
Abstract
The selective incorporation of single atoms into molecular frameworks remains a fundamental challenge in synthetic chemistry. While atom transfer reactions offer promising solutions, they require reagents with precisely tunable reactivity and excellent chemoselectivity. Herein, we report pyridinium ylides as photocatalytic oxygen atom and nitrogen group transfer reagents that combine a unique reaction mode with practical advantages including bench stability and operational simplicity. These ylides form reactive triplet diradicals via reversible photocatalytic activation, enabling selective formation of aziridines, epoxides, and oxo- and imino-transfer products at room temperature in open air. Their controlled reactivity allows selective transformations across diverse substrates, proving particularly valuable for late-stage functionalization. Mechanistic investigations combining experimental and computational methods reveal a triplet-triplet energy transfer pathway proceeding through a carbocation intermediate.
Similar content being viewed by others
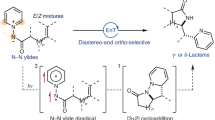
Introduction
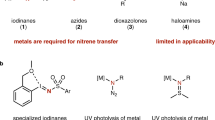
The strategic manipulation of atoms—their installation, utilization, and removal—is fundamental to constructing complex molecular targets. In this context, atom transfer reactions serve as essential transformations in organic synthesis, enabling the selective incorporation of atoms into molecular frameworks. The success of these reactions critically depends on developing reagents that strike a balance between sufficient reactivity, broad applicability, and chemoselectivity. Traditional approaches utilize highly reactive intermediates such as nitrenes and oxenes; however, these methods often require external oxidants and harsh conditions while suffering from poor atom economy, limiting their practical utility1,2. Traditional approaches to these challenges have centered on transition metal catalysis (Fig. 1A), particularly Fe and Mn systems, which offer precise control over reactivity and selectivity3. While these metals enable efficient reactions through reversible formation of reactive intermediates at their core, their toxicity, high cost, and demanding reaction conditions have motivated the search for alternative methods.

A Schematic representation of the atom transfer reactions and strategies. B This work: development of pyridinium ylides as photocatalytic O-atom and N-group transfer reagents enabled by triplet–triplet energy transfer.
Recent advances in photocatalysis have provided an alternative platform for atom transfer under mild and environmentally benign conditions, circumventing many limitations associated with metal-based methods3. Current photochemical strategies utilize a range of precursors, including hydroxamates, azides, and iminoiodanes for nitrogen-group transfer4,5,6, and thiophene-oxides and periodates for oxygen-atom transfer7,8. Despite their utility, these systems often suffer from poor thermodynamic stability, require excess coupling partners to trap transient intermediates, and raise safety concerns. Additionally, controlling the reactivity of free reactive intermediates is inherently challenging, often leading to unselective reactions. Consequently, developing stable, selective, and practical reagents for photocatalytic atom transfer remains a significant challenge.
Inspired by transition metal catalysis, we sought to develop an organic atom transfer reagent capable of reversible activation under photocatalytic conditions. Previous studies of triplet-state reactivity in alkenes and ketones have primarily explored [n + 2] cycloadditions9,10,11. However, these systems lack a crucial requirement for efficient atom transfer: the combination of photoinduced reversible radical formation with an effective leaving group. N-functionalized pyridinium salts emerged as promising candidates for redox-triggered transformations due to their bench stability and synthetic accessibility. These compounds enable functional group transfer through single-electron reduction-mediated release of radical fragments12,13,14. Nevertheless, the irreversible nature via single-electron reduction inherently leads to substrate limitations, such as being restricted to styrene derivatives, poor functional group tolerance, and low efficiency.
Our previous studies revealed an alternative approach using pyridinium ylides, which form triplet diradicals via reversible photocatalytic energy transfer. In these diradicals, the unpaired electrons delocalize between the amide and pyridine moieties, enabling selective pyridyl lactamization15. Inspired by this reversible activation mode, which resembles transition metal catalysis, and the pyridine group’s ability to act as a leaving group, we hypothesized that pyridinium ylides would serve as effective precursors for controlled atom transfer reactions. Herein, we report pyridinium ylides as a versatile photocatalytic platform for O-atom and N-group transfer through triplet-triplet energy transfer rather than an irreversible redox pathway, enabling selective formation of aziridines, epoxides, and products of oxo and imino transfer under ambient conditions (Fig. 1B). This strategy exhibits broad substrate scope, enabling selective transformations from simple feedstocks to complex bioactive molecules. Additionally, pyridine derivatives form as readily separable byproducts, simplifying purification and enhancing synthetic practicality. Mechanistic investigations, integrating experimental and computational studies, elucidate the role of triplet-state activation, providing fundamental insights into next-generation photocatalytic atom transfer methodologies.
Results and Discussion
Reaction development
Our initial reaction design was guided by the established photoreactivity of pyridinium ylides. Previous studies demonstrated photochemical conversion under UV irradiation with a mercury vapor lamp (Fig. 2A)16,17. However, direct excitation at short wavelengths yielded poor product formation, even with excess alkenes. Mechanistic analysis of related photochemical transformations suggested that high-energy UV light promotes excited singlet-state reactivity, leading to undesired side products through radical coupling between the pyridine C2 position and heteroatom-centered radical.

A Literature precedents and challenges in conventional UV photolysis of pyridinium ylides. B Proposed mechanism supported by DFT calculations, illustrating the triplet-triplet energy transfer (TTEnT) pathway for aziridination and epoxidation. C Optimization of aziridination across diverse photosensitizers. Yields were determined by ¹H NMR spectroscopy. *Reactions performed using 5 mol% of catalyst.
To circumvent these side pathways, we postulated that leveraging the triplet state of pyridinium ylides could facilitate selective atom transfer reactions (Fig. 2B). Computational studies were conducted to evaluate the triplet-state behavior of pyridinium ylides. For nitrogen group transfer, N-pyridinium ylide 1a was selected for its synthetic accessibility and thermal stability. Analysis revealed a triplet-state energy of 54.3 kcal mol⁻¹ for 1a, indicating feasible triplet energy transfer. The N–N bond length decreased from 1.406 to 1.365 Å in the triplet state, indicating bond retention. We speculated that the amidyl radical, generated in situ via reversible triplet-state activation, would undergo selective alkene addition, followed by exergonic intersystem crossing (ISC) and pyridine elimination, ultimately driving aziridine formation. Building on the nitrogen group transfer strategy, we next explored oxygen atom transfer capabilities. Recognizing that pyridine N-oxides readily undergo oxidation by photocatalysts, we anticipated that electron-deficient pyridines could enhance reactivity18,19. Computational screening revealed 4-cyano-substituted pyridine as the optimal candidate, exhibiting a favorable triplet-state energy of 46.0 kcal mol⁻¹ for efficient photocatalytic activation.
To validate our mechanistic hypothesis, we examined photocatalytic reactivity using cyclohexene as a model substrate and tosyl-protected pyridinium ylide 1a as the nitrogen source under blue light irradiation (Fig. 2C). Screening of photocatalysts revealed a direct correlation between reactivity and triplet-state energy, with successful transformations occurring only when the photocatalyst’s triplet energy exceeded that of ylide 1a. Optimization of reaction conditions identified dichloromethane and 5 mol% of Q1 as optimal, providing the desired product in 87% yield. The versatility of this system was demonstrated using Ir(dF(CF₃)ppy)₂(dtbbpy)PF₆ as an alternative photocatalyst, which afforded the product in comparable yield. Notably, the transformation proceeded efficiently under ambient air, demonstrating operational simplicity and practical scalability.
Substrate scope
To evaluate the generality of the atom transfer reaction, we investigated diverse alkyl- and aryl-substituted alkene substrates for aziridination, employing various ylides (1) under blue light irradiation (Fig. 3A). Mechanistic insights emerged from several key observations. Notably, common competing pathways associated with nitrenes, such as allylic amination20, were suppressed. The substrate scope investigation began with cyclic alkenes, which underwent efficient conversion to corresponding multi-ring aziridine products (2a and 2b). Acyclic unactivated alkenes, including those bearing phenyl rings and bromides, reacted smoothly (2c–2 f). The methodology also accommodated activated alkenes such as styrene derivatives, with α-methylstyrene providing the desired aziridine (2 g). While triplet nitrenes typically facilitate C–H amidation through hydrogen atom transfer (HAT)21, our system exhibited distinct reactivity. Furthermore, β-methylstyrene yielded trans-aziridine products selectively, supporting a stepwise mechanism (2 h). Next, we examined the reactivity of substrates with competing functional groups. Notably, the aziridination reaction proceeded exclusively at the activated site, even in substrates containing potentially reactive heterocycles, sulfide moieties, and unactivated alkenes, demonstrating its high selectivity and functional group tolerance (2i–2 m). Studies with diolefin substrates revealed unexpected chemoselectivity. Rather than undergoing Pauson-Khand-type cyclization22, these substrates yielded mono-aziridination products predominantly (2n and 2o), further demonstrating the method’s selective nature. The versatility of aziridination was further demonstrated using various functionalized pyridinium ylides with cyclohexene as a model substrate. Sulfonyl derivatives with diverse substituents proved effective, with small alkyl chain precursors showing high reactivity while avoiding instability issues common to azide-based reagents (2p)23. Benzyl, heteroaryl, and naphthyl groups were well-tolerated (2q–2t). In addition to sulfonyl groups, carbonyl-functionalized precursors also proved viable for this transformation, although they required increased alkene concentration for optimal yields (2u–2w). Among them, carbamate derivatives, including N-Troc and N-Cbz ylides (2x and 2 y), reacted efficiently and enabled subsequent facile deprotection to free NH aziridines. Selective aziridine formation from phenethyl carbonyl ylide, without oxazolidinone byproducts, suggested reaction progression via pyridine-bound intermediates rather than free nitrenes (2z)4. In addition, pyrazole carbonyl derivatives demonstrated broad functional group compatibility (2aa). Late-stage aziridination proved successful across a range of structurally diverse substrates, including camphor, α-ionone, cholesterol and boldenone undecylenate, further demonstrating the broad synthetic utility of this methodology (2ab–2ae).

A Substrate scope of alkenes and N-amine substituents in aziridination. B Alkene scope of epoxidation. Reaction conditions: ylide (0.05 mmol), alkene (0.05 mmol) and photocatalyst in dichloromethane (0.05 M), irradiated with a Penn PhD Photoreactor M2 or Kessil LED under air at r.t. for 2 h. Isolated yields are given. *Reactions performed using PC7 (1 mol%) under 450 nm irradiation. †Reactions performed using PC8 (1 mol%) under 450 nm irradiation. ‡Reactions performed with CsF (1 equiv) as an additive. §Reactions performed with 2 equiv of alkene. #Reactions performed with 10 equiv of alkenes. The yield in parentheses was determined by 1H NMR analysis of the crude reaction mixture using 1,1,2,2-tetrabromoethane as an internal standard.
Next, an investigation of epoxidation was conducted using various styrene substrates and pyridine N-oxide. Terminal styrenes exhibited excellent reactivity, with styrene and its derivatives bearing both electron-donating and electron-withdrawing substituents at the para position showing high efficiency (Fig. 3B, 3a–3d). Notably, substrates containing Bpin groups were well-tolerated, enabling subsequent cross-coupling transformations (3e). Interestingly, the reaction exhibited high chemoselectivity, achieving selective epoxidation at the alkene even in the presence of an easily oxidizable sulfide moiety (3 f). This selectivity remained consistent even with pyridine-containing substrates, which are typically prone to oxidation by conventional oxidants like m-CPBA, highlighting the robustness of this approach (3 g). Similar to aziridination, α-substituted styrene underwent efficient epoxidation without HAT or ring-opening side reactions (3 h and 3i). In a significant advance, β-methylstyrene exhibited trans-selective epoxidation, allowing selective isomer synthesis from E/Z mixtures—a distinct advantage over conventional methods (3k). The scope extended to cyclic styrene derivatives, which afforded epoxides in high yields (3j and 3 l). When the reaction was conducted using β-dimethyl-substituted styrene, a slight decrease in reactivity was observed due to steric hindrance (3 m). Interestingly, a ketone product resulting from a 1,2-methyl shift was detected as a side product (Supplementary Fig. 12). This suggests that the product was formed via a semipinacol rearrangement, indicating the possible involvement of a polar intermediate. Finally, when substrates containing multiple reactive sites (alkenes and alkynes) were used, the reaction proceeded selectively at the activated alkene, demonstrating its practical utility and high chemoselectivity (3n and 3o). Collectively, aziridination and epoxidation proceeded with controlled reactivity and excellent tolerance to functional groups, including traditionally challenging moieties, such as boron, sulfur, amine, and carbonyls.
The broad substrate scope and functional group tolerance prompted exploration of divergent atom transfer pathways (Fig. 4A). Under the original standard conditions, carbonyl ylide and styrene selectively formed the aziridine product (60%). Remarkably, the addition of acid additives redirected reactivity toward selective oxazoline formation. This oxazoline formation proved general across various substituted styrenes (4a–4c), establishing a complementary transformation to our aziridination protocol. Encouraged by these results with alkenes, we next explored the potential of pyridinium ylides to engage in atom transfer reactions with other classes of substrates. The oxo and imino transfer reactivity proved remarkably general across diverse substrates. Sulfides, sulfoxides, and phosphines afforded the corresponding oxo and imino transfer products in high yields (5a–7e), while silyl enol ethers efficiently generated α-amidated ketones (8a and 8b)6,24. The reaction also transformed heteroarenes into imine products through a unique mechanistic pathway (9a and 9b)25.

A Various reaction modes of pyridinium ylides. B Application to bioactive compounds. *Reactions performed using PC8 (1 mol%) under 450 nm irradiation. †Reactions performed with 10 mol% of acid (CAS 1010799-98-0) as an additive. ‡Reactions performed using PC7 (1 mol%) under 450 nm irradiation. §Reactions performed with 2 equiv of coupling partners. ∥Reactions performed with 2 equiv of ylides. #Reactions performed with CsF (1 equiv) as an additive.
To demonstrate the synthetic utility of photocatalytic atom transfer via pyridinium ylides, we investigated the selective modification of alkene-containing drug scaffolds and natural products. Gratifyingly, various bioactive molecules, including fenofibrate, isoeugenol, estrone, and zaltoprofen, underwent efficient conversion to the corresponding epoxides and N-tosyl aziridines (Fig. 4B, 10a–10 h). The consistent high efficiency across such structurally diverse substrates—from simple molecules to complex pharmaceutical derivatives—highlights the broad applicability of this transformation in both synthetic and medicinal chemistry.
Mechanistic investigation
To elucidate the mechanistic pathway, we conducted comprehensive experimental and computational studies, focusing on reaction initiation and excited-state interactions. Initial Stern-Volmer quenching experiments revealed strong interactions between photocatalysts and pyridinium ylides, while alkenes (styrene and cyclohexene) showed minimal quenching under identical conditions. These results established pyridinium ylides as the primary excited-state quenchers in reaction media (Supplementary Fig. 6). Electrochemical analysis of ylides (1a and 1b) showed reduction potentials of -1.61 and -1.74 V versus SCE, respectively. These values lie outside the excited photocatalyst’s redox window, precluding a photo-induced electron transfer (PET) pathway (Supplementary Fig. 7). Investigation of the triplet-triplet energy transfer mechanism through control experiments (Fig. 5A) required characterization of the Q1 photocatalyst. While its redox potential was previously reported26, Raman spectroscopic analysis revealed a triplet energy of 59.1 kcal mol⁻¹, sufficient for triplet energy transfer (Supplementary Fig. 9). UV-Vis absorption studies showed strong absorption below 400 nm for 1a, enabling direct UV excitation (Supplementary Fig. 4). Direct irradiation with 390 nm LEDs without Q1 yielded 9% of desired product 2a, alongside 60% of 1,2-diazepine byproduct. These results indicate that triplet state access via TTEnT suppresses undesired singlet-state reactivity27. Mechanistic studies with triplet sensitizers and quenchers provided additional support for TTEnT initiation. Addition of benzil (10 mol%) as triplet sensitizer afforded 32% product yield, while trans-stilbene (1 equiv) as triplet quencher completely suppressed product formation, corroborating the TTEnT pathway. Taken together, these results indicate that while minor SET contributions cannot be excluded, TTEnT is the predominant productive pathway under our conditions.

A Control experiments supporting triplet–triplet energy transfer as the reaction initiation mechanism. B Experimental evidence for the formation of a carbocation intermediate. C The proposed reaction mechanism based on experimental and computational insights. D DFT-computed energy profile at (u)B3LYP-D3/def2qzvpp//6-311 + G** level of theory with self-consistent reaction field approach for solvation with ε = 9.08 for dichloromethane was used. Values are in kcal mol-1. ISC intersystem crossing, TS transition state. E Molecular orbital analysis.
Distinct reactivity patterns compared to previous reports suggested involvement of a carbocation intermediate. Initial radical clock experiments showed 35% retention of the closed-ring structure without detectable byproducts, indicating deviation from open-shell radical pathways (Supplementary Fig. 8). To validate carbocation formation, we designed strategic trapping experiments (Fig. 5B). A substrate bearing an intramolecular nucleophile afforded both the aziridine-opened product (2af, 53%) and a rearranged product (2ag, 27%) via a 1,2-hydride shift of the carbocation intermediate. Further evidence for this mechanism was obtained when the N-oxide reaction under identical conditions yielded epoxide-opened product (3p, 34%) and rearranged product (3q, 21%), consistent with carbocation intermediate formation (Supplementary Fig. 10, 11). Density functional theory (DFT) calculations (Supplementary Data1 and Data2) provided detailed insights into reaction energetics and mechanistic pathways (Fig. 5C, D). The reaction initiates with visible-light excitation, followed by triplet-triplet energy transfer from excited Q1 to ¹I-N, generating triplet diradical (³I-N) with an excited-state energy of 54.3 kcal mol⁻¹. The resulting amidyl radical proceeds through a C–N bond-forming transition state (³TS-I-N) with 18.5 kcal mol⁻¹ barrier. The triplet diradical intermediate (³II-N) undergoes spontaneous ISC to form open shell singlet intermediate (¹II-N). Final product (2a) formation occurs via intramolecular electron transfer, driven by pyridine re-aromatization, with 12.5 kcal mol⁻¹ barrier. To gain deeper insight, we conducted a molecular orbital analysis (Fig. 5E). Spin density calculations of ¹II-N revealed unpaired electrons localized on both the alkyl radical and pyridine moiety, indicating distinct diradical centers. The α- and β-spin singly occupied molecular orbitals (SOMOs) were localized on the pyridine and alkyl groups, respectively. Analysis of the transition state TS-SET-N showed that the convergence of the α- and β-spin SOMOs results in a molecular orbital identical to the HOMO. This orbital convergence, combined with pyridine’s exceptional leaving group ability, supports our proposed mechanism where electron density redistribution between alkyl and pyridine moieties in the transition state drives pyridine formation and subsequent carbocation generation. Alternative C2 functionalization pathways previously proposed28 were deemed unfavorable, requiring higher transition state energy (19.6 kcal mol⁻¹). This trend was consistently observed with N-oxide, further reinforcing the proposed mechanism.
In conclusion, we have established pyridinium ylides as effective photocatalytic O-atom and N-group transfer reagents through triplet-triplet energy transfer rather than an irreversible redox pathway. These bench-stable ylides operate through triplet-state activation, enabling selective formation of aziridines, epoxides, and products of oxo and imino transfer under ambient conditions. Their controlled reactivity enables selective, atom-economical transformations with excellent tolerance to various functional groups. The scalability and exceptional stability of these ylides, coupled with their operational simplicity, offer significant practical advantages over existing methodologies. This strategy demonstrates broad applicability, including late-stage functionalization of complex molecules, highlighting its potential impact in pharmaceutical synthesis and materials science.
Methods
General procedure
Reactions were conducted in the vial (4 mL) sealed with a white cap. Pyridinium ylide (0.05 mmol), alkene (0.05 mmol) and photocatalyst and dichloromethane (1.0 mL) were combined under open-air conditions. The vial was sonicated for 10 s and placed at a Penn PhD Photoreactor M2 or Kessil LEDs. The resulting mixture was stirred at room temperature for 2 h. Isolation was carried out by merging the products from two reactions. The resulting mixture was concentrated under reduced pressure and purified by short silica filter or flash column with (diethyl ether or ethyl acetate: n-hexane = 1:9) solvent to obtain the desired aziridine compound.
Data availability
Experimental procedure and characterization data of new compounds are available within the Supplementary Information files. Data supporting the findings of this manuscript are also available from the corresponding author upon request.
References
Hirokami, S. & Cvetanovic, R. J. Reaction of oxygen atoms, O(3P), with olefins in liquid nitrogen solution at 770K. J. Am. Chem. Soc. 96, 3738–3746 (1974).
Lee, K. et al. Topics in Heterocyclic Chemistry. pp. 313–377 (Springer International Publishing, 2023)
Empel, C. & Koenigs, R. M. Visible-light-mediated amination reactions via nitrene intermediates. Chem. Catal. 2, 2506–2514 (2022).
Jung, H., Keum, H., Kweon, J. & Chang, S. Tuning triplet energy transfer of hydroxamates as the nitrene precursor for intramolecular C(sp3)-H amidation. J. Am. Chem. Soc. 142, 5811–5818 (2020).
Meyer, A. R. et al. Combined synthetic, spectroscopic, and computational insights into a general method for photosensitized alkene aziridination. ACS Catal. 14, 12310–12317 (2024).
Guo, Y., Pei, C., Empel, C., Jana, S. & Koenigs, R. M. Photochemical nitrene transfer reactions of iminoiodinanes with sulfides. ChemPhotoChem 6, 0293 (2022).
Maness, P. F. & McCulla, R. D. Insights into the mechanism of O(3P)-mediated oxidation of alkenes through kinetic isotope effects and computational modeling. J. Org. Chem. 89, 1458–1464 (2024).
Basistyi, V. S. & Frederich, J. H. Pyridazine N-oxides as photoactivatable surrogates for reactive oxygen species. Org. Lett. 24, 1907–1912 (2022).
Zou, Y.-Q. et al. Visible light induced intermolecular [2+2]-cycloaddition reactions of 3-ylideneoxindoles through energy transfer pathway. Tetrahedron 68, 6914–6919 (2012).
Rykaczewski, K. A. & Schindler, C. S. Visible-light-enabled paternò–Büchi reaction via triplet energy transfer for the synthesis of oxetanes. Org. Lett. 22, 6516–6519 (2020).
Zhu, M., Zheng, C., Zhang, X. & You, S.-L. Synthesis of cyclobutane-fused angular tetracyclic spiroindolines via visible-light-promoted intramolecular dearomatization of indole derivatives. J. Am. Chem. Soc. 141, 2636–2644 (2019).
Rössler, S. L. et al. Pyridinium salts as redox-active functional group transfer reagents. Angew. Chem. Int. Ed. 59, 9264–9280 (2020).
Yu, W.-L., Chen, J.-Q., Wei, Y.-L., Wang, Z.-Y. & Xu, P.-F. Alkene functionalization for the stereospecific synthesis of substituted aziridines by visible-light photoredox catalysis. Chem. Commun. 54, 1948–1951 (2018).
Greulich, T. W., Daniliuc, C. G. & Studer, A. N-aminopyridinium salts as precursors for N-centered radicals-direct amidation of arenes and heteroarenes. Org. Lett. 17, 254–257 (2015).
Lee, W., Koo, Y., Jung, H., Chang, S. & Hong, S. Energy-transfer-induced [3+2] cycloadditions of N-N pyridinium ylides. Nat. Chem. 15, 1091–1099 (2023).
Albini, A. & Alpegiani, M. The photochemistry of the N-oxide function. Chem. Rev. 84, 43–71 (1984).
Strub, H., Strehler, C. & Streith, J. Photoinduced nitrene, carbene, and atomic oxygen transfer reactions starting from the corresponding pyridinium N-, C-, and O- ylides. Chem. Ber. 120, 355–363 (1987).
Ang, H. T., Miao, Y., Ravelli, D. & Wu, J. Pyridine N-oxides as hydrogen atom transfer reagents for site-selective photoinduced C(sp3)–H functionalization. Nat. Synth. 3, 568–575 (2024).
Schlegel, M., Qian, S. & Nicewicz, D. A. Aliphatic C-H functionalization using pyridine N-oxides as H-atom abstraction agents. ACS Catal. 12, 10499–10505 (2022).
Knecht, T., Mondal, S., Ye, J.-H., Das, M. & Glorius, F. Intermolecular, branch-selective, and redox-neutral Cp*IrIII -catalyzed allylic C-H amidation. Angew. Chem. Int. Ed. 58, 7117–7121 (2019).
Guo, Y., Pei, C. & Koenigs, R. M. A combined experimental and theoretical study on the reactivity of nitrenes and nitrene radical anions. Nat. Commun. 13, 86 (2022).
Li, F. et al. Photosensitization enables Pauson-Khand-type reactions with nitrenes. Science 383, 498–503 (2024).
Kolb, H. C., Finn, M. G. & Sharpless, K. B. Click chemistry: diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 40, 2004–2021 (2001).
He, S. et al. Photochemical α-amination of carbonyl groups with iodinanes. Chem. Commun. 60, 10128–10131 (2024).
Brachet, E., Ghosh, T., Ghosh, I. & König, B. Visible light C-H amidation of heteroarenes with benzoyl azides. Chem. Sci. 6, 987–992 (2015).
Kim, I. et al. Visible-light-induced pyridylation of remote C(sp3)-H bonds by radical translocation of N-alkoxypyridinium salts. Angew. Chem. Int. Ed. 57, 15517–15522 (2018).
Boudry, E., Bourdreux, F., Marrot, J., Moreau, X. & Ghiazza, C. Dearomatization of pyridines: Photochemical skeletal enlargement for the synthesis of 1,2-diazepines. J. Am. Chem. Soc. 146, 2845–2854 (2024).
Moon, Y., Lee, W. & Hong, S. Visible-light-enabled ortho-selective aminopyridylation of alkenes with N-aminopyridinium ylides. J. Am. Chem. Soc. 142, 12420–12429 (2020).
Acknowledgements
This research was supported financially by Institute for Basic Science (IBS-R010-A2).
Author information
Authors and Affiliations
Contributions
Experiment, W.L., C.K., A.J., and S.H.; Computation, W.L.; Manuscript, W.L., C.K., A.J., and S.H.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jia-Rong Chen, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, W., Kim, C., Jang, A. et al. Pyridinium ylides as photocatalytic atom transfer reagents. Nat Commun 16, 10716 (2025). https://doi.org/10.1038/s41467-025-65740-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65740-w
