
Abstract
Light-driven methanol synthesis from CO2 provides a sustainable fuel source and approach to carbon neutralization. Mimicking natural photosynthesis could improve gas-solid photocatalytic efficiency, but it remains highly challenging due to the absence of well-organized mass and charge transfer networks in artificial materials. Herein, we report a chlorophyll-mimicking, nano-pigment nickel gallium oxide, which facilitates discrete light/dark reactions and proton-mediated charge transfer for efficient photocatalytic hydrogenation of CO2 to methanol. This nano-pigment features surface frustrated Lewis pairs, enabling heterolytic hydrogen splitting into H- and H+. The H- acts analogously to nicotinamide adenine dinucleotide phosphate in natural photosynthesis, with Ni(II)/Ni(III) and OH(-I) respectively serving as conduits for ion transport of H- and H+ to the Ni site, where they subsequently react with CO2, mimicking natural carbon fixation. This approach establishes a chlorophyll-mimetic structure for photocatalytic stepwise CO2 hydrogenation, achieving 3.0% quantum efficiency, 3.20 mmol·h-1·g-1 methanol activity, and 79.6% selectivity.
Similar content being viewed by others
Introduction
Photocatalytic hydrogenation of CO2 into methanol offers a promising approach to address today’s energy and environmental issues and promote the global circular carbon-neutral energy system1,2,3,4. However, achieving high reaction activity and quantum efficiency remains challenging due to the inefficient separation, migration, and transport of photogenerated electrons and holes5,6,7. Additionally, limited light absorption and poor active site selectivity further hinder overall performance8,9,10. Thus, optimizing the spatial distribution of active sites and enhancing charge separation through advanced interface and material design are essential for improving photocatalytic efficiency.
Mimicking natural systems offers a potential strategy for enhancing quantum yield by spatially separating oxidation and reduction sites, similar to the division of light and dark reactions in plants11,12. Green leaves from plants are nature’s opto-chemical photosynthetic machines. Their hierarchical architectures are engineered to capture and transport incident sunlight to the chloroplasts, within which chlorophyll pigments photochemically transform carbon dioxide and water into carbohydrate energy vectors13,14. During sunlight exposure, chloroplasts split water to produce equivalents of [H] and oxygen [O2]15,16,17, which are subsequently used to produce reduced nicotinamide adenine dinucleotide phosphate (NADPH) and adenosine triphosphate (ATP) through sequential electron (e−) and proton (H+) transfers. Under dark, CO2 can be stepwise converted into carbohydrates with the aid of NADPH, H+, and ATP. Inspired by the leaf, artificial photosynthetic analogues have been proposed as a feasible approach to convert solar energy into storable chemical energy18,19. However, replicating the complex electron transport chain of natural systems, which facilitates stepwise electron transfer and energy conversion, is significantly challenging in gas-solid photocatalytic systems due to the lack of well-organized mass and charge transfer networks in artificial materials.
Surface frustrated Lewis pairs (SFLPs), composed of spatially adjacent yet sterically hindered Lewis acid and base sites, have emerged as highly effective active centers for activating small molecules including CO2, H2, alkenes, dienes, and alkynes. Typically, these bifunctional sites are engineered by creating isolated oxygen vacancies (Ov) or by designing Ov-OH configurations on metal oxide surfaces20. A prominent example is the In2O3-x(OH)y catalyst, where unsaturated In³+ cations (Lewis acid, generated by Ov) cooperatively interact with proximal In-OH groups (Lewis base) to enable efficient CO2 hydrogenation to methanol as validated by both theoretical and experimental studies21,22. However, the lower methanol production rate and lower quantum yield endow the challenge to find more efficient SFLP-based catalyst for photocatalytic CO2-to-methanol production. Thus, to find an alternative that circumvents this problem yet retains the attributes of defect engineered indium oxide, we turn to gallium oxide as it has been extensively studied as a promoter or support for methanol synthesis though it suffers from the poor light absorption due to its wide band gap.
Combining the natural process and the important role of OH and Ov during CO2 hydrogenation, we have developed a chlorophyll-mimicking green nano-pigment, nickel gallium oxide (NiGa2O4-x(OH)y), which consists of Ov and hydroxyl groups (OH) and mixed-valence Ni(II,III) sites within an inverse spinel structure (space group Fd3m; inversion parameter of 92%)23,24. Notably, edge-sharing Ni(II,III) and Ga(III) octahedra endow the material with a high electrical conductivity favorable for electron transfer and ion diffusion25,26. Notably, Ga(III) and adjacent Ni(II)/Ni(III) sites function as Lewis acid sites, while the O(-II) serve as Lewis base sites for the formation of surface frustrated Lewis pairs (SFLPs). In this situation, the nano-pigment NiGa2O4-x(OH)y not only mimics the optical response of chlorophyll, but also emulates the sequential dark and light reactions in natural photosynthesis (Fig. 1a,b). Specifically, light interacts with the SFLPs sites to promote heterolytic splitting of hydrogen, forming hydride (H-) and proton (H+), where H- acts as a redox mediator similar to the role of NADPH. Ni(II)/Ni(III) and OH(-I) respectively serve as conduits for the ion-transport of H- and H+ to the Ni site, where they react with CO2, mimicking the dark reaction in nature photosynthesis. Consequently, the equivalent transfer of electrons (e-) and protons (H+) can occur within such a chlorophyll-mimicking structure, promoting the conversion of CO2 to methanol. This nano-pigment achieved an impressive methanol production rate of 3.20 mmol·h-1·g-1 and a methanol selectivity of 79.6% with the assistance of light. Additionally, a quantum efficiency of 3.0% was achieved during wavelength-dependent studies, with 92.3% photons contributing to methanol synthesis. These findings demonstrate significant progress in photocatalytic hydrogenation and provide a nature-inspired strategy for designing highly efficient photocatalysts. Ultimately, these insights form a solid foundation for the development of solar-driven refineries, paving the way for scalable and sustainable methanol synthesis.

a Graphical representation of a plant leaf and NG. b Absorbance spectra of Chlorophylls (Chl a, Chl b, Chl d, Chl f) and NG. c PXRD patterns of NG and NG-300. d HRTEM images of NG (left) and NG-300 (right). e XPS spectra of O 1 s for NG and NG-300. f Measured water contact angles of NG and NG-300. g XPS spectra of Ni 2p for NG and NG-300. h, i Ni L-edge and O K-edge EELS spectra of NG and NG-300, respectively. j Proposed reconstruction mechanism by deprotonation of NG (up) to NG-300 (down). Color code: blue, Ga; green, Ni; red, O; grey, H.
Results
Design scheme and crystal structure characterization
The nano-pigment NiGa2O4-x(OH)y samples containing abundant oxygen vacancies (Ov) and hydroxyl groups (OOH) were prepared using the hydrothermal method. To determine the effect of surface OOH groups on the SFLP site, samples of NiGa2O4-x(OH)y calcined at 300 °C and not calcined were compared. The calcinated and uncalcined samples were denoted as NG-300 and NG, respectively. As displayed in Fig. 1c, the diffraction peaks of the as-prepared NG and NG-300 are identical and are consistent with those from standard cubic inverse spinel structure (Fd-3m, PDF no. 78-0546)27. High-resolution transmission electron microscopy (HRTEM) image also displays the same inverse spinel (311) planes over NG and NG-300, with a lattice parameter of 0.249 nm (Fig. 1d)24,27. Transmission electron microscopy (TEM) images (Supplementary Fig. 1a,b) revealed that NG has a particle size of ~9 nm, while NG-300 exhibits particles with an average diameter of 11 nm, resulting in a slight decrease in specific surface area for the latter (94.2 m2 g-1 versus 83.0 m2 g-1, Supplementary Fig. 2 and Table 1). The uniform distributions of Ni, Ga, and O in NG were confirmed from high angle annular dark-field (HAADF) imaging combined with energy dispersive X-ray (EDX) element mapping (Supplementary Fig. 3). The atomic ratio of Ni with Ga was measured to be 0.47 from inductively coupled plasma optical emission spectrometry (ICP-OES) (Supplementary Table 2), which is close to the theoretical value of 0.5 in the inverse spinel structure.
A series of characterizations were used to identify the oxygen defects and surface hydroxyls. Xray photoelectron spectroscopy (XPS) analysis on NG revealed a wide range of oxygen species conducive to SFLP-enabled hydrogenation. The relative percentages of lattice oxygens (Olatt), Ov, and OOH were 42.4%, 23.3% and 34.4% (Fig. 1e), respectively24. NG-300 was found to have lower OOH (20.5%), more Ov (27.5%), and more Olatt (52.0%) than that of NG (Fig. 1d and Supplementary Table 3). The decreased number of surface hydroxyls were further confirmed by the greater wettability of NG compared to NG-300 (Fig. 1f), more positive Zeta-potential (Supplementary Fig. 4)28, and higher FTIR peak intensity at 3420 cm-1 (OOH) (Supplementary Fig. 5). After calcination, NG-300 showed a slightly lower percentage of Ni(III), which consequently affects Ga oxidation state in the Ga-O-Ni sites, as shown in Fig. 1g and Supplementary Fig. 6. However, the electron energy loss spectroscopy (EELS) analysis (Fig. 1h,i) indicated a higher Ni oxidation state accompanied by more Ov following calcination. The valence states of Ni were determined by calculating its white-line ratio (L3/L2) (See Methods and Supplementary Table 4). The discrepancy between the XPS and STEM-EELS results may arise from variations in sampling depth between the two techniques and changes in Ni valence between the surface and subsurface layers29. Deprotonation likely releases H2O via coupling between the OH group of a bridging hydroxyl linked to Ni-Ga and the H atom of an adjacent terminal OH on Ni or Ga site, thereby generating additional Ov and exposing more Olatt, as supported by the Raman results (Supplementary Fig. 7)30,31. Consequently, different Ni valences were observed at the surface and in the subsurface. Based on the above analysis, the changes resulting from the reconstruction of NG to NG-300 are proposed as shown in Fig. 1j. Changes in the Lewis acidic and basic sites between NG and NG-300 were characterized by pyridine-IR spectroscopy and CO2-TPD, as shown in Supplementary Fig. 8.
The collected results demonstrate the formation of oxygen vacancies, hydroxide, and Ga(III) accompanied by redox-active Ni(II,III) sites in NG, providing rational and systematic control over the physicochemical properties of SFLPs. Light excitation of the SFLP system can tune the Lewis acidity and Lewis basicity of structures compared to the ground state32,33. The photogenerated electron can rapidly converse the Ni in the octahedral sites between Ni(II) and Ni(III) in the octahedral sites, which endows NG with high electrical conductivity that is favored for electron transfer, boosts the population of Ga-H under light, and promotes subsequent CO2 hydrogenation34.
Light-driven CO2 hydrogenation performance
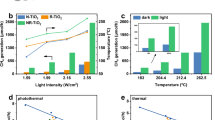
The catalytic performance of nano-pigment NG for CO2 hydrogenation, with or without light irradiation, was investigated at different temperatures in a Harrick Cell reactor under 0.7 MPa pressure. Under irradiation, CO and CH3OH production rates increased with temperature from 260 °C to 300 °C, while CH3OH selectivity linearly decreased with increasing temperature for all tested conditions (Fig. 2a and Supplementary Table 5). CO and CH3OH production rates at 300 °C were 0.29 and 0.77 mmol·h-1·g-1, respectively, corresponding to a CH3OH selectivity of 72.8%. When the catalyst was irradiated with white LED light, significant increases in CO and CH3OH production were observed across the investigated temperature range. Remarkably, an high CH3OH production rate of 2.95 mmol·h-1·g-1 and a selectivity of 78.3% were achieved at 300 °C (Fig. 2a), demonstrating the advantage of light in methanol production. The performance of photocatalytic CO2 hydrogenation to methanol over NG was excellent compared to the prior state-of-the-art, both in thermal and photocatalytic reactivity at low pressure (Supplementary Table 6). The stability of NG was studied at 300 °C (Fig. 2b) for 150 h, during which the CH3OH production rate continuously increased over the first 60 h, reaching a maximum of 3.20 mmol·g-1·h-1 with the selectivity of 79.6%, and then decreased thereafter.

a CO and CH3OH production rates of as a function of reaction temperature, with and without light (white LED light, catalyst amount: 15 mg, light intensity: 1.87 W cm-2, pressure: 0.7 MPa). b Stability test for NG under light off and on conditions. c–f Different LED light irradiation intensities (main peak at 365 nm, 460 nm, 525 nm, and 625 nm; catalyst amount: 7.88 mg) for CO and CH3OH production and selectivity of NG. g Total external quantum yield (EQE) of NG for CO2 hydrogenation with different levels of light irradiation. h Absorption spectrum (solid line) and EQE results obtained under illumination at 0.83 and 1.26 W cm-2. Mean values were derived from quadruplicate measurements and their standard deviation shown as error bars.
We speculate that the spinel structure of NG plays a vital role in its performance, which was verified by comparing the activities of NG with control samples including Ga2O3, a mixture of NiO/Ga2O3 (Ni:Ga molar ratio of 1:2), and reduced NiGa2/SiO2 (see Supplementary Notes 1 for synthesis). From Supplementary Fig. 9, all control samples showed low methanol production and selectivity compared to the spinel structure of NG, confirming the latter’s key role in CO2 hydrogenation to methanol. This could be attributed to the edge-sharing nature between Ni and Ga sites in the crystal lattice, endowing the system with high electrical conductivity and favorable electron transfer dynamics35. Furthermore, to corroborate the efficacy of surface hydroxyls and multiple Ni oxidation states in catalysis, the activities of NG-300, NG-N2 (treated in N2), NG-H2 (treated in H2), and NG-H2O (treated in H2O) were also studied (Supplementary Figs. 9,10). All CO and methanol production rates were substantially lower than that of NG, highlighting the importance of having Ov, OH, and/or multiple Ni oxidation states simultaneously during methanol synthesis. NG-N2 showed similar activity to NG-300, while NG-H2 and NG-H2O displayed comparable levels activity. This difference in activity can be attributed to the surface structural changes caused by different treatments. H2 and H2O atmospheres can partially retain OH. In contrast, N2 or air atmospheres may lead to the removal of OH, thereby reducing the density or effectiveness of SFLP sites and leading to lower methanol production. These findings support the hypothesis that OH acts as proton (H+) transport conduit, facilitating the hydrogenation of CO2 to methanol. Further, to investigate the synergistic effect of SFLP sites on CO2 hydrogenation activity, quenching experiments were conducted using pyridine and pyrrole to selectively block the Lewis acid and base sites, respectively. The results presented in Supplementary Fig. 11 demonstrate that deactivation of either the acidic or basic site leads to a significant decrease in the CO2 hydrogenation activity. Specifically, methanol production activity decreased to approximately 75% upon acidic site poisoning and dropped to near zero upon basic site quenching. These observations indicate that both sites are required to work cooperatively for effective CO2 hydrogenation to methanol, with basic sites playing a more critical role in this process.
To distinguish the photochemical versus photothermal roles of illumination, the following experiments were conducted. First, the surface temperatures were measured using an infrared (IR) camera under both dark and light conditions. As shown in Supplementary Fig. 12 and Table 7, the surface temperature can increase about ~20 °C under light, confirming the photothermal contributions. Yet the CH3OH production rates under light remain higher compared to those in the dark even when the temperature was increased by 30 °C for the latter condition (Fig. 2a, Supplementary Table 4). This suggests a prominent photochemical contribution to CH3OH production from the excited carriers, distinct from the photothermal effect. The activation energies (Ea) were calculated using the surface temperatures under both dark and light conditions, as shown in Supplementary Fig. 13. A much lower Ea was measured under light than in the dark, indicating the contribution of solar light excitation in reducing energy barriers compared to the purely thermal ground-state pathway. Normally, light has been shown to have the capacity to serve dual roles, both in photothermal and photochemical processes32. Differentiating the contribution of photothermal and photochemical effects is essential in the photocatalytic systems.
Irradiation wavelength and photon flux can influence photocatalytic activity and products36,37. Thus, four LED light sources (UV, blue, green, and red lights with center peaks at 365 nm/3.4 eV, 460 nm/2.7 eV, 525 nm/2.4 eV, and 625 nm/2.0 eV) with tunable intensities were used to systematically explore the photochemical role of light in CO2 hydrogenation at 300 °C. The spectrum of each light source is shown in Supplementary Fig. 14. From Fig. 2c–f, Supplementary Fig. 15 and Table 8, an obvious increase of CO and CH3OH production was observed with increasing light intensity in the range of 0.1 to 0.83 W cm-2 for all four light sources. We were pleasantly surprised to find that the selectivity of CH3OH is not affected by light intensity, indicating that CH3OH production is independent of variations in both light intensity and wavelength. In addition, the CH3OH production rate is in the order of UV > red > blue > green for NG, which aligns with NG’s absorption spectra, suggesting a direct correlation between enhanced photoactivity and the light absorption properties of NG. Above results further confirm the existence of a consistent charge transfer route with photo-induced carriers under each light irradiation condition.
The near linear correlation between photocatalytic activity of methanol and irradiation intensity provides compelling evidence that excited charge carriers actively participate in the photochemical reaction rather than solely undergoing relaxation to produce photothermal heating in CO2-to-methanol.
Next, the external quantum efficiency (EQE) of CO2 hydrogenation to form either CO or CH3OH was calculated using the rate difference between the dark and light reactions for different LED wavelengths (see Supplementary Note 3 for details). As shown in Fig. 2g and Supplementary Fig. 16, the EQE linearly increased with greater light intensity under UV and blue light but decreased with greater light intensity under green and red light. With an incident intensity of 0.83 W cm-2, measured EQEs are 2.7%, 1.6%, 1.1% and 1.4% under UV, blue, green, and red light, respectively. When these EQEs were separated into EQECO and EQEmethanol contributions, we found that the EQEmethanol value exhibited a similar trend to that of total EQE, whereas a decreasing EQECO was observed with increasing light intensity for all wavelengths used (Supplementary Fig. 17). By calculating the selectivity for EQEmethanol in EQE at the light intensity of 0.83 W cm-2, as shown in Supplementary Fig. 18, it was found that the EQEmethanol are 92.3%, 92.6%, 91.3%, and 92.6% at UV, blue, green and red LED lights, respectively. That is to say, a predominant fraction of the excited charge carriers actively engages in the production of methanol. Notably, the observed consistency between the optical absorption of NG and its EQE at different wavelengths originates from the absorption coefficient of NG, further suggesting that the CO2 hydrogenation reaction is driven by a photochemical process, as shown in Fig. 2h.
Conventionally, a positive correlation between EQE and incident photon flux is a distinct feature of light-induced, charge carrier-driven photocatalysis38. This is because increased carrier density at higher light intensities reduces recombination and enlarges the difference between electron and hole quasi-Fermi levels39. Furthermore, UV and blue light fall within NG’s interband absorption. Thus, increased CO and CH3OH productions with higher light intensity in this range further confirm the photochemical role of light in reactivity (Fig. 2h). Green light’s decreasing EQE with intensity can be attributed to NG’s lower absorption in the excitation wavelength centered at 525 nm. The pattern observed for red light, with an excitation wavelength centered at 625 nm and measured increase and subsequent decrease in EQE with light intensity, can be attributed to the saturation of NG’s long-wavelength photon utilization under higher intensity. Thus, the above results illustrate the important photochemical role of light for CO2-to-methanol production over NG.
Considering the linear increase of EQEs as the intensity increases under UV and blue light, we further increased the light intensity to 1.26 W cm-2 and found the highest EQEs are 3.0% and 1.9% for UV and blue light, respectively (Fig. 2h), which surpasses the majority of reported analogical photocatalytic CO2 hydrogenation to methanol using solar energy (Supplementary Table 6).
Identification of active sites
A series of spectroscopic characterizations were performed to probe the catalytic active site and its reaction pathway in the catalytic CO2 methanation process. The structure and composition of spent NG and NG-300 were first investigated with PXRD and HRTEM (Supplementary Figs. 19–21) and no significant changes were observed between the spent and fresh samples, indicative of the catalysts’ structural stability. These findings also necessitate in situ characterizations under reaction conditions. First, in situ X-ray photoelectron spectroscopy (in situ XPS) tests were conducted in the dark under Ar, H2, and CO2 + H2 reaction conditions to investigate the surface electronic properties specified in Fig. 3a–c. When moving from Ar to H2 atmosphere, an obvious decrease in the intensity of OH from 28.5% to 8.5% was observed, followed by an increase to 16.0% in a CO2 + H2 atmosphere. This behavior indicates that H2 undergoes heterolytic dissociation on the surface of NG and consumes surface OH. In the presence of CO2 this OH removal is partially inhibited. (Fig. 3a and Supplementary Table 9). Concurrently, the atomic contribution of Ni(II) peaks increased from 63.2% to 73.1% when switching from Ar to H2 and shifted to lower binding energies, and was partially restored under CO2/H2 gas compared to when under Ar (Fig. 3b and Supplementary Table 10). Meanwhile, the percentage of Ga-O-Ni changes from 36.7% (H2) to 39.0% (CO2 + H2), which is close to the 39.8% observed at RT under Ar. This small variation can be attributed to the CO2 reaction at Ga-O-Ni sites (Fig. 3c and Supplementary Table 11). These results above indicate that Ni contributed to H2 heterolytic splitting and the subsequent CO2 hydrogenation reaction along with Ga sites, which agrees with DFT results that both Ni and Ga donate electrons for H2 splitting (as further discussed in the following section).

In situ XPS spectra of O (a), Ni (b) and Ga (c) of NG obtained at 300 °C with different reaction conditions of Ar, H2 and CO2 + H2 (CO2/H2 = 1/3), respectively. d In situ high-resolution TEM images of NG nanoparticle. e, f In situ STEM-EELS spectrum of Ni-L edge and O-K edge. These tests were conducted at room temperature, 300 °C with H2 and then 300 °C with CO2 to form a CO2 + H2 atmosphere, respectively.
The structure and oxidation changes during the reaction were further explored by in situ scanning transmission electron microscopy with electron energy loss spectroscopy (STEM-EELS) in different gaseous environments, as shown in Fig. 3e, f. In H2 environment, the surface d-spacing of (311) plane expands to 2.59 Å compared to 2.49 Å at room temperature, and remains constant when CO2 is added to form a CO2 + H2 atmosphere. This lattice expansion can be attributed to Ni reduction from Ni(III) to Ni(II) and corresponding increased ionic radius40. Moreover, Ni L-edge and O K-edge EELS spectra confirmed the increase of Ni2+ and oxygen vacancies under H2 and the decrease under CO2, thereby further corroborating the formation of Ni-O bonding resulting from CO2 hydrogenation (Fig. 3e and Supplementary Table 12).
Elucidation of the role of light
The photo-thermal catalytic CO2 hydrogenation involves both photothermal and photochemical roles. Thus, we conducted a detailed investigation into their effects on the reaction. First, NG and NG-300 band structures were measured using the transformed Kubelka–Munk function from ultraviolet-visible (UV-vis) diffuse reflectance spectra (DRS) and UV photoelectron spectroscopy (UPS) (Supplementary Figs. 22,23). Conduction band (CB) and valence band (VB) edge potentials of NG and NG-300 were found to fulfill the thermodynamic requirements for converting CO2 to methanol41, as shown in Supplementary Fig. 24.
Subsequently, to investigate the hydrogen heterolytic splitting behaviors, we conducted the diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements under dark and light conditions in H2 with varying temperature, as shown in Fig. 4a and Supplementary Figs. 25,26. The prominent appearance of the peaks at 1825 cm-1 and 3682 cm-1 were attributed to Ga-H and Ga-OH2+/Ga-OH species42. Notably, under light conditions, the intensities of these two species increased, demonstrating that light illumination enhances heterolytic H2 splitting at SFLP sites. To identify the hydrogen species from heterolytic H2 splitting in H2-DRIFTS tests, D2-DRIFTS was also conducted (Fig. 4b). Absorptions at 3682 and 1825 cm-1 were shifted to 2602 and 1185 cm-1, which matched the expected (νH2/νD2)0.5 ratio43,44. Moreover, H2 splitting on NG was confirmed by solid-state 1H nuclear magnetic resonance (NMR) spectroscopy by detecting the formation of H-related species (Fig. 4c). Without H2, only one peak, with a chemical shift at −2.98 ppm, assigned to surface OH was observed45. After exposing the sample to H2, a peak with the chemical shift at 4.72 ppm was observed and assigned to Ga-H46,47. Furthermore, the peak at −2.98 ppm showed a positive shift and increased area, as shown in Supplementary Table 13. This can be attributed to the formation of more Ga-OH2+ or Ga-OH during H2 heterolytic splitting. The decrease in H2 desorption intensity under light compared to dark conditions in the H2 temperature-programmed desorption (H2-TPD) further confirmed light promotion of H2 splitting (Supplementary Fig. 27)48,49.

a In situ DRIFTS spectra for the activation of H2 on NG with and without light at 300 °C. b In situ DRIFTS spectra for the activation of D2 on NG with light. c Ex situ solid-state 1H MAS-NMR spectra of NG before and after light irradiation at room temperature and 300 °C. d In situ photocurrent of NG under H2, CO2, H2 + CO2 and vacuum. In situ XANES spectra of Ni L-edge (e) and Ga K-edge (f) over NG. In situ Fourier transform (FT) of EXAFS spectra of Ni L-edge (g) and Ga K-edge (h) over NG. i, j In situ DRIFTS spectra for the activation of CO2 + H2 (CO2/H2 = 1/3) on NG with or without light at 300 °C. k In situ DRIFTS spectra of NG with CO2 + D2 (CO2/D2 = 1:3) under light at 300 °C.
To study the dynamics of photoexcited electrons, in-situ photoconductivity measurements under vacuum, H2, CO2 and CO2 + H2 conditions were performed on NG, as shown in Fig. 4d. Accordingly, NG displayed photocurrent saturation and decay process lifetimes on the order of minutes. A slower increase in rate of photocurrent flow under H2 atmosphere was observed compared to that under CO2, which indicates the formation of hydrogen species (e.g., Ga-H) can trap more electrons and promote photocarrier excitation from the valence band to the conduction band. While the photocurrent under CO₂ increased faster compared to that under vacuum conditions, their decay behaviors were similar, suggesting that CO₂ adsorption removes pre-existing surface species which delay photoexcitation and relaxation. A similar increase in photocurrent between H2 + CO2 and vacuum conditions suggests that the reaction in H2 + CO2 proceeds quickly, which means that the photoelectrons are generated at the same rate as they are consumed during the CO2 hydrogenation reaction, and the surface is almost identical to that of the vacuum surface state. These results indicate that NG is sensitive to both acidic and basic gases as they influence photo-generated electron flow by either trapping or promoting it when adsorbed on the catalyst surface. An environment with both types of gases neutralizes their individual effects, restoring the catalytic surface to its original state and enhancing catalyst stability during repeated use. Additionally, the structure of SFLPs facilitates the formation of both Lewis acidic and basic adsorbates close to each other, which rapidly drives the desired chemical reaction and adsorbate removal under light. This forms the basis of the photochemical role of light in CO2-to-methanol production.
In order to further characterize the charge transfer dynamics and the changes to the Ni and Ga species in NG during the light illuminated reaction processes, in situ X-ray absorption fine-structure spectroscopy (XAFS) measurements were conducted under dark and light conditions in H2 + CO2 (3:1) atmosphere (Fig. 4e–h). Upon illumination, the white line of the Ni L-edge in its XANES spectrum shifts to higher energy, while the Ga K-edge shifts to lower energy, indicating electron transfer from Ni to Ga. This electron transfer modulates the charge state of adsorbed intermediates and enhances catalytic activity, which could be attributed to a faster combination of ligand effects and the dissociation of reactants or reaction intermediates under light (Fig. 4e,f, Supplementary Fig. 28)50. The results are further verified by corresponding Fourier-transform extended X-ray absorption fine structure (FT-EXAFS) (Fig. 4g,h). Peak I at ~1.5 Å was assigned to the metal-oxygen bond distance. Peaks II and III at 2.4 Å and 3.1 Å, respectively, were assigned to the radial distances of MOh (metal in octahedral site) and MTd (metal in tetrahedral site) to their neighboring metal atoms51. Strongly enhanced scattering at peak II was observed in Ni FT-EXAFS under light conditions compared to dark conditions, which was attributed to the faster adsorption and conversion of reactants at NiOh sites in the presence of light30. Meanwhile, only minor changes were observed in the Ga FT-EXAFS between light and dark conditions, which is attributed to reaction intermediates bound to Ga transferring more quickly to Ni in the presence of light. The peak positions in our samples closely match those of NiO and Ga2O3 standards and no correspondence was observed with the spectra of their respective metal foils, further supporting the structural interpretation.
Next, in situ DRIFTS was employed in CO2 + H2 atmosphere to analyze the faster light promoted conversion of key intermediates during the reactions. As shown in Fig. 4i,j, formate bands52,53 at 1594, 1349, 1381, 2881, 2906, 2715, and 2739 cm-1 decreased under light irradiation compared to their intensity in the dark. Meanwhile, two bands at 2924 and 2843 cm-1, attributed to the δas(CH3) and δs(CH3) modes of methoxy species (*H3CO)54,55, increase under light. The downshift of the band at 2848 cm-1 under dark to 2843 cm-1 under light can be explained by the increased hydrogen bond strength of *H3CO under light56. CO2 + D2 in situ DRIFTS experiments were conducted to further verify the surface intermediate species (Fig. 4k). No obvious peaks like the ones measured in CO2 + H2 experiments were observed between 2700 to 3000 cm-1. Two new peaks appeared at 2175 and 2142 cm-1 and increased moderately over time, which can be attributed to DCOO* and D3CO* species57, verifying the formation of formate and methoxy intermediates during CH3OH production. In situ DRIFTS results were constant with the in situ XAFS results and further support the enhanced conversion of the reaction intermediates under light.
DFT calculations
Density functional theory (DFT) calculations were performed to understand the H2 heterolysis and reaction pathway over NG. The (311) surface of NG, with one surface OH site and one Ov site, was used for the simulation, as shown in Fig. 5a, Supplementary Data 1. From the optimized configuration, Ga and Ni sites adjacent to the Ov and O sites were spatially separated by 3.330 and 3.337 Å, respectively. The sufficient distance between SFLP provides unquenched Lewis acid and base sites, allowing for flexible polarization of H-H bonds and heterolytically dissociation of H258. Bader charge analysis confirmed the SFLP property of Lewis acid-base (Ga/Ni)•••O sites, with the related Ga with Ni and O involving local atomic charges of +1.300 e, +0.835 e and −1.059 e, respectively. The energy barriers of H2 splitting on the SFLP sites showed that ΔGbar = 0.43 eV for the formation of surface OH and (Ga/Ni)-H, as shown in Fig. 5b. After H2 splitting, one proton attaches to the Lewis basic O and the other hydride bridges Ga and Ni. Analyzing the charge distribution, we see the differential charge density map of H+δ by the O and H-γ by the Ga/Ni (Fig. 5c), and the Bader charges show that the hydrogen in the OH moiety exhibited a charge of +0.658 e, whereas in the metal hydride, it endowed a charge of −0.361 e, thus reinforcing the evidence for heterolytic splitting of H2.

a Optimized structure of nano-pigment NG and c corresponding 2D charge density difference plots of H2 splitting at SFLP sites. b kinetic energy barriers of H2 splitting on the SFLP sites, where TS denotes the transition state. d Computed absorption spectra obtained from linear response time dependent DFT (LR-TDDFT) for the NG, NG with H2 physical adsorption (NG_H2), and NG with the split H2 (NG_2H), respectively. e, f Charge density difference and the corresponding orbital energy diagram for a representative transition involved in H2 splitting (H = highest occupied band/orbital, L = lowest unoccupied band/orbital). g Reaction process of CO2 hydrogenation to methanol. Color code: green, Ga; gray, Ni; red, O; pink, H; brown, C.
The projected density of states (PDOS) demonstrated that once H2 approaches the surface-active site, Ga \({p}_{y}\) sub-gap states disappear, supporting the role of Ga acting as a Lewis acid (Supplementary Fig. 29). In addition, noticeable shifts were also observed in the states around the Fermi level for Ni, Ga, and O (Supplementary Fig. 30a-c). As H2 splits to form a proton and hydride on the surface, a clear additional peak in the PDOS that can be attributed to the hydride bonding interaction. Additionally, some of the electron density from the hydride shifted up to the Fermi level, while there is a slight splitting in the peak for the O-H bond, indicating the presence of two oxygen-attached hydrogens. A new surface OH forms from the proton of H2, and the original surface OH remains (Supplementary Fig. 30d).
We also employed linear response time dependent DFT (LR-TDDFT) under a hybrid functional approach (details in the Methods) to study the role of light in facilitating H2 splitting. Figure 5d shows the computed absorption spectra for the bare NG, NG with H2 physical adsorption, and NG with the split H2 (named as NG, NG_H2, and NG_2H, respectively). The spectrum for bare NG, to a first approximation, is quite comparable to that from experiment (Fig. 1b), with major peaks in the correct regions. Upon H2 approach, the absorption intensity between 425 and 525 nm (a trough in the bare NG spectrum) and 600-700 nm increases significantly, indicating that H2 in proximity to the surface can be involved in electronic excitations coupled with the surface states of NG. Among all excitations with an oscillator strength greater than 0.1, we find several transitions that are conducive to charge polarization on H2, as demonstrated by difference densities between the relevant bands with the largest contribution to specific excitations (Fig. 5e,f). For example, we see an excitation of around 530 nm that involves an accumulation of electron density on the hydrogen closer to Ga and a depletion of electron density on the hydrogen closer to O. Another around 550 nm (Supplementary Fig. 31) that shows charge depletion around the Ga and charge accumulation around the O across the vacancy site (for the latter, the additional charge accumulation on an oxygen is consistent with previous findings)59. These results demonstrate that light can create a polarizing environment around the vacancy site that heterolytically splits H2 to produce H-γ by the Ga and H+δ by the O. In the absence of oxygen vacancies, one is typically limited to the dominant ligand to metal charge transfer from O to Ga. However, in the existence of oxygen vacancies, light can induce polarization around the vacancy site. With the atoms fixed in their lattice positions, this polarization contributes to the frustration of surface Lewis acid-base sites, facilitating the splitting of H2.
After H2 splits, we also see multiple transitions that lead to significant electron accumulation around the hydride and Ga, as demonstrated by the corresponding charge density difference plots (Supplementary Fig. 32). Since these transitions show up across the visible region (from around 400 to 570 nm) and are also among the ones with the largest oscillator strengths out of all transitions, this indicates that light can play a further role in making the hydride more hydridic following H2 splitting, thereby facilitating the hydrogenation of CO2.
Further, DFT calculations were conducted to determine the reaction pathway and associated energetics. As shown in Fig. 5g, a CO2 molecule is the physical adsorbed on the Ni near the Ga-H group with the ΔEad-CO2 of −0.228 eV. Subsequently, the transfer of hydride to the carbon of CO2 leads to HCOO* formation with a free energy barrier of 0.794 eV, which agrees with in situ DRIFTS and in situ XAFS results. For further hydrogenation, the HCOO* has to be flipped. Fortunately, this flipping is an exothermic process (−0.857 eV) with a small free energy barrier of 0.138 eV, indicating the favourable flipping process. After that, the second H diffuses from OH to the O terminal of HCOO* with the free energy barrier of 0.365 eV, resulting in the formation of HCOOH*. The C atom of the HCOOH* group could then be hydrogenated to H2COOH* group with a free energy barrier of 0.945 eV and a reaction free energy of 0.335 eV. The H2COOH* could further break the C–O bond and further hydrogenate to form H2CO* and a water molecule almost spontaneously with a free energy barrier of 0.049 eV. H2CO* could stepwise react with neighbouring hydrogen atoms with a third heterolytic H2 dissociation to form H3CO* and CH3OH*. The free energy barriers for the former hydrogenation steps were 0.650 eV, in which the last formation of CH3OH is spontaneous with the reaction energies being −0.854 eV. Note that all H+ species resulting from the heterolytic cleavage of H2 react with O atoms in reaction intermediates, while the H- species selectively attack the positively charged C atoms in these intermediates, as shown in Supplementary Figs. 33–38. It is noteworthy that the preexisting OH group acts as a transporter for the subsequently split H+, which promotes the entire process of CO2 hydrogenation to methanol. This suggests that the designed SFLP primarily facilitates the heterolysis of H2, directing the generated H+ and H- to selectively bond with O and C atoms in the reaction intermediates, respectively. Such orchestrated bonding significantly accelerates the hydrogenation of CO2, ultimately resulting in the formation of CH3OH. Combining the experimental results, it can be illustrated that light interacts with the SFLP sites to promote the heterolytic splitting of hydrogen, forming a hydride (H-) and a proton (H+), where the hydride acts as a redox mediator, similar to the role of NADPH. Ni(II)/Ni(III) and OH(-I) respectively serve as conduits for the ion-transport of H- and H+ to the Ni site, where they react with CO2, mimicking the dark reaction in biologically-mediated photosynthesis. Consequently, the equivalent transfer of electrons (e-) and protons (H+) can occur within the nano-pigment and promote the photocatalytic conversion of CO2 to methanol. To investigate the influence of CO presence during the reaction process on NG, seven possible adsorption sites for CO* on the NG surface were considered, as shown in Supplementary Fig. 39. Among these, the Ni-4 site exhibited the most stable adsorption energy, confirming it as the preferred configuration. Consequently, all subsequent calculations modeling CO co-adsorption utilized the Ni-4 site. Given that heterolytic H2 splitting and CO2 activation were the key steps in CO2 hydrogenation to methanol over NG, we focused our analysis on these processes under CO presence (Supplementary Fig. 40). The calculated reaction free energies for H2 splitting and the first CO2 hydrogenation step are 0.58 eV and 0.69 eV, respectively, with co-adsorbed CO. These values are slightly different from those without CO (0.43 eV and 0.79 eV), indicating minor influence from CO adsorption. Notably, while CO presence modestly increases the energy barrier for H2 splitting (0.15 eV), it facilitates CO2 hydrogenation (−0.10 eV). This acceleration of the CO2 activation step is particularly advantageous for methanol synthesis.
Discussion
In summary, this work describes a chlorophyll-mimicking and defect-decorated green colored nano-pigment NG inverse-spinel material that features surface-frustrated Lewis pairs (SFLPs) that enables high performance broadband photocatalytic hydrogenation of CO2 to green methanol. Oxygen vacancies and surface hydroxyls were introduced to create Ga(III) with mixed-valence Ni(II, III) to establish the frustrated Lewis acid-base sites. The high methanol production (3.20 mmol·h-1·g-1) and selectivity (79.6%), impressive quantum efficiency (3.0%), and excellent photon-to-methanol selectivity (>90.0%) endow NG as a potential practical candidate for industrial photocatalytic CO2 hydrogenation to methanol. In situ experimental techniques including photocurrent measurements, XPS, STEM-EELS, XAFS, and DRIFTS measurements, combined with DFT and TD-DFT simulations, provided evidence that the photochemical role of NG originates from d-d transitions of Ni(II, III) and the promotion of Ga-H formation, favored as the active species in the proposed reaction pathway for the conversion of CO2 to methanol. The coexistence of mixed valence states of Ni(II)/Ni(III) as well as OH(-I) serves as conduits for the ion-transport of hydrides and protons, H- and H+, respectively, mimicking the functionality of chlorophyll for the efficient CO2 to methanol conversion. The performance of NiGa2O4-x(OH)y opens exciting avenues for the development of green chlorophyll mimics using heterogeneous photocatalytic systems and provides valuable opportunities for the light-driven synthesis of sustainable commodity chemicals from greenhouse gases. Future studies are expected to benefit from more efficient photon-capturing photocatalyst designs through active site engineering, as well as from careful optical and chemical optimization of photoreactors based on catalyst shape and morphology. These advancements aim to further enhance and improve the activity, selectivity, and quantum efficiency of photo-to-methanol conversion60.
Methods
Synthesis of NiGa2O4-x(OH)y photocatalyst
1.0 mmol of nickel nitrate (Ni(NO3)2·6H2O, Sigma ≥97.0%) and 2.0 mmol of gallium nitrate (Ga(NO3)3·xH2O, Sigma 99.9%) were dissolved in 15 mL deionized water and stirred for 30 min. Subsequently, ammonium hydroxide (NH3·H2O; Anachemia 28%) was added to adjust the pH to 8-9 under continuous stirring. After aging for 30 min, the obtained suspension was transferred into a 30 mL stainless-steel Teflon lined autoclave and kept at 180 °C for 16 h before being cooled to room temperature. Next, the obtained products were collected after being centrifuged and washed with deionized water several times and finally dried at 60 °C for 12 h in a vacuum oven. The NG-300 sample was prepared by treating the NG sample under air for 4 h at 300 °C with a heating rate of 5 °C min−1.
Material characterization
High-resolution transmission electron microscopy (HRTEM) image and electron energy loss spectroscopy (EELS) measurements were performed using a Hitachi HF-3300 environmental TEM/STEM. The EELS results were recorded using an EELS collection angle of 149 mrad. An energy dispersion of 0.25 eV/channel was used to acquire Ni L-edge, Ga L-edge and O K-edge, respectively. The energy resolution in EELS, as measured by the full-width at half-maximum of the zero-loss peak, was 1.0 to 1.3 eV for 0.25 eV/channel. All pre-peaks (labelled as peak a) of O-K edge are aligned to 532 eV. Gaussian function was used to fit the Ni-L2,3 edges for white-line ratio calculations. Powder X-ray diffraction (PXRD) was conducted on a Bruker D2-Phaser X-ray diffractometer, using Cu Kα radiation at 30 kV. X-ray photoelectron spectroscopy (XPS) was measured using a Perkin Elmer Phi 5500 ESCA spectrometer. The spectrometer uses an Al Kα X-ray source to generate X-rays with an energy of 1486.7 eV. Diffuse reflectance spectra (UV-Vis-NIR) of the samples were recorded using a Lambda 1050 UV/VIS/NIR spectrometer from Perkin Elmer equipped with an 150 mm diameter integrating sphere. FTIR spectra were collected on a Thermo Scientific iS50 series. The surface areas of samples were measured on an Autosorb-1 system (Quantachrome, USA) from N2 adsorption isotherms obtained via the BET method. The thermal properties of the samples were evaluated using a TA Instruments SDT Q600 thermogravimetric analyzer. The temperature was steadily increased from 30 °C to 800 °C at N2 a rate of 10 °C min-1. Contact angle (CA) measurements on pelletized samples were done by JC2000C1 goniometer to determine hydrophilicity. The zeta potential was measured by Malvern Zetasizer Nano ZS90 via dispersing samples in water. Raman spectra were acquired on a Bruker SENTERRA Dispersive Raman Microscope with an excitation wavelength of 532 nm. The H2 temperature-programmed desorption (H2-TPD) was recorded with an infrared spectrometer (ThermoFisher Nicolet iS 50) equipped with a Deuterated Triglycine Sulfate (DTGS) detector. Before CO2-TPD, catalyst samples (~100 mg) were pretreated in Ar at 50 mL min-1 to 200 °C for 1 h with a ramp rate of 5 °C min-1. After the sample was cooled down to 40 °C, it was exposed to CO2 (40 kPa, diluted in Ar) for adsorption until saturation. Then, CO2-TPD was performed in 30 mL min-1 Ar with a heating ramp of 5 °C min-1 from 40 to 700 °C. Pyridine infrared absorption spectroscopy (pyridine-lR) was performed using a Bruker Vertex 70 spectrometer, equipped with CaF windows. Each spectrum was collected at a resolution of 2 cm-1 using an average of 64 scans. Before spectrum collection, catalyst was pressed into a self-supporting wafer (~30 mg cm-2) and placed into a customized cell. After heating under Ar (50 mL min-1) at 200 °C for 2 h, a background spectrum was acquired. Pyridine (~0.1 kPa) was then introduced until saturation. Finally, the sample was evacuated to remove weakly adsorbed pyridine before spectroscopic measurements61,62.
In situ photocurrent measurement
The photocurrent measurements were carried out in a modified cryostat with atmospheric pressure control and temperature control. 1 cm by 1 cm gold electrodes of 300 nm thicknesses were evaporated onto a glass slide with a 1 mm gap between the two electrodes. The catalytic powder was then deposited over the gap and pressed down firmly with a glass pipette. Tungsten probe needles were pressed in the gold electrodes on the areas where no powder was present, and current measurements were taken with a Kiethley 4200 instrument. The sample was heated to 200 °C for 3 h under a vacuum pressure of 4E-2 mtorr before current measurements were conducted. Photo-illumination was provided using a Xenon-mercury lamp with 250 W power input. After the first current measurement, the lamp was turned on for 100 minutes. Gases were added until to 1.5 atm pressure and the sample was allowed to sit for 2 h in the gas atmosphere before measurements began. For the mixed atmosphere, H2 and CO2 gases were introduced at a 3:1 pressure ratio.
In situ TEM test
In situ TEM was performed with a single-tilt Hitachi Blaze heating holder on a Hitachi HF-3300 environmental TEM operated at 300 kV with a cold field-emission gun. EELS spectra were acquired with a 149 mrad collection angle. An energy dispersion of 0.25 eV channel-1 was used to record the Ni-L, Ga-L, and O-K edges. The zero-loss peak full width at half maximum indicated an energy resolution of 1.0–1.3 eV under these conditions. Experiments began at room temperature and then proceeded at 300 °C under H2 before switching to CO2 at the same temperature.
In situ X-ray photoelectron spectroscopy
In situ XPS spectra were obtained using Thermo Scientific ESCALAB XI + XPS Microprobe. A high pass energy (100 eV) and low point density (1 point eV-1) were used for photoelectron detection, and all results were calibrated to C 1 s (284.8 eV). The tests were conducted at room temperature under vacuum, 300 °C under Ar, 300 °C under H2 and 300 °C under CO2 + H2 (H2/CO2 = 3/1), respectively.
In situ DRIFTS measurements
DRIFT spectra were performed using a Thermo Scientific iS50 FT-IR equipped with a Harrick Praying Mantis DRIFT accessory. Spectra were collected at 4 cm-1 resolution by averaging 32 scans. Undiluted powder samples were placed directly in the sample holder. Before measurements, the cell was purged with He at 300 °C for 2 h with the flow rate of 20 mL min-1 to obtain the background. Then, H2 or D2 was introduced for 20 min at different temperatures, followed by a 3:1 H2:CO2 or D2/CO2 for 20 min at 300 °C the total gas flow was maintained at 20 mL min-1.
DFT calculations
Spin-polarized density function theory (DFT) calculations were performed using the Vienna ab initio simulation package (VASP) in this work. Projector-augmented wave (PAW) pseudopotentials were applied to treat the core electrons63. The generalized gradient approximation (GGA) with the Perdew-Burke-Ernzerhof functional (PBE) was adopted in geometric optimization and exploration of reaction pathway for CO2 reduction reaction64. The kinetic energy cutoff for the wave-function calculations was set to 500 eV. The Fermi smearing function was applied with a smearing width of 0.05 eV. The (311) surface of NiGa2O4 was used as the active surface, including 84 atoms with the lattice of a = 10.11 Å and b = 11.68 Å. The vacuum gap of ~15 Å was used to avoid interactions between the system and its mirror images. The geometric relaxation was stopped when the incremental changes in total energy and forces were smaller than 1 × 10-5 eV and 0.05 eV/Å, respectively. The van der Waals interaction was considered through the DFT-D3 method proposed by Grimme65. All the transition states were obtained using the climbing image nudged elastic band (CI-NEB) method with a convergence force smaller than 0.05 eV/Å66.
The reaction free energy (ΔG) was corrected by considering the zero-point energy and entropy, as shown below:
where ΔE, ΔZPE, and TΔS denote the reaction energy (or energy barrier), zero-point energy change, and entropy change, respectively.
For studying the role of light in H2 splitting, spin-polarized electronic structure calculations for bare NG, NG_H2, and NG_2H were further carried out for the PBE-relaxed geometries using the Heyd–Scuseria–Ernzerhof (HSE) functional67, with dipole and dispersion corrections included. The latter was done at the DFT-D3(BJ) level68, with the parameters for Becke-Johnson damping selected based on those for PBE069. Density of states were evaluated at 1000 grid points with Gaussian smearing, and Bader charges were evaluated using the program of Henkelman70. The ground state HSE wavefunction was then used as the input for TD-DFT calculations. The frequency-dependent dielectric matrix was first calculated with a total of 800 bands (512 occupied, 288 unoccupied), and the result was used to evaluate the Casida equation under the Tamm-Dancoff Approximation. For the latter, 20 occupied and unoccupied bands were used to obtain the imaginary dielectric function (simulating the absorption coefficient) and the oscillator strengths (simulating the probability of excitations). For all calculations at the HSE level, the plane wave cutoff was set to 600 eV, SCF convergence achieved at 10-5 eV, and the Brillouin zone sampled at the Γ point only. The GW version of the PAW pseudopotentials with the extensions sv (i.e. the s and p semi-core states are treated as valence states) were used for all elements.
Excitations with an oscillator strength greater than 0.1 and between 1.5 and 3.5 eV were selected for further analysis. Within linear-response TDDFT71, each excitation (i.e. each oscillator strength that gets printed) corresponds to a linear combination of all possible 1-electron interband transitions, within the number of occupied and unoccupied bands specified-often, a few of these transitions contribute a lot and most do not contribute much (on the other hand, an individual transition can also contribute to multiple excitations across several wavelengths). The contribution of an individual transition from an occupied band/orbital i to an unoccupied band/orbital j is given by the interband coupling constant \({X}_{{ij}}\) for that pair of orbitals. For each selected excitation, we filtered out all origin and destination bands/orbitals with \({X}_{{ij}}\) < 0.1, and the remaining were selected for analysis. For the selected 1-electron interband transitions, we plotted their charge density differences by subtracting the partial charge density of the origin orbital from the destination orbital (i.e. destination orbital minus origin orbital), and after visualization, we down-selected the ones based on the oscillator strengths of the excitations they contribute to and/or ones that have visible shifts in electron density on the adsorbate and NG surface (at an isosurface level of 10-4 e/Bohr3 unless otherwise indicated). Filtering and parsing of the VASP TDDFT outputs were done using an in-house written code. Charge density differences between the relevant bands were generated using VASPKIT72 and visualized with VESTA73.
Catalytic tests
The catalytic activity of the NG for CO2 hydrogenation with and without light under different temperatures were investigated in a Harrick Cell reactor at different temperatures with a H2:CO2 ratio of 3:1 (6 sccm H2 + 2 sccm CO2). The reaction was conducted at a pressure of 0.7 MPa under white LED light irradiation at an intensity of 1.87 W cm-2. The reactor cell volume was ~13.0 mL, the catalyst loading was ~15 mg, and the photoirradiation area was ~28.3 mm2. Catalyst temperature was controlled by copper heating block under the catalyst bed, the temperature of which was controlled by a temperature controller equipped with a OMEGA k-type thermocouple, resistive heating cartridge, and water-cooling37. CO2 and H2 flow rates were set to 2 and 6 sccm by two mass flow controllers (Alicat MC-2SCCM-D/5 M). The concentrations of the products in the effluent gas were periodically sampled and analyzed by gas chromatography equipped with a flame Ionization detector (GC-FID) using Ar as the carrier gas. The quenching experiments on the SFLPs were carried out with pyrrole and pyridine. 50 mg NG sample was dispersed in 2 mL of pyrrole or pyridine and kept in the dark for 1 h to achieve adsorption-desorption equilibrium. The treated samples were then evacuated and used for the activity tests under the same photocatalytic conditions as other samples.
Data availability
The data related to the figures in the paper are provided as Excel files in the source data. Source data are provided with this paper.
References
Wang, L. et al. Photocatalytic hydrogenation of carbon dioxide with high selectivity to methanol at atmospheric pressure. Joule 2, 1369–1381 (2018).
Jiang, X., Nie, X., Guo, X., Song, C. & Chen, J. G. Recent advances in carbon dioxide hydrogenation to methanol via heterogeneous catalysis. Chem. Rev. 120, 7984–8034 (2020).
Navarro-Jaén, S. et al. Highlights and challenges in the selective reduction of carbon dioxide to methanol. Nat. Rev. Chem. 5, 564–579 (2021).
Miller, T. E. et al. Light-powered CO2 fixation in a chloroplast mimic with natural and synthetic parts. Science 368, 649–654 (2020).
Li, X.-B., Tung, C.-H. & Wu, L.-Z. Semiconducting quantum dots for artificial photosynthesis. Nat. Rev. Chem. 2, 160–173 (2018).
Takata, T. et al. Photocatalytic water splitting with a quantum efficiency of almost unity. Nature 581, 411–414 (2020).
Bi, W. et al. Molecular co-catalyst accelerating hole transfer for enhanced photocatalytic H2 evolution. Nat. Commun. 6, 8647 (2015).
Dong, C. et al. Size-dependent activity and selectivity of carbon dioxide photocatalytic reduction over platinum nanoparticles. Nat. Commun. 9, 1252 (2018).
Cestellos-Blanco, S., Zhang, H., Kim, J. M., Shen, Y.-X. & Yang, P. Photosynthetic semiconductor biohybrids for solar-driven biocatalysis. Nat. Catal. 3, 245–255 (2020).
Guan, X. et al. Maximizing light-driven CO2 and N2 fixation efficiency in quantum dot-bacteria hybrids. Nat. Catal. 5, 1019–1029 (2022).
Lv, J. et al. Solar utilization beyond photosynthesis. Nat. Rev. Chem. 7, 91–105 (2023).
Bonchio, M. et al. Hierarchical organization of perylene bisimides and polyoxometalates for photo-assisted water oxidation. Nat. Chem. 11, 146–153 (2019).
Li, Y. et al. Boosting thermo-photocatalytic CO2 conversion activity by using photosynthesis-inspired electron-proton-transfer mediators. Nat. Commun. 12, 123 (2021).
Janssen, P. J. et al. Photosynthesis at the forefront of a sustainable life. Front. chem. 2, 36 (2014).
Gust, D., Moore, T. A. & Moore, A. L. Solar fuels via artificial photosynthesis. Acc. Chem. Res. 42, 1890–1898 (2009).
Ulmer, U. et al. Fundamentals and applications of photocatalytic CO2 methanation. Nat. Commun. 10, 3169 (2019).
Kozuleva, M. A. & Ivanov, B. N. The mechanisms of oxygen reduction in the terminal reducing segment of the chloroplast photosynthetic electron transport chain. Plant Cell Physiol. 57, 1397–1404 (2016).
Tadepalli, S., Slocik, J. M., Gupta, M. K., Naik, R. R. & Singamaneni, S. Bio-optics and bio-inspired optical materials. Chem. Rev. 117, 12705–12763 (2017).
Xie, Y. et al. Advancement of renewable energy technologies via artificial and microalgae photosynthesis. Bioresour. Technol. 363, 127830 (2022).
Xie, Y. et al. Frustrated Lewis pairs boosting low-temperature CO2 methanation performance over Ni/CeO2 nanocatalysts. ACS Catal. 12, 10587–10602 (2022).
Zhang, Z. et al. New black indium oxide-tandem photothermal CO2-H2 methanol selective catalyst. Nat. Commun. 13, 1512 (2022).
Wang, L. et al. Room-temperature activation of H2 by a surface frustrated Lewis pair. Angew. Chem. Int. Ed. 58, 9501–9505 (2019).
Liu, S. et al. An asymmetric supercapacitor with excellent cycling performance realized by hierarchical porous NiGa2O4 nanosheets. J. Mater. Chem. A 5, 19046–19053 (2017).
He, Z.-H., Gao, J.-F. & Kong, L.-B. NiGa2O4 nanosheets in a microflower architecture as anode materials for Li-Ion capacitors. ACS Appl. Nano Mater. 2, 6238–6248 (2019).
Zhang, J. et al. Modulation of inverse spinel Fe3O4 by phosphorus doping as an industrially promising electrocatalyst for hydrogen evolution. Adv. Mater. 31, 1905107 (2019).
Guan, D. et al. Exceptionally robust face-sharing motifs enable efficient and durable water oxidation. Adv. Mater. 33, 2103392 (2021).
Li, J., Gao, J.-F., He, Z.-H., Hou, J.-F. & Kong, L.-B. Nanoflower architecture NiGa2O4 with a spinel structure modified by 2D layered RGO for enhanced li-ion battery anode performance. Energ. Fuels (2022).
Cao, R., Yang, H., Zhang, S. & Xu, X. Engineering of Z-scheme 2D/3D architectures with Ni(OH)2 on 3D porous g-C3N4 for efficiently photocatalytic H2 evolution. Appl. Catal. B 258, 117997 (2019).
Lin, R. et al. Hierarchical nickel valence gradient stabilizes high-nickel content layered cathode materials. Nat. Commun. 12, 2350 (2021).
Wu, T. et al. Iron-facilitated dynamic active-site generation on spinel CoAl2O4 with self-termination of surface reconstruction for water oxidation. Nat. Catal. 2, 763–772 (2019).
García-Alonso, J., Maestre, D., Nogales, E., Cremades, A. & Méndez, B. Study of NiGa2O4 microneedles grown by a thermal-evaporation method. J. Alloy. Compd. 919, 165718 (2022).
Song, R. et al. Ethylene production via photocatalytic dehydrogenation of ethane using LaMn1-xCuxO3. Nat. Energy 9, 1–11 (2024).
Li, Z. et al. Engineered disorder in CO2 photocatalysis. Nat. Commun. 13, 7205 (2022).
Wang, N. et al. Electrocatalytic hydrogen evolution with gallium hydride and ligand-centered reduction. Chem. Sci. 10, 2308–2314 (2019).
Sun, S. et al. Shifting oxygen charge towards octahedral metal: a way to promote water oxidation on cobalt spinel oxides. Angew. Chem. Int. Ed. 131, 6103–6108 (2019).
Kim, C. et al. Energy-efficient CO2 hydrogenation with fast response using photoexcitation of CO2 adsorbed on metal catalysts. Nat. Commun. 9, 1–8 (2018).
Zhang, X. et al. Product selectivity in plasmonic photocatalysis for carbon dioxide hydrogenation. Nat. Commun. 8, 14542 (2017).
Robatjazi, H. et al. Plasmon-induced selective carbon dioxide conversion on earth-abundant aluminum-cuprous oxide antenna-reactor nanoparticles. Nat. Commun. 8, 1–10 (2017).
Zou, H. et al. Alternating current photovoltaic effect. Adv. Mater. 32, 1907249 (2020).
Ni, L. et al. Atomical reconstruction and cationic reordering for nickel-rich layered cathodes. Adv. Energy Mater. 12, 2103757 (2022).
Li, X., Yu, J., Jaroniec, M. & Chen, X. Cocatalysts for selective photoreduction of CO2 into solar fuels. Chem. Rev. 119, 3962–4179 (2019).
Collins, S. E., Baltanás, M. A., Fierro, J. L. G. & Bonivardi, A. L. Gallium-hydrogen bond formation on gallium and gallium-palladium silica-supported catalysts. J. Catal. 211, 252–264 (2002).
Wang, L. et al. Room-temperature activation of H2 by a surface frustrated Lewis pair. Angew. Chem. Int. Ed. 58, (2019).
Collins, S. E., Baltanás, M. A. & Bonivardi, A. L. Infrared spectroscopic study of the carbon dioxide adsorption on the surface of Ga2O3 polymorphs. J. Phys. Chem. B 110, 5498–5507 (2006).
Hayashi, K., Sushko, P. V., Hashimoto, Y., Shluger, A. L. & Hosono, H. Hydride ions in oxide hosts hidden by hydroxide ions. Nat. Commun. 5, 1–8 (2014).
Zhu, Z. et al. Addition of Hydrogen or Ammonia to a Low-Valent Group 13 Metal Species at 25 °C and 1 Atmosphere. Angew. Chem. Int. Ed. 48, 2031–2034 (2009).
Tan, G., Szilvási, T., Inoue, S., Blom, B. & Driess, M. An elusive hydridoaluminum (I) complex for facile C-H and C-O bond activation of ethers and access to its isolable hydridogallium (I) analogue: syntheses, structures, and theoretical studies. J. Am. Chem. Soc. 136, 9732–9742 (2014).
Ning, S. et al. Co0-Coδ+ Interface Double-Site-Mediated C-C Coupling for the Photothermal Conversion of CO2 into Light Olefins. Angew. Chem. Int. Ed. 135, e202302253 (2023).
He, R. et al. Tailoring the CO2 hydrogenation performance of Fe-based catalyst via unique confinement effect of the carbon shell. Chem. -eur. J. 29, e202301918 (2023).
Lan, J. et al. Efficient electrosynthesis of formamide from carbon monoxide and nitrite on a Ru-dispersed Cu nanocluster catalyst. Nat. Commun. 14, 2870 (2023).
Song, R. et al. Solar hydrocarbons: single-step, atmospheric-pressure synthesis of C2-C4 alkanes and alkenes from CO2. Angew. Chem. Int. Ed. 135, e202304470 (2023).
Wu, C. et al. Inverse ZrO2/Cu as a highly efficient methanol synthesis catalyst from CO2 hydrogenation. Nat. Commun. 11, 1–10 (2020).
Yan, T. et al. Bismuth atom tailoring of indium oxide surface frustrated Lewis pairs boosts heterogeneous CO2 photocatalytic hydrogenation. Nat. Commun. 11, 1–10 (2020).
Rhodes, M. D. & Bell, A. T. The effects of zirconia morphology on methanol synthesis from CO and H2 over Cu/ZrO2 catalysts: Part I. Steady-state studies. J. Catal. 233, 198–209 (2005).
Tountas, A. A. et al. Direct CO2-to-renewable methanol: Outlook, performance and optimization approach. Sustain. Mater. Technol. 36, e00630 (2023).
Noguchi, T. Light-induced FTIR difference spectroscopy as a powerful tool toward understanding the molecular mechanism of photosynthetic oxygen evolution. Photosynth. Res. 91, 59–69 (2007).
Rezvani, A. et al. CO2 reduction to methanol on Au/CeO2 catalysts: mechanistic insights from activation/deactivation and SSITKA measurements. ACS Catal. 10, 3580–3594 (2020).
Yan, T. et al. Polymorph selection towards photocatalytic gaseous CO2 hydrogenation. Nat. Commun. 10, 1–10 (2019).
Ghuman, K. K. et al. Photoexcited surface frustrated Lewis pairs for heterogeneous photocatalytic CO2 reduction. J. Am. Chem. Soc. 138, 1206–1214 (2016).
Kant, P., Liang, S., Rubin, M., Ozin, G. A. & Dittmeyer, R. J. J. Low-cost photoreactors for highly photon/energy-efficient solar-driven synthesis. Joule 7, 1347–1362 (2023).
Cai, G. & Chin, Y.-H. C. Catalytic sequences and kinetic requirements for C–O bond scission and formation in methanol and trioxane on solid polyoxometalate clusters. ACS Catal. 13, 14828–14844 (2023).
Cai, G. & Chin, Y.-H. C. Thermochemical and kinetic correlations of redox and Lewis sites on cobalt–molybdenum oxides: illustrated with alkanol-O2 catalysis. ACS Catal. 15, 11673–11694 (2025).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132 (2010).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Heyd, J., Scuseria, G. E. & Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 118, 8207–8215 (2003).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Bokdam, M., Lahnsteiner, J., Ramberger, B., Schäfer, T. & Kresse, G. Assessing density functionals using many body theory for hybrid perovskites. Phys. Rev. Lett. 119, 145501 (2017).
Tang, W., Sanville, E. & Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 21, 084204 (2009).
Gajdoš, M., Hummer, K., Kresse, G., Furthmüller, J. & Bechstedt, F. Linear optical properties in the projector-augmented wave methodology. Phys. Rev. B Condens. Matter 73, 045112 (2006).
Wang, V., Xu, N., Liu, J.-C., Tang, G. & Geng, W.-T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 267, 108033 (2021).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011).
Acknowledgements
This work supported by the National Key R&D Program of China (2021YFF0502000 L.H.), the National Natural Science Foundation of China (52172221, L.H.; 52025061, D.J.; W2412063, L.H.; 52272229, C.L.; 22302137, X.A.; 52573339, C.L.), the Natural Science Foundation of Jiangsu Province (BK20220027, L.H.), Jiangsu Key Laboratory for Carbon-Based Functional Materials and Devices (ZZ2201, L.H.), Suzhou Key Laboratory of Advanced Photonic Materials, Collaborative Innovation Center of Suzhou Nano Science & Technology, the Ontario Ministry of Research and Innovation (G.A.O.), the Ontario Ministry of Economic Development, Employment and Infrastructure (G.A.O.), the Ontario Ministry of the Environment and Climate Change (G.A.O.); the Ontario Ministry of Research Innovation and Science Low Carbon Innovation Fund (G.A.O.), the Ontario Centre of Excellence Solutions 2030 Challenge Fund (G.A.O.), the Alexander von Humboldt Foundation (G.A.O.), the University of Toronto Connaught Innovation Fund (G.A.O.), the Connaught Global Challenge Fund and the Natural Sciences and Engineering Research Council of Canada (G.A.O.). We also acknowledge Greg Vezina of Hydrofuel Canada Inc. for financial support from June 2023. Thanks go to Darcy Burns from Chemistry department of University of Toronto (darcy.burns@utoronto.ca) for NMR test, and Zheng-qing Huang and Xi-yang Yu from Xi’an Jiaotong University (huangzq@xjtu.edu.cn, xiyangyu@stu.xjtu.edu.cn) for helping analyzing and reviewing DFT calculation results. Special thanks go to Miao Wang and Xiyang Wang for assistance with XPS and XAS data analysis.
Author information
Authors and Affiliations
Contributions
R.S. and G.A.O. conceived and designed the experiments. G.A.O., L.H. X.Z. and C.V.S. supervised the project. R.S. prepared the materials and performed the XRD, FT-IR, NMR, in situ DRIFTS and photocatalytic characterizations. C.Q. did the in situ STEM-EELS and TEM tests. X.Y. did the in situ XPS tests, L.W. and C.X. helped the data analysis. J.L. and A.M. conducted the in situ photocurrent tests. Y.X. and Y.F. helped with the activity tests and data analysis. C.J.V. performed the TGA test and helped the reactor design. R.S., C.A., Zhijie Chen, J.G., J.H., C.L., X.A. G.C., W.Q. and D.J. performed the UV-Vis, PL, zeta potential, UPS, and H2-TPD. Z.L. and Z.Z. helped the samples synthesis. Zhiwen Chen, A.W., A.A.G. and C.V.S. performed the DFT calculations. P.D., E.M. and J.Z. did the XAFS tests. G.C. performed the CO2-TPD and pyridine-lR. R.S., Zhiwen Chen, A.W., A.A.T., J.Y., L.H. and G.A.O. co-wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Weibin Fan, Lingzhi Wang, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Song, R., Chen, Z., Qiu, C. et al. A nickel gallium oxide chlorophyll mimic for green methanol synthesis. Nat Commun 16, 10542 (2025). https://doi.org/10.1038/s41467-025-65560-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65560-y
