
Abstract
Electrocatalytic oxygen reduction is an attractive alternative for sustainable H2O2 production. However, the electrocatalyst still suffers from low H2O2 efficiency due to unsuitable intermediate binding, sluggish active hydrogen (*H) generation in neutral/alkaline solutions and high interfacial proton concentration in acid. Meanwhile, the modulation mechanism remains insufficiently understood. Here we report efficient pH-universal H2O2 electrosynthesis at ampere-level current densities by modulating interfacial microenvironment via sulfonic acid (SO3H)-functionalization of carbon nanotubes (SCNT). Experimental and theoretical results show that SO3H-functionalization accelerates *H generation from water dissociation for neutral/alkaline H2O2 electrosynthesis while creating more alkaline microenvironment in acid. Moreover, it not only optimizes *OOH binding energy and facilitates *OOH generation, but also reduces the energy barrier for *HOOH desorption (rate-determining step). It exhibits good H2O2 electrosynthesis performance with Faradaic efficiencies of 81.7–97.2% and H2O2 concentrations of 834–1537 mM (0.8 min) at pH 0.7–13 and 1.0–1.5 A cm−2. The estimated cost for H2O2 electrosynthesis is 28.5% of industrial anthraquinone process. The on-site application of SCNT has been demonstrated by efficient pollutant degradation and sterilization.
Similar content being viewed by others
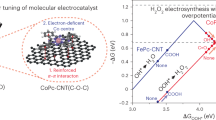


Introduction
Hydrogen peroxide (H2O2) is a versatile and eco-friendly oxidizer with ever-growing need in industry and daily life1. The industrial H2O2 production is dependent on the energy-intensive anthraquinone process with massive organic wastes and safety concerns in H2O2 transportation2. Electrocatalytic two-electron oxygen reduction reaction (2e-ORR) offers a sustainable alternative for on-demand H2O2 production3. Currently, carbon4,5,6 and transition metals-based7,8,9 materials have been developed for 2e-ORR. Although significant progress have been made in alkaline H2O2 production, neutral and acidic H2O2 electrosynthesis still have poor selectivity under industrial-level currents10,11. However, acidic or neutral H2O2 is well-suited for various applications such as wastewater treatment, organic synthesis, and disinfection12. Therefore, it is urgent to develop efficient catalysts for pH-universal H2O2 electrosynthesis.
The 2e-ORR involves proton-coupled electron transfer (PCET) steps to generate *OOH and *HOOH. H2O2 electrosynthesis is highly dependent on active hydrogen (*H) supply, *OOH generation, and *HOOH desorption13,14. A suitable energy for *OOH binding and *HOOH desorption is favorable for enhancing activity and selectivity for H2O2 production. Meanwhile, *H is crucial for *OOH formation and conversion. The sluggish *H generation kinetics in neutral and alkaline media leads to low H2O2 yields15. In acidic media, the proton accumulated on catalyst surface could facilitate hydrogen evolution and oxygen reduction to water (4e-ORR), leading to low H2O2 selectivity16. Therefore, modulating *H generation, *OOH, and *HOOH binding energy are imperative for enhancing ampere-level H2O2 electrosynthesis under pH-universal conditions.
Carbon nanotubes (CNTs) are appealing for H2O2 electrosynthesis owing to their good reduction activity and corrosion resistance in acid17. However, their H2O2 selectivity and activity are unsatisfied in neutral and acidic media. The introduction of electron-withdrawing sulfonic acid (SO3H) group can modulate their electronic structure, thereby tailoring the interfacial microenvironment while simultaneously optimizing the binding affinities of *OOH and *HOOH. The hydrophilic SO3H could also enhance water adsorption to construct water-hydronium network18, which is expected to facilitate *H generation from water dissociation during neutral 2e-ORR. As SO3H group has higher affinity for alkali metal ions than proton19, SO3H-functionalized CNTs (SCNT) may enhance the modulation effect of alkali metal ions on interfacial proton under acid condition. However, modulating interfacial *H/H+ availability under neutral and acid solutions has been rarely reported for carbon materials. Optimizing *OOH generation and *HOOH desorption, along with the modulation mechanism, remains insufficiently understood.
Herein, SCNT is designed for enhancing ampere-level H2O2 electrosynthesis under pH-universal conditions. Experiments and theoretical calculations were used to probe the modulatory effect of SO3H-functionalization on interfacial *H/H+ availability and intermediate binding during 2e-ORR. The results show that SO3H-functionalization promotes *H generation from water dissociation for neutral and alkaline H2O2 electrosynthesis, while it creates local alkaline microenvironment for acidic H2O2 electrosynthesis. Furthermore, the *OOH generation and *HOOH desorption are optimized by SO3H-functionalization. The resulted SCNT achieves high Faradaic efficiencies (FEsH2O2) of 81.7–97.2% at pH 0.7–13 and 1.0–1.5 A cm−2, surpassing the recently reported catalysts. Its on-site application is demonstrated by efficient degradation of organic pollutants and sterilization.
Results
Catalyst design and characterization
Tailoring *OOH binding and interfacial *H/H+ dynamics are crucial for efficient H2O2 electrosynthesis (Fig. 1a). Specifically, a suitable *OOH binding energy is favorable for improving 2e-ORR activity and selectivity. For neutral/alkaline 2e-ORR, the limited *H supply can be addressed by promoting *H generation from water dissociation via constructing water-hydronium network. In contrast, the inhibited acidic 2e-ORR caused by excessively high H+ concentration can be enhanced by creating local alkaline microenvironment. Given the strong electron-withdrawing property of SO3H, SO3H-functionalized CNTs are expected to tailoring intermediate binding and interfacial *H/H+ availability for 2e-ORR. The effect of SO3H-functionalization on 2e-ORR was explored by theoretical calculations, in which the models of SO3H-functionalized carbon, pristine carbon network, S-doped carbon, and defect-modified carbon are constructed (Fig. 1b, Supplementary Figs. 1–5 and Supplementary Data 1). The Gibbs free energy for *OOH adsorption (ΔG*OOH) is a valid descriptor for 2e-ORR. Figure 1c shows the volcano plot of the limiting potential and ΔG*OOH. The strong *OOH adsorption located at the left side of volcano plot tends to 4e-ORR20. In contrast, weak *OOH adsorption on the right side implies low ORR activity. SO3H-functionalized carbon approaches the peak of volcano plot with an optimal ΔG*OOH value of 4.26–4.41 eV, indicating its good activity and selectivity for 2e-ORR. However, the other catalysts are positioned on the right side of the volcano plot. The SO3H group of SO3H-functionalized carbon has high electron density (Supplementary Fig. 6), which can enhance *OOH adsorption (Supplementary Table 1). Thus, SO3H-functionalization optimizes *OOH binding energy, which facilitates 2e-ORR.

a Schematic diagram of *OOH binding and interfacial *H/H+ modulation for 2e-ORR on SCNT. b Top views of model configuration. c Volcano plot for 2e-ORR on different models. d Free energy profiles of *H generation from water dissociation. Source data for the figure are provided as a Source data file.
Neutral/alkaline 2e-ORR is usually hindered by sluggish *H generation for PCET process. Herein, the free energy for *H generation via water dissociation was calculated (Fig. 1d, Supplementary Figs. 7–11 and Supplementary Data 1). Notably, water molecules can easily dissociate to *H on SO3H-functionalized carbon with free energy of 0.76–0.93 eV, much lower than those of S-doped carbon (1.11 eV), defect-modified carbon (1.69 eV), and original carbon (1.79 eV). The electron-enriched SO3H sites increase the length of O-H bond in H2O to make O-H bond more easily broken (Supplementary Table 2), which significantly reduces the energy barrier for water dissociation to *H, consequently boosting H2O2 electrosynthesis. Although S-doping can also enhance *H generation in comparison to pristine carbon, SO3H-functionalization is superior to S-doping in *H generation. Therefore, SO3H-functionalized carbon has suitable *OOH binding energy and enhanced *H generation, which is conducive to H2O2 electrosynthesis.
Guided by the theoretical findings, xSCNT (x = 0.9 and 0.4, representing SO3H content) was prepared by etching CNTs in alkaline solution and pyrolyzing the mixture of etched CNTs and sulfur (Supplementary Fig. 12). As comparison, S-doped CNTs (0SCNT) and etched CNTs without sulfur (denoted as CNT) were also prepared. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images show their intertwined tubular structures with diameters of ~50 nm (Supplementary Figs. 13 and 14). The diffraction peaks for (002) and (101) planes of graphitic carbon can be observed (Supplementary Fig. 15). Both D band (~1350 cm−1) and G band (~1570 cm−1) appear in their Raman spectra (Supplementary Fig. 16), which originate from defects and graphitic carbon, respectively21. Their intensity ratio (ID/IG) is 1.28 for 0.9SCNT and 1.23 for 0.4SCNT, slightly higher than those of 0SCNT (1.11) and CNT (1.15), indicating more defects in 0.9SCNT and 0.4SCNT. As 0.9SCNT was obtained by pyrolyzing CNT and sulfur, the increased defect density in 0.9SCNT relative to CNT could be due to the introduction of sulfur. Fourier transform infrared (FTIR) spectra of 0.9SNT and 0.4SCNT show distinct vibration peaks at 1110 cm−1 (Fig. 2a), which are attributed to SO3H group22,23. In the X-ray photoelectron spectra (XPS, Supplementary Fig. 17), sulfur peak appears at 172–160 eV for 0SCNT, 0.4SCNT, and 0.9SCNT. The S 2p XPS spectra could be deconvoluted into three peaks at 163.9 eV, 165.1 eV, and 168.8 eV (Fig. 2b), corresponding to C = S, C-S, and SO3H, respectively24,25,26. The SO3H content is 0.9 at% for 0.9SCNT, higher than 0.4 at% for 0.4SCNT (Fig. 2c). Only C=S and C-S can be observed for 0SCNT. It is noted that the contents of C = S and C-S are similar for 0.9SCNT, 0.4SCNT, and 0SCNT (Supplementary Table 3).

a FTIR spectra of CNT, 0SCNT, 0.4SCNT, and 0.9SCNT. b S 2p XPS spectra and c atomic contents of S species for 0SCNT, 0.4SCNT, and 0.9SCNT. SECM 3D-maps showing the distribution of catalytic sites on 0SCNT (d), 0.4SCNT (e), and 0.9SCNT (f) at 0.1 M K2SO4 and 0.2 V vs. RHE (All the potentials have not been iR-corrected). Source data for the figure are provided as a Source data file.
The induced SO3H group (acidic group) of 0.9SCNT was further validated by Boehm titration and ammonia temperature-programmed desorption (NH3-TPD). 0.9SCNT has higher acid density (1.1 mmol g−1) than 0SCNT (0.05 mmol g−1, Supplementary Fig. 18a), indicating its significantly higher content of acidic group relative to 0SCNT due to the introduction of SO3H group. The NH3-TPD curve shows that no peak appears on 0SCNT (Supplementary Fig. 18b). However, 0.9SCNT shows a distinct desorption peak at 280–350 °C, which can be attributed to the chemical desorption of NH3 from the acid sites (SO3H groups). These results confirm that SO3H group has been successfully modified on 0.9SCNT.
To examine the local electrochemical activity of catalyst towards 2e-ORR, scanning electrochemical microscope (SECM) technology with platinum ultramicroelectrode was employed to in situ monitor the current gradient difference on catalyst surface. The substrate-generation/tip-collection (SG/TC) mode was used. The scanning images show the outlines of conductive regions for 0.9SCNT, 0.4SCNT, and 0SCNT (Fig. 2d–f). The conductive region of 0.4SCNT exhibits a feedback current of −6.0 to −19.6 nA for 2e-ORR, higher than −5.0 to −14.3 nA of 0SCNT, indicating the better catalytic activity of 0.4SCNT. With the increase of SO3H group, the feedback current on 0.9SCNT (−10.0 to −31.1 nA) is further increased. It suggests that the catalytic activity of 0.9SCNT is improved in comparison with 0SCNT and 0.4SCNT, which can be attributed to its more SO3H groups.
Modulating interfacial *H/H+ availability and intermediate binding for H2O2 electrosynthesis
The *H plays an essential role in 2e-ORR. To investigate the contribution of *H for H2O2 electrosynthesis, tert-butyl alcohol (t-BuOH) was added as the quencher for *H during 2e-ORR on 0.9SCNT, 0.4SCNT 0SCNT and CNT (1.0 A cm−2, Fig. 3a and Supplementary Fig. 19). Their H2O2 concentrations and FEsH2O2 all decrease after the addition of t-BuOH at 0.1 M K2SO4, and the decrease is more remarkable on 0.9SCNT than on other catalysts, indicating that *H generation may be enhanced on 0.9SCNT. Its 2e-ORR selectivity decreases with the presence of t-BuOH, while its 4e-ORR selectivity increases (Supplementary Fig. 20). The characteristic peaks of *H can be observed on their electron paramagnetic resonance spectra (Fig. 3b). Impressively, the *H signal intensity of 0.9SCNT is much stronger than those of 0SCNT and CNT, which confirms that the presence of SO3H group significantly promotes *H generation. Besides, *H generation is slightly enhanced on 0SCNT compared to CNT. At neutral solution, *H is usually produced from water dissociation. The ability of *H generation from water dissociation was further verified by the kinetic isotope effect (KIE) of H/D (H2O/D2O). It was determined by comparing the current densities obtained from linear sweep voltammograms in H2O and D2O (Supplementary Fig. 21). The KIE value being close to 1 suggests an acceleration of water dissociation27. As shown in Fig. 3c, 0.4SCNT has a lower KIE value (1.5) than 0SCNT (1.8), demonstrating that water dissociation is accelerated on 0.4SCNT. Notably, 0.9SCNT exhibits the lowest KIE value (1.2) among the three electrocatalysts. These results confirm that SO3H-functionalization enhances *H generation from water dissociation during neutral 2e-ORR, and the increased SO3H group is more favorable for *H generation.

a FEsH2O2 of CNT, 0SCNT, 0.4SCNT, and 0.9SCNT in 0.1 M K2SO4 with and without 0.5 M t-BuOH (Error bars represent the standard deviation from three independent measurements). b Their electron paramagnetic resonance spectra for *H (0.1 M K2SO4, 0.5 A cm−2). c KIE and corresponding current densities over 0.9SCNT, 0.4SCNT and 0SCNT at 0 V vs.RHE. d Schematic illustration of 0.9SCNTCH and its FEsH2O2 in 0.1 M K2SO4 (1.0 A cm−2). e S 2p XPS spectra of 0.9SCNT and 0.9SCNTCH. In situ ATR-SEIRAS spectra for 0SCNT (f), 0.4SCNT (g), and 0.9SCNT (h) in 0.1 M K2SO4. All the potentials have not been iR-corrected. Source data for the figure are provided as a Source data file.
To identify the role of SO3H group for H2O2 electrosynthesis, cyclohexanol that can selectively bind to SO3H28 was used for poisoning the SO3H sites of 0.9SCNT (0.9SCNTCH, Fig. 3d). The S 2p XPS spectra show that its peak for SO3H group is shifted about 0.6 eV after reacting with cyclohexanol (Fig. 3e), indicating the successful bind between SO3H and cyclohexanol. Both H2O2 concentration and FEH2O2 of 0.9SCNT are significantly decreased after poisoning the SO3H sites (Fig. 3d and Supplementary Fig. 22). The FEH2O2 of 0.9SCNTCH decreases to 57.7% at 0.1 M K2SO4, much lower than 94.8% for 0.9SCNT (1.0 A cm−2), indicating the indispensable role of SO3H group for 2e-ORR. The FEH2O2 of 0.9SCNTCH is further decreased after the addition of t-BuOH (Supplementary Fig. 23), and its decrement is much lower than that of 0.9SCNT, revealing that SO3H group promotes *H generation. To evaluate the influence of SO3H modification on O2 transport, the diffusion layer thickness was calculated. 0.9SCNT exhibits a slightly increased diffusion layer thickness (13.3 µm) compared to 0SCNT (12.7 µm, Supplementary Fig. 24), demonstrating that the introduction of SO3H groups has a negligible effect on oxygen diffusion. In addition, oxygen temperature-programmed desorption curves reveal that the total oxygen adsorption capacities of 0.9SCNT and 0SCNT are almost the same (Supplementary Fig. 25). 0.9SCNT exhibits larger peak area than 0SCNT for chemically adsorbed oxygen (200–350 °C), while its peak area for physically adsorbed oxygen (50–120 °C) is smaller than that of 0SCNT. These results indicate that SO3H-functionalization has negligible effect on oxygen adsorption capacity. Polarization curves of 0SCNT and 0.9SCNT were tested under varying O2 flow rates (Supplementary Fig. 26). At overpotential of 0.30–0.45 V, O2 reaction order is 0.76–0.71 for 0.9SCNT, while it is 0.63–0.42 for 0SCNT (Supplementary Fig. 27). The opposite trend between overpotential and O2 reaction order confirms that oxygen adsorption is not the rate-determining step for 0SCNT and 0.9SCNT. Upon modification with SO3H group, 0.9SCNT shows higher electrochemically active surface area than 0SCNT and CNT (Supplementary Figs. 28–31). Its increased active sites also contribute to the high 2e-ORR performance of 0.9SCNT. In addition, the contribution of K+ for neutral H2O2 electrosynthesis can be ruled out by comparing its performance with those in electrolytes without K+ or alkali metal ions (Supplementary Fig. 32). 0.9SCNT also exhibits high FEH2O2 (92–95.7%) in 0.1 M (NH4)2SO4, Na2SO4, Li2SO4, and Cs2SO4, suggesting that the impact of K+ on its superior performance is negligible in neutral electrolyte under the applied conditions.
The intermediates of ORR were probed by in situ attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS). As shown in Fig. 3f–h, the characteristic peak at ~1100 cm−1 is attributed to the stretching of O-O bond from *OOH adsorbed on the catalyst surface. The *OOH peak intensity of 0.9SCNT is much stronger than those of 0.4SCNT and 0SCNT. It reveals that *OOH generation is facilitated on 0.9SCNT, thus attaining high H2O2 yields29,30. The peaks at 1670 cm−1 and 3000–3700 cm−1 can be attributed to the bending vibration of *H2O and O-H stretching. 0.9SCNT exhibits stronger signals for *H2O and O-H than 0.4SCNT and 0SCNT, suggesting that more interfacial water may adsorb on 0.9SCNT surface to construct water-hydronium network and decompose to produce *H31. In addition, the signal intensities of *OOH, *H2O, and O-H are significantly decreased after poisoning SO3H sites (Supplementary Fig. 33), indicating that SO3H group can promote water dissociation and *OOH generation. The O-H signal can be deconvoluted into four-coordinated hydrogen-bonded water (4-HB-H2O), two-coordinated hydrogen-bonded water (2-HB-H2O), and free H2O. Among them, 2-HB H2O is more prone to water dissociation towards *H generation32,33,34,35. As the potential shifts from 0.5 to −0.3 V (vs. RHE), the proportion of 2-HB H2O on 0.9SCNT increases markedly from 46% to 57% (Supplementary Fig. 34). This increase is accompanied by a decrease in 4-HB-H2O content, indicating the reconstruction of water-hydronium network on 0.9SCNT surface. 2-HB H2O content is increased with the increase of SO3H group, indicating that SO3H group can accelerate water dissociation. Therefore, 0.9SCNT is more favorable for interfacial water adsorption and water dissociation than 0SCNT. These results prove that 0.9SCNT effectively enhances *OOH and *H generation, which further facilitates neutral/alkaline 2e-ORR.
To investigate the effect of SO3H group on proton regulation for acidic 2e-ORR, the local pH at electrode surface was probed by rotating ring-disk electrode (RRDE, Supplementary Fig. 35). As depicted in Fig. 4a, the local pH for H2O2 electrosynthesis in 0.1 M H2SO4 is increased to 7.8–9.0 at 0.9SCNT surface (0.5–1.0 A cm−2). Impressively, the pH increment on 0.9SCNT is 1.2 and 2.0 times as high as that on 0.4SCNT and 0SCNT, demonstrating that 0.9SCNT is more effective than 0.4SCNT and 0SCNT for modulating local pH. The pH increment is more significant at high current density due to its improved 2e-ORR rate with faster *H consumption. The local alkaline microenvironment on 0.9SCNT can suppress 4e-ORR or hydrogen evolution, resulting in the superior H2O2 selectivity. It is noted that the local pH is further increased with the presence of K+, and the increment on 0.9SCNT surface is more remarkable than that on 0.4SCNT and 0SCNT surface. Phenolphthalein method was used to validate the local pH change at electrode surface. When phenolphthalein indicator is added into the acidic electrolyte for 2e-ORR (1.0 A cm−2), the color of 0.9SCNT surface turns into pink while 0SCNT surface keeps colorless (Supplementary Fig. 36), which confirms the local alkaline microenvironment on 0.9SCNT. As expected, 0.9SCNT also creates more alkaline microenvironment than 0SCNT in 0.1 M K2SO4 (Supplementary Fig. 37). Therefore, SO3H modification can create a more alkaline microenvironment on the electrode surface to enhance H2O2 electrosynthesis.

Local pH value (a) and adsorbed-K+ concentration (b) on 0SCNT, 0.4SCNT, and 0.9SCNT surfaces during acidic 2e-ORR. c FEsH2O2 of 0SCNT, 0.4SCNT and 0.9SCNT at 0.1 M H2SO4 or 0.1 M H2SO4 + 0.1 M K2SO4 (1.5 A cm−2). d Representative snapshots for H2O, H+ and K+ distribution on SO3H-functionalized carbon surface at 0.1 M H2SO4 and 0.1 M H2SO4 + 0.1 M K2SO4. e Radial distribution function of K+ and H2O on SO3H-functionalized carbon surface at 0.1 M H2SO4 + 0.2 M K+. f H2O density on SO3H-functionalized carbon surface at 0.1 M H2SO4 and 0.1 M H2SO4 + 0.2 M K+. g Schematic diagram of acidic 2e-ORR on 0SCNT and 0.9SCNT. h Free energy profiles of 2e-ORR on carbon network, S-doped carbon, and SO3H-functionalized carbon. i Energy barriers for 2e-ORR and 4e-ORR. Error bars represent the standard deviation from three independent measurements. All the potentials have not been iR-corrected. Source data for the figure are provided as a Source data file.
To probe into the better pH regulation effect of 0.9SCNT in the presence of K+, K+ adsorption capacity of 0.9SCNT, 0.4SCNT, and 0SCNT was tested in 0.1 M H2SO4 + 0.1 M K2SO4 electrolyte with electrode transfer method (Supplementary Fig. 38). Remarkably, 0.9SCNT exhibits 1.3 times and 1.8 times higher K+ adsorption capacity than 0.4SCNT and 0SCNT (Fig. 4b), respectively. It can be attributed to the high K+ affinity of SO3H group. By adding K+ into 0.1 M H2SO4, the FEH2O2 increment of 0.9SCNT (13.4%) is higher than that of 0SCNT (6.6%) under 1.5 A cm−2 (Fig. 4c and Supplementary Fig. 39). These results reveal that 0.9SCNT has higher K+ affinity than 0SCNT, which can construct a more alkaline microenvironment at 0.9SCNT surface.
Ab initio molecular dynamics simulations were executed to analyze the dynamic behavior of H2O, K+, and H+ on SO3H-functionalized carbon surface (Supplementary Data 1). In 0.1 M H2SO4, SO3H-functionalized carbon prefers to adsorb H2O rather than H+ (Fig. 4d), facilitating H2O dissociation and moderating local pH. In the presence of K+, K+ preferentially moves toward the electrode surface, while H+ is repelled away from electrode surface due to the cation shielding effect of K+, resulting in higher local pH than that without K+. Radical distribution function result (Fig. 4e) and in situ Raman spectra (Supplementary Fig. 40) reveal the formation of K+-H2O in 0.1 M H2SO4 + 0.2 M K+, leading to an increased water density on SO3H-functionalized carbon surface than that in 0.1 M H2SO4 (Fig.4f). This increased water density accelerates *H supply from water dissociation while creating more alkaline microenvironment than that in 0.1 M H2SO4. Its more alkaline microenvironment is favorable for promoting acidic H2O2 electrosynthesis (Fig. 4g).
The free energy for ORR was further calculated to get insight into the modulation effect of SCNT on H2O2 electrosynthesis (Supplementary Data 1). In the process of 2e-ORR, O2 is adsorbed on the catalyst surface to form *O2 and converted to *OOH via PCET. Subsequently, *OOH is reduced to *HOOH, followed by *HOOH desorption to produce H2O2. The steps for O2 adsorption and conversion to *HOOH are downhill processes (Fig. 4h), which demonstrates that O2 → *HOOH is thermodynamically favorable. The SO3H-functionalized carbon exhibits lower free energy for *OOH generation (−1.05 and −0.91 eV) than S-doped carbon (−0.61 eV) and pristine carbon (−0.42 eV, Supplementary Fig. 41), confirming the accelerated *OOH generation on SO3H-functionalized carbon. However, *HOOH desorption is an uphill process, which is the rate-determining step (RDS) for all catalysts. The energy barrier for this RDS is 0.10–0.39 eV on SO3H-functionalized carbon, significantly lower than those of other catalysts (1.25 eV and 0.69 eV). Therefore, SO3H-functionalization promotes H2O2 electrosynthesis with reduced energy barrier for the RDS. This phenomenon can be explained by that the electron-withdrawing SO3H group can optimize the adsorption of intermediates on catalyst surface. It reduces the energy for *HOOH desorption (Supplementary Table 1). The free energy diagrams reveal 2e-ORR is more thermodynamic feasible compared to 4e-ORR on SO3H-functionalized carbon, S-doped carbon, and pristine carbon network (Supplementary Figs. 42 and 43). However, SO3H-functionalized carbon has the highest energy gap between 2e-ORR and 4e-ORR among the three catalysts. It exhibits a much higher energy barrier for *OOH → *O + H2O (1.63–1.92 eV) relative to *OOH → H2O2 (0.38–0.69 eV, Fig. 4i), indicating that SO3H-functionalization effectively stabilizes the key intermediate of *OOH to facilitate 2e-ORR and suppress 4e-ORR.
Performance for H2O2 electrosynthesis
The 2e-ORR performance of xSCNT was evaluated at pH 0.7–13 (0.1 M H2SO4, 0.1 M K2SO4, and 0.1 M KOH). As revealed by RRDE data, the 2e-ORR activity and selectivity of 0.9SCNT are significantly higher than those of 0SCNT and 0.4SCNT in all electrolytes (Fig. 5a and Supplementary Figs. 44–46). Compared to the reported electrocatalysts, 0.9SCNT is still one of the best-performing catalysts for pH-universal 2e-ORR (Supplementary Table 4). To further explore their 2e-ORR activity, H2O2 electrosynthesis was conducted in a flow cell at 1.0 A cm−2 through chronopotentiometry (Supplementary Figs. 47 and 48). The H2O2 concentration of 0.9SCNT is 1120 mM (3.8 wt%, 0.8 min) at 0.1 M K2SO4 (Fig. 5b and Supplementary Fig. 49), which can be used for medical-level disinfection (~3 wt%). The corresponding FEH2O2 of 0.9SCNT is 94.8%, which is higher than those of 0.4SCNT (84.3%), 0SCNT (61.9%), and CNT (47.7%). The structure and composition of 0.9SCNT exhibit no obvious change after H2O2 electrosynthesis at 1.0 A cm−2 (Supplementary Fig. 50). Significantly, 0.9SCNT exhibits the highest H2O2 concentration and FEH2O2 among the four electrocatalysts over a wide pH range (0.7–13, Fig. 5c, d). The FEH2O2 of 0.9SCNT is increased to 97.2% at 0.1 M KOH. Although its FEH2O2 is slightly declined to 81.7% at 0.1 M H2SO4, it is still 1.2–2 times higher than other electrocatalysts. The 0.9SCNT, 0.4SCNT, and 0SCNT have similar C=S and C-S contents. Compared with 0SCNT, 0.4SCNT with SO3H group shows remarkably higher H2O2 concentration and FEH2O2. The H2O2 electrosynthesis performance is further significantly improved on 0.9SCNT with higher SO3H content, highlighting the significant role of SO3H-functionalization in enhancing 2e-ORR. By further increasing the SO3H content (1.3SCNT), H2O2 electrosynthesis performance exhibits insignificant increment (Supplementary Fig. 51). In addition, 0SCNT exhibits higher H2O2 selectivity than CNT, indicating that S-doping (C=S and C-S) can also promote H2O2 production. However, the promoting effect is more pronounced for SO3H group than S-doping under the same S content, as evidenced by the enhanced performance of 0.4SCNT and 0SCNT relative to CNT. The worst performance of CNT suggests that the impact of defects on H2O2 electrosynthesis is not significant here. Therefore, SO3H group is the most important factor responsible for the superior 2e-ORR of xSCNT under pH-universal conditions.

a H2O2 selectivity of CNT, 0SCNT, 0.4SCNT and 0.9SCNT in 0.1 M K2SO4. Their H2O2 concentrations (mM) and FEsH2O2 in 0.1 M K2SO4 (b), KOH (c), and H2SO4 (d) at 1.0 A cm−2. H2O2 concentrations (mM) and FEsH2O2 of 0.9SCNT in 0.1 M K2SO4 (e) and KOH (f) at 0.1–1.5 A cm−2. g Its FEsH2O2 in 0.1 M H2SO4 and 0.1 M H2SO4 + 0.1 M K2SO4 at 0.1–1.5 A cm−2. h Comparison of FEsH2O2 and H2O2 yields between 0.9SCNT and other reported electrocatalysts. Error bars represent the standard deviation from three independent measurements. All the potentials have not been iR-corrected. Source data for the figure are provided as a Source data file.
Efficient H2O2 electrosynthesis at a wide range of current densities can meet the requirements of various application scenarios. Here H2O2 electrosynthesis performance of 0.9SCNT was further explored at 0.1–1.5 A cm−2 and flow rate of 18.0 mL h−1 (Hydraulic retention time of 0.8 min). As shown in Fig. 5e, f, its H2O2 concentration almost increases linearly with the increase of current density at 0.1 M K2SO4 or KOH. A maximum H2O2 concentration of 1489 mM (~5.1 wt%, 0.8 min) is achieved on 0.9SCNT at 1.5 A cm−2 and 0.1 M K2SO4 (Supplementary Fig. 52a), corresponding to H2O2 yield of 911.6 mg h−1 cm−2. It maintains high FEsH2O2 (86.2%-97.7%) across a wide range of current densities (0.1–1.5 A cm−2). At 0.1 M KOH, its H2O2 concentration is 1537 mM (~5.2 wt%, 941.0 mg h−1 cm−2) with FEH2O2 above 90% at 1.5 A cm−2 (Supplementary Fig. 52b). The H2O2 concentration can be modulated by varying the catholyte flow rate (Supplementary Fig. 53). When the flow rate decreases from 18.0 mL h−1 to 5.4 mL h−1, the H2O2 concentration gradually increases. At 0.1 M K2SO4, the highest H2O2 concentration is 2313 mM (~7.9 wt%) at 5.4 mL h−1. These results indicate that 0.9SCNT is active and selective for neutral and alkaline H2O2 production.
The performance of 0.9SCNT for H2O2 electrosynthesis was also evaluated at 0.1 M H2SO4 and 0.1–1.5 A cm−2. Its H2O2 concentration increases under higher current density (Fig. 5g and Supplementary Fig. 54). As expected, 0.9SCNT shows superior FEsH2O2 of 92.4–81.7% at 0.1–1.0 A cm−2, further increased current density leads to a slightly declined FEH2O2. In the presence of 0.2 M K+, both H2O2 yield and FEH2O2 of 0.9SCNT are improved. Its FEH2O2 increases from 81.7–69.5% to 86.0–82.9% under 1.0–1.5 A cm−2, and the increase is more pronounced under higher current density (Supplementary Fig. 55). It is well known that the excessive proton in acidic media is prone to H2O2 reduction or hydrogen evolution, and the alkali metal ions can regulate interfacial proton concentration36,37. The good K+ affinity of 0.9SCNT leads to its more alkaline microenvironment at interface. Therefore, 0.9SCNT is a pH-universal electrocatalyst for H2O2 production. Interestingly, 0.9SCNT outperforms the reported electrocatalysts with higher H2O2 production rates and FEsH2O2 under neutral, alkaline, and acid solutions (Fig. 5h and Supplementary Table 5), which reveals that 0.9SCNT is promising for sustainable H2O2 production under pH-universal conditions and industrial-level current densities.
On-site application
To demonstrate its practical applications, the H2O2 produced on 0.9SCNT was used in Fenton reaction for pollutant degradation (Supplementary Fig. 56). FeOCl loaded on carbon cloth (CC-FeOCl) was used as heterogeneous catalyst for H2O2 activation (Supplementary Fig. 57). The H2O2 was synthesized in a flow cell at 0.5 A cm−2 and acidic electrolyte, which was flowed into the heterogeneous Fenton cell for degrading methylene blue, rhodamine B and phenol. After 30 min of reaction, methylene blue and rhodamine B are decolorized (Fig. 6a). The phenol removal efficiency in Fenton system is up to almost 95.8% within 30 min (Fig. 6b), significantly higher than the adsorption removal of phenol by CC-FeOCl (10.5%). The corresponding TOC removal rate is 81.9% (1.5 h, Supplementary Fig. 58). Meanwhile, the H2O2 produced in neutral electrolyte (0.5 A cm−2) was used for disinfection. The Escherichia coli was chosen to evaluate the sterilization performance. When the synthesized H2O2 is flowed into Escherichia coli solution (6 × 108 and 2 × 108 cfu mL−1, ~1.6 wt% H2O2), the bacterial colonies disappear within 1 min (Fig. 6c, d), demonstrating the effective sterilization performance of 0.9SCNT. These results demonstrate the applicability and feasibility of onsite H2O2 electrosynthesis on 0.9SCNT for pollutant degradation and disinfection.

Degradation curves of dyes (a) and phenol (b) in CC-FeOCl-catalyzed Fenton process with H2O2 produced on 0.9SCNT (0.5 A cm−2). c Colony number and sterilization rate. d photographs of Escherichia coli before and after sterilization. e H2O2 electrosynthesis on 0.9SCNT with air source. f Stability of 0.9SCNT for H2O2 electrosynthesis in 0.1 M K2SO4. Error bars represent the standard deviation from three independent measurements. All the potentials have not been iR-corrected. Source data for the figure are provided as a Source data file.
H2O2 electrosynthesis on 0.9SCNT was also performed by using air as oxygen source. It exhibits high FEsH2O2 of 91.8–97.8% at 0.1–0.5 A cm−2 (Fig. 6e). The long-term stability of 0.9SCNT for H2O2 production was investigated at neutral electrolyte. As shown in Fig. 6f, its outflow H2O2 concentration (~3.1 wt%) and FEH2O2 (~90%) exhibit no obvious difference during 72 h of continuous reaction at 0.5 A cm−2 and single-pass flow rate of 9.0 mL h−1 (Hydraulic retention time of 1.6 min). Meanwhile, the cell potential (Ecell) presents insignificant change (Supplementary Fig. 59). After the stability test, 0.9SCNT retains the initial intertwined tubular structures (Supplementary Fig. 60). XPS and FTIR spectra prove its well-maintained SO3H group after 2e-ORR (0.5 A cm−2, 72 h). These results suggest that 0.9SCNT is stable for H2O2 electrosynthesis under industrial-level current density. The energy consumption of 0.9SCNT for H2O2 electrosynthesis at pH 0.7–13 is 5.62–6.90 kWh kg−1 at 0.3 A cm−2 and 7.55–8.76 kWh kg−1 at 0.5 A cm−2 (Supplementary Table 6), lower than those of the reported electrocatalysts. The H2O2 cost breakdown has been evaluated as a function of current densities, which is calculated by normalizing capital investment and operating costs according to a 20-year lifespan. The H2O2 production cost of 0.9SCNT decreases from $0.602 kg−1 at 0.1 A cm−2 to $0.437 kg−1 at 1.0 A cm−2 (Supplementary Table 7, Fig. 61 and Note 1), including electricity cost and O2 cost, labor cost, equipment cost, separation cost, and H2O cost. The H2O2 production cost at 1.0 A cm−2 is only 28.5% of the cost of the anthraquinone process ($1.5 kg−1). Techno-economic analysis shows that H2O2 electrosynthesis on 0.9SCNT is superior to traditional industrial processes.
Discussion
SO3H-functionalization on CNTs was an attractive strategy for enhancing pH-universal H2O2 electrosynthesis at ampere-level current densities. SCNT facilitated *H generation from water dissociation for neutral/alkaline 2e-ORR and generated more alkaline local microenvironment in acidic media relative to CNTs and S-doped CNTs, thereby improving H2O2 production efficiency. It had good affinity to K+, which enhanced the promotion effect of K+ on acidic H2O2 electrosynthesis. SCNT also promoted the generation of *OOH with suitable binding energy while simultaneously lowered the energy barrier for *HOOH desorption (RDS). The 0.9SCNT was highly efficient for neutral/alkaline H2O2 electrosynthesis with FEsH2O2 of 94.8–97.2% at 1.0–1.5 A cm−2, and it also exhibited high FEH2O2 of 81.7–86.0% at pH 0.7 and 1.0 A cm−2. The applicability of SCNT has been proved by its good performance for organic pollutant degradation and disinfection. This work provides new insights into designing efficient catalyst and modulating interfacial microenvironment for pH-universal 2e-ORR.
Methods
Chemicals
Carbon nanotubes (CNTs) with an average diameter of 40–60 nm and a tube length of <2 µm were purchased from Shenzhen Nanotech Port Co., Ltd. The H2SO4 (98%) was bought from Sinopharm Chemical Reagent Co., Ltd. The isopropanol (>99.7%), KOH (>95%), K2SO4 (99.9%), Ce(SO4)2 (99.9%), and Sulfur (99.95%) were purchased from Aladin Co., Ltd. The carbon papers (HCP122) were obtained from Hesen Electrical Co., Ltd. Nafion 117 membrane and Nafion solution (5 wt%) were purchased from Du Pont Co., Ltd. Ultrapure deionized water (18 MΩ cm) was used for all experiments.
Catalyst preparation and characterization
The CNTs were etched with alkaline solution (mKOH: mCNT = 1: 1) under 80 °C, and the obtained CNTs were denoted as CNT. The CNT powder (100 mg) was fully mixed with 32 mg or 64 mg sulfur by grinding in a mortar, and annealed at temperature of 800 °C under Ar to obtain 0.4SCNT and 0.9SCNT. As comparison, the CNTs without etching were mixed with sublimated sulfur by grinding in a mortar, which was annealed at 800 °C under Ar to obtain 0SCNT. The catalyst morphology was examined using scanning electron microscope (Hitachi S-4800) and transmission electron microscope (Tecnai G2 F30 S-Twin). Its chemical composition was analyzed by X-ray photoelectron spectroscopy (ESCALAB250Xi) and Fourier transform infrared spectrometer (Thermo Nicolet IS5). The structure was identified by X-ray diffraction (D8 ADVANCE) and Raman spectroscopy (inVia Qontor).
The acidic group content was tested by Boehm titration38. 0.05 g catalyst was dispersed in 30 mL of 2 mol L−1 NaCl solution and stirred for 24 h to fully exchange the H+ in solid acid catalyst with Na+. The resulting mixture was filtered, and the filtrate was collected. Phenolphthalein was used as indicator, and 20 mmol L−1 NaOH solution was used as standard solution to add into 10 mL filtrate.
where C is the content of acidic group (mol g−1), CNaOH is the NaOH concentration (mol L−1), VNaOH is the consumed volume of NaOH solution (L), mcatalyst is the catalyst mass (g).
The NH3-TPD was tested by temperature-programmed desorption on chemisorption analyzer (Micromeritics AutoChem II 2920, USA). 100 mg catalyst was primarily pretreated at 100 °C for 1 h under He atmosphere, followed by cooling to room temperature and exposing to NH3/He at 50 °C for 1 h. The NH3-TPD was carried out by increasing the temperature from 50 °C to 550 °C at 10 °C min−1 under He atmosphere.
Electrochemical measurements
The 2e-ORR selectivity of catalysts was tested by RRDE setup (AFMSRCE type, Pine). The RRDE speed was set to 1600 rpm. The potentials of disk electrode were 0–1.0 V vs. RHE (All the potentials have not been iR-corrected in this work). The catalyst loading was ~48 µg cm−2. All potentials were measured against Ag/AgCl reference electrode and converted to the reversible hydrogen electrode (RHE) based on Eq. (2):
where E0(Ag/AgCl) (saturated KCl) is 0.197 V (25 °C).
The H2O2 selectivity and the number of transferred electrons (n) were calculated according to Eqs. (3) and (4):
where IR is the ring current, ID is the disk current, and N is the collection efficiency (N = 0.37).
The SECM experiment was carried out on VersaSCAN. The substrate was xSCNT-loaded Au plate. A platinum wire and Ag/AgCl electrode were used as the counter and reference electrodes, respectively. A platinum ultramicroelectrode (Pt UME) with diameter of 25 μm was used as the probe electrode (Supplementary Fig. 62). During the SECM measurement, the working electrode was fixed at the bottom of cell, while the Pt UME could be moved freely in the X, Y, and Z directions using a stepper motor. The probe current was recorded as a function of the traveling distance by moving the probe electrode in an XY plane parallel to the working electrode panel at a scan rate of 100 μm s−1.
The electrochemical double-layer capacitance (Cdl) was measured using cyclic voltammetry (CV) in a non-Faradic potential region at scan rates of 20–100 mV s−1 and Ar-saturated electrolytes. The oxygen diffusion layer can be calculated from the equivalent impedance in the low-frequency region of electrochemical impedance spectroscopy (EIS)39. The EIS was tested in O2-saturated electrolytes. The diffusion layer thickness was calculated according to Eq. (5):
where Rd and Cd are the equivalent resistance and capacitance of the diffusion layer, respectively. Do is the O2 diffusion coefficient (1.73 × 105 cm2 s−1) under normal pressure and temperature.
The H2O2 electrosynthesis via 2e-ORR was conducted in a double-chamber flow cell at room temperature (20–25 °C). The anode and cathode chambers were separated by Nafion 117 membrane (DuPont). The Nafion 117 membrane was soaked in 5 wt% H2O2 solution at 80 °C for 1 h, followed by immersing in deionized water for 30 min. It was then treated with 0.5 M H2SO4 at 80 °C for 1 h, and rinsed with deionized water for another 30 min before use. The distance between anode and cathode was 0.5 cm. In this three-electrode system, Pt mesh and Ag/AgCl electrode were employed as counter electrode and reference electrode. The current was applied by Chronopotentiometry with an electrochemical workstation (CHI1140). The catalyst ink was sprayed onto carbon paper with catalyst loading of 0.4 mg cm−2 (1 cm2). 0.1 M K2SO4 (7.1 ± 0.3), 0.1 M KOH (13.0 ± 0.4), or 0.1 M H2SO4 (0.9 ± 0.3) were used as the cathodic electrolytes with one-pass flow of 18 mL h−1 to bring out the generated H2O2, while the anodic electrolyte was cycled. The electrolytes were prepared before experiments using H2SO4, K2SO4, or KOH with ultrapure water, which were stored in sealed bottles. The time to reach the measured H2O2 concentration was 0.8 min at 18 mL h−1, and H2O2 concentration was obtained from the average value of samples in 30 min.
In situ ATR-SEIRAS measurements were conducted on a PerkinElmer Spectrum 3 infrared spectrometer equipped with a mercury cadmium telluride (MCT) detector (Supplementary Fig. 63). An Au layer (~20 nm) was plated on the ATR crystal (germanium crystal for this experiment). The working electrode was prepared by drop-coating 90 µL of catalyst solution (5 mg catalyst powder added into 500 µL solution containing 30 µL 5 wt% Nafion, 170 µL ethyl alcohol, and 300 µL isopropanol) on the Au layer. The cell was assembled after drying, and the electrode was inserted into the electrolyte (0.1 M K2SO4). The signal under open-circuit potential was used as background.
H2O2 concentration quantification
The H2O2 concentration was measured by the cerium sulfate titration method based on the stoichiometry of 2Ce4++H2O2 = 2Ce3++2H+ + O2. In a typical operation, the H2O2 solution outflowed from the reactor was added into a certain amount of Ce4+ solution to reduce Ce4+ to Ce3+. The Ce4+ concentration can be detected by spectrophotometry at 316 nm. Thus, we calculated H2O2 concentration via the Eq. (6):
The FE (%) and the H2O2 productivity were calculated using Eqs. (7) and (8):
where v is H2O2 flow rate (mL h−1), F is faraday constant (96485 C mol−1), A is electrode area (1 cm2).
Evaluation of local pH at electrode surface
RRDE technique with a Pt ring and pH-sensing material (iridium oxide, IrOx) was used to detect local pH changes. IrOx electrode was prepared by electrochemically depositing IrOx on the Pt ring using the documented method40. The electrodeposition was conducted via cycling potential between 0.6 to 0.5 V vs Ag/AgCl for 300 cycles at a scan rate of 1 V s−1 (All the potentials have not been iR-corrected in this work). The prepared RRDE was stored in ultrapure water overnight to stabilize the catalyst surface. The Pt ring’s open-circuit potential (OCP) was monitored, which had a linear relationship with the pH near the disk (catalyst surface). The Pt ring electrode can detect the local pH changes as the rotation brings the local environment of the disk to the ring. The pH response of ring was calibrated using a series of standard pH electrolytes. It exhibited a good linear fit (R2 = 0.97) between OCP and known pH when the Pt ring stabilized for 1 min before making a final reading (Supplementary Figs. 64 and 65). The catalyst-coated RRDE was applied with different currents for 30 s. Subsequently, the OCP of the ring electrode was tested to determine the local pH value. The phenolphthalein acid-base indicator test was carried out at 1.0 A cm−2 with 300 μL of phenolphthalein reagent (2 wt% in ethanol).
Measurement of K+ adsorbed on electrode surface
The electrode transfer method was used to detect the concentration of K+ adsorbed on the catalyst surface41. In detail, the electrode was operated at 0.5 A cm−2 at 0.1 M H2SO4 + 0.1 M K2SO4 for 1 min and then transferred to the deionized water (5 mL), which was repeated three times. The K+ concentration in deionized water was tested by ion chromatography.
Synthesis of FeOCl-loaded carbon cloth (FeOCl-CC)
30 g of FeCl3·6H2O was dissolved in 30 mL ethanol. Subsequently, the carbon cloth (CC) was immersed in the above solution and sonicated for 1 h. The FeCl3·6H2O-loaded CC was dried in an oven at 60 °C for 2 h. Then, FeCl3·6H2O loaded CC was heated at 240 °C for 90 min.
Pollutant degradation and sterilization
The pollutant degradation experiments were conducted in a 50 mL reactor at room temperature. FeOCl loaded on carbon cloth (4 cm2) was used as Fenton catalyst. H2O2 in situ synthesized on 0.9SCNT for 20 min (0.5 A cm−2) was gradually introduced into the Fenton reactor and mixed with the pollutant solution. Pollutant concentration was determined by high-performance liquid chromatography via sampling 1 mL of the pollutant solution at a specific time interval. The phenol adsorption was investigated under the same conditions but without adding H2O2. In the sterilization experiments, H2O2 in situ synthesized on 0.9SCNT for 20 min (0.5 A cm−2) was collected and added into 25 mL of Escherichia coli solution (6 × 108 and 2 × 108 cfu mL−1), and samples were taken after sterilization for 1 min.
DFT calculations
All the calculations were performed in the framework of the density functional theory with the projector-augmented plane-wave method, as implemented in the Vienna ab initio simulation package42. The generalized gradient approximation from Perdew-Burke-Ernzerhof (PBE) was selected for the exchange-correlation potential43. The cut-off energy for the plane wave was set to 480 eV. The energy criterion was set to 10−4 eV in the iterative solution of the Kohn-Sham equation. All the structures were relaxed until the residual forces on the atoms have declined to less than 0.02 eV/Å. To avoid interlaminar interactions, a vacuum spacing of 20 Å was applied perpendicular to the slab. *OOH decomposition processes on carbon network model, S-doped carbon model, SO3H-functionalized carbon (1) model, and SO3H-functionalized carbon (2) model were shown in Supplementary Figs. 66–69.
Here, differences in Gibbs free energy (ΔG) for intermediates in ORR are defined as:
where ΔG is the total energy difference between the slab and respective terminations computed by DFT-PBE. ΔEZPE and TΔS denote differences in zero-point energy and entropy between adsorbed states of reaction intermediates and gap phase, respectively. T is the room temperature (298.15 K). ΔGU = −eU, whereby U is the electrode potential.
Data availability
The data supporting the conclusions of this study are presented in the paper and the supplementary information. Source data are provided with this paper.
References
Campos-Martin, J. M., Blanco-Brieva, G. & Fierro, J. L. G. Hydrogen peroxide synthesis: an outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 45, 6962–6984 (2006).
Xia, C., Xia, Y., Zhu, P., Fan, L. & Wang, H. T. Direct electrosynthesis of pure aqueous H2O2 solutions up to 20% by weight using a solid electrolyte. Science 366, 226–231 (2019).
Lu, Z. Y. et al. High-efficiency oxygen reduction to hydrogen peroxide catalysed by oxidized carbon materials. Nat. Catal. 1, 156–162 (2018).
Zhang, Q. Z. et al. Highly efficient electrosynthesis of hydrogen peroxide on a superhydrophobic three-phase interface by natural air diffusion. Nat. Commun. 11, 1731 (2020).
Long, Y. D. et al. Tailoring the atomic-local environment of carbon nanotube tips for selective H2O2 electrosynthesis at high current densities. Adv. Mater. 35, 2303905 (2023).
Lee, K. et al. Structure-controlled graphene electrocatalysts for high-performance H2O2 production. Energy Environ. Sci. 15, 2858–2866 (2022).
Xue, S. et al. Hydrogen radical enabling industrial-level oxygen electroreduction to hydrogen peroxide. Angew. Chem. Int. Ed. 64, e202420063 (2025).
Liu, W. et al. A generalized coordination engineering strategy for single-atom catalysts toward efficient hydrogen peroxide electrosynthesis. Adv. Mater. 36, 2406403 (2024).
Wang, Y. M. et al. Charge-polarized selenium vacancy in nickel diselenide enabling efficient and stable electrocatalytic conversion of oxygen to hydrogen peroxide. Adv. Sci. 10, 2205347 (2023).
Xia, Y. et al. Highly active and selective oxygen reduction to H2O2 on boron-doped carbon for high production rates. Nat. Commun. 12, 4225 (2021).
Ding, S. et al. An abnormal size effect enables ampere-level O2 electroreduction to hydrogen peroxide in neutral electrolytes. Energy Environ. Sci. 16, 3363–3372 (2023).
Liu, L. X. et al. Atomically dispersed asymmetric cobalt electrocatalyst for efficient hydrogen peroxide production in neutral media. Nat. Commun. 15, 4079 (2024).
Jing, L. Y. et al. Unveiling favorable microenvironment on porous doped carbon nanosheets for superior H2O2 electrosynthesis in neutral media. Adv. Energy Mater. 14, 2304418 (2024).
Xu, X. M. et al. Revealing *OOH key intermediates and regulating H2O2 photoactivation by surface relaxation of Fenton-like catalysts. Proc. Natl. Acad. Sci. USA 119, e2205562119 (2022).
Bu, Y. F. et al. Carbon-based electrocatalysts for efficient hydrogen peroxide production. Adv. Mater. 33, 2103266 (2021).
Zhang, X. et al. Electrochemical oxygen reduction to hydrogen peroxide at practical rates in strong acidic media. Nat. Commun. 13, 2880 (2022).
Hu, C. G. & Dai, L. M. Doping of carbon materials for metal-free electrocatalysis. Adv. Mater. 31, 1804672 (2019).
Shi, B. B. et al. Short hydrogen-bond network confined on COF surfaces enables ultrahigh proton conductivity. Nat. Commun. 13, 6666 (2022).
Kusoglu, A. & Weber, A. Z. New insights into perfluorinated sulfonic-acid ionomers. Chem. Rev. 117, 987–1104 (2017).
Tang, C. et al. Tailoring acidic oxygen reduction selectivity on single-atom catalysts via modification of first and second coordination spheres. J. Am. Chem. Soc. 143, 7819–7827 (2021).
Liang, J., Jiao, Y., Jaroniec, M. & Qiao, S. Z. Sulfur and nitrogen dual-doped mesoporous graphene electrocatalyst for oxygen reduction with synergistically enhanced performance. Angew. Chem. Int. Ed. 51, 11496–11500 (2012).
Tu, J. W. et al. Modification of porous N-doped carbon with sulfonic acid toward high-ICE/capacity anode material for potassium-ion batteries. Adv. Funct. Mater. 32, 2204991 (2022).
Thushari, I. & Babel, S. Sustainable utilization of waste palm oil and sulfonated carbon catalyst derived from coconut meal residue for biodiesel production. Bioresour. Technol. 248, 199–203 (2018).
Hu, C. G. & Dai, L. M. Multifunctional carbon-based metal-free electrocatalysts for simultaneous oxygen reduction, oxygen evolution, and hydrogen evolution. Adv. Mater. 29, 1604942 (2017).
Zou, W. W. et al. Hierarchically macro-microporous covalent organic frameworks for efficient proton conduction. Adv. Funct. Mater. 33, 2213642 (2023).
Shen, J. G. C., Herman, R. G. & Klier, K. Sulfonic acid-functionalized mesoporous silica: Synthesis, characterization, and catalytic reaction of alcohol coupling to ethers. J. Phys. Chem. B 106, 9975–9978 (2002).
Li, Y. et al. Single-atom iron catalyst with biomimetic active center to accelerate proton spillover for medical-level electrosynthesis of H2O2 disinfectant. Angew. Chem. Int. Ed. 62, e202306491 (2023).
Chen, F. D. et al. Blocking the sulfonate group in Nafion to unlock platinum’s activity in membrane electrode assemblies. Nat. Catal. 6, 392–401 (2023).
Chen, S. Y. et al. Identification of the highly active Co-N4 coordination motif for selective oxygen reduction to hydrogen peroxide. J. Am. Chem. Soc. 144, 14505–14516 (2022).
Vacque, V., Sombret, B., Huvenne, J. P., Legrand, P. & Suc, S. Characterisation of the O-O peroxide bond by vibrational spectroscopy. Spectrochim. Acta A Mol. Biomol. Spectrosc. 53, 55–66 (1997).
Huang, L. S., Bao, D. Y., Jiang, Y. L., Zheng, Y. & Qiao, S. Z. Electrocatalytic acetylene hydrogenation in concentrated seawater at industrial current densities. Angew. Chem. Int. Ed. 63, e202405943 (2024).
Du, H. et al. Identifying highly active and selective cobalt X-ides for electrocatalytic hydrogenation of quinoline. Adv. Mater. 36, 2411090 (2024).
Zhao, R. et al. Pd single atoms guided proton transfer along an interfacial hydrogen bond network for efficient electrochemical hydrogenation. Sci. Adv. 11, eadu1602 (2025).
Liu, H. M. et al. Observation on microenvironment changes of dynamic catalysts in acidic CO2 reduction. J. Am. Chem. Soc. 8, 5333–5342 (2024).
Wei, K. et al. Hydrophilic single-atom interface empowered pure formic acid fuel cells. J. Am. Chem. Soc. 147, 15490–15498 (2025).
Ni, W. P. et al. Molecular engineering of cation solvation structure for highly selective carbon dioxide electroreduction. Angew. Chem. Int. Ed. 62, e202303233 (2023).
Li, X. Y. et al. Mechanism of cations suppressing proton diffusion kinetics for electrocatalysis. Angew. Chem. Int. Ed. 62, e202218669 (2023).
Yao, P. C. et al. Greener and higher conversion of esterification via interfacial photothermal catalysis. Nat. Sustain. 5, 348–356 (2022).
Yang, C. W. et al. Interfacial O2 accumulation affects microenvironment in carbon-based electrocatalysts for H2O2 production. ACS Energy Lett. 7, 4398–4407 (2022).
Anjana, J. & Muthukrishnan, A. Effect of local pH change on non-PGM catalysts-a potential-dependent mechanistic analysis of the oxygen reduction reaction. Catal. Sci. Technol. 12, 6246–6255 (2022).
Wang, A. Q. et al. Polyelectrolyte additive-modulated interfacial microenvironment boosting CO2 electrolysis in acid. Angew. Chem. Int. Ed. 64, e202412754 (2025).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 78, 1396–1396 (1997).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22222601 and 22076019, Y.L.) and the Fundamental Research Funds for the Central Universities (DUT23LAB611, Y.L.).
Author information
Authors and Affiliations
Contributions
Y.Y. and Y.L. conceived the idea. Y.Y., X.F., and Y.L. designed the experiments. Y.Y. performed the experiments. X.F., B.S., and X.Q. contributed to analytic tools. Y.Y., X.F., B.S., G.Z., Y.X., and X.Q. analyzed the data and discussed the results. Y.Y. and Y.L. co-wrote the manuscript. All authors commented on the manuscript. Y.L. supervised the whole project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yu, Y., Fan, X., Shan, B. et al. Regulating active hydrogen supply and intermediate binding for pH-universal H2O2 electrosynthesis at ampere-level current density. Nat Commun 16, 10784 (2025). https://doi.org/10.1038/s41467-025-65830-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65830-9
