
Abstract
Quantum tunneling is known to be mass dependent. Here we report an unexpected mass effect on tunneling reactions in isocyanic acid radical anions (HNCO•−), which exhibit negative electron affinities. The cis- and trans-isomers of HNCO•− were generated in a solid neon matrix and their interconversion and electron-detachment tunneling kinetics were investigated using infrared absorption spectroscopy. The results reveal that electron tunneling, which involves some degree of nuclear motion of HNCO•− to neutral HNCO, occurs at a slower rate than the cis-to-trans isomerization of HNCO•−, with the latter following a distinct bond angle inversion pathway driven by the heavy carbon atom. This contrasts to the typical dominance of light hydrogen atom tunneling in cis-trans isomerization systems. These findings are rationalized by instanton theory calculations. Our model, which explicitly includes the neon matrix via a QM/MM approach, reveals that the carbon-driven pathway is favored by a lower barrier and shorter tunneling distance.
Similar content being viewed by others
Introduction
Quantum tunneling is a fundamental phenomenon that allows particles, such as electrons or atoms, to pass through energy barriers that, classically, they would be unable to overcome. It is well established that the probability of a particle tunneling through a barrier decreases exponentially with the square-root of the effective mass of the particle involved in tunneling1, making it most prominent for low-mass particles like electrons and protons. Since its introduction in chemistry, quantum tunneling has been recognized as playing a crucial role in various hydrogen/proton transfer reactions2,3,4,5,6,7,8. Heavy-atom tunneling, involving elements like carbon, nitrogen, and oxygen, has also been observed in certain reactions, especially under low-temperature conditions9,10,11,12,13,14,15,16.
Isomerization is one area where tunneling plays a key role, especially in molecular systems undergoing cis-trans transformations17. While more complex mechanisms, such as bond-angle inversion (BAI), have been identified in many thermally or photochemically induced isomerization processes18,19,20,21, cis-trans isomerization reactions governed by tunneling are primarily driven by simple rotation about single bonds involving light hydrogen atoms. This phenomenon has been observed in high-lying conformers of various molecular systems, including carboxylic acids and their derivatives (such as amino acids)22,23, diacids like oxalic acid24, phenol derivatives25, hydroxycarbenes26,27, metaphosphorous acid (HOPO)28, and carboxyl radical (HOCO)29.
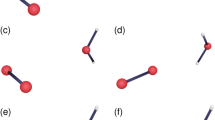
In this work, the isocyanic acid radical anion offers a fascinating system for studying mass-dependent tunneling dynamics, owing to its unique electronic structure and the presence of multiple potential tunneling mechanisms. The radical anion exhibits negative electron affinity, enabling it to release an electron and form neutral HNCO through electron tunneling. The cis- and trans- isomers of HNCO•− exhibit distinct stabilities and can interconvert via two possible tunneling pathways: one involves N−H bond rotation through hydrogen tunneling, while the other involves NCO BAI via the tunneling of the heavier carbon atom (Fig. 1). In addition to its fundamental interest, the HNCO•− anion holds practical significance in astrochemistry. Isocyanic acid is a key molecule in understanding the evolution of prebiotic chemistry, including the formation of peptides, amino acids, and proteins30,31.

Two possible tunneling pathways, namely, the N − H bond rotation and the NCO bond angle inversion are shown for the cis- to trans-HNCO•− isomerization reaction. Red dashed line: rotation axis of the N − H bond; red dotted arrow: direction of carbon motion during NCO bond-angle inversion.
Results and Discussion
The HNCO•− anions were produced through high-frequency discharge of neutral HNCO in the gas phase and then trapped in a solid neon matrix (See “Methods” for details). After 30 min of sample deposition at 3 K, two distinct groups of product absorptions, labeled as a and b, were detected. Group a displayed five absorptions at 2883.3, 2718.9, 1637.2, 1198.8 and 1090.8 cm−1, while group b showed six absorptions at 3267.4, 1645.4, 1218.8, 1120.2, 654.8 and 636.1 cm−1. The IR difference spectrum presented in Fig. 2A (comparing the spectrum acquired after the sample kept in the dark for a period of time with that obtained immediately after sample deposition) demonstrates a decline in the absorptions of group a, while those of group b and HNCO neutral (c) increased. Over time, the absorptions of group a almost disappeared, group b absorptions started to decrease, and the absorptions of neutral HNCO continued to increase (Fig. 2B). These observations suggest spontaneous conversions of species a to species b and to neutral HNCO in the dark at 3 K. Notably, as illustrated in Supplementary Fig. 1, species b can convert back to species a upon red light (625 nm) irradiation, while both groups of absorptions disappear under blue light irradiation, accompanied by an increase in neutral HNCO absorptions (Supplementary Fig. 2). These results suggest that species a and b are charged forms of HNCO32,33,34. Experiments with isotopically labeled H15NCO, HN13CO and DNCO samples were also conducted, and their IR difference spectra in selected regions are presented in Supplementary Figs 4–6. The band positions of both groups and their isotopic counterparts are listed in Table 1.

Difference spectra in the regions of 2300 − 2240, 1680 − 1600, 1240 − 1180, and 1140 − 1060 cm−1 reflecting the spontaneous isomerization of cis-HNCO•− (a) to trans-HNCO•− (b), and to HNCO (c) in a Ne matrix at 3 K. A The spectrum obtained after the sample kept in the dark for 16 h to that taken immediately after sample deposition; B The spectrum obtained after the sample had stood in the dark for 65 h to that taken after the sample had stood in the dark for 16 h. For clarity, the spectra in the ranges of 1240 − 1180, and 1140 − 1060 cm−1 are expanded 5 times along the absorbance axis.
The strongest absorption at 1637.2 cm−1 for species a and for species b at 1645.4 cm−1 exhibited significant carbon-13 isotopic frequency shifts of −42.1 cm−1, alongside minor shifts for deuterium (−8.0 and −8.9 cm−1) and nitrogen-15 (−2.1 and −2.9 cm−1), suggesting that these absorptions are due to an NCO antisymmetric stretching vibration. The band positions (1637.2 and 1645.4 cm−1) are notably lower than that of the neutral HNCO molecule at 2268.5 cm−1, suggesting that species a and b are HNCO•− anion isomers. This is comparable to the isoelectronic CO2•− anion, which also exhibits a lower OCO antisymmetric stretching vibration mode (1658.3 cm−1, Ne matrix) than that of neutral CO2 (2348.2 cm−1, Ne matrix)33.
Quantum chemistry calculations using the XYGJ-OS functional, an accurate doubly hybrid density functional, with the jul-cc-pVTZ basis set, were performed to validate the experimental assignments (See “Methods” for details). The neutral HNCO molecule exhibits a single conformer, which has a quasi-linear N = C = O structure. In contrast, the HNCO•− anion is predicted to be bent, with two isomeric forms: cis and trans. The trans-isomer is 17.5 kJ mol−1 more stable than the cis-isomer, confirming that the group a absorptions correspond to the less stable cis-isomer, while the group b absorptions are associated with the more stable trans-isomer (Table 1). The calculated vibrational frequencies and isotopic frequency shifts are listed in Table 1, which provides strong support for the experimental assignments. For the cis-isomer, the band at 1637.2 cm−1 aligns well with the theoretically calculated IR frequency of 1689.5 cm−1 for the NCO antisymmetric stretching mode (ν2). Compared to neutral HNCO (2141.0 cm−1), this mode shows a significant red-shift of 631.3 cm−1, consistent with the calculated shift of 612.5 cm−1. The 2883.3 cm−1 absorption observed in cis-isomer is attributed to the N − H stretching mode (ν1)35. The absorption at 1198.8 cm−1 is assigned to the in-plane HNC bending mode (ν3). A similar mode of the isoelectronic HNNO molecule is observed at 1166.9 cm−1 in the Xe matrix36. The absorption at 1090.8 cm−1 is attributed to the in-plane N − H bending mode (ν4). The weak absorption at 2718.9 cm−1 is assigned to a combination mode (ν2 + ν4) based on its band position and isotopic shifts. For the trans-isomer, the N − H stretching mode is observed at 3267.4 cm−1, and the 1218.8 and 1120.2 cm−1 absorptions are assigned to the in-plane HNC bending and in-plane N − H bending modes, respectively, both of which occur at slightly higher frequencies than those observed in the cis-isomer.
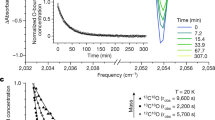
The spontaneous cis-to-trans and anion-to-neutral conversions observed in the dark at 3 K suggest that the cis-to-trans isomerization and electron detachment reactions possibly proceed via quantum mechanical tunneling, as the available energy (kBT ≈ 0.25 meV) is several orders of magnitude lower than the calculated barrier for cis-to-trans isomerization (see below) and is insufficient to drive these processes thermally. Kinetics for these reactions were investigated by monitoring the intensity changes of the IR bands corresponding to the three species over time (Fig. 3). The strongest absorption associated with the NCO antisymmetric stretching mode was used for each species. The experimental data were fitted using exponential decay and/or growth equations (See “Methods” for details), with the resulting rate constants summarized in Supplementary Tables 1, 2. The cis-to-trans isomerization reaction has a rate of 8.5 ± 0.4 × 10−5 s−1, which is larger than those of the electron detachment reactions, determined to be 5.0 ± 0.2 × 10−5 and 4.9 ± 0.2 × 10−6 s−1 for cis- and trans-HNCO•−, respectively. To assess whether the observed processes are stimulated by the IR radiation from the spectrometer, additional comparative experiments with IR filter or under distinct conditions were performed. The results are shown in Supplementary Figs 8–10, respectively, which demonstrate that globar IR radiation has negligible influence on both the cis-to-trans HNCO•− isomerization and the cis-HNCO•− → HNCO electron-detachment kinetics, while it promotes the trans-HNCO•− → HNCO detachment process.

The kinetics (squares in black) of the cis-to-trans isomerization of HNCO•− and the electron detachment involving nuclear motion to neutral HNCO measured in a Ne matrix at 3 K. The solid lines in red represent the best fits obtained using exponential decay and/or growth equations, Eqs. (4–6) shown in “Method” section. All fitted curves yielded coefficients of determination (R2 ≥ 0.999). A cis-HNCO•−, B trans-HNCO•−, and C HNCO.
To elucidate the tunneling reaction mechanism for the cis-to-trans isomerization of HNCO•− and the electron-detachment of HNCO•− to HNCO, rate constants for the isotopically labeled H15NCO•−, HN13CO•−, and DNCO•− species were measured (Supplementary Figs 4–6), and kinetic isotope effects (KIEs) were determined. The KIE of nitrogen was determined to be 1.1 ± 0.1, indicating minimal nitrogen involvement in the tunneling process. The KIE of H/D was determined to be 9.9 ± 1.0, which is significantly lower than those typically observed for hydrogen-atom tunneling-controlled cis-trans isomerization reactions in cryogenic matrices under similar conditions (for oxalic acid, kH/kD > 103)24,37. In contrast, the KIE of carbon (1.7 ± 0.1) is substantially larger than those observed in typical carbon-atom tunneling reactions38,39. For instance, the KIE of carbon in the ring-opening of cyclopropylcarbinyl radical at −100 °C was determined to be only 1.238. This KIE value suggests significant involvement of the carbon atom in the tunneling process, indicating that the isomerization reaction follows the BAI mechanism rather than the N − H bond rotation mechanism. It’s interesting to note that electron detachment tunneling reactions also exhibit notable kinetic isotope effects for carbon. For instance, the cis-HNCO•− to HNCO reaction shows a KIE of 1.3 ± 0.1, while the trans-HNCO•− to HNCO reaction has a KIE of 1.2 ± 0.1. These values suggest that the electron detachment tunneling processes involve nuclear motion of the carbon atom, which drives the significant structural changes from the bent HNCO•− to neutral HNCO with a quasi-linear N = C = O moiety.
Quantum chemistry calculations predicted that the BAI pathway for cis-to-trans isomerization features a quasi-linear intermediate with a barrier of 21.3 kJ mol−1 (Fig. 4A). For both the cis- and trans-HNCO•− isomers, electron detachment, concurrent with nuclear rearrangement, is exothermic but requires passage through the crossing between the negatively charged and the neutral quasi-diabatic states. Two crossing points were identified on the potential energy surface (Fig. 4A): one for the cis-HNCO•− anion isomer, located ~14.3 kJ mol−1 above cis-HNCO•−, and another for the trans-HNCO•− isomer, positioned ~9.2 kJ mol−1 above trans-HNCO•−. Notably, both crossing points lie below the energy of the transition state (TS) in the BAI pathway. The observed electron detachment rates, which are lower than the cis-to-trans isomerization rates, suggest that curve crossing occurs with low probability—possibly due to matrix effects localizing the electron. However, the complexity of the matrix environment makes it challenging to compute this probability.

A Potential energy profile of the negatively charged diabatic surface (blue curve, for HNCO•−) and the neutral diabatic surface (orange curve, for neutral HNCO) along the bond inversion angle (∠NCO) at the XYGJ-OS/jul-cc-pVTZ level. Key structures are shown, including the transition states (TS1 and TS2). B Potential energy profiles along the gas-phase C-inversion (blue solid line) and the H-rotation (black solid line) instanton pathways. The dotted lines represent a downhill intrinsic reaction coordinate (IRC) calculation connecting the instanton endpoint to the trans minimum. In the unit of the (mass-weighted) tunneling path length, amu stands for atomic mass unit. The two deep tunneling instanton trajectories are shown. The numbers indicate the percentage contribution of each atom to the total squared mass-weighted tunneling path length for the respective gas-phase pathways. C Comparison of the potential energy profiles for the C-inversion instanton pathways in the gas phase (blue solid line) and within the QM/MM model (dashed magenta line). D A cross-sectional view of the C-inversion instanton pathway embedded in the QM/MM model simulating the neon matrix. The surrounding Ne atoms are partitioned into an inner shell (light blue) flexible in the instanton calculations and a fixed outer shell (dark blue). Atomic contributions to the tunneling path within the matrix are shown.
To understand why the conventional N − H bond rotation isomerization mechanism was not observed, we extensively performed TS searches for both in-plane and out-of-plane N − H rotation mechanisms40. No TSs were found for either mechanism. The in-plane N − H rotation has a second order stationary point rather than a TS. Meanwhile, we show through a 2D scan (Supplementary Fig. 13) that out-of-plane N − H rotation is energetically unfavorable compared to BAI, which might be rationalized as follows. Orbital analysis reveals that in the planar cis- and trans-HNCO•− isomers, the nitrogen atom adopts sp² hybridization. If the isomerization were to occur via N − H bond rotation, the hybridization would need to transition to sp3 hybridization at the pseudo-TS, where the hydrogen atom is oriented perpendicular to the NCO plane. This change in hybridization necessitates the breaking of the N − C π bond, which is energetically unfavorable. In contrast, along the BAI pathway, no change in bonding occurs. Furthermore, the unpaired electron of the HNCO•− radical anion occupies an NCO anti-bonding π* orbital, which weakens both the N − C and C − O bonds compared to those in neutral HNCO, making this pathway energetically favorable. The combination of these two factors makes the isomerization mechanism via N − H bond rotation in HNCO•− quite unusual.
Nevertheless, a reaction with an energy barrier of 21.3 kJ mol−1 would not occur classically at cryogenic temperatures without the assistance of tunneling. To explore tunneling in HNCO•−, we performed ring-polymer instanton theory41,42,43 calculations (see Supplementary Materials for the full computational details and discussions). Instantons reveal significant carbon tunneling on the BAI pathway (Fig. 4B). Specifically, carbon contributes about 64.7% of the squared mass-weighted tunneling path length (defined as \({\sum }_{i,j}{m}_{j}{({x}_{{ij}}-{x}_{i+1,j})}^{2}\,\), where i and j are indices for the instanton beads and atoms, respectively), while hydrogen, nitrogen, and oxygen contribute relatively small percentages of 11.5%, 8.5% and 15.3%, respectively. Interestingly, due to corner-cutting, instantons for the N − H bond rotation pathway also exist (Fig. 4B), despite that the classical TS does not exist. Although the barrier of the BAI pathway via carbon tunneling is less than half of that of the N − H bond rotation pathway via hydrogen tunneling, carbon’s mass is 12 times larger than that of hydrogen. According to the Jeffreys-Wentzel-Kramers-Brillouin (JWKB) semiclassical picture of tunneling, the tunneling probability depends not only on the barrier height, but also on the effective mass and tunneling distance of the atoms involved1,9. Carbon tunnels only 0.54 Å in the BAI process, which is considerably shorter than the hydrogen tunneling distance of 1.43 Å in the N − H bond rotation process. As a result, as shown in Fig. 4B, the mass-weighted tunneling path lengths (\(\sqrt{{\sum }_{i,j}{m}_{j}{\left({x}_{{ij}}-{x}_{i+1,j}\right)}^{2}\,}\)) for both pathways are similar (at ~2 amu1/2 Å), with the shorter tunneling distance of carbon compensating for its greater mass.
The calculated reaction rate constants using the ring-polymer instanton theory and the classical transition state theory, for temperatures ranging from 30 to 100 K, are compared in Supplementary Fig. 14. The instanton rate constants for the isomerization reaction via carbon atom tunneling along the NCO BAI pathway remain near-temperature-independent below 50 K (Supplementary Tables 7, 8). These rates are many orders of magnitude higher than those calculated using classical transition state theory, indicating that tunneling dominates the reaction at low temperatures. Instanton theory predicts that the tunneling rate constant of BAI is nearly three orders of magnitude faster than that of the N − H bond rotation, suggesting that BAI is the more favorable pathway. Furthermore, instanton theory estimates a very large H/D KIE (~105) for the N − H bond rotation pathway (Supplementary Tables 7–9), which contrasts sharply with the experimentally observed value of only 9.9 ± 1.0. In contrast, the KIEs predicted by instanton theory for the BAI mechanism align semi-quantitatively with the experimental values (Table 2).
Despite the good semi-quantitative agreement between the theoretical and experimental KIEs, the instanton rates calculated in the gas phase at the DFT level exceed the observed rates by six orders of magnitude. Two primary factors may account for this discrepancy: the accuracy of the electronic structure and the neglect of the solid neon matrix environment. To understand their impact, firstly, we performed multireference calculations. As detailed in Supplementary Materials, while the cis- and trans-HNCO•− isomers do not exhibit multireference characteristics, strong multireference effects appear along the isomerization pathway. Supplementary Fig. 12 shows that multireference methods predict higher barriers than both DFT and CCSD(T). After applying a correction at the MRCI level using the dual-level approach44, the instanton rates decrease by over three orders of magnitude, bringing them much closer to the experimental results (Table 2). Secondly, to quantitatively assess the influence of the matrix environment on tunneling, we performed instanton calculations that explicitly model the HNCO•− anion embedded within a neon lattice using a QM/MM setup45 (see Supplementary Materials for full details). The results show that the presence of the neon matrix dramatically suppresses the tunneling rate to 2.2 × 10−3 s−1, a value significantly closer to the experimental observation. Analysis shows that this substantial rate reduction is caused by the matrix environment altering the instanton pathway, elongating the tunneling path meanwhile increasing the potential energy along the path (Fig. 4C). This finding provides direct theoretical evidence for the key role of the neon matrix and is consistent with the established theory of dissipative quantum tunneling, which predicts that the environment can suppress tunneling rates46,47. The small remaining discrepancy can be attributed to a combination of the aforementioned multi-reference character, the idealizations within the matrix model, and perhaps the approximations in instanton theory.
This study uncovers a counterintuitive mass dependence in quantum tunneling during the cis-trans isomerization and electron detachment of isocyanic acid radical anions. Contrary to conventional expectations, electron tunneling involving some degree of nuclear motion proceeds more slowly than pure nuclear tunneling. Nuclear tunneling follows an unconventional BAI pathway driven by the movement of the carbon atom, rather than the anticipated N − H bond rotation dominated by hydrogen tunneling. Quantum chemistry calculations combined with instanton theory reveal that the carbon-driven pathway has a lower barrier and a shorter tunneling distance than the N − H bond rotation pathway. The energetic and geometric advantages compensate for carbon’s higher mass, enhancing tunneling probability. Instanton calculations explicitly modeling the matrix environment using a QM/MM approach quantitatively demonstrate the crucial role of the matrix in slowing down tunneling, bridging the gap between the rates predicted in the gas-phase and the experiment. These findings challenge the conventional light-particle dominance in quantum tunneling, underscoring the critical role of molecular geometry and electronic structure in determining tunneling efficiency. This study sheds light on the competing tunneling mechanisms of heavy and light particles, with implications for astrochemical environments where HNCO-derived species may drive low-temperature molecular evolution via previously unrecognized heavy-atom tunneling pathways.
Methods
Experimental details
Matrix-isolation spectroscopy
The isocyanic acid anion (HNCO•−) was prepared by high-frequency discharge of the HNCO/Ne mixtures. The HNCO/Ne gas stream was subjected to discharge from a Tesla coil. The tip of the Tesla coil was connected to a copper cap on one end of a quartz tube extending into the vacuum chamber. The other end of the quartz tube was connected to a copper tube with ground potential. Discharge takes place between the cap and the copper tube. The discharged products were deposited onto a cryogenic gold-plated copper block, which was maintained at 3 K with a closed-cycle helium refrigerator. After 30 min of sample deposition at 3 K, IR absorption spectra in the mid-infrared region (4000 − 450 cm−1) were recorded with a Bruker Vertex 70 V spectrometer with a 0.5 cm−1 resolution using a liquid nitrogen cooled broad band HgCdTe (MCT) detector. Bare mirror backgrounds, recorded prior to sample deposition, were used as references for processing the sample spectra. The spectra were subjected to baseline corrections to compensate for infrared light scattering and interference patterns. For selected samples, photoexcitation (625 nm) was performed through a quartz window mounted on the assembly. The HNCO/Ne mixtures were prepared in a stainless-steel vacuum line using a standard manometric technique.
Sample preparation
HNCO was prepared by heating a mixture of stearic acid (0.57 g, 2 mmol) and sodium cyanate (0.13 g, 2 mmol) at 90 °C in a glass vessel (25 mL), which was connected to a dynamic vacuum line (0.1 Pa) through a poly (tetrafluoroethylene) Young-valve48. All of the volatile products were condensed in a liquid nitrogen trap and then purified by fractional distillation through three successive cold U-traps at −80, −135, and −196 °C. Pure isocyanic acid was retained in the middle trap, and its quality was checked with gas-phase IR spectroscopy (Bruker, Tensor 27). Isotopically-labeled HN13CO and H15NCO were prepared from 13C and 15N-labelled potassium cyanate, which were synthesized as a precipitate by heating 13C and 15N-labeled urea with potassium phenoxide in 4-methyl-2-pentanol at 135 °C for 2 h, followed by filtration, washing with cold ethanol, and drying49. DNCO was prepared by allowing a saturated solution of potassium cyanate in D2O to drop onto a saturated aqueous solution of phosphoric acid (85% D3PO4 in D2O) under vacuum50.
Kinetic measurement
For the kinetic measurements, the matrix was carefully shielded from visible light and maintained at the lowest achievable temperature of the cryostat (~3 K). IR spectra were recorded at 30-min intervals over a total period of 90 h. The integrated areas of the absorption bands corresponding to the NCO antisymmetric stretching modes were extracted from the corresponding IR difference spectra (ΔA) and used for the kinetic analysis. To ensure clarity, the integrated band intensities were normalized to the integral of the corresponding peaks.
The experimental results show that the cis-HNCO•− anion (a) can spontaneously convert to the more stable trans-HNCO•− anion isomer (b) via nuclear tunneling, as described in reaction (1). Additionally, both anion isomers can undergo electron detachment to form the neutral HNCO molecule (c), as represented in reactions (2) and (3).
The entire process can be described by three rate constants: k1, k2, and k3 corresponding to reactions (1) to (3), respectively. Since each species participates in two independent reactions, the experimentally measured kinetic curves for each species were fitted using double exponential decay/growth equations, as shown in Eqs. (4–6):
We first fitted the experimental data for trans-HNCO•− (b) with the double exponential decay equation (Eq. 4) to determine the rate constants k1 and k2. Using k1, the rate constant k3 for the conversion from cis-HNCO•− (a) to HNCO (c) was obtained by fitting the experimental kinetic decay curve of cis-HNCO•− with Eq. (5). To verify the accuracy of k2 and k3, the coefficients of determination (R2) were obtained by fitting the kinetic growth curve of HNCO (c) using Eq. (6), where k2 and k3 had already been determined from Eqs. (4) and (5), respectively. All fitted curves yielded coefficients of determination (R2 ≥ 0.999), confirming the reliability of the kinetic rate constants.
Kinetic measurements for the isotopologues HNCO•−, H15NCO•− and HN13CO•− were conducted in duplicate. The rate constants for each species from the different experiments are listed in Supplementary Tables 1, 2, with the average values used to calculate the kinetic isotope effect (KIE) values.
Computation details
Electronic structure calculations
All quantum chemistry calculations were performed with the Gaussian 09 program51, locally modified to implement the XYGJ-OS doubly hybrid density functional52, and the Molpro 2019 package53. Unless specified otherwise, geometry optimizations were carried out using the XYGJ-OS functional with the jul-cc-pVTZ basis set54, as this functional showed the best agreement with experimental results among several tested double-hybrids. Harmonic and anharmonic vibrational frequencies were calculated at this level.
Instanton rate calculations
Quantum tunneling rates were computed using ring-polymer instanton theory41,42,43, which provides a practical path-integral formulation of semiclassical instanton theory55. Instanton pathways, which represent the optimal tunneling paths, are first-order saddle points on the ring-polymer potential energy surface. This theory inherently accounts for zero-point energy and corner-cutting effects.
Ab initio instanton calculations were performed using a Python program developed based on the instanton program of Prof. J. O. Richardson's. The instanton rates were converged with respect to the number of beads, and all the instanton pathways were optimized to a convergence criterion of 2.6 kJ mol−1 Å−1 on the total gradient norm. For gas-phase calculations, a dual-level approach44 was employed: the pathway was optimized at the XYGJ-OS level, and the action was subsequently refined with single-point energy corrections at the CCSD(T) and MRCI levels. The temperature-independent plateau rates below 50 K were used for comparison with experimental results.
Modeling of the neon matrix environment
To quantitatively assess the influence of the neon matrix, a quantum mechanics/molecular mechanics (QM/MM) approach was adapted for instanton calculations.
QM/MM Crystal Model Construction: The model was constructed from 88 Ne atoms surrounding the HNCO•− anion within a face-centered cubic (fcc) lattice. This set of atoms was partitioned into a flexible inner shell of 22 Ne atoms and a fixed outer shell of 66 Ne atoms to simulate the bulk lattice while allowing for local relaxation.
QM/MM Energy Decomposition Scheme: The total potential energy of the system was calculated as a sum of two components, \({E}_{{{\rm{total}}}}={E}_{{{\rm{QM}}}+{{\rm{Pauli}}}}+{E}_{{{\rm{vdW}}}}\). The primary term, \({E}_{{{\rm{QM}}}+{{\rm{Pauli}}}}\), was calculated with Gaussian 09 and includes the full quantum mechanical energy of the HNCO•− anion (XYGJ-OS/AVTZ) and its electrostatic and Pauli repulsion interactions with the 88 Ne atoms, which were represented by repulsive effective core potentials (ECPs). The ECP parameters were benchmarked against a small full-quantum cluster model to ensure accuracy. The missing long-range van der Waals dispersion interactions, \({E}_{{{\rm{vdW}}}}\), were computed separately using the LAMMPS package56 with a classical Lennard-Jones potential.
Data availability
The data supporting the findings of this study are available in the ZENODO databases (https://doi.org/10.5281/zenodo.17066317) and (https://doi.org/10.5281/zenodo.17393768). Further data are available from the corresponding author upon request.
References
Razavy, M. et al. Quantum Theory of Tunneling. (World Scientific, 2003).
Cha, Y., Murray, C. J. & Klinman, J. P. Hydrogen tunneling in enzyme reactions. Science 243, 1325–1330 (1989).
Kohen, A., Cannio, R., Bartolucci, S. & Klinman, J. P. Enzyme dynamics and hydrogen tunnelling in a thermophilic alcohol dehydrogenase. Nature 399, 496–499 (1999).
Hiraoka, K., Sato, T. & Takayama, T. Tunneling reactions in interstellar ices. Science 292, 869–870 (2001).
Schreiner, P. R. et al. Methylhydroxycarbene: tunneling control of a chemical reaction. Science 332, 1300–1303 (2011).
Meisner, J. & Kästner, J. Atom tunneling in chemistry. Angew. Chem. Int. Ed. 55, 5400–5413 (2016).
Constantin, T. et al. Halogen-atom and group transfer reactivity enabled by hydrogen tunneling. Science 377, 1323–1328 (2022).
Wild, R., Nötzold, M., Simpson, M., Tran, T. D. & Wester, R. Tunnelling measured in a very slow ion–molecule reaction. Nature 615, 425–429 (2023).
Borden, W. T. Reactions that involve tunneling by carbon and the role that calculations have played in their study. WIREs Comput. Mol. Sci. 6, 20–46 (2016).
Castro, C. & Karney, W. L. Heavy-atom tunneling in organic reactions. Angew. Chem. Int. Ed. 59, 8355–8366 (2020).
Nunes, C. M., Knezz, S. N., Reva, I., Fausto, R. & McMahon, R. J. Evidence of a nitrene tunneling reaction: Spontaneous rearrangement of 2-formyl phenylnitrene to an imino ketene in low-temperature matrixes. J. Am. Chem. Soc. 138, 15287–15290 (2016).
Wu, Z. et al. Fast heavy-atom tunneling in trifluoroacetyl nitrene. Angew. Chem. Int. Ed. 56, 15672–15676 (2017).
Nunes, C. M., Eckhardt, A. K., Reva, I., Fausto, R. & Schreiner, P. R. Competitive nitrogen versus carbon tunneling. J. Am. Chem. Soc. 141, 14340–14348 (2019).
Choudhury, A. et al. Condensed-phase isomerization through tunnelling gateways. Nature 612, 691–695 (2022).
Zhou, Y. et al. Quantum tunneling in peroxide O–O bond breaking reaction. J. Am. Chem. Soc. 145, 8817–8821 (2023).
Zhou, Y., Fan, W., Tang, J., Fang, W. & Zhou, M. Heavy-atom tunneling in ring-closure reactions of beryllium ozonide complexes. J. Am. Chem. Soc. 146, 26719–26725 (2024).
Fausto, R., Ildiz, G. O. & Nunes, C. M. IR-induced and tunneling reactions in cryogenic matrices: the (incomplete) story of a successful endeavor. Chem. Soc. Rev. 51, 2853–2872 (2022).
Canfield, P. J. et al. A new fundamental type of conformational isomerism. Nat. Chem. 10, 615–624 (2018).
Greb, L., Eichhöfer, A. & Lehn, J.-M. Synthetic molecular motors: thermal N inversion and directional photoinduced C=N bond rotation of camphorquinone imines. Angew. Chem. Int. Ed. 54, 14345–14348 (2015).
Adam, A. & Haberhauer, G. Switching process consisting of three isomeric states of an azobenzene unit. J. Am. Chem. Soc. 139, 9708–9713 (2017).
Wu, Z. et al. Photoinduced sulfur–nitrogen bond rotation and thermal nitrogen inversion in heterocumulene OSNSO. J. Am. Chem. Soc. 140, 1231–1234 (2018).
Pettersson, M., Maçôas, E. M. S., Khriachtchev, L., Fausto, R. & Räsänen, M. Conformational isomerization of formic acid by vibrational excitation at energies below the torsional barrier. J. Am. Chem. Soc. 125, 4058–4059 (2003).
Bazsó, G., Najbauer, E. E., Magyarfalvi, G. & Tarczay, G. Near-infrared laser induced conformational change of alanine in low-temperature matrixes and the tunneling lifetime of its conformer VI. J. Phys. Chem. A 117, 1952–1962 (2013).
Schreiner, P. R. et al. Domino tunneling. J. Am. Chem. Soc. 137, 7828–7834 (2015).
Roque, J. P. L. et al. Switching on H-tunneling through conformational control. J. Am. Chem. Soc. 143, 8266–8271 (2021).
Bernhardt, B., Ruth, M., Eckhardt, A. K. & Schreiner, P. R. Ethynylhydroxycarbene (H–C≡C–C–OH). J. Am. Chem. Soc. 143, 3741–3746 (2021).
Quanz, H. et al. Identification and reactivity of s-cis,s-cis-dihydroxycarbene, a new [CH2O2] intermediate. J. Am. Chem. Soc. 142, 19457–19461 (2020).
Qian, W. et al. Hydrogen-atom tunneling in metaphosphorous acid. Chem. Eur. J. 26, 8205–8209 (2020).
Ryazantsev, S. V., Feldman, V. I. & Khriachtchev, L. Conformational switching of HOCO radical: selective vibrational excitation and hydrogen-atom tunneling. J. Am. Chem. Soc. 139, 9551–9557 (2017).
Haupa, K. A., Tarczay, G. & Lee, Y.-P. Hydrogen abstraction/addition tunneling reactions elucidate the interstellar H2NCHO/HNCO ratio and H2 formation. J. Am. Chem. Soc. 141, 11614–11620 (2019).
Zhong, J. et al. Solvation and hydrolysis reaction of isocyanic acid at the air–water interface: a computational study. J. Am. Chem. Soc. 144, 5315–5322 (2022).
Zhou, M., Andrews, L. & Bauschlicher, C. W. Spectroscopic and theoretical investigations of vibrational frequencies in binary unsaturated transition-metal carbonyl cations, neutrals, and anions. Chem. Rev. 101, 1931–1962 (2001).
Thompson, W. E. & Jacox, M. E. The vibrational spectra of CO2+, (CO2)2+, CO2−, and (CO2)2− trapped in solid neon. J. Chem. Phys. 111, 4487–4496 (1999).
Jiang, X. et al. Spectroscopic identification and bonding properties of HNCOCa+: a matrix isolation and computational study. Chin. J. Chem. Phys. 37, 361–368 (2024).
Thompson, C. A., Andrews, L., Martin, J. M. L. & El-Yazal, J. Infrared spectra of boron-ammonia reaction products in solid argon. J. Phys. Chem. 99, 13839–13849 (1995).
Laursen, S. L., Delia, A. E. & Mitchell, K. Reaction of NH (X3Σ−) with NO in xenon matrix: Infrared detection of the HNNO intermediate. J. Phys. Chem. A 104, 3681–3692 (2000).
Maçôas, E. M. S., Khriachtchev, L., Pettersson, M., Fausto, R. & Räsänen, M. Rotational isomerization of small carboxylic acids isolated in argon matrices: Tunnelling and quantum yields for the photoinduced processes. Phys. Chem. Chem. Phys. 7, 743–749 (2005).
Gonzalez-James, O. M. et al. Experimental evidence for heavy-atom tunneling in the ring-opening of cyclopropylcarbinyl radical from intramolecular 12C/13C kinetic isotope effects. J. Am. Chem. Soc. 132, 12548–12549 (2010).
Nunes, C. M., Reva, I., Kozuch, S., McMahon, R. J. & Fausto, R. Photochemistry of 2-formylphenylnitrene: A doorway to heavy-atom tunneling of a benzazirine to a cyclic ketenimine. J. Am. Chem. Soc. 139, 17649–17659 (2017).
Wang, X.-G. & Carrington, T. Jr. A variational calculation of vibrational levels of vinyl radical. J. Chem. Phys. 152, 204311 (2020).
Richardson, J. O. & Althorpe, S. C. Ring-polymer molecular dynamics rate-theory in the deep-tunneling regime: Connection with semiclassical instanton theory. J. Chem. Phys. 131, 214106 (2009).
Kästner, J. Theory and simulation of atom tunneling in chemical reactions. WIREs Comput. Mol. Sci. 4, 158–168 (2014).
Richardson, J. O. Ring-polymer instanton theory. Int. Rev. Phys. Chem. 37, 171–216 (2018).
Meisner, J. & Kästner, J. Dual-level approach to instanton theory. J. Chem. Theory Comput. 14, 1865–1872 (2018).
Marefat Khah, A., Reinholdt, P., Olsen, J. M. H., Kongsted, J. & Hättig, C. Avoiding electron spill-out in QM/MM calculations on excited states with simple pseudopotentials. J. Chem. Theory Comput. 16, 1373–1381 (2020).
Rips, I. & Pollak, E. Quantum Kramers model: solution of the turnover problem. Phys. Rev. A 41, 5366–5382 (1990).
Ankerhold, J. & Pollak, E. Dissipation can enhance quantum effects. Phys. Rev. E 75, 041103 (2007).
Drozdoski, W. S., Baronavski, A. P. & McDonald, J. R. Photodissociation of HNCO at 193 nm. NH(a1Δ) internal energy distribution and reaction rate with O2. Chem. Phys. Lett. 64, 421–425 (1979).
Filer, C. N. & Peng, C. T. An alternative and simple synthesis of [14C]potassium cyanate. J. Label. Compd. Radiopharm. 63, 240–242 (2020).
Sanov, A., Droz-Georget, T. H., Zyrianov, M. & Reisler, H. Photofragment imaging of HNCO decomposition: Angular anisotropy and correlated distributions. J. Chem. Phys. 106, 7013–7022 (1997).
Gaussian 09, Revision D.01 Frisch, M. J. et al. Gaussian 09, Revision A. 02, Gaussian. Inc, Wallingford CT. (2013).
Zhang, I. Y., Xu, X., Jung, Y. & Goddard, W. A. A fast doubly hybrid density functional method close to chemical accuracy using a local opposite spin ansatz. Proc. Natl Acad. Sci. 108, 19896–19900 (2011).
Werner, H.-J., Knowles, P. J., Knizia, G., Manby, F. R. & Schütz, M. Molpro: a general-purpose quantum chemistry program package. WIREs Comput. Mol. Sci. 2, 242–253 (2012).
Papajak, E. & Truhlar, D. G. Convergent partially augmented basis sets for post-Hartree−Fock calculations of molecular properties and reaction barrier heights. J. Chem. Theory Comput. 7, 10–18 (2011).
Miller, W. H. Semiclassical limit of quantum mechanical transition state theory for nonseparable systems. J. Chem. Phys. 62, 1899–1906 (1975).
Thompson, A. P. et al. LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 271, 108171 (2022).
Acknowledgements
The authors thank Prof. D. H. Zhang, K. P. Liu, Eli Pollak, and D. Q. Xie for very helpful discussions and suggestions, and Prof. J. O. Richardson for sharing the software for instanton calculations. This work is supported by the National Natural Science Foundation of China (22321003 (M. Z.)).
Author information
Authors and Affiliations
Contributions
J.T., L.W., X.J. and Y. Z. did the matrix isolation experiments. X.Z. contributed to data collection and analysis of the experimental results. L.W. and M.Z. contributed to conception and design of experiments, data analysis and interpretation, and drafting the manuscript. W. Fan, B.S. and W. Fang carried out the calculations. W. Fan and W. Fang contributed to conception and analysis of the theoretical results and drafting the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors have no competing interests.
Peer review
Peer review information
Nature Communications thanks Gereon Niedner-Schatteburg, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tang, J., Fan, W., Wang, L. et al. Competing quantum tunneling processes of heavy and light particles in isocyanic acid radical anions. Nat Commun 16, 10687 (2025). https://doi.org/10.1038/s41467-025-65720-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65720-0
