions were collected and averaged. The homemade cell for Raman measurement is shown in Supplementary Fig. 18.
PVC electrolysis
The electrolysis of PVC is carried out in both divided and undivided cells. Firstly, the conditions for an undivided cell paired-electrolysis are shown below. The experiments are carried out under constant-voltage and on a 40 mL scale. The PVC (1000 mg, 12.8 mmol (repeat unit),), NBu4BF4 (1056 mg, 3.20 mmol), target reactant (10 mmol) and tridecane internal standard (0.050 mL) were mixed. All reagents were dissolved (or swelled) by stirring for at least 15 min, then the reaction mixture was subjected to constant-voltage electrolysis (1.4 V) with alternating polarity (15 min) at room temperature. During the reaction, we kept stirring at 400 rpm for 24 h. We also carried out a divided cell paired-electrolysis, which was performed under constant current. The divide reaction vessel was assembled using graphite electrodes and a 5 μm frit separator layered with a PiperION anion-exchange membrane (pre-soaked in 0.1 M NBu4BF4 in DMF). The substrate (5 mmol), NBu4BF4 (1100 mg), and DMF (50 mL) were added into the working (anodic) chamber; PVC47k (1000 mg), NBu4BF4 (1100 mg), and DMF (7 mL) were added into the counter (cathodic) chamber. All reagents were added under air and then dissolved by stirring for at least 15 min. Then, the reaction mixture was subjected to constant-current electrolysis (10 mA) at room temperature. The anodic reaction was monitored by gas chromatography and stirred until all the substrate was consumed. The working (anodic) chamber was poured into ~250 mL of ethyl acetate and washed with H2O (5 × 40 mL) and brine (50 mL) to remove DMF. The organic layer was dried with MgSO4, filtered, and concentrated under reduced pressure. The crude reaction was further purified by column chromatography and the final product was dried under reduced pressure.
Solar-driven PVC electrolysis
We set up a solar-driven (solar cell: 19.5 × 15.8 cm) electrochemical apparatus, with parallel circuits for magnetic stirring and electrolysis. Before carrying out the reaction, the storage battery attached to the solar cells was discharged to zero. We used in a BUCK circuit board to tune the volts between the anode and cathode. With the same electrolysis cell, the electrodes were replaced with the 10 cm* 10 cm ones. By exposing the device to sunlight for three days, the solution was detected to investigate the conversion of the substrate.
DFT calculations
The structural optimization was performed using first-principal calculations within the framework of density functional theory (DFT), as implemented in the plane wave set Vienna ab initio Simulation Package (VASP) code. The exchange correlation energy was modeled using Perdew-Burke-Ernzerhof (PBE) functional within the generalized gradient approximation (GGA). The projector augmented wave (PAW) pseudo-potentials were used to describe ionic cores. The cutoff energy of 500 eV was adopted after a series of tests. The self-consistent-field (SCF) was set to be 10-5 eV, and that for geometry optimization by BFGS algorithm was set to be 0.01 eV/Å on maximum force component. The k-point sampling of the Brillouin zone was obtained using a 7 × 7 × 1 grid for unit by Monkhorst-Pack Scheme. Denser k-points (14 × 14 × 1) were used for the electronic structure calculations. A large vacuum slab of 15 Å was inserted in z direction for surface isolation to prevent interaction between two neighboring surfaces. Spin-polarization was considered in all calculations. The vibrational frequencies of free molecules and adsorbates, which are needed to determine zero-point energies and vibrational entropies, were calculated using the phonon modules.
The surfaces were constructed based on the optimized bulk structure. After the test, a slab containing seven atomic layers was chosen to model the TiN (100) surface. The models of TiN and Ru1@TiN with a 5 × 5 × 7 supercell were first chosen for the simulation of the charge density distribution and the DOS. The bottom three layers were kept frozen to the bulk positions, and the other atoms were relaxed. We compared the stabilities of Ru located at different sites on the TiN (100) surface. The DFT calculations indicate that the most possible site for a single Ru atom on the TiN (100) surface is the four-fold hollow site, where Ru atom is coordinated by four surface titanium atoms and one nitride atom. This configuration is consistent with the results of the EXAFS fitting results. Further free geometry optimizations and energy calculations were performed based on the Ru1@TiN model. And the model for RuO2 is a slab containing eight atomic layers. The model of RuO2 with a 3 × 3 × 8 supercell was first chosen for the absorption energy calculation. The bottom three layers were kept frozen to the bulk positions, and the other atoms were relaxed. The free energy of the adsorbed state was calculated based on the adsorption energy:
where ΔE is the adsorption energy of hydrogen and ΔEZPE is the difference corresponding to the zero-point energy between the adsorbed state and the gas phase. The adsorption site of hydrogen was confirmed by comparing the adsorption energy of hydrogen at different positions on the catalyst surface after free geometry optimizations with an SCF tolerance level of 1.0 × 10−5.
Reaction mechanism
The oxygen evolution reaction (OER) pathways on the SAC were calculated according to electrochemical framework developed by Nørskov and co-workers. The OER occurs in an acidic electrolyte through elementary steps44:
where the * represent the adsorption site.
The CER pathways on the SAC were calculated according to the electrochemical framework developed by Rossmeisl and co-workers45. Firstly, we investigated the mechanism involving ClOc:
The potential at which all steps are neutral or downhill is:
where Ueq is the equilibrium potential for chlorine evolution, in this case 1.36 vs. RHE. Secondly, we investigated the mechanism involving Cl(Oc)2:
The potential at which all steps are neutral or downhill is:
Thirdly, we proposed a mechanism involving Clc adsorbed directly at the metal cus site.
The potential where all steps are neutral or downhill is:
Reaction free energy calculation
For the proton-transfer step, the reaction free energy is regarded as an approximate value of the activation barrier. Because the calculation has proven the neglect of over over-barrier for the transfer of a solvated proton to adsorbed OH- for a situation where the proton transfer is downhill in energy, this approximation may result in a slight overestimation of activity for a given proton-transfer elementary step, but can still qualitatively represent the relative energetic ordering of the various proton-transfer elementary steps. Therefore, we took reactions (5) - (8) to derive the thermodynamics for OER. The reversible hydrogen electrode (RHE) model developed by Nørskov and co-workers was used to obtain Gibbs reaction free energy of these electrochemical elementary steps46. In this model, we set up RHE as the reference electrode, which allows us to replace chemical potential (μ) of the proton-electron pair with that of half a hydrogen molecule: μH+ + μe- = 1/2μH2, at conditions of U = 0 V and PH2 = 1 bar.
The chemical potential of each adsorbate is defined as:
where the E is the total energy obtained from DFT calculations, EZPE is zero-point energy and S is the entropy at 298 K. In order to obtain the reaction free energy of each elementary step of OER, we calculated the adsorption free energy of O*, OH* and OOH*. Since it is difficult to obtain the exact free energy of OOH, O and OH radicals in electrolyte, the adsorption free energy ΔGOOH*, ΔGO* and ΔGOH*, are relative to the free energy of stoichiometrically appropriate amounts of H2O (g) and H2 (g), as defined by:
For each elementary step, the Gibbs reaction free energy ΔG is defined as the difference between free energies of the initial and final states and is given by the expression:
where ΔE is the reaction energy of reactant and product molecules adsorbed on catalyst surface, obtained from DFT calculations. ΔEZPE and ΔS are the change in zero-point energy and entropy due to the reaction. The free energy for gas phase water is calculated at 0.035 bars because this is the equilibrium pressure in contact with liquid water at 298 K. The free energy of gas-phase water at these conditions is equal to the free energy of liquid water.
The reaction free energy of (5) − (8) for OER can be calculated using the following equations:
For OER, the onset potential is calculated by
In the same way, the reaction free energy of (9) and (13) for CER can be calculated using the following equations:
The calculation of energy consumption
The calculation of electric consumption during the chlor-alkali process uses the following equation:
In the equation, Wg is the practical consumption of DC energy for one ton of 100% NaOH, with unit of kW∙h/tNaOH. V is the cell voltage of the electro bath with unit of V. η is the efficiency of the cathode with unit of %. 1.492 is the electrochemical equivalent of NaOH, and its unit is g/A ∙ h.
In this work, Ru1@TiN exhibits a selectivity of over 99.5% towards CER at room temperature. The applied Pt-based cathode catalyst exhibits a selectivity of 99.8% and 99.7% towards HER and ORR, respectively. The η is thus set to be 99.7 (%) during the oxygen depolarized cathode (ODC) system. We carried out the stability test for 2 h under 0.1, 4, and 10 kA m-2 to obtain the cell voltage for the energy consumption calculation. It can be seen from the equation that the cell voltage plays a main role in determining the final energy consumption, which is affected by the catalyst’s efficiency, ohmic resistance, and so on.
The details for the synthesis with the electricity supply from photovoltaic solar panels
The solar energy was also collected by photovoltaic solar panels (35 × 45 cm, output voltage: 0 ~ 6 V) and batteries to storage and supply the energy during the dark period. The synthesis process is carried out at Beijing (40.006°N, E 116.336°E).
The contact angle measurement of the Ru1@TiN
The static sessile drop contact angle is measured using a LAUDA Scientific LSA100S-T goniometer. After instrument warm-up (≥30 mins) on a vibration-free surface and verification of environmental controls (if used), a solvent-rinsed high-precision syringe filled bubble-free with pure test liquid (e.g., ultrapure water, ≥99.9%), is mounted into the dosing unit. Solvent-cleaned, air-dried substrate samples are securely leveled on the stage. System calibration is performed using the supplied standard (e.g., precision ball bearing) within the LSA Image software, inputting the known dimension and verifying accuracy. For measurement, the motorized stage positions the sample horizontally beneath the needle tip (1–3 mm above surface). A liquid droplet is deposited automatically via software-defined parameters. Immediately post-deposition/needle retraction, a high-resolution image of the static drop is captured; ambient temperature/humidity are recorded if uncontrolled. The contact angle between the bubble and the Ru1@TiN is detected by the same equipment with a single bubble pumped to the interface in the solution of 5 M NaCl (pH = 2).
Data availability
All data supporting the findings of this study are presented within the article (and Supplementary Information files), and the atomic coordinates of the optimized computational models are provided in Supplementary Data 1. Additional data are available from the corresponding authors upon request. Source data are provided with this paper.
References
Chlorine Industry Review 2022-2023 Euro Chlor https://www.chlorineindustryreview.com/wp-content/uploads/2023/10/Chlor-Alkali-Industry-Review-CORRECTED-2023-10-06.pdf (2024).
P. Schmittinger et al. “Chlorine” in Ullmann’s Encyclopedia of Industrial Chemistry, B. Elvers Ed. (Wiley, 2011).
Leow, W. R. et al. Chloride-mediated selective electrosynthesis of ethylene and propylene oxides at high current density. Science 368, 1228–1233 (2020).
Shao, X. et al. Coordination Environment and Distance Optimization of Dual Single Atoms on Fluorine-Doped Carbon Nanotubes for Chlorine Evolution Reaction. Angew. Chem. Int. Edit. 63, e202406273 (2024).
Vos, J. G. et al. Selectivity trends between oxygen evolution and chlorine evolution on iridium-based double perovskites in acidic media. ACS Catal. 9, 8561–8574 (2019).
Lim, H. W. et al. Rational design of dimensionally stable anodes for active chlorine generation. ACS Catal. 11, 12423–12432 (2021).
Tong, W. et al. Electrolysis of low-grade and saline surface water. Nat. Energy 5, 367–377 (2020).
Xie, H. et al. A membrane-based seawater electrolyser for hydrogen generation. Nature 612, 673–678 (2022).
Trasatti, S. Electrocatalysis: understanding the success of DSA. Electrochim. Acta 45, 2377–2385 (2000).
Beer, H. B. Electrode with a titanium core and porous protective layer of noble metal. U.S. 3096272 (1961).
Lim, T. et al. Atomically dispersed Pt–N4 sites as efficient and selective electrocatalysts for the chlorine evolution reaction. Nat. Commun. 11, 412 (2020).
Yang, J. et al. Regulating the tip effect on single-atom and cluster catalysts: forming reversible oxygen species with high efficiency in chlorine evolution reaction. Angew. Chem. Int. Edit. 61, e202200366 (2022).
Yang, J., Zhu, C. & Wang, D. A simple organo-electrocatalysis system for the Chlor-related Industry. Angew. Chem. Int. Edit. 63, e202406883 (2024).
Zackasee, J. L. S. et al. Electrocatalytic grafting of polyvinyl chloride plastics. Chem. 11, 102298 (2024).
Liang, Y. et al. Efficient electrocatalysis for the preparation of (hetero) aryl chlorides and vinyl chloride with 1, 2-dichloroethane. Angew. Chem. Int. Edit. 58, 4566–4570 (2019).
Choi, C. et al. CO2-Promoted electrocatalytic reduction of chlorinated hydrocarbons. J. Am. Chem. Soc. 146, 8486–8491 (2024).
Fagnani, D. E. et al. Using waste poly (vinyl chloride) to synthesize chloroarenes by plasticizer-mediated electro (de) chlorination. Nat. Chem. 15, 222–229 (2023).
Liu, M., Wu, X. & Dyson, P. J. Tandem catalysis enables chlorine-containing waste as chlorination reagents. Nat. Chem. 16, 700–708 (2024).
Choi, C. et al. Efficient electrocatalytic valorization of chlorinated organic water pollutant to ethylene. Nat. Nanotech. 18, 160–167 (2023).
Ha, M. et al. High-performing atomic electrocatalyst for chlorine evolution reaction. Small 19, 2300240 (2023).
Wang, Q. et al. Operando XAFS reveals dynamic structural evolution of pt single-atom catalysts for efficient chlorine electrosynthesis. Angew. Chem. Int. Edit. 64, e202513656 (2025).
Kötz, R. & Stucki, S. Stabilization of RuO2 by IrO2 for anodic oxygen evolution in acid media. Electrochim. Acta 31, 1311–1315 (1986).
Finke, C. E. et al. Enhancing the activity of oxygen-evolution and chlorine-evolution electrocatalysts by atomic layer deposition of TiO2. Energy Environ. Sci. 12, 358–365 (2019).
Exner, K. S. et al. Controlling selectivity in the chlorine evolution reaction over RuO2-based catalysts. Angew. Chem. Int. Edit. 53, 11032–11035 (2014).
Chen, R. Y. et al. Microstructural impact of anodic coatings on the electrochemical chlorine evolution reaction. Phys. Chem. Chem. Phys. 14, 7392–7399 (2012).
Zhu, G. et al. Rechargeable Na/Cl2 and Li/Cl2 batteries. Nature 596, 525–530 (2021).
Gao, R. T. et al. Photoelectrochemical production of disinfectants from seawater. Nat. Sustain 8, 672–681 (2025).
Zhang, C. et al. Stable and high-yield hydrogen peroxide electrosynthesis from seawater. Nat. Sustain 8, 542–552 (2025).
Trasatti, S. Progress in the understanding of the mechanism of chlorine evolution at oxide electrodes. Electrochim. Acta 32, 369–382 (1987).
Yang, J. et al. Organocatalyst supported by a single-atom support accelerates both electrodes used in the Chlor-Alkali industry via modification of non-covalent interactions. Angew. Chem. Int. Edit. 63, e202314382 (2024).
Hansen, H. A. et al. Electrochemical chlorine evolution at rutile oxide (110) surfaces. Phys. Chem. Chem. Phys. 12, 283–290 (2010).
MacMillan, D. W. C. The advent and development of organocatalysis. Nature 455, 304–308 (2008).
Nicewicz, D. A. & MacMillan, D. W. C. Merging photoredox catalysis with organocatalysis: the direct asymmetric alkylation of aldehydes. Science 322, 77–80 (2008).
Yang, J. et al. CO2-mediated organocatalytic chlorine evolution under industrial conditions. Nature 617, 519–523 (2023).
Song, X.-J. et al. Bistable state of protons for low-voltage memories. J. Am. Chem. Soc. 142, 9000–9006 (2020).
Shinde, S. S. et al. Ampere-hour-scale zinc–air pouch cells. Nat. Energy 6, 592–604 (2021).
I. Moussallem, J., Jörissen, U., Kunz, S., Pinnow, T. & Turek, T. Chlor-related electrolysis with oxygen depolarized cathodes: history, present status and future prospects. J. Appl. Electrochem. 38, 1177–1194 (2008).
Chen, P. & Zhan, X. Chem. Soc. Rev. 45, 2544–2582 (2016).
Arora, N. et al. Perovskite solar cells with CuSCN hole extraction layers yield stabilized efficiencies greater than 20%. Science 358, 768–771 (2017).
Exner, K. S. et al. Full kinetics from first principles of the chlorine evolution reaction over a RuO2 (110) model electrode. Angew. Chem. Int. Edit. 55, 7501–7504 (2016).
Zhang, H. et al. Standardized protocols for evaluating platinum group metal-free oxygen reduction reaction electrocatalysts in polymer electrolyte fuel cells. Nat. Catal. 5, 455–462 (2022).
Wang, J. et al. Engineering the coordination environment of ir single atoms with surface titanium oxide amorphization for superior chlorine evolution reaction. J. Am. Chem. Soc. 146, 11152–11163 (2024).
Newville, M. IFEFFIT: interactive XAFS analysis and FEFF fitting. J. Synchrotron Radiat. 8, 322–324 (2001).
Huang, Z. F. et al. Chemical and structural origin of lattice oxygen oxidation in Co–Zn oxyhydroxide oxygen evolution electrocatalysts. Nat. Energy 4, 329–338 (2019).
Svane, K. L. & Rossmeisl, J. Theoretical optimization of compositions of high-entropy oxides for the oxygen evolution reaction. Angew. Chem. Int. Edit. 61, e202201146 (2022).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886–17892 (2004).
Acknowledgements
This work was supported by the Key R&D Program of Zhejiang (2024SSYS0081, J.S.). This work was financially supported by the National Key Research and Development Program of China (2021YFB4000601, J.S.), the National Natural Science Foundation of China (223B2902, J. Y.; U21A20328, J.S.; 22225903, J.S.). The authors acknowledge the use of XAS facilities of Beijing Synchrotron Radiation Facility at beamline 1W1B and thank Lirong Zheng (Institute of High Energy Physics) for assistance in the XAS measurements. We also acknowledge the funding support for the Global STEM Professorship from the Innovation, Technology and Industry Bureau (“ITIB”) and Education Bureau (“EDB”) of Hong Kong (X.W.L.). Open Access made possible with partial support from the Open Access Publishing Fund of the City University of Hong Kong.
Author information
Authors and Affiliations
Contributions
J.Y., X.W.L. and J.S. conceived the idea. J.Y. performed the synthesis, electrochemical tests and characterizations. J.Y. performed XAS measurement and analysis. J.X.S. and J.Y. provided the structural models for XAS analysis and carried out the DFT calculations. X.W.L. and J.S. supervised the project and participated in the planning of the research. J.Y., X.W.L. and J.S. co-wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hao Tan and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, J., Shang, J., Lou, X.W.D. et al. Sustainable chlorine cycle enabled by single atom catalysis. Nat Commun 16, 10525 (2025). https://doi.org/10.1038/s41467-025-65505-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65505-5
