
Abstract
Cryogenic focused ion beam (Cryo-FIB) milling has become a standard step in the cryogenic electron tomography (Cryo-ET) workflow and is required to thin cells to electron-semitransparency. However, this destructive process removes the vast majority of the cellular material and raises a critical question: what thin section should be preserved for Cryo-ET analysis? Using a tri-coincident cryogenic FIB-SEM-LM system, we identify an interferometric optical response that can be used for targeting lamella production to fluorescently labeled structures with accuracy beyond the diffraction limit. Here we demonstrate this approach using synthetic samples of fluorescent beads embedded in micron-scale droplets of amorphous ice. We then apply the approach to capture virions inside host cells. Successful targeting is confirmed by Cryo-ET revealing clusters of virions in intracellular vesicles. The method does not require any fluorescent fiducials or axial registration and can be performed on any fluorescently labeled structure that is visible in widefield fluorescence microscopy.
Similar content being viewed by others

Introduction
Cryogenic electron tomography (Cryo-ET) is a proven approach for uncovering subcellular architecture in situ1,2,3. However, due to the limited penetration of electrons through biological material, the approach is limited to samples far thinner than typical eukaryotic cells4. To amend this, Cryogenic Focused-Ion-Beam (Cryo-FIB) milling uses an ion beam to ablate cellular material and produce sections of a desirable thickness (lamella), typically on the order of 200 nm thick5. Capturing small and rare biological targets with this destructive approach in the final thin lamella has proven challenging. Targeting the milling process via fluorescence has provided a promising route to capture structures of interest. Recently, commercial options for the integration of light microscopy into the Cryo-FIB vacuum chamber have emerged; however, these systems place the light microscope and FIB focal positions in different locations within the chamber6. This makes guidance challenging by requiring a registration process to align the fluorescence and Cryo-FIB images, as well as making real-time response impossible. Image registration requires a high density of objects that are observable in both the optical and ion imaging modalities. Typically, micron-scale fluorescently labeled polystyrene beads are used for this purpose7,8,9, but these can obscure labeled structures of interest. Furthermore, the accuracy of guided milling in this manner suffers from registration errors10,11,12, localization errors due to refractive index mismatch13,14,15, and shifts of the sample during milling9. These effects limit the accuracy of guidance in the axial dimension. Furthermore, the target lamella thickness is approximately 1/10th the axial diffraction limit, so even small errors generated by these effects can mean missing a target completely. An alternative configuration is to match the focal planes of the optical, electron, and ion microscopes. These tri-coincident systems16, such as the ENZEL used here17, can monitor fluorescence in real-time while milling (Supplementary Fig. S1). This ability removes the axial registration problem. So long as the object of interest is significantly larger in axial extent than the target lamella thickness, the object can be captured by halting milling from one direction as fluorescence dims due to ablation of the target18,19. However, this is partially destructive and does not permit the capture of objects on the order of the lamella thickness or smaller. Here, we will show how this limitation can be overcome using an additional interferometric signal to direct milling with no loss of the target structure. Exploiting this effect, we can regularly capture small and rare structures in the final lamella for visualization by Cryo-ET.
Results
Observation of optical interferogram during milling
We begin with a synthetic system to remove complications associated with biological samples and to cleanly observe optical effects during milling. Using a modified plunge freezer20, fluorescent polystyrene beads in micron-scale water droplets were deposited on the surface of EM grids as the grids were transiting towards cryogenic liquid ethane, Fig. 1a and Methods. Frozen droplets were then milled in cleaning cross-section mode (i.e., a line of ions was scanned repeatedly, and that line was slowly advanced through the sample). The advance of the ion beam either comes from the bottom of the grid upwards, termed “bottom-up” milling, or from the top of the droplet downwards, termed “top-down” milling, Fig. 1b. In bottom-up milling, the fluorescence intensity increases as the support film, which lies between the excitation source and the fluorescent object, is removed, Fig. 1c. This enhancement is quite prominent due to the significant optical density associated with carbon support films21 and the fact that the excitation source and the emitted fluorescence are both attenuated. More interestingly, in top-down milling, prominent oscillations in fluorescence intensity are observed as the top surface approaches a fluorescent target. These oscillations grow in amplitude until a rapid drop is seen due to milling through the target, Fig. 1d. Given that the observed interferogram arises as a fluorescent object approaches the surface of a lamella, we hoped to exploit the effect for the guidance of FIB milling. However, we first needed to characterize the interferogram and understand its physical basis.

a Fluorescent beads embedded in droplets were sprayed and frozen on a grid. Cartoon sample depiction, along with an ion and fluorescent image of one droplet. b Cartoon depiction of either bottom-up or top-down milling that can be performed while monitoring fluorescence. c Single-bead fluorescence brightness trace when milled bottom-up. An enhancement in the fluorescence can be seen as the absorptive support film is removed21. Corresponding ROI’s can be seen below to show this enhancement at different times indicated by the colored asterixis. d Single-bead fluorescence brightness trace when milled top-down (black solid and dotted lines). This milling results in oscillations in brightness as the surface of the lamella approaches the embedded fluorescent beads. The oscillations were then fit to a model function (red), and their periodicity is consistent with the excitation wavelength.
Characterization of interferogram
To isolate fluorescence of individual beads from out-of-focus background and autofluorescence, the fluorescent point-spread-function (PSF) of individual beads was fit to a two-dimensional symmetric Gaussian. The integrated intensity of the resulting fit more clearly exhibits oscillations as a function of the distance between the top interface and the object of interest than the simpler integration of the raw intensity in a region of interest. Assuming the object originates from a point source, the intensity of oscillations, I, as a function of position below the milling surface, d, can heuristically be fit to the following function:
where A is the peak amplitude of the oscillations, \(\phi\) is the phase, \({d}_{0}\) is an offset fit parameter, \(k\) is the rate of enhancement in fringe contrast related to the coherence length of the LED-based excitation source, and \(\lambda\) is the wavelength of the oscillation. The recovered wavelength in Eq. 1 is corrected to the optical wavelength in vacuum by accounting for the refractive index of the material, n, and the milling geometry through the following equation:
where the index of refraction is taken to be that of amorphous ice22 and \(\theta\) is the angle of incidence of the FIB relative to the optical imaging plane of the light microscope. In our system, this angle is ~ 10 ± 2 degrees.
Source and physical basis of interference
The origin of the interferogram was initially unclear, with the primary question being: is it derived from the excitation light or the emitted light? To distinguish between these two possibilities, polystyrene fluorescent beads of 200 nm and 40 nm diameters were respectively doped with either small Stokes shift (ThermoFisher F8811) or large Stokes shift (ThermoFisher T10711) dyes and were milled in the top-down direction. The sample with a large Stokes shift dye provides a clear difference between excitation and emission wavelengths and makes assignment of the interference to the excitation source obvious. Figure 2 shows a large sampling of fluorescent beads where the fluorescence intensity was monitored with a constant top-down milling rate. The oscillations in the traces were then aligned, averaged, and fit to the model function described in Eq. 1 and the corrected wavelength was determined with Eq. 2. The small Stokes shift beads were excited at 470 ± 10 nm and emission collected at 515 ± 15 nm. This yielded a fit with a corresponding periodicity of 484 ± 3.62 nm. The large Stokes shift beads were excited at 470 ± 10 nm and emission collected at 698 ± 45 nm. Again, the averaged trace was fit to the same function, and a wavelength of 486 ± 7.27 nm was measured. Figure 2 demonstrates that common excitation wavelengths, but different emission wavelengths, still led to consistent oscillation frequencies. Supplementary Fig. S2 shows the results of a similar experiment that used different excitation wavelengths and common emission wavelengths, which led to different oscillation frequencies that again matched the excitation wavelengths. Taken together, these results suggest the source of the interference is the excitation light. The slight shift of the recovered periodicity from the true value of 470 ± 10 nm is likely due to uncertainties in the index of refraction, milling angle, and the convolution of the excitation spectrum of the fluorescent beads and the excitation spectrum of the LED. Interestingly, in the traces of the 40 nm beads, a deterioration is observed in the amplitude of the last oscillation. We attribute this to damage and/or charging from the milling process as the milling surface approaches the 40 nm diameter fluorescent object.

Beads were doped with small or large Stokes shift dyes. a The integrated fluorescence brightness from individual 200 nm diameter small Stokes shift fluorescent beads (505 nm maximum excitation/515 nm maximum emission). Beads were excited at 470 nm while performing top-down milling. Traces have been aligned by the loss of fluorescence that occurs when the beads are milled through. The beads exhibited consistent oscillations in brightness as a function of the milling. b Average brightness traces were fit to a model function to determine the periodicity of the interferogram to be 484 ± 3.6 nm. c Similarly, 40 nm beads with a larger Stokes shift (488 nm maximum excitation/645 nm maximum emission) were excited at 470 nm and exhibited consistent oscillations. d The traces were again aligned, averaged, and fit to the model function. The resulting periodicity of 486 ± 7.3 nm attributes the source of the interference to the incoming excitation light and not the emitted fluorescence. The cause of the dimming observed in the final oscillation of the 40 nm diameter beads—but not in the 200 nm diameter beads—remains unknown. It may be related to the proximity of dye molecules to the lamella surface, which varies with bead size, or to differential dye sensitivity to charging, with the large Stokes shift dye potentially being more susceptible in this example.
The physical basis for this interferogram is illustrated in Fig. 3a. The incoming excitation light will reflect off the top milling surface due to the mismatch of refractive indices at that interface23. The returning light will then interfere with the incoming light, leading to constructive and destructive interference as a function of the distance from the interface. A fluorescent object near the interface will be excited more or less, depending on the interference being constructive or destructive, respectively. To guide the milling process using the interferogram, it is critical to know the expected amplitude and phase of the oscillation as we approach the target. For this, we turn to the Fresnel equation for reflection. Our relatively low NA (0.85) objective leads to excitation being largely P-polarized at the interface. Thus, we can describe the reflection coefficient, which is the fraction of reflected incident light intensity, as23,24,25,26,27
where \({\theta }_{i}\) is the angle of incidence of the incoming light, \({\theta }_{t}\) is the angle of refraction of the transmitted light, \({n}_{1}\) is the index of refraction of the amorphous ice, and \({n}_{2}\) is the refractive index of material at the milling surface interface, see Fig. 3a. Snell’s law gives the relation between incident angle and the angle of refraction as
which can be rearranged to determine \({\theta }_{t}\) as

a Diagram of the interface of reflection showing the necessary geometry to recreate the interferometric signal. As excitation enters the sample at incident angle \({\theta }_{i}\) and phase \(\phi\) the index of refraction differences at the surface, \({n}_{1}\) and \({n}_{2}\), will cause a reflection. The outcoming reflection will travel with an extra phase factor \(\phi+\pi\) at the same angle \({\theta }_{i}\). Along this, we denote the transmission path as well, along an angle \({\theta }_{t}\). b Simulated interference with high to low index interface (blue) or low to high (red), generating interferograms that differ in phase by π. (black) A fluorescence intensity trace from milling through 40 nm large Stokes-shifted beads to match the simulated data. The experimental result is consistent with the interface being low to high index.
Substituting Eq. 5 into Eq. 3 yields
Here, the incident angle is well known from the geometry of the setup, 10° ± 2°; the index of refraction of amorphous ice is ~ 1.26 at 550 nm22 with the key unknown variable in Eq. 6 being \({n}_{2}\). We will return to Eq. 6 later when computing expected amplitudes of the interferogram.
From here, we turn to interferometry, where the field at a position d below the lamella at time t is given as24,28
Here, \(\tau\) is the time delay due to the round-trip travel from the fluorescent object and back, while \(\phi\) is an additional phase factor induced by reflection. If \({n}_{1}\) < \({n}_{2}\) then \(\phi\) gives a phase delay of π; otherwise \(\phi=0\). The intensity of this field is given as
which expanded gives
where c is the speed of light and \({\epsilon }_{o}\) is the permittivity in a vacuum. The time-average of the two leading homodyne components of Eq. 9 lead to a constant intensity as a function of d, but the cross-term results in oscillations in intensity as the fields \({E}_{i}\) and \({E}_{r}\) constructively and destructively interfere as a function of d. This oscillatory component is
This can be written as
where \({\lambda }_{{vac}}\) is the excitation wavelength and \(\phi\) is either \(\pi\) or 0, depending on the relative indices of refraction. Using a product to sum identity, Eq. 11 can be expanded as:
Considering the time-averaged field, the second term goes to zero and can be further simplified to:
Equation 13 shows that the intensity of the field at position d below the lamella surface will oscillate as a function of d, Supplementary Fig. S3. However, this assumes an infinitely coherent source. To account for the limited spatial and temporal coherence of the LED-based excitation source, we have an additional exponential to artificially dampen the signal where k describes this finite coherence. This value can be rigorously calculated from the spectral characteristics of the light or taking the autocorrelation of the cross-term in Eq. 925,26,29.
Equation 14 directly relates to Eq. 1.
The interferograms expected from Eq. 14 are simulated in Fig. 3b and compared to experimental results from the large Stokes shift beads. In this simulation, the fluorescent object was a uniformly fluorescent sphere of 40 nm in diameter. The phase factor \(\phi\) is 0 when we consider the interface of reflection to be amorphous ice and vacuum, creating a positive amplitude and ending on a peak. Conversely, a phase of \(\pi\) is added when assuming the dominating reflective interface to be the damaged layer and amorphous ice, with the damaged layer having a higher index of refraction, creating a negative amplitude and ending on a trough. The final oscillations from the 40 nm diameter large Stokes shift beads are overlayed for comparison, which clearly demonstrates the phase of the interferogram, and the reflection dominated by the damage layer. The two likely possibilities for the material at this interface, leading to the increased refractive index, include gallium ions from the milling process and residual organo-platinum complex from the gas injection system (GIS) deposited before milling to protect the leading edge of the lamella. Another indication that the reflection is not due simply to the amorphous ice–vacuum interface is seen in the fringe contrasts observed. Using the indices of refraction of amorphous ice and vacuum in Eq. 4, one would expect a consistent reflection coefficient of ~ 0.013. This coefficient describes the intensity, or the field squared. Therefore, the fringe contrast, which scales linearly with the field, would be ~ 11%. Because we observe contrasts varying from sample to sample and being as high as 34%, it becomes more likely that reflections are dominated by a damaged contamination layer rather than an amorphous ice to vacuum interface. Given this understanding, we anticipate the same interferences will be present in the new plasma-based FIB systems, but their amplitude and phase may differ from those observed here.
Guidance of FIB milling
Because the amplitude of the reflected wave depends on the damage layer, it varies from sample to sample as a function of milling parameters such as voltage. Therefore, to guide the milling process with this effect, we first mill through a sacrificial target of interest to record the amplitude of the interferogram. Then, using the recovered amplitude and the same milling parameters, subsequent targets could be milled with the oscillations fit to Eq. 1, and the appropriate trough at which to halt milling could be determined in real-time before milling the target of interest. This information is relayed to the user by a custom Python-based GUI running on the acquisition computer, Supplementary Fig. S4.
Capturing virions with sub-diffraction fluorescent signal
With these new capabilities, we set out to target rare and sub-diffraction-limited structures in cellular samples. As a challenging target, we turned to adeno-associated virus (AAV) infecting human cells30. AAV is a parvovirus with icosahedral capsids 26 nm in diameter that is most notable for its use in human gene therapy31. Like all parvoviruses, it is internalized by the human cell in endosomes but eventually escapes membrane-bound organelles in a process that (for AAV) involves the phospholipase activity of the sometimes sequestered N-terminus of capsid protein VP132. Researchers have postulated that Cryo-ET-based observation of AAV as it moves through, and out of, the endomembrane system will unlock mechanistic questions that cannot be reliably recapitulated in vitro33. Unfortunately, in contrast to the process of viral assembly, wherein virions are abundant in the infected cell and optical targeting is not typically required34, viral trafficking and genome delivery processes necessitate precise ion beam targeting because the small virion in the large mammalian cell constitutes a “needle in a haystack.”
As we had done for fluorescent beads, we first performed top-down milling through several fluorescently labeled AAV-transduced samples (see “Methods” and Supplementary Fig. S5) to determine the amplitude of oscillations before the virions were ablated. Then, using the measured fringe contrast of ~ 20% and the real-time Python GUI, we directed milling to virion clusters in the main cell body. Fluorescence collected post-milling verified the presence of the target in the final thin lamella and could be registered with the low magnification montage of the lamella to direct tomographic data collection. Tomography of optically-targeted lamellae in HeLa cells at 8 h post-infection (h.p.i.) revealed clusters of closely packed viruses, seen predominantly in membranous vesicles; some of these vesicles had a dozen or more concentric bilayers, indicating a lysosomal subtype. Figure 4 and Supplementary Fig. S6 show representative examples of interferometrically guided FIB milling. Lamella generation using the interferometric approach took approximately 40 min each. Of the twelve attempts performed here, ~ 80% successfully captured virions in the final lamella.

a Integrated PSF brightness of target fluorescent punctum while milling. The sample was excited at 625 ± 10 nm and fluorescence was collected at 698 ± 45 nm. The oscillations are fit to Eq. 1 in real-time, and the software predicts the stop point for milling. The green dashed line indicates when milling was stopped, and the black dashed line is the center of mass of the virion cluster determined by annotated tomography reconstructions. b Montage EM showing entire lamella with the corresponding fluorescence post-milling overlaid. The region where tomography was measured is shown with the boxed ROI in white. c High magnification overlay of the final fluorescent image and a slice from the tomographic reconstruction. d Annotation of virions and encapsulating membrane from the tomogram in (c). e Histogram of virions in (d) as a function of distance from the top of the lamella with an average distance of ~ 80 nm.
Discussion
Here, we have demonstrated the ability to use fluorescence-based interferometric effects to guide Cryo-FIB milling of biological samples far beyond the axial diffraction limit. The interferometric approach requires no external fiducials and minimal registration of fluorescence and FIB images. Instead, it uses real-time fluorescence monitoring of a structure of interest to direct the milling process beyond the diffraction limit of light.
To use this approach, two criteria must be met. First, the fluorescent object of interest must be a distance greater than half the excitation wavelength from the top surface of the lamella. Second, the object must be distinguishable using widefield fluorescence microscopy. The first criterion is straightforward: the object must be far enough from the surface so that top-down milling can estimate the amplitude of oscillation. Without this, it is impossible to determine when to stop milling. The second criterion is difficult to quantify in terms of brightness and the copy number of the fluorescent label. In widefield microscopy, especially of thick samples like a mammalian cell, the ability to distinguish a fluorescent punctum from background will depend on both the levels of background from autofluorescence and out-of-focus fluorescence, as well as the brightness of the labeled structure. With low enough backgrounds, single molecules can be observed, and under cryogenic conditions, have been demonstrated to be photostable for tens of minutes, potentially long enough to direct the production of a lamella35. While this background is difficult to achieve, it is not inconceivable for certain samples in certain wavelength regimes. As a guiding principle, if the object can be seen in a stand-alone cryogenic light microscope in widefield, then this approach should be able to direct lamella production to preserve the structure for subsequent Cryo-ET. Based on the requirements, it is reasonable to deduce that any marker with suitable brightness may be used with the technique. To name a few common examples, dyes, quantum dots, or genetically encoded fluorescent proteins are all suitable candidates. This approach opens new possibilities to observe small and rare structures previously out of reach for Cryo-FIB milling and Cryo-ET approaches. While it is outside of the scope of this paper, future work will investigate how milling parameters—such as accelerating voltage and beam current—influence the observed interferometric effect36. These settings are expected to affect the abundance and depth of ion implantation and GIS redeposition37,38, which in turn will alter the Fresnel reflection and potentially damage the fluorescent target within the sample. In addition, the use of alternative ion species, such as those available in plasma FIB systems, represents a promising direction for further exploration within the tri-coincident FIB milling framework.
Method
Sample preparation
Spray frozen fluorescent bead samples
A gas dynamic virtual nozzle (GDVN) previously described in Yoniles et al.20 was used to aerosolize a 0.1% solids aqueous sample of 200 nm yellow-green (505/515 nm) polystyrene beads (FluoSpheresTM F8811). Briefly, the GDVN was placed ~7 cm away from– and perpendicular to– the grid’s path of plunging, allowing for the aerosolized sample to be sprayed onto the grid before freezing. A total flow rate of 0.14 ml/min and intentionally non-glow discharged copper Quantifoil grids (EMS Q2100CR2) were used to produce 10 – 20 µm droplets with high contact angles. The same was done to aerosolize the large Stokes-shift 0.002% solids aqueous sample of 40 nm yellow-red (488/645 nm) Streptavidin-Labeled Microspheres (TransFluoSpheresTM T10711), but at a lower percentage of solids to keep the density of fluorescent objects nearly constant between samples.
AAV-infected host cells
AAV2 non-replicating virions containing the GFP gene in lieu of rep/cap30 were labeled in 1 × borate buffer (1 M, pH 8.5) in a 15:1 dye to protein ratio with either Alexa Fluor 488 (ThermoFisher) or Janelia Fluor 669 (Tocris) covalently linked NHS ester dye. Stochastic lysine labeling of parvoviruses has minimal impact on infectivity at low multiplicities of labeling but affects the virus as the number of dyes per capsid increases. Preliminary room-temperature confocal microscopy, using Alexa Fluor 488 NHS ester labeled non-replicating AAV2 virions, indicated that the brightness of the dye and wavelength difference from the cellular autofluorescence peak were necessary to detect single virions inside cells. The high autofluorescence detected in the ~ 500 nm emission spectrum warranted the switch to the fluorescent dye in the far-red spectrum for the cryogenic fluorescent experiments.
The labeled virions were re-purified on a sucrose gradient in 1 × TNTM pH 8 (50 mM Tris pH 8, 100 mM NaCl, 0.2% Triton X-100, 2 mM MgCl2) and dialyzed into 1 × PBS. For confocal microscopy, 0.3 × 106 HeLa cells (ATCC via the lab of Prof. Paul Copeland) were seeded on a glass coverslip and incubated overnight. The culture was pre-chilled for 20 min at 4 °C prior to infection with fluorescently labeled AAV2 at the MOI of 104. Following 30 min of incubation at 4 °C to allow the virus particles to attach without being endocytosed, the culture was placed back into the incubator for 20 h at 37 °C with 5% CO2. Infected cells were stained with Hoechst stain in 1:2000 concentration and fixed in 2% paraformaldehyde for 30 min. The coverslips were mounted and imaged by a Leica SP8 confocal microscope (Leica Microsystems), Supplementary Fig. S5. For cryogenic imaging, glow-discharged Quantifoil gold grids with a holey SiO2 support film were disinfected in 70% isopropanol. Following their placement into a 35 mm culturing dish and three washes in 1 × PBS, 0.5 × 106 HeLa or HEK293A (Life Technologies cat. no. R70507) cells were seeded onto the disinfected grids and incubated overnight. The cells were pre-chilled and infected with JF-669 labeled AAV2 at the MOI of 105 and incubated as above. Following Hoechst staining, the grids were back-blotted and plunge frozen by a Leica EM GP plunge freezer in liquid ethane.
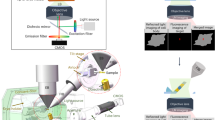
Tri-coincident FIB milling system
The Cryo-FIB milling system used for all studies was the ENZEL system, initially developed by Delmic, installed on an Aquilos 2 FIB-SEM system from Thermo Fisher Scientific. Within the ENZEL system, the stage remains in the same imaging plane while maintaining the ability to simultaneously take FIB, SEM, and light microscopy data. A recreated cartoon depiction of this is seen in Supplementary Fig. S1a along with an image of the inside of the ENZEL, Supplementary Fig. S1b. The FIB is equipped with a gallium ion source. In addition, a gas injection system (GIS) of organo-platinum was layered on all samples prior to milling. More information on the optics employed can be found in a previous publication17.
Software interface with ENZEL system
The ENZEL system from Delmic has a tri-coincident geometry, allowing for simultaneous fluorescence and FIB to be performed. To integrate these operations, a Python-based GUI was developed to interface with existing Delmic software. A sample view can be seen in Supplementary Fig. S4. Common controls such as commands to the camera, recording images, LED and filter control, and objective stage movement are integrated into the software. Newly added is the ability to define regions of interest (ROI), monitor the average fluorescence, and—if desired—fit the PSF to a 2D Gaussian and integrate and record that fitting. Initial parameters are adjusted to optimize fitting routines, and images are continuously saved. As milling progresses, the software will actively fit intensity traces to the model function and extrapolate to when a desired contrast and phase will be reached. Once that condition is reached, the software indicates it is time to stop milling to capture the target. The milling is then stopped manually by simultaneously operating the ion beam using the xT UI interface (ThermoFisher Scientific). The lamella is then cleaned to reach an optimal thickness of ~ 200 nm by bottom-up milling.
Cryogenic electron tomography
Tomograms were collected using a 300 keV transmission electron microscope (G2 Titan KriosTM) with images recorded using a direct electron detection camera Falcon4i with a Selectris Imaging filter (Thermo Fisher Scientific) and a 10 eV slit width. Images were taken with an exposure time of 2.5 s corresponding to 3.5e-/Å2 dose per projection and a pixel size of 2.5 Å. The integrated images were saved in the mrc format, and individual frames were saved in the eer format39. The tilt series were taken with a dose-symmetric scheme40, starting at a − 10 degrees to compensate for the physical pre-tilt of the lamella, imaging with a range of −60 to 40 degrees, incremented at 2.5-degree steps. The resulting total dose was approximately 144 e-/Å2. Reconstruction was done using IMOD, and the resulting image binned by 4, reducing pixel size to 9.8 Å.
The EER frames were converted to a tif format (Relion command - convert to tif)41, corrected for local and global motion (MotionCor2)42 using an option to split even and odd frames that were subsequently used for denoising (CryoCare)43. The aligned tilt series were reconstructed to a volume (IMOD)44, denoised with CryoCare, and annotated using Dragonfly.
Image registration
Fluorescence microscopy was registered to the Cryo-ET data using a series of affine transformations following methods described previously21,45,46. First, the optical images were registered to a low magnification EM image using any features identifiable in both imaging modalities, such as ice or defects in the lamella. These control point pairs were used to compute a projective transformation carrying the optical data into the low-magnification EM space. This low-magnification EM space was then registered with the Cryo-ET space by identifying unique features, such as subcellular structures and ice contamination, visible in both the low-magnification image and the z-projection of the Cryo-ET reconstructions. From these control point pairs, a similar transformation was computed to carry the low-magnification EM micrograph to the Cryo-ET space. Application of the projective and similar transformations sequentially to the optical data carries the fluorescence micrographs into the Cryo-ET space.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are included in the manuscript and its supplementary materials, including the raw fluorescence micrographs from top-down milling of 200 nm diameter beads (505/515 nm Ex/Em) available through the Stanford Digital Repository at https://purl.stanford.edu/qk257rq6365 and tomographic reconstructions and fluorescence micrographs for AAV infected cells are available on the Electron Microscopy Data Bank (accession no. EMD-47204). Additional data are available from the corresponding author upon request.
Code availability
The code used to monitor fluorescence while milling and fit the interferogram can be found here: https://github.com/pdahlb/trico_softwaresuite. This software used is used in conjunction with the existing Odemis software developed by Delmic, available at https://github.com/delmic/odemis.
References
Asano, S., Engel, B. D. & Baumeister, W. In situ cryo-electron tomography: a post-reductionist approach to structural biology. J. Mol. Biol. 428, 332–343 (2016).
Quemin, E. R. J. et al. Cellular electron cryo-tomography to study virus-host interactions. Annu. Rev. Virol. 7, 239–262 (2020).
Oikonomou, C. M. & Jensen, G. J. The atlas of bacterial & archaeal cell structure: an interactive open-access microbiology textbook. J. Microbiol. Biol. Educ. 22, https://doi.org/10.1128/jmbe.00128-21 (2021).
Guaita, M., Watters, S. C. & Loerch, S. Recent advances and current trends in cryo-electron microscopy. Curr. Opin. Struct. Biol. 77, 102484 (2022).
Wagner, F. R. et al. Preparing samples from whole cells using focused-ion-beam milling for cryo-electron tomography. Nat. Protoc. 15, 2041–2070 (2020).
Gorelick, S. et al. PIE-scope, integrated cryo-correlative light and FIB/SEM microscopy. ELife 8, e45919 (2019).
Bieber, A., Capitanio, C., Wilfling, F., Plitzko, J. & Erdmann, P. S. Sample preparation by 3D-correlative focused ion beam milling for high-resolution cryo-electron tomography. J. Vis. Exp. https://doi.org/10.3791/62886 (2021).
Allegretti, M. et al. In-cell architecture of the nuclear pore and snapshots of its turnover. Nature 586, 796–800 (2020).
Klumpe, S. et al. A modular platform for streamlining automated cryo-FIB workflows. eLife 10, e70506 (2021).
Wu, G.-H. et al. Multi-scale 3D cryo-correlative microscopy for vitrified cells. Structure 28, 1231–1237 (2020).
Spehner, D. et al. Cryo-FIB-SEM as a promising tool for localizing proteins in 3D. J. Struct. Biol. 211, 107528 (2020).
Arnold, J. et al. Site-specific cryo-focused ion beam sample preparation guided by 3D correlative microscopy. Biophys. J. 110, 860–869 (2016).
DeRosier, D. J. Where in the cell is my protein?. Q. Rev. Biophys. 54, e9 (2021).
Petrov, P. N. & Moerner, W. E. Addressing systematic errors in axial distance measurements in single-emitter localization microscopy. Opt. Express 28, 18616–18632 (2020).
Loginov, S. V., Boltje, D. B., Hensgens, M. N. F., Hoogenboom, J. P. & Wee, E. B. van der. Depth-dependent scaling of axial distances in light microscopy. Optica 11, 553–568 (2024).
Li, S. et al. HOPE-SIM, a cryo-structured illumination fluorescence microscopy system for accurately targeted cryo-electron tomography. Commun. Biol. 6, 1–12 (2023).
Boltje, D. B. et al. A cryogenic, coincident fluorescence, electron, and ion beam microscope. ELife 11, e82891 (2022).
Wang, J. et al. Human NLRP3 inflammasome activation leads to formation of condensate at the microtubule organizing center. Preprint at https://doi.org/10.1101/2024.09.12.612739 (2024).
Li, S. et al. ELI trifocal microscope: a precise system to prepare target cryo-lamellae for in situ cryo-ET study. Nat. Methods 20, 276–283 (2023).
Yoniles, J. et al. Time-resolved cryogenic electron tomography for the study of transient cellular processes. Mol. Biol. Cell 35, mr4 (2024).
Dahlberg, P. D., Perez, D., Hecksel, C. W., Chiu, W. & Moerner, W. E. Metallic support films reduce optical heating in cryogenic correlative light and electron tomography. J. Struct. Biol. 214, 107901 (2022).
Kofman, V., He, J., Kate, I. L. ten & Linnartz, H. The refractive index of amorphous and crystalline water ice in the UV–vis. Astrophys. J. 875, 131 (2019).
Crew, H., Huygens, C., Young, T., Fresnel, A. J. & Arago, F. (François). The Wave Theory of Light Memoirs of Huygens, Young and Fresnel. (New York, Cincinnati American Book Company, 1900).
Loudon, R. The Quantum Theory of Light. (OUP Oxford, 2000).
Born, M., Wolf, E. & Bhatia, A. B. Principles of Optics: Electromagnetic Theory of Propagation, Interference and Diffraction of Light. (Cambridge University Press, 1999).
Hecht, E. Optics. (Pearson Education, Incorporated, 2017).
Sick, B., Hecht, B. & Novotny, L. Orientational imaging of single molecules by annular illumination. Phys. Rev. Lett. 85, 4482–4485 (2000).
Sica, A. V. et al. Spectrally selective time-resolved emission through fourier-filtering (STEF). J. Phys. Chem. Lett. 14, 552–558 (2023).
Mandel, L. & Wolf, E. Optical Coherence and Quantum Optics. Cambridge University Press, Cambridge, (1995).
Grimm, J. B. et al. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat. Methods 12, 244–250 (2015).
Yost, S. A. et al. Characterization and biodistribution of under-employed gene therapy vector AAV7. J. Virol. 97, e0116323 (2023).
Harbison, C. E., Chiorini, J. A. & Parrish, C. R. The parvovirus capsid odyssey: from the cell surface to the nucleus. Trends Microbiol. 16, 208–214 (2008).
Stagg, S. M., Yoshioka, C., Davulcu, O. & Chapman, M. S. Cryo-electron microscopy of adeno-associated virus. Chem. Rev. 122, 14018–14054 (2022).
Shah, P. N. M. et al. Characterization of the rotavirus assembly pathway in situ using cryoelectron tomography. Cell Host Microbe 31, 604–615.e4 (2023).
Dahlberg, P. D. et al. Identification of PAmKate as a red photoactivatable fluorescent protein for cryogenic super-resolution imaging. J. Am. Chem. Soc. 140, 12310–12313 (2018).
Yang, Q. et al. The reduction of FIB damage on cryo-lamella by lowering energy of ion beam revealed by a quantitative analysis. Structure 31, 1275–1281.e4 (2023).
Giannuzzi, L. A. Introduction to Focused Ion Beams: Instrumentation, Theory, Techniques and Practice, Springer, Boston, (2005).
Yang, Z., Kim, J., Zhang, G., Aronova, M. A. & Leapman, R. D. Biological volume EM with focused Ga ion beam depends on formation of radiation-resistant Ga-rich layer at block face. Preprint at https://doi.org/10.1101/2024.09.16.613321 (2024).
Guo, H. et al. Electron-event representation data enable efficient cryoEM file storage with full preservation of spatial and temporal resolution. IUCrJ 7, 860–869 (2020).
Hagen, W. J. H., Wan, W. & Briggs, J. A. G. Implementation of a cryo-electron tomography tilt-scheme optimized for high resolution subtomogram averaging. J. Struct. Biol. 197, 191–198 (2017).
Scheres, S. H. W. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Buchholz, T.-O. et al. Content-aware image restoration for electron microscopy. Methods Cell Biol. 152, 277–289 (2019).
Kremer, J. R., Mastronarde, D. N. & McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116, 71–76 (1996).
Dahlberg, P. D. et al. Cryogenic single-molecule fluorescence annotations for electron tomography reveal in situ organization of key proteins in Caulobacter. Proc. Natl. Acad. Sci. USA 117, 13937–13944 (2020).
Perez, D. et al. Exploring transient states of PAmKate to enable improved cryogenic single-molecule imaging. J. Am. Chem. Soc. 146, 28707–28716 (2024).
Acknowledgements
The authors would like to thank Chensong Zhang and Lydia-Marie Joubert from the SCSC NIH tomography center and members of Delmic for support throughout the project, Jessica Shivas for technical support, Ash Sueh Hua for clarifying edits, and Maud Dumoux for helpful discussions. Funding was provided in part by REGENXBIO Inc. (to J.T.K.) and NIGMS grant no. R21GM140345 (J.T.K.), NIAID grant no. RO1AI127401 (G.J.J.), Department of Energy, Office of Science, Office of Biological and Environmental Research, under Contract No. DE-AC02-76SF00515 FWP 100883 (PDD), and grant 2021-234593 from the Chan Zuckerberg Initiative DAF, an advised fund of Silicon Valley Community Foundation.
Author information
Authors and Affiliations
Contributions
A.V.S. and P.D.D. identified and interpreted the interferometric effect. G.J.J., J.T.K., and P.D.D. conceptualized and supervised the research. A.V.S. with support from D.B.B. wrote the real-time fluorescence monitoring software into the current ENZEL system. A.V.S. and M.Z. carried out all FIB milling and collection of Cryo-ET data. C.A. provided synthetic test samples. J.J.P., L.M.M., and J.T.K. provided the virion samples along with preliminary work and helpful discussions. P.D.D. and A.V.S. wrote the manuscript, and all authors reviewed the text.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Fei Sun and the other anonymous reviewer(s) for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sica, A.V., Zaoralová, M., Antolini, C. et al. Optical interference for the guidance of cryogenic focused ion beam milling beyond the axial diffraction limit. Nat Commun 17, 481 (2026). https://doi.org/10.1038/s41467-025-65548-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65548-8
