
Abstract
Sepsis-induced liver injury is common, but the underlying mechanisms remain poorly understood. Given the critical role of gut-liver crosstalk in sepsis, we hypothesize that gut-trained neutrophils, migrating via the portal vein, release neutrophil extracellular traps (NETs) to activate Kupffer cells, thereby exacerbating hepatic injury during sepsis. Here we show that iNOS expression in Kupffer cells increases in septic wild type mice but decreases in PAD4-/- mice. In vitro, NETs stimulate Kupffer cell IL-6 and TNF release, while conditioned media from NET-treated Kupffer cells induces hepatocyte death. Inhibition of neutrophil elastase and protease-activated receptor-1 (PAR-1) mitigates IL-6 and TNF secretion by Kupffer cells. Ex vivo, portal vein neutrophils from septic mice produce more NETs and induce greater Kupffer cell activation than systemic neutrophils, with this effect attenuated in PAD4-/- neutrophils. Furthermore, gut intraepithelial lymphocytes (IELs) interact with neutrophils during sepsis and facilitate NETosis, and IEL-primed neutrophils also induce Kupffer cell activation in vitro and in vivo. Our data thus suggest that IEL-facilitated, gut-derived neutrophil NETs activate Kupffer cells to contribute to sepsis-induced liver injury.
Similar content being viewed by others


Introduction
Sepsis, a severe inflammatory disorder due to infection, is a major global health problem, affecting approximately 50 million patients worldwide annually1,2. Pathogen-associated molecular patterns (i.e., LPS) stimulate pattern-recognition receptors, such as toll-like receptor 4 (TLR4), to initiate aberrant immune responses to cause organ dysfunction in sepsis3. Liver failure frequently complicates sepsis and is linked to disease severity, even in the absence of direct liver infection4,5. While pivotal for metabolic and immune homeostasis, the liver is vulnerable to inflammation. During sepsis, hepatic dysfunction impairs detoxification, alters metabolism, and dysregulates coagulation4,6. Furthermore, the liver generates proinflammatory mediators, exacerbating tissue damage in sepsis4. Kupffer cells are macrophages residing in the lumen of the liver sinusoids. They eliminate bacteria, bacterial endotoxins, and microbial debris entering the liver from the gastrointestinal system through the portal vein7. However, the aberrant activation of Kupffer cells by pathogen insults, hypoxia, or endogenous inflammatory molecules leads to the release of proinflammatory mediators, such as cytokines, via active secretion or cell death to affect the surrounding hepatocytes, leading to liver injury8,9,10.
Neutrophils, the major leukocytes in the human blood, are the first responders against infection11. Neutrophil extracellular traps (NETs), which are generated via the activation of protein-arginine deiminase type-4 (PAD4), are released from neutrophils primarily to eliminate pathogens. NETs contain a plethora of inflammatory molecules, e.g., citrullinated histone H3 (cit-H3), myeloperoxidase (MPO), damage-associated molecular patterns (DAMPs), as well as serine proteases, such as neutrophil elastase (NE)3. Hence excessive NETs cause tissue injury by inducing aberrant activities in the affecting immune cells, including, but not limited to, macrophages3. Protease-activated receptor-1 (PAR-1), a ubiquitously expressed protease receptor, is involved in innate immune responses12. In macrophages, activation of PAR-1 has shown to promote M1 polarization, as evidenced by increased expression of inducible nitric oxide synthase (iNOS)13. We have recently found that Kupffer cells became proinflammatory, possessing an M1 macrophage-like phenotype in sepsis9. These M1 Kupffer cells expressed iNOS and produced proinflammatory cytokines, including IL-6 and TNF9. Thus, we hypothesized that NETs activate Kupffer cells via PAR-1, leading to liver injury during sepsis. Given the close portal venous connection between the gut and liver, gut-liver crosstalk is implicated in exacerbating hepatic injury during sepsis, though the underlying mechanisms remain poorly understood. We have recently shown that gut intraepithelial lymphocytes (IELs) became proinflammatory in septic mice and that CD112-expressing neutrophils interacted with lymphocytes in a proinflammatory manner during sepsis14,15. Therefore, we further hypothesize that neutrophils interact with gut IELs via CD112 to overproduce NETs, which then migrate to the liver via the portal vein and activate Kupffer cells.
Our study aims to elucidate the mechanisms of gut-driven hepatic injury in sepsis from the perspective of cell biology. We reveal that neutrophils primed by IELs via CD112 in the gut become NETotic and migrate to the liver, where they activate Kupffer cells through PAR-1 to cause liver injury. This study sheds light on the concept of organ-to-organ crosstalk, which could lead to the identification of potential therapeutic targets.
Results
NETs activate Kupffer cells in sepsis
We first examined the status and impact of NETs in the liver during sepsis by utilizing CLP-induced septic mice. We found that NETs+ neutrophil frequency and number were both significantly increased in the liver of septic mice compared to sham mice (Fig. 1A–C). We also observed increased deposition of NETs in the liver tissue histologically (Supplementary Fig. 1). It is known that PAD4−/− neutrophils are defective in forming NETs upon stimulation due to the inability of catalytic histone citrullination. Therefore, PAD4−/− mice can be used as a negative control for NET-mediated experiments16. Thus, we assessed Kupffer cell activation in WT and PAD4−/− mice by using the M1 marker, i.e., iNOS. iNOS expression in Kupffer cells was significantly increased in WT mice subjected to sepsis. However, Kupffer cell iNOS expression was significantly decreased in PAD4−/− septic mice compared to WT septic mice, indicating that NETs accounted for the activation of Kupffer cells in sepsis (Fig. 1D, E). We then assessed the impact of NETs on Kupffer cells directly in vitro. Kupffer cells were treated by various doses of NETs for different durations. NETs increased the release of IL-6 and TNF from Kupffer cells in a time- and dose-dependent manner (Fig. 1F–I). We further evaluated the impact of NET-induced Kupffer cell activation on hepatocytes. Hepatocytes were cultured in the conditioned media of NET-challenged Kupffer cells and cell death was assessed in hepatocytes. The conditioned media of NET-challenged Kupffer cells significantly increased cell death in hepatocytes compared to PBS, NETs, or the conditioned media of PBS-challenged Kupffer cells, indicating that hepatocytes were injured by NET-activated Kupffer cells rather than directly by NETs (Fig. 1J, K). Taken together, NETs are increased in the liver during sepsis to activate Kupffer cells, leading to hepatocyte injury.

Sepsis was induced in mice by CLP, and the liver was harvested 20 h after the surgery to evaluate NETs and iNOS+ Kupffer cells by flow cytometry. A Representative dot plots, B frequency, and C number (no.) of neutrophils (PMNs) forming NETs in sham and CLP mice. Experiments were performed 3 times, and all data were used for analysis. Data are expressed as mean ± SEM (n = 6 samples/group) and compared by paired two-tailed Student’s t test. P values: 1.06E-2 (B); 1.97E-2 (C). *p < 0.05 vs. Sham. D Representative dot plots and histograms and E frequency of iNOS+ Kupffer cells in sham, WT CLP, and PAD4−/− CLP mice. Experiments were performed 3 times, and all data were used for analysis. Data are expressed as mean ± SEM (n = 6 samples/group) and compared by one-way ANOVA and SNK test. P values based on the order of appearance: 5.62E-5, 5.01E-4. *p < 0.05 vs. Sham, #p < 0.05 vs. WT CLP. Kupffer cells were treated with (F, G) different doses of NETs for 20 h or 1000 ng/mL NETs for (H, I) different durations, and (F, H) IL-6 and (G, I) TNF levels in the supernatants were assessed by ELISA. Experiments were performed 2 times, and all data were used for analysis. Data are expressed as mean ± SEM (n = 4 samples/group) and compared by one-way ANOVA and SNK test. P values based on the order of appearance: 1.17E-6, 3.07E-6, 1.53E-5 (F); 4.27E-4, 1.23E-3, 7.30E-3 (G); 2.72E-2, 3.66E-3, 2.11E-6, 1.35E-4, 7.76E-4 (H); 2.22E-5, 5.88E-7, 5.24E-9, 3.18E-2, 5.38E-6, 4.07E-4 (I).*p < 0.05 vs. PBS, #p < 0.05 vs. NETs 10 ng/mL or 1 h, †p < 0.05 vs. NETs 100 ng/mL or 4 h. Hepatocytes were treated with PBS or 100 ng/mL NETs or incubated in the conditioned media (CM) of Kupffer cells subjected to 20-h treatment with PBS or 100 ng/mL NETs. After hepatocytes were cultured in the respective media for 20 h, cell death in hepatocytes was evaluated by Live/Dead staining using flow cytometry. J Representative histograms and K frequency of cell death in hepatocytes are shown. Experiments were performed 3 times, and all data were used for analysis. Data are expressed as mean ± SEM (n = 7 samples/group) and compared by one-way ANOVA and SNK test. P values based on the order of appearance: 2.38E-4, 9.18E-4, 1.01E-4. *p < 0.05 vs. PBS, #p < 0.05 vs. NETs, †p < 0.05 vs. PBS Kupffer cell CM. CLP cecal ligation and puncture, WT wild type, PAD4 protein-arginine deiminase type-4.
NETs activate Kupffer cells via PAR-1
To determine how NETs activate Kupffer cells, we conceived the notion that proteases contained in NETs have the potential to activate protease receptors expressed on Kupffer cells. NETs induced IL-6 and TNF release from Kupffer cells, while protease inhibition significantly decreased IL-6 and TNF release (Fig. 2A, B). We then evaluated the molecular interaction between NE, one of the proteases contained in NETs, and PAR-1, a ubiquitously expressed protease receptor. Computational modeling predicted that NE interacts with the propeptide region (Asp35-Leu55) of PAR-1, indicating that NE cleaves and proteolytically activates PAR-1 (Fig. 2C). Treatment with an NE inhibitor significantly decreased IL-6 and TNF release from NET-challenged Kupffer cells in a dose-dependent manner (Fig. 2D, E). A PAR-1 inhibitor significantly attenuated NET-induced IL-6 and TNF from Kupffer cells by 67% and 46%, respectively, at the high dose (Fig. 2F, G). These data indicate that NETs via NE stimulate PAR-1 to activate Kupffer cells.

Kupffer cells were treated with 100 ng/mL NETs in the presence and absence of protease inhibitor (Inh.) for 20 h. A IL-6 and B TNF levels in the supernatants were assessed by ELISA. Experiments were performed 2 times, and all data were used for analysis. Data are expressed as mean ± SEM (n = 4 samples/group) and compared by one-way ANOVA and SNK test. P values based on the order of appearance: 6.43E-7, 3.68E-2, 5.09E-6 (A); 1.14E-6, 1.21E-2, 2.60E-5 (B). *p < 0.05 vs. PBS, #p < 0.05 vs. NETs alone. C Three-dimensional (3D) computational prediction of molecular binding between NE and PAR-1. NE binds the propeptide region (Asp35-Leu55) of PAR-1. Lower binding energy (∆iG) and higher free energy of dissociation (ΔGdiss) indicate higher-affinity interaction. Kupffer cells were treated with 100 ng/mL NETs and different doses of (D, E) NE inhibitor or (F, G) PAR-1 inhibitor for 20 h. D, F IL-6 and E, G) TNF levels in the supernatants were assessed by ELISA. Experiments were performed 2 times, and all data were used for analysis. Data are expressed as mean ± SEM (n = 4/group for (D, E); n = 5/group for (F, G) and compared by one-way ANOVA and SNK test. P values based on the order of appearance: 3.34E-12, 2.13E-11, 4.99E-11, 5.21E-9, 9.9E-3, 1.05E-6 (D); 1.98E-12, 1.72E-11, 1.37E-10, 3.72E-10, 2.14E-4, 2.64E-5 (E); 6.08E-9, 8.49E-7, 6.75E-5, 1.46E-2, 5.95E-4, 3.46E-6 (F); 9.86E-7, 1.54E-6, 3.21E-5, 5.54E-3, 5.19E-3 (G). *p < 0.05 vs. PBS, #p < 0.05 vs. NETs alone. NE neutrophil elastase, PAR-1 protease-activated receptor-1.
Gut-derived neutrophils produce NETs to activate Kupffer cells in sepsis
We then pursued the origin of NETs in the liver during sepsis. We compared NETs in the systemic blood and portal vein blood taken simultaneously. Neutrophils isolated from the portal vein blood of septic mice produced increased levels of NETs than neutrophils isolated from sham mice or the systemic blood, indicating that gut-derived neutrophils produce excessive NETs (Fig. 3A–C). We cocultured systemic or portal vein neutrophils of septic mice with Kupffer cells and assessed cytokine production. Interestingly, neutrophils isolated from the portal vein of septic mice induced significantly more IL-6 and TNF production in Kupffer cells compared to the systemic neutrophils of septic mice (Fig. 3D, E). The involvement of NETs in this Kupffer cell activation was evaluated using PAD4−/− mice. Portal vein neutrophils of PAD4−/− mice induced significantly less IL-6 and TNF production in Kupffer cells compared to those of WT mice, indicating that NETs accounted for the Kupffer cell activation by gut-derived neutrophils (Fig. 3F, G). Moreover, we adoptively transferred WT or PAD4−/− portal vein septic neutrophils into WT septic mice and evaluated Kupffer cell activation. mRNA expressions of an M1 marker iNOS and proinflammatory cytokines IL-6 and TNF were significantly lower in the Kupffer cells of septic mice transferred with PAD4−/− portal vein septic neutrophils compared to WT portal vein septic neutrophils (Fig. 3H–J). Taken together, gut-trained neutrophils produce excessive NETs to activate Kupffer cells in sepsis.

Mice were subjected to CLP and the blood was drawn from the heart (systemic or Sys.) or portal vein (P.V.) to isolate neutrophils (PMNs). Neutrophils were cultured for 12 h and stained with SYTOX green (100 nM) to evaluate NETs by microscopy. A Microscopic images of NETs in systemic and portal vein neutrophils of sham and CLP mice. Original magnification, ×200; scale bar: 100 μm. Experiments were performed 3 times, and representative images are shown. B Representative dot plots following the gating strategy of NETs in Fig. 1A, C frequency of NETs+ (Cit-H3+MPO+) neutrophils assessed by flow cytometry. Experiments were performed 3 times, and all data were used for analysis. Data are expressed as mean ± SEM (n = 5 samples/group) and compared by one-way ANOVA and SNK test. P values based on the order of appearance: 1.80E-3, 1.62E-3, 1.92E-2. *p < 0.05 vs. Sham Sys., #p < 0.05 vs. Sham P.V., †p < 0.05 vs. CLP Sys. Kupffer cells were cultured with systemic or portal vein neutrophils of CLP mice for 12 h to assess D IL-6 and E TNF levels in the supernatants by ELISA. Experiments were performed 3 times, and all data were used for analysis. Data are expressed as mean ± SEM (n = 8 samples/group) and compared by one-way ANOVA and SNK test. P values based on the order of appearance: 3.06E-3, 7.43E-6, 4.27E-2 (D); 1.44E-2, 1.91E-5, 2.50E-2 (E). *p < 0.05 vs. PBS, #p < 0.05 vs. Sys. PMNs. Kupffer cells were cultured with portal vein neutrophils of WT CLP mice or PAD4−/− CLP mice for 20 h to assess F IL-6 and G TNF levels in the supernatants by ELISA. Experiments were performed 3 times, and all data were used for analysis. Data are expressed as mean ± SEM (n = 5 samples/group) and compared by one-way ANOVA and SNK test. P values based on the order of appearance: 2.83E-4, 1.36E-2 (F); 2.55E-3, 2.82E-2 (G). *p < 0.05 vs. PBS, #p < 0.05 vs. WT P.V. PMNs. Portal vein neutrophils were isolated from WT or PAD4−/− CLP mice and i.v. injected into CLP mice. 20 h after the procedure, the liver was harvested to isolate Kupffer cells. mRNA expressions of H iNOS, I IL-6, and J TNF in Kupffer cells are shown. Experiments were performed 3 times, and all data were used for analysis. Data are expressed as mean ± SEM (n = 7 samples/group) and compared by one-way ANOVA and SNK test. P values based on the order of appearance: 3.48E-6, 2.05E-4 (H); 1.02E-12, 2.38E-5, 9.44E-10 (I); 7.18E-6, 7.68E-4 (J). *p < 0.05 vs. Sham, #p < 0.05 vs. WT PMNs. CLP cecal ligation and puncture, WT wild type, PAD4 protein-arginine deiminase type-4.
Neutrophils interacting with gut IELs produce NETs via CD112
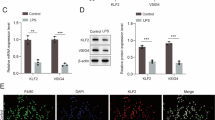
Next, we investigated how NETotic neutrophils are formed in the gut. We found an increased number of neutrophils in the gut epithelium of septic mice (Fig. 4A, Supplementary Fig. 2A). Interestingly, neutrophils were in contact with IELs in the septic gut epithelium (Fig. 4B). Therefore, we focused on the interaction between neutrophils and gut IELs for NET formation. Neutrophils were cocultured with or without IELs in the presence and absence of LPS, and NETs were then assessed. We found that neutrophils cocultured with IELs produced increased levels of NETs in the presence of LPS (Fig. 4C–E). We have previously found that neutrophils expressed CD112 upon TLR4 stimulation to interact with lymphocytes in sepsis. Thus, we investigated whether the NETs induction by neutrophil-IEL interaction is mediated via CD112. We found increased CD112+ neutrophils in the gut epithelium of septic mice (Fig. 4F, Supplementary Fig. 2B). In vitro, the expression of CD112 was increased by LPS stimulation in a dose-dependent manner (Supplemental Fig. 3). When neutrophils were cocultured with IELs and LPS, anti-CD112 Ab treatment significantly attenuated NETs compared to the vehicle and isotype control (Fig. 4G, H). Thus, our data reveal that neutrophils interact with gut IELs to produce NETs via CD112.

WT mice were subjected to CLP and the small intestines were harvested after 20 h. A The number (no.) of neutrophils in the gut epithelium. Experiments were performed 2 times, and all data were used for analysis. Data are expressed as mean ± SEM (n = 4 samples/group) and compared by paired two-tailed Student’s t test. P value: 1.11E-2. *p < 0.05 vs. Sham. B Representative microscopic images of the gut epithelium (original magnification, ×630; scale bar: 10 μm). Experiments were performed 3 times, and representative images are shown. Bone marrow-derived neutrophils (PMNs) were treated with or without LPS in the presence and absence of intraepithelial lymphocytes (IELs) at 1:1 ratio for 12 h to evaluate NETs. C Representative microscopic images from 3 independent experiments are shown (original magnification, ×200; scale bar: 100 μm). Ly6G (purple) serves as a neutrophil marker. D Representative dot plots following the gating strategy of NETs in Fig. 1A and E frequency of NETs+ (Cit-H3+MPO+) neutrophils assessed by flow cytometry are shown. Experiments were performed 3 times, and all data were used for analysis. Data are expressed as mean ± SEM (n = 6 samples/group) and compared by one-way ANOVA and SNK test. P values based on the order of appearance: 3.02E-7, 1.42E-6, 4.15E-5. *p < 0.05 vs. PBS, #p < 0.05 vs. LPS, †p < 0.05 vs. PBS+IELs. F The number of CD112+ neutrophils in the gut epithelium of sham and CLP mice. Experiments were performed 2 times, and all data were used for analysis. Data are expressed as mean ± SEM (n = 4 samples/group) and compared by paired two-tailed Student’s t test. P value: 1.11E-2. *p < 0.05 vs. Sham. Neutrophils were incubated with IELs and LPS in the presence of a vehicle (PBS, Veh.), isotype control (Iso.), or 10 μg/mL anti-CD112 Ab for 12 h to evaluate NETs by flow cytometry. G Representative dot plots following the gating strategy of NETs in Fig. 1A and H frequency of NETs+ neutrophils are shown. Experiments were performed 2 times, and all data were used for analysis. Data are expressed as mean ± SEM (n = 5 samples/group) and compared by one-way ANOVA and SNK test. P values based on the order of appearance: 6.21E-3, 2.52E-2. *p < 0.05 vs. Veh., #p < 0.05 vs. Iso. CLP cecal ligation and puncture.
Neutrophils interacting with IELs activate Kupffer cells and cause liver injury in sepsis
We further investigated the impact of neutrophil-IEL interaction on Kupffer cells, considering that gut-primed neutrophils migrate to the liver via the portal vein. Neutrophils pretreated with IELs and LPS were isolated by FACS and cocultured with Kupffer cells or adoptively transferred into septic mice (Fig. 5A). Interestingly, neutrophils pretreated with IELs and LPS significantly increased the release of IL-6 and TNF from Kupffer cells compared to the control groups (Fig. 5B, C). We then investigated the involvement of PAR-1 in this setting. A PAR-1 inhibitor significantly attenuated IL-6 and TNF release from Kupffer cells cocultured with neutrophils pretreated with IELs and LPS (Fig. 5D, E). These data demonstrate that neutrophils interacting with IELs activate Kupffer cells via PAR-1. In septic mice, the adoptive transfer of neutrophils pretreated with IELs and LPS significantly increased iNOS, IL-6, and TNF levels in Kupffer cells compared to neutrophils treated with LPS alone (Fig. 5F–H). Furthermore, the serum levels of liver enzymes AST and ALT were significantly higher in septic mice that received neutrophils with IEL pretreatment than in those that received neutrophils without IEL pretreatment (Fig. 5I, J). These Kupffer cell activation markers and liver enzymes were decreased when transferred neutrophils were pretreated with IELs in the presence of anti-CD112 Ab (Supplementary Fig. 4). Taken together, it is indicated that neutrophils interacting with IELs activate Kupffer cells to cause liver injury in sepsis.

A Neutrophils (PMNs) were pretreated with LPS alone or in the presence of IELs (IEL preTx) for 12 h and sorted by fluorescence-activated cell sorting (FACS) following the neutrophil population gating strategy in Fig. 1A. Sorted neutrophils were cocultured with Kupffer cells for 20 h or adoptively transferred into CLP mice via i.v. to collect the liver and blood after 20 h. Created in BioRender. Murao, A. (2025) https://BioRender.com/k5a1i7dB IL-6 and C TNF levels in the supernatants were assessed by ELISA. Experiments were performed 3 times, and all data were used for analysis. Data are expressed as mean ± SEM (n = 6 samples/group) and compared by one-way ANOVA and SNK test. P values based on the order of appearance: 3.99E-14, 4.10E-14, 4.63E-14, 2.73E-13 (B); 3.28E-14, 3.30E-14, 3.28E-14, 3.30E-14 (C). *p < 0.05 vs. Kupffer cells alone, #p < 0.05 vs. IEL-untreated PMNs alone, †p < 0.05 vs. IEL-pretreated PMNs alone, ‡p < 0.05 vs. Kupffer cells+IEL-untreated PMNs. Kupffer cells were incubated with neutrophils pretreated with IELs and LPS in the presence and absence of PAR-1 inhibitor (Inh.) for 20 h. D IL-6 and E TNF levels in the supernatants were assessed by ELISA. Experiments were performed 2 times, and all data were used for analysis. Data are expressed as mean ± SEM (n = 4 samples/group) and compared by one-way ANOVA and SNK test. P values based on the order of appearance: 2.85E-6, 3.61E-3, 1.65E-4 (D); 8.32E-4, 1.06E-2 (E). *p < 0.05 vs. PBS, #p < 0.05 vs. PAR-1 Inh(-). mRNA expressions of F iNOS, G IL-6, and H TNF in Kupffer cells and I AST J ALT levels in the serum of neutrophil-injected CLP mice. Experiments were performed 3 times, and all data were used for analysis. Data are expressed as mean ± SEM (n = 7 samples/group) and compared by one-way ANOVA and SNK test. P values based on the order of appearance: 3.91E-2, 4.96E-10, 2.04E-8 (F); 4.06E-2, 2.41E-5, 7.06E-3 (G); 4.59E-2, 1.49E-4, 3.97E-2 (H); 2.86E-2, 2.14E-6, 6.74E-4 (I); 8.28E-5, 4.73E-4 (J). *p < 0.05 vs. Sham, #p < 0.05 vs. IEL-untreated PMNs. CLP cecal ligation and puncture.
Discussion
In the present study, we have demonstrated that NETs, which were increased in septic liver, activated Kupffer cells via PAR-1, causing hepatic failure. We have further revealed that neutrophils interacted with gut IELs via CD112 to produce excessive NETs, and NET-forming neutrophils migrate from the gut into the liver via the portal vein to activate Kupffer cells. We discovered that a distinct cell type (IELs) in a remote organ (intestines) induces neutrophils to become hyper NETotic. These hyper NETotic neutrophils then home to the liver, interact with resident macrophages (Kupffer cells), and trigger excessive release of inflammatory cytokines, ultimately causing hepatocyte death. These findings provide novel insights into the mechanism of sepsis-induced hepatic failure from the aspect of inflammatory gut-liver crosstalk mediated by multiple immune cell types (Fig. 6). Because neutrophils perform immunosurveillance by patrolling the circulation, our data further demonstrate that neutrophil phenotype and function can be modulated by the tissue microenvironment. The concept of inter-organ crosstalk mediated by cellular interaction could broaden the perspective beyond conventional approach focusing on soluble mediators or microorganisms.

During sepsis, neutrophils interact with IELs via CD112 in the gut, resulting in increased NETosis via PAD4-mediated histone citrullination (Cit). NET-forming neutrophils migrate from the gut into the liver and activate PAR-1 on Kupffer cells by NET-contained proteases, such as NE. NET-activated Kupffer cells polarize to an iNOS-expressing M1 phenotype and produce proinflammatory mediators, such as IL-6 and TNF. This gut-liver crosstalk mediated by immune cells leads to sepsis-induced liver injury. PAD4, protein-arginine deiminase type-4; NET, neutrophil extracellular trap; PAR-1, protease-activated receptor-1; NE, neutrophil elastase; iNOS, inducible nitric oxide synthase. Created in BioRender. Murao, A. (2025) https://BioRender.com/fhkfis3.
Neutrophils are regarded to significantly contribute to the pathophysiology of sepsis. They not only act as first responders to swiftly reach to the site of infection but also subsequently reverse transendothelial migrate back to the circulation and further infiltrate into remote organs17. It has been shown that the reverse transendothelial migration of neutrophils aggravated acute lung injury in sepsis18, but little has been explored in the context of gut-liver crosstalk. Here, we have identified an underappreciated role of neutrophils in this inter-organ crosstalk in sepsis. It would also be possible that the liver neutrophils further reverse transendothelial migrate into the circulation and cause tissue injury in remote organs, such as the lungs. It has been known that the microbiome plays a significant role not only in the gut environment but also in the liver, mediating the interaction between these two organs19. The development and phenotype of IELs are affected by the gut microbiome. For instance, commensal Bacteroidetes have been shown to derive IEL differentiation in the gut, regulating the surrounding environment20. Here, we used LPS as a stimulus to mimic septic condition in vitro and demonstrated inflammatory interaction between IELs and neutrophils. It would be of interest to explore the impact of the gut microbiome on this cellular interaction and subsequent inter-organ crosstalk.
NETs are implicated in multiple organ dysfunction during sepsis. In the septic liver, NETs have been linked to platelet aggregation and tissue damage21. A previous work has also demonstrated that NETs regulate Kupffer cell M1 polarization during acute liver transplant rejection22. Consistent with these findings, we observed that NETs activated Kupffer cells, inducing M1 polarization and cytokine production in sepsis. We assessed IL-6, TNF, and iNOS expression as representative markers of Kupffer cell activation. Given the increasing use of proteomics and transcriptomics for comprehensive cellular profiling, we recently performed single-cell RNA-sequencing on septic mice, identifying significantly altered genes and pathways across various cell types23. Such comprehensive approaches will further elucidate the detailed cellular mechanisms by which septic neutrophils and NETs affect Kupffer cells.
In the present study, we focused on the role of NET-associated proteases in Kupffer cell activation. NE is known to proteolytically activate PAR-1 via N-terminal cleavage24, and our data similarly show that inhibiting NE and PAR-1 significantly reduced Kupffer cell cytokine release. While PAR-1 inhibition yielded a greater reduction than NE inhibition, suggesting contributions from other neutrophil proteases (e.g., proteinase 3, cathepsin G), neither completely abrogated cytokine production. This suggests that additional NET components, such as nuclear molecules like DNA and histones, which can activate pattern-recognition receptors, may also contribute to Kupffer cell activation. Further investigation of NET components, e.g., using DNase, and Kupffer cell sensors will provide greater insight into this inflammatory crosstalk. We revealed that the conditioned media of NET-challenged Kupffer cells induced cell death in hepatocytes. It is known that cell death can be induced by TNF25, which was found in our conditioned media. Nevertheless, Kupffer cells have the capacity to produce other cytotoxic molecules, such as reactive oxygen and nitrogen species9,10. Thus, it would be of importance to dissect the contents released from NET-challenged Kupffer cells and evaluate their effect on hepatocyte viability. Furthermore, the hepatotoxic impacts of NET-activated Kupffer cells should be tested in vivo by evaluating hepatic tissue injury and liver dysfunction in animals with a conditional knockout of hepatotoxic content in Kupffer cells.
Here, we identified a novel mechanism of NETs formation induced by neutrophil-IEL interaction via CD112. We have recently demonstrated that a novel DAMP, extracellular cold-inducible RNA-binding protein (eCIRP), upregulated CD112 expression in neutrophils via TLR415. As LPS is a potent ligand for TLR4, it is likely that, in the present study, LPS upregulated CD112 expression on neutrophils through the similar mechanism. In the previous study, we focused on the effect of neutrophils on lymphocytes and found that CD112+ neutrophils promoted Th1 polarization15. Considering that the CD112-mediated cellular interaction is known to be bidirectional, we investigated the impact of IELs, resident lymphocytes in the gut, on neutrophils. CD112 has multiple ligands, including DNAX accessory molecule-1 (DNAM-1), T cell immunoreceptor Ig tyrosine (TIGIT), and CD112R. DNAM-1 serves as an activator, and TIGIT and CD112R are suppressors26,27. Sepsis consists of early proinflammatory and later immunosuppressive phases28. Since we studied on the acute phase of septic condition, it is presumable that CD112-mediated NETs were induced by the engagement with DNAM-1. Even though CD112 blockade significantly decreased IEL-induced NETosis, it did not result in a complete inhibition. Exploring other factors could provide a more comprehensive understanding of this cellular interaction mechanism.
We revealed in the present study that neutrophils isolated from the portal vein of septic mice or cultured with IELs in vitro produced excessive NETs, suggesting NET-forming neutrophils facilitate gut-liver crosstalk. However, whether these NETotic neutrophils can retain mobility to migrate actively into the liver could be a question since NETosis is considered as a form of cell death. We evaluated NETs after culturing the portal vein-derived neutrophils for 12 h, at which time the NETs became visible. This implies that neutrophils which already released NETs were eliminated or remain in the organ due to cell death, and migrating neutrophils are the ones prone to NETosis but are still alive. In vitro, we cocultured neutrophils and IELs in closed environment and evaluated NETs after 12 h. In the physiological condition, it is presumable that neutrophils primed by IELs in the gut are prone to become NETotic, migrate through the portal vein while they are still alive, and release NETs once they arrive at the liver.
In this study, we primarily focused on the mechanism of liver inflammation and investigated the impact of neutrophil-IEL interaction and subsequent NETosis on Kupffer cells. Nonetheless, it is plausible that not all neutrophils migrate out of the gut after interacting with IELs, and some would remain in the gut to affect the local microenvironment. Similar to the liver, the gut also contains resident macrophages in the lamina propria29. Thus, NETs generated by the neutrophil-IEL interaction could also activate the gut macrophages in the same manner as Kupffer cells to induce cytokine production, causing cell death in gut epithelial cells to damage the crypt or villus. Indeed, it has been shown that serious tissue injury was observed in the gut epithelium during sepsis14. This sepsis-induced gut injury could be, at least in part, due to NETs released through the mechanism revealed in the present study.
Currently, the treatment of patients with sepsis-induced liver injury in the clinical setting is mostly limited to supportive care. This includes the supplementation of liver proteins and transfusion of fresh frozen plasma, along with respiratory and circulatory support4. Targeting the newly discovered inflammatory mechanism of sepsis-induced hepatic failure in this study could be a novel approach to overcome the current clinical situation. We have recently developed a small peptide which inhibits detrimental neutrophil-B-1a cell crosstalk by targeting NE-Siglec-G interaction. This peptide was generated based on the sequence of the interaction site between NE and Siglec-G to act as a decoy peptide30. In preclinical studies, this drug has shown promising beneficial effects on sepsis, as indicated by improved systemic inflammation and survival rates30. Similar approach could be applied to develop therapeutics for sepsis-induced liver injury by targeting CD112-mediated IEL-neutrophil interaction as well as PAR-1-mediated NET-Kupffer cell interaction, which were demonstrated in the current study.
We have demonstrated in septic mice that the adoptive transfer of IEL-interacting neutrophils activates Kupffer cells and causes liver injury. While intervening on subpopulations like IELs or directly observing cell migration in vivo is challenging, techniques such as conditional knockout or cellular tracking would strengthen our findings. In addition, unintended or off-target effects of interventions, such as inhibitors and antibodies, should be carefully considered when conducting in vivo studies. This study used the CLP model of sepsis, a widely used model that primarily mimics intra-abdominal sepsis induced by commensal bacteria. To ensure the generalizability of our findings to other forms of sepsis, future studies should utilize diverse models, including LPS administration, bacterial infection via various routes, and viral infection. Furthermore, factors such as sex and age should be considered.
In summary, we have demonstrated that neutrophils interacting with gut IELs produced excessive NETs to cause hepatic injury by activating Kupffer cells in sepsis. Targeting the inflammatory inter-cellular and inter-organ crosstalk could be potential therapy for sepsis-induced hepatic failure.
Methods
Animals
8–12-week-old male wild type (WT) C57BL/6 J mice (stock no. 000664) and PAD4−/− (B6.Cg-Padi4tm1.1Kmow/J) mice (stock no. 030315) were purchased from The Jackson Laboratory (Bar Harbor, ME). PAD4−/− mice were generated by inserting a loxP site followed by a frt-flanked neomycin resistance (neo) cassette upstream of exon 9, and a second loxP site downstream of exon 10 of the peptidyl arginine deiminase, type IV (Padi4) gene. Only male mice were used to minimize deviations based on sex, which could require an inhumane number of animals. Both WT and PAD4−/− mice were bred at The Jackson Laboratory and housed in the same specific pathogen-free environment at the Feinstein Institutes for Medical Research until experiments were performed. Mice were housed in a temperature-controlled room (20–26 °C), with a humidity level of 30–70%, under 12-h intermittent light and dark cycles and fed a standard mouse chow diet with water. Mice were euthanized rapidly and painlessly using CO₂ inhalation after experiments or at the time of sample collection. All animal experiments were performed following the National Institutes of Health guidelines for the care and use of laboratory animals. The present studies using live animals were reviewed and approved by the Feinstein Institutes for Medical Research’s Institutional Animal Care and Use Committee (IACUC) in Manhasset, NY.
Mouse model of sepsis
Intra-abdominal sepsis was induced in 8–12-week-old male WT or PAD4−/− mice by cecal ligation and puncture (CLP). Mice were anesthetized with isoflurane, and a midline abdominal incision was created. The cecum was ligated with a 4–0 silk suture 1 cm proximal from its distal extremity and punctured twice with a 22-gauge needle. The wound was then closed in layers. Sham animals underwent laparotomy without CLP. At the end of the procedure, 1 mL of normal saline was subcutaneously (s.c.) injected to avoid surgery-induced dehydration and 0.05 mg/kg buprenorphine was s.c. injected as an analgesic. 20 h after the surgery, the blood was drawn from the heart and portal vein, and the liver was harvested.
Isolation of Kupffer cells and hepatocytes
Kupffer cells were isolated from 8 to 12-week-old male mouse liver using the collagenase digestion method described below. After laparotomy, the portal vein was cannulated, and the liver was perfused with HBSS (Cat. No.: 14025092; Thermo Fisher Scientific, Waltham, MA) containing 0.5 mM EGTA (Cat. No.: E3889; MilliporeSigma, Burlington, MA) pre-warmed at 37 °C, followed by perfusion with pre-warmed HBSS-CaCl2 (1 mM) containing 0.5 mg/mL collagenase type 4 (Cat. No.: NC9919937; Worthington Biochemical, Lakewood, NJ). Perfused liver tissue was gently dispersed in a 100 mm cell culture dish using a pair of ophthalmic forceps, and the cell suspension was filtered through a 70 μm cell strainer. After centrifugation of the cell suspension at 50 × g for 2 min at 4 °C, the pellet was collected as hepatocytes and the supernatant was centrifuged at 450 × g for 5 min at 4 °C to obtain non-parenchymal cells (NPCs). NPCs were suspended in 25% Percoll (Cat. No.: 17089101; Cytiva, Marlborough, MA), layered onto 50% Percoll, and centrifuged at 850 × g for 20 min at 15 °C for gradient separation. The interface containing Kupffer cells was collected and further purified by selective adherence to cell culture plates for 3 h in RPMI 1640 medium (Cat. No.: 11875093; Thermo Fisher Scientific) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Cat. No.: A5669801, Thermo Fisher Scientific), 2 mM glutamine (Cat. No.: A2916801, Thermo Fisher Scientific), and 1% penicillin-streptomycin (Cat. No.: 15140122, Thermo Fisher Scientific).
Assessment of NETs and iNOS+ Kupffer cells in liver by flow cytometry
Single cell suspension of liver tissues was obtained by the digestion method described earlier to assess NETs and Kupffer cell iNOS expression by flow cytometry. NETs are web-like DNA structures decorated with cit-H3 and MPO. The bases of these structures remain in contact with neutrophils during their release and can be detected on the cell surface31. Cells were stained without permeabilization with APC anti-Ly6G (clone: 1A8; BioLegend, San Diego, CA), PerCP-Cy5.5 anti-CD11b (clone: M1/70; BioLegend), FITC anti-MPO (clone: 2D4; Abcam, Cambridge, MA), anti-Histone H3 (citrulline R2 + R8 + R17; cit-H3; Cat. No.: ab5103; Abcam), and PE anti-rabbit IgG (clone: Poly4064; BioLegend) antibodies (Abs). This NET assessment follows an established and validated protocol31,32. For the assessment of iNOS expression in Kupffer cells, cells were stained with LIVE/DEAD Fixable Violet Dead Cell Stain Kit (Cat. No.: L34955; Thermo Fisher Scientific), PerCP-Cy5.5 anti- CD11b Ab, and APC anti-F4/80 Ab (clone: BM8; BioLegend), and then fixed in Fluorofix buffer (Cat. No.: 422101; BioLegend), followed by intracellular staining with PE anti-iNOS Ab (clone: C-11; Santa Cruz Biotechnology, Dallas, TX) in Permeabilization Wash Buffer (Cat. No.: 421002; BioLegend). Acquisition was performed using a BD LSR Fortessa (BD Biosciences, San Jose, CA), and data were analyzed by FlowJo software (ver. 10; Tree Star, Ashland, OR). Cell numbers were calculated by using Precision Count Beads (Cat. No.: 424902; BioLegend).
Treatment of Kupffer cells with NETs
Neutrophils were isolated from the bone marrow of 8–12-week-old male WT mice using the EasySep Mouse Neutrophil Enrichment Kit (Cat. No.: 19762; STEMCELL, Vancouver, BC). Neutrophils were treated with 100 nM PMA for 12 h. Supernatants were replaced with PBS and adhered cells were detached from the well bottom by scraping and pipetting. Samples were centrifuged at 300 × g for 10 min at 4 °C to precipitate cells, leaving NETs in the supernatants. NET-containing supernatants were centrifuged at 18,000 × g for 10 min at 4 °C, and precipitated NETs were resuspended in PBS. DNA concentration of NETs was measured using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific). Kupffer cells were treated with 10, 100, 1000 ng/mL of NETs for 1, 4, 20 h in the presence of EDTA-free Protease Inhibitor Cocktail (Cat. No.: 11836170001, MilliporeSigma), 5, 50, 500 μM NE inhibitor, sivelestat sodium salt (Cat. No.: 3535; STEMCELL), or 1,10, 100 nM PAR-1 inhibitor, Parmodulin 2 (Cat. No.: HY-13965; MedChemExpress, Monmouth Junction, NJ).
Assessment of cell death in hepatocytes
Hepatocytes were treated with PBS or 1000 ng/mL NETs, which were isolated by the method described above, or incubated in the conditioned media of Kupffer cells subjected to 20-h treatment with PBS or 1000 ng/mL NETs. After being cultured in these conditions for 20 h, cell death in hepatocytes was assessed by LIVE/DEAD Fixable Violet Dead Cell Stain Kit using flow cytometry.
Computational modeling
The amino acid sequences of NE (Q3UP87) and PAR-1 (P30558) were retrieved from the UniProt database. The structural models of NE and PAR-1 were generated by using Iterative Threading ASSEmbly Refinement (I-TASSER) server [https://zhanggroup.org/I-TASSER/] based on templates identified by the threading approach to maximize percentage identity, sequence coverage and confidence33. For the generation of structure models of NE and PAR1 the AlphaFold best templates were selected. The model was refined by short molecular dynamics simulations for mild (0.6 ps) and aggressive (0.8 ps) relaxations with a 4 fs time step after structure perturbations. The refinement of the protein structure model enhanced certain parameters including Rama favored residues and decrease in poor rotamers. The protein-protein docking approach was employed to generate NE-PAR-1 complex by using the ATTRACT and InterEvDock [https://bioserv.rpbs.univ-paris-diderot.fr/services/InterEvDock3/] tools34,35. The interaction between NE and PAR-1 was calculated using the PDBePISA tool, and the complex structure was visualized using PyMOL (ver. 3.0)36.
Treatment of Kupffer cells with septic neutrophils
Neutrophils were isolated from the blood of heart and portal vein of WT and PAD4−/− mice using the EasySep Mouse Neutrophil Enrichment Kit. These neutrophils were cultured with Kupffer cells at 1:1 ratio for 20 h, the supernatants were collected to assess cytokine levels.
Isolation of intraepithelial lymphocytes (IELs)
IELs were isolated from mice following the protocol described below. The small intestines were harvested from 8 to 12-week-old male WT mice and opened immediately after the removal of fat tissues and Peyer’s patches. The contents were gently washed out with cold PBS. Small intestine tissues were incubated in RPMI 1640 medium supplemented with 2% FBS, 5 mM EDTA (Cat. No.: AM9260G; Thermo Fisher Scientific), and 1 mM DTT (Cat. No.: P2325; Thermo Fisher Scientific) at room temperature for 20 min to isolate the epithelial layer. After samples were filtered through a 100 μm mesh, density gradient centrifugation was performed. 75% Percoll was layered onto the cells suspended in 40% Percoll, and samples were centrifuged at 800 × g for 20 min at 20 °C. The interphase layer was collected to obtain IELs.
Coculture of neutrophils and IELs
Bone marrow-derived neutrophils (BMDNs) were treated with or without 100 ng/mL LPS (MilliporeSigma) in the presence and absence of intraepithelial lymphocytes (IELs) at 1:1 ratio. Neutrophils incubated with IELs and LPS were also treated with a vehicle (PBS), isotype control, or 10 μg/mL anti-CD112 Ab (Cat. No.: MAB3869; R&D Systems, Minneapolis, MN). After 12 h of coculture, NETs were evaluated by microscopy and flow cytometry, and neutrophils were sorted by fluorescence-activated cell sorting (FACS) using a BD FACSAria (BD Biosciences). Sorted neutrophils were cultured with Kupffer cells in the presence and absence of 100 nM PAR-1 inhibitor to assess cytokine production.
Assessment of neutrophil count in the gut epithelium
Cells isolated from the epithelial layer of the small intestines without gradient separation were stained with APC anti-Ly6G and BV421 anti-CD112 (clone: 829038; BD Biosciences) Abs and analyzed by flow cytometry as described above. Cell numbers were calculated by using Precision Count Beads.
Adoptive transfer of neutrophils into septic mice
8–12-week-old male WT mice were subjected to CLP and injected intravenously (i.v.) via the retro-orbital vein with neutrophils isolated from the portal vein of septic WT or PAD4−/− mice. WT CLP mice were also i.v. injected with neutrophils treated with LPS alone or cultured with IELs and LPS and FACS-sorted. 20 h after the surgery, the blood was drawn from the heart to isolate serum, and the liver was harvested to isolate Kupffer cells.
Assessment of cytokines and liver enzymes
Cell-culture supernatants were analyzed by ELISA kits for IL-6 and TNF (Cat. No.: 555240, 560478; both from BD Biosciences). Serum levels of AST and ALT were determined using specific colorimetric enzymatic assays (Cat. No.: A7561450, A7526150; Pointe Scientific, Canton, MI). Absorbance was measured on a Synergy Neo2 (Agilent Technologies, Santa Clara, CA) according to the manufacturers’ instructions.
Real-time quantitative reverse transcription PCR
RNA extraction was performed following the manufacturer’s instructions with the Illustra RNAspin Mini RNA Isolation kit (Cat. No.: 25050072; Cytiva, Marlborough, MA, USA). cDNA was synthesized using MLV reverse transcriptase (Cat. No.: 28025013; Thermo Fisher Scientific), and PCR was performed with forward and reverse primers and SYBR Green PCR Master Mix (Cat. No.: 4309155; Thermo Fisher Scientific) using a Step One Plus real-time PCR machine (Thermo Fisher Scientific). All primers were designed in-house using the Primer-BLAST tool (National Center for Biotechnology Information) and synthesized by Eurofins Genomics (Louisville, KY). Primer sequences are provided in Supplementary Table 2.
Microscopy
Cultured neutrophils were incubated with 100 nM of SYTOX Green (Cat No: S7020; Thermo Fisher Scientific) for 30 min, and then assessed under a fluorescence microscope, EVOS FL Auto Imaging System (Thermo Fisher Scientific). Gut tissue sections were stained with APC anti-Ly6G Ab and DAPI (4′,6-diamidino-2-phenylindole) and observed using an LSM900 confocal microscope (ZEISS, Oberkochen, Germany).
Statistical analysis
Data represented in the figures are expressed as mean ± SEM. Analysis of variance (ANOVA) was used for one-way comparison among multiple groups, and the significance was determined by the Student Newman-Keuls (SNK) test. The paired two-tailed Student t test was applied for two-group comparisons. Significance was considered for p ≤ 0.05 between study groups. Data analyses were carried out in GraphPad Prism graphing and statistical software (ver. 8; GraphPad Software, San Diego, CA).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are included in the Supplementary Information or available from the authors, as are unique reagents used in this Article. The raw numbers for charts and graphs are available in the Source Data file whenever possible. Source data are provided with this paper.
References
Singer, M. et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315, 801–810 (2016).
Rudd, K. E. et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the Global Burden of Disease Study. Lancet 395, 200–211 (2020).
Denning, N. L., Aziz, M., Gurien, S. D. & Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 10, 2536 (2019).
Strnad, P., Tacke, F., Koch, A. & Trautwein, C. Liver - guardian, modifier and target of sepsis. Nat. Rev. Gastroenterol. Hepatol. 14, 55–66 (2017).
Savio, L. E. B. et al. CD39 limits P2X7 receptor inflammatory signaling and attenuates sepsis-induced liver injury. J. Hepatol. 67, 716–726 (2017).
Stravitz, R. T. & Lee, W. M. Acute liver failure. Lancet 394, 869–881 (2019).
Guilliams, M. & Scott, C. L. Liver macrophages in health and disease. Immunity 55, 1515–1529 (2022).
Hirao, H., Nakamura, K. & Kupiec-Weglinski, J. W. Liver ischaemia-reperfusion injury: a new understanding of the role of innate immunity. Nat. Rev. Gastroenterol. Hepatol. 19, 239–256 (2022).
Shimizu, J., Murao, A., Lee, Y., Aziz, M. & Wang, P. Extracellular CIRP promotes Kupffer cell inflammatory polarization in sepsis. Front. Immunol. 15, 1411930 (2024).
Ju, C. & Tacke, F. Hepatic macrophages in homeostasis and liver diseases: from pathogenesis to novel therapeutic strategies. Cell Mol. Immunol. 13, 316–327 (2016).
Herro, R. & Grimes, H. L. The diverse roles of neutrophils from protection to pathogenesis. Nat Immunol. 25, 2209–2219 (2024).
Künze, G. & Isermann, B. Targeting biased signaling by PAR1: function and molecular mechanism of parmodulins. Blood 141, 2675–2684 (2023).
Wilkinson, H. et al. PAR-1 signaling on macrophages is required for effective in vivo delayed-type hypersensitivity responses. iScience 24, 101981 (2021).
Akama, Y., Murao, A., Aziz, M. & Wang, P. Extracellular CIRP induces CD4CD8αα intraepithelial lymphocyte cytotoxicity in sepsis. Mol. Med. 30, 17 (2024).
Murata, K., Murao, A., Aziz, M. & Wang, P. Extracellular CIRP Induces Novel Nectin-2+ (CD112+) neutrophils to promote Th1 differentiation in sepsis. J. Immunol. 210, 310–321 (2023).
Sørensen, O. E. & Borregaard, N. Neutrophil extracellular traps - the dark side of neutrophils. J. Clin. Investig. 126, 1612–1620 (2016).
Filippi, M. D. Neutrophil transendothelial migration: updates and new perspectives. Blood 133, 2149–2158 (2019).
Jin, H., Aziz, M., Ode, Y. & Wang, P. CIRP induces neutrophil reverse transendothelial migration in sepsis. Shock 51, 548–556 (2019).
Hsu, C. L. & Schnabl, B. The gut-liver axis and gut microbiota in health and liver disease. Nat. Rev. Microbiol. 21, 719–733 (2023).
Bousbaine, D. et al. A conserved Bacteroidetes antigen induces anti-inflammatory intestinal T lymphocytes. Science 377, 660–666 (2022).
McDonald, B. et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 129, 1357–1367 (2017).
Liu, Y. et al. Neutrophil extracellular traps regulate HMGB1 translocation and Kupffer Cell M1 polarization during acute liver transplantation rejection. Front. Immunol. 13, 823511 (2022).
Murao, A., Jha, A., Aziz, M. & Wang, P. Transcriptomic profiling of immune cells in murine polymicrobial sepsis. Front. Immunol. 15, 1347453 (2024).
Mihara, K., Ramachandran, R., Renaux, B., Saifeddine, M. & Hollenberg, M. D. Neutrophil elastase and proteinase-3 trigger G protein-biased signaling through proteinase-activated receptor-1 (PAR1). J. Biol. Chem. 288, 32979–32990 (2013).
van Loo, G. & Bertrand, M. J. M. Death by TNF: a road to inflammation. Nat. Rev. Immunol. 23, 289–303 (2023).
Xu, Z. & Jin, B. A novel interface consisting of homologous immunoglobulin superfamily members with multiple functions. Cell Mol. Immunol. 7, 11–19 (2010).
Zeng, T. et al. The CD112R/CD112 axis: a breakthrough in cancer immunotherapy. J. Exp. Clin. Cancer Res. 40, 285 (2021).
Cao, C., Yu, M. & Chai, Y. Pathological alteration and therapeutic implications of sepsis-induced immune cell apoptosis. Cell Death Dis. 10, 782 (2019).
Viola, M. F. & Boeckxstaens, G. Niche-specific functional heterogeneity of intestinal resident macrophages. Gut 70, 1383–1395 (2021).
Tan, C. et al. Neutrophils disrupt B-1a cell homeostasis by targeting Siglec-G to exacerbate sepsis. Cell Mol. Immunol. 21, 707–722 (2024).
Gavillet, M. et al. Flow cytometric assay for direct quantification of neutrophil extracellular traps in blood samples. Am. J. Hematol. 90, 1155–1158 (2015).
Jin, H. et al. Antigen-presenting aged neutrophils induce CD4+ T cells to exacerbate inflammation in sepsis. J. Clin. Investig. 133, e164585 (2023).
Yang, J. et al. The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8 (2015).
Schindler, C. E., de Vries, S. J. & Zacharias, M. iATTRACT: simultaneous global and local interface optimization for protein-protein docking refinement. Proteins 83, 248–258 (2015).
Quignot, C. et al. InterEvDock3: a combined template-based and free docking server with increased performance through explicit modeling of complex homologs and integration of covariation-based contact maps. Nucleic Acids Res. 49, W277–W284 (2021).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Acknowledgements
This work was supported by National Institutes of Health (NIH) grants R35GM118337 (P.W.), R01GM129633 (M.A.), and R01HL076179 (P.W.). BioRender software was used to prepare the protocol and finding schema.
Author information
Authors and Affiliations
Contributions
A.M. and M.A. outlined the experiments. A.M., A.J., T.K., J.S. and Y.A. performed in vitro and in vivo experiments. A.J. did computational modeling. A.M. and M.A. analyzed the data and wrote the manuscript. P.W. critically reviewed and edited the manuscript. M.A. and P.W. conceived the idea. P.W. and M.A. supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Paolo Dellabona, Gustavo Menezes and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Murao, A., Jha, A., Kato, T. et al. Gut-primed neutrophils activate Kupffer cells to promote hepatic injury in mouse sepsis. Nat Commun 16, 9657 (2025). https://doi.org/10.1038/s41467-025-65572-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65572-8
