
Abstract
Electrocatalytic CO2 reduction reaction (CO2RR) using membrane electrode assembly (MEA) systems requires complex regulation of protons, hydroxyls, carbonate ions and alkali-metal ions across both electrodes to efficiently produce multicarbon products. In acid-fed CO₂RR MEAs, excessive proton migration and accumulation on the catalyst surface suppress CO₂ adsorption and promote hydrogen evolution, leading to low Faradaic and energy efficiencies. Sluggish hydroxide transport further triggers carbonate precipitation, undermining system stability. Here we report an acid-fed membrane electrode assembly system for highly efficient CO2RR by integrating hydrazone-linked covalent organic framework (COF) and catalyst on the anion-exchange membrane to enable bidirectional pathway for hydroxide and potassium ions diffusion, while enhancing transport of CO2 to the catalyst surface. As a result, the scaled-up MEA operates at a full-cell voltage of ~4.5 V under a total current of 10 A (current density of 204 mA cm⁻²), delivering a Faradaic efficiency of ~50% for CO₂-to-C₂H₄ conversion and maintaining stability for over 300 hours.
Similar content being viewed by others

Introduction
CO2RR using renewable electricity holds promise for ethylene (C2H4) production1. The use of MEA systems has emerged as the most promising approach to both achieve high Faradaic efficiency (FE) and energy efficiency (EE) because of its low ohmic resistance and enhanced reaction kinetics compared to conventional flow-cell systems2. In MEAs, the close proximity of the cathode/ion exchange membrane interface and gas diffusion electrode (GDE) facilitates faster electron transfer and ion conduction. This enables much higher current operation than flow-cell systems with voluminous electrolyte, which are essential for improving the overall efficiency of CO2 reduction reactions. Other advantages of the MEA systems include their ease of scalability compared to traditional flow reactors, and their modular design allows for easier integration into larger systems without significant losses in efficiency, making them commercially viable options for industrial applications aimed at CO2 conversion.
Alkaline and neutral electrolytes have been employed in CO2RR MEA systems. However, these configurations are challenged by bicarbonate formation due to the reaction of CO2 with hydroxide ions (OH−) at the cathode. This parasitic reaction not only depletes CO2 but also destabilizes system operation and reduces the MEA’s operational lifespan (Supplementary Fig. 1). To address these challenges, mitigation strategies such as periodic pulsing of water and solvents3, adoption of bipolar membranes (BPMs)4 or integrated anion exchange membrane (AEM)/proton exchange membrane (PEM) in MEA systems5, and voltage cycling between operational and regenerative modes6, have been developed to enhance system stability. However, some of these approaches require frequent activation processes and suffer from ionic conductivity issues or short-term stabilities owing to electrode flooding. Another deleterious effect is that carbonate (CO32−) and bicarbonate (HCO3−) ions migrate through the AEM, acidified by protons (H+), and converts to CO2 on the anode side, leading to low carbon efficiency7. She. X et al. demonstrated that an alkali cathode MEA using pure water (alkali cation-free) could prevent carbonate precipitation, enabling sustained ethylene production (~50% FE at 1000 h)8. However, for a cell stack comprising six MEA cells, a high operation voltage is needed (25–27 V at 10 A), indicating the need for strategies to improve overall energy efficiency (EE) for CO₂ reduction.
In acidic electrolyzers, the abundance of H+ converts locally formed carbonate anions back to CO2 within the diffusion layer, mitigating CO2 crossover and salt precipitation to some extent, thus offering a carbon-efficient platform for CO2RR7,9,10. The main problem for CO2RR with acidic electrolyte is the occurrence of severe HER at elevated current densities. The performance of acid-fed flow cells suggests that the catalytic microenvironment plays an important part in the CO2-to-C2H4 conversion. By establishing a high-concentration of alkali cations or anions in the catalyst microenvironment, significant enhancement in multicarbon production and overall system energy efficiency have been achieved11,12,13.
The local concentration of ions and molecules plays an important role in determining the CO2RR performance in a zero-gap acidic MEA system. A strongly acidic electrolyte flows exclusively through the anode compartment, this setup creates competition between alkali cation ions and protons in the anolyte as they traverse the membrane to reach the cathode side14. The origin of low FEC2H4 and EEC2H4 in the zero-gap acid-fed CO2RR MEA electrolyzer can be traced to the transportation of protons from anode to cathode and their accumulation on the catalyst surface. This limits CO2 adsorption and leads to severe HER, despite adding a high concentration of alkali cations in the acidic electrolyte. Such a scenario differs markedly from H-cells and flow cells, where the cathode catalyst operates in a relatively stable acidic environment. In addition, the sluggish migration of locally generated OH− from cathode to anode induces bicarbonate precipitation on the cathode during CO2 reduction, despite the low pH of the bulk electrolyte, which hinders the sustainability of CO2 reduction15. Consequently, the mechanisms governing cation effects in acidic CO2RR within MEA systems warrant further investigation, as the local microenvironment is complicated by the presence of various charged ions (e.g., H+, OH−, K+, HCO3−) and molecules (e.g., CO2 and H2O)16.
To address challenges facing MEA systems operating under acidic CO₂RR conditions, we developed a strategy to selectively attract K⁺ while repelling locally generated OH−. This approach improves gas transport efficiency and sustains robust C₂H₄ selectivity. We used hydrazone-linked COFs functionalized with amine and ether groups, and coated it over the catalysts on the cathode to modulate the kinetics of K+ and OH−. This strategy enabled a high FEs of C2H4 of 54% and 82% for C2+ operating at 600 mA cm−2, with partial current densities of 311 mA cm−2 and 490 mA cm−2, respectively, when operating at pH~1 in the MEA cell. Finally, we demonstrated a scale-up MEA cell system with a full-cell voltage of ~4.5 V with a Faradaic efficiency of (50 ± 3) % for CO2-to-C2H4 conversion at the current of 10 A (current density = 204 mA cm−2) with long-term stability over 300 h.
Results
Analysis and the design of acid-fed MEA system
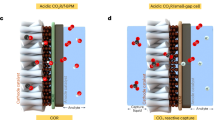
The use of the cation exchange membrane (CEM) is predicated upon its high cation conductivity and selectivity. However, acidic CO2RR using a CEM-MEA becomes unstable at high current densities. This instability arises from the sluggish migration of electrogenerated hydroxide ions (Fig. 1a), leading to bicarbonate precipitation in the cathode chamber and resulting in significant degradation of cell stability. The introduction of an anion exchange Membrane (AEM) in conjunction with the CEM creates a bipolar interface that aids in CO2 regeneration for recycling, thereby partially alleviating salt precipitation issues. However, the continuous generation of gas molecules between the AEM and CEM membranes forms gas pockets that increase the Ohmic impedance and fluctuating overpotentials for reactions (Fig. 1b). This adversely affects the Faradaic and energy efficiency of ethylene production. To tackle these challenges, we propose revisiting the acid-fed MEA system utilizing an AEM, as depicted in Fig. 1c. The AEM facilitates the transport of OH− ions via an electromigration process to the anode, reducing the concentration of OH− near the cathode and enhancing stability by mitigating salt precipitation. While the Donnan exclusion effect of the AEM limits cation migration, K⁺ and H₃O⁺ ions may accumulate near the AEM due to potential drops at the catalyst-membrane interface. Driven by an electric field, these ions migrate from the AEM to the cathode, where they participate in CO2 regeneration and conversion processes19.

a Acidic CEM MEA (b) Acidic AEM/CEM MEA system (c) Acidic AEM-based MEA system.
When using an acid-fed AEM-based system for CO₂-to-C₂H₄ conversion, the high HER (FEH₂ > 30%) resulted in a low C₂H₄ selectivity (FEC₂H₄, ~ 25%), this is despite the fact that a K⁺ concentration of 1 M was added in the bulk anolyte to suppress HER (Supplementary Fig. 2). Increasing the interfacial K⁺ concentration by pre-spraying electrode with potassium salt can enhance C₂H₄ selectivity, prolonged operation however led to salt precipitation (details in “Methods”, Supplementary Fig. 3). These findings underscore the critical interplay between localized K⁺ and OH− concentrations in the acidic CO₂RR MEA system. Optimizing this balance is vital for sustaining high C₂₊ product selectivity while avoiding parasitic HER and salt deposition.
Synthesis and characterization of the Th-TF COF modified Cu3N catalyst
To achieve a high C₂₊ product in an acid-fed CO2RR AEM MEA system, we constructed a hierarchical layer-by-layer electrode configuration consisting of Cu3N/COFs/anion-conducting ionomer (Sustainion), which is abbreviated as CNCP; see details in methods. Copper nitrite (Cu3N) catalyst is selected for its high CO2-to-C2H4 conversion17,18,19. The Cu3N nanoparticles were synthesized via a one-step pyrolysis method17. Powder X-ray diffraction (PXRD) was used to prove the pure phase of Cu3N (Supplementary Fig. 4). Annular dark-field scanning transmission electron microscopy (ADF-STEM) revealed the cubic morphology of the pristine Cu₃N catalyst. Energy-dispersive X-ray spectroscopy (EDS) mapping confirmed the uniform distribution of Cu and N across the particle (Fig. 2a, Supplementary Fig. 5). The atomic-scale ordering of Cu and N was further resolved using atomic-resolution ADF-STEM and annular bright-field (ABF) STEM imaging (Fig. 2b). Due to the atomic number (Z)-contrast nature of ADF imaging, the heavier Cu atoms appeared bright, while ABF-STEM—sensitive to light elements—clearly resolved the N sublattice as light dark contrast, supported by the corresponding intensity line profile (Fig. 2b). The observed lattice fringes with an interplanar spacing of 3.5 Å align with the (010) plane of Cu₃N, reflecting its characteristic lattice arrangement. The valence state and coordination environment of Cu3N were examined by X-ray absorption spectroscopy (XAS) and X-ray photoelectron spectroscopy (XPS). XPS of Cu atom in Cu3N catalyst showed spin orbit coupling-split peaks at 952.3 eV and 932.5 eV in the Cu2p spectrum (Supplementary Fig. 6a, b), which can be attributed to positively charged Cu species (Cu1+)17. In addition, XPS of nonmetal elements was also used to probe the chemical state of nitrogen atoms. For Cu3N, the peak of covalent N was detected at a binding energy of 397.3 eV (Supplementary Fig. 6c). Similar to the results of XPS, the Cu K-edge X-ray absorption near-edge structure (XANES) spectrum of Cu3N was collected to reveal its coordinated state. Extended X-ray absorption fine structure (EXAFS) spectroscopy confirmed the existence of the Cu-N bonding located at 1.34 Å in Cu3N catalyst, and the Cu-Cu coordination at 2.33 Å was observed in the Fourier transform-EXAFS spectra in Supplementary Fig. 7. These results indicate that the formation of phase-pure Cu3N catalyst.

a EDS mapping of a pristine Cu3N catalyst. Scale bars: 20 nm. b atomic-resolution ADF-STEM and ABF-STEM images of Cu3N reflecting its lattice arrangement. Scale bars: 5 Å. c Schematic showing structure of the Th-TF COF and (d) CO2R product distribution in the acid-fed COF AEM MEA system at current density ranging from 100 to 400 mA cm−2 in 0.5 M K2SO4/H2SO4 (pH~2) conditions. No iR correction was applied. Source data for Fig. 2d are provided as a Source Data file. e Schematic of the zero-gap COF-based acid-fed CO2RR MEA system. f Photograph of scale-up electrolyzer cell (a total electrode area of 100 cm2).
Zhao Y. et al. utilized amphoteric COF and perfluorinated sulfonic acid ionomer (PFSA) to establish a proton-blocking, K+-enriched environment and achieved high efficiency for multicarbon products (Faradaic efficiency; FEC2+ ~ 75%; FEC2H4 ~ 40%) in a flow system11. To achieve better performance suited to an acid-fed MEA system, we synthesized and screened acid-resistant COF with tailored reticular backbones and functional groups (Supplementary Fig. 8). We identified a triformylbenzene-derived COF (denoted as Th-TF COF) as the top performer (Fig. 2c). Th-TF COF has a hydrazone-linked (R₁R₂C = N-NH-R₃) framework, functionalized with an amine group and oxygen-containing alkyl chains. The synthetic procedures for monomers are provided in Methods and Supplementary Fig. 9. Ex-situ PXRD and FTIR confirmed the formation of the hydrazone framework, revealing a (100) reflection at 2θ ≈ 3.67° and fingerprint C = N, C = O, and N–H vibrations with red-shifts relative to the precursors (Supplementary Fig. 10). We found that Th-TF COF could effectively regulate local ion concentration (K+ and OH−) and interaction with CO2 molecules, thereby mitigating competitive proton transport and carbonate precipitation in the acidic MEA system. The Cu3N catalyst was coated with Th-TF COF and loaded onto the gas diffusion layer (GDL) to be used as CNCP electrodes in the acid-fed MEA system, achieving a FE of 55% for C₂H₄ and 75% for C₂+ products at 400 mA cm−² (Fig. 2d), rivaling the highest reported values under similar pH conditions7,9,11,12,20,21. When integrated with an AEM acid-fed MEA system, the Th-TF COF facilitated migration and adsorption of local anions (OH−) and cations (K⁺), critical for stabilizing CO₂ activation and reduction pathways (Fig. 2e).
Efficient C2H4 electrosynthesis in the acid-fed Th-TF COF AEM MEA system
We assessed the CO2RR performance of the CNCP electrodes in the acid-fed AEM MEA electrolyzer setup with 0.5 M K2SO4 /H2SO4 (pH ~ 1) anolyte and an iridium oxide as the anode. In Fig. 3a and Supplementary Fig. 11, over the full current density range (100-600 mA cm−2), the use of Th-TF COF greatly suppressed the competing HER (<20%) and improved C2H4 selectivity. At 600 mA cm−2, the FEC2H4 and FEC2+ (53% towards C2H4, 25% towards C2H5OH and 4% towards C3H7OH) values showed peaks of (52 ± 2) % and (80 ± 2) %, respectively, with a C₂H₄ partial current density (jC2H4) of 311 mA cm−2 and a C2+ partial current density (jC2+) of 490 mA cm−2. The CNCP electrode achieved a peak of EEC2H4 of ~20% with low full-cell voltage of ~3.5 V at 400 mA cm−2 (without iR; i, current; R, resistance compensation; Supplementary Fig. 12). In contrast, the highest C₂H₄ selectivity of Cu3N electrode (Sus/Cu3N) is limited to (36 ± 4) % with a low C₂H₄ partial current density of (64 ± 5) % at 200 mA cm−2. The FE of C2H4 then dropped dramatically when current densities increased, the C₂H₄ FE is only 15% at 400 mA cm−2 due to the severe HER (FEH2: ~60%, Supplementary Fig. 13). The value of FEC2H4 of CNCP electrode remained nearly constant as the electrolyte acidity was reduced from pH 1.2 to 6.3 (Supplementary Fig. 14). This evidences that the CNCP electrode could maintain high catalyst activity even with changing surface pH during high current densities CO2 reduction.

a C2H4 selectivity on CNCP and Sus/Cu3N electrodes tested in various current densities in H2SO4 solution with 0.5 M K2SO4 (pH ~ 1). Values are means, and error bars indicate s.d. (n = 3 replicates). b CO2R product distribution and (c) C2H4 production current of CNCP electrodes at 10 A in a scale-up acid-fed MEA system. d FE and single-pass carbon efficiency of CO2-to-C2H4 on 49 cm−2 CNCP and Sus/Cu3N electrodes under total current of 10 A (current density = 204 mA cm−2) with different flow rate of CO2. e The system stability performance of CO2R to C2H4 on CNCP GDEs in a scale-up MEA system at a constant current of 10 A. f Comparison of this work with previous studies on the acidic electrocatalytic CO2 to multi-carbon products at a similar electrolyte pH. No iR correction was applied. Source data for Fig. 3 are provided as a Source Data file.
The superior CO2-to-C₂H₄ conversion performance of CNCP in the acid-fed AEM MEA system prompted us to scale the MEA cell to operate at 10 A in a 100 cm−² electrolyzer MEA cell (Fig. 2f), where both C₂H₄ selectivity (FEC₂H₄) and specific productivity (SPCEC₂H₄) in acidic CO₂RR were strongly enhanced. As shown in Fig. 3b, c, the CNCP electrode achieved a C₂H₄ faradaic efficiency (FE) of ~53% with a total C₂H₄ production current of 5.5 A (current density: 204 mA cm−²) across a 49 cm² reaction area. Single-pass carbon efficiency (SPCE) measurements revealed a peak CO₂-to-C₂H₄ utilization of 16% at 10 A when reducing the CO₂ feedstock flow rate to 60–100 sccm (Fig. 3d), ranking among the highest reported values for acidic CO2RR scale-up MEA systems. The acid-fed AEM MEA system demonstrated stable operation for over 300 h at 10 A with a full-cell voltage of 4.2–5.0 V (no iR compensation) and an average C₂H₄ production rate of 25.91 mmol h⁻¹, with the ~50% Faradaic efficiency benchmark sustained during the first ~100 h (Fig. 2e, Supplementary Table 1). In contrast, a Sustainion coated-Cu3N (Sus/Cu3N) AEM MEA system with 0.5 M K₂SO₄/H₂SO₄ anolyte showed unstable performance within 30 h due to dominant hydrogen evolution (FEH₂: ~ 60%) over C–C coupling (Supplementary Fig. 15), underscoring the critical role of Th-TF COF in stabilizing ion transport and interfacial reactions.
The enhanced CO₂-to-C₂H₄ performance stems from the synergistic effects of the CNCP architecture and Th-TF COF’s ion-regulation capabilities. Post-electrolysis SEM analysis confirmed the preserved microstructure of the catalyst and Th-TF COF (Supplementary Fig. 16). To elucidate the origin of the gradual performance decay, we conducted a series of post-reaction characterizations. XRD and operando XAFS reveal that the characteristic reflections of Cu₃N disappear after extended operation, while the Cu K-edge XANES confirm a phase transformation into Cu0. In contrast, Raman spectra show that the characteristic vibrational bands of the Th-TF COF remain unchanged after 300 h, thereby excluding oxidative degradation of the framework. Complementary TEM/EDS and cross-sectional SEM-EDS analyses demonstrate progressive nitrogen depletion of Cu₃N and electrolyte infiltration into the GDL, which together account for the steady decline in FE and cell voltage (Supplementary Figs. 17, 18). In addition, gradual flooding of hydrophobic domains by liquid CO₂ reduction products (e.g., C₂H₅OH), which increases electrolyte penetration, likely contributed to performance decline after 300 h. Further optimization of gas diffusion electrodes (GDEs) will be essential to improve long-term stability. Overall, the Th-TF COF-modified Cu3N system delivers competitive C2H4 electroproduction performance metrics compared to prior reports on acidic C2 electrosynthesis systems (Fig. 3f, Supplementary Table 2)7,9,11,12,13,20,22,23,24,25, highlighting its potential for scalable acidic CO₂ electroreduction.
COF’s role as ion transport regulator
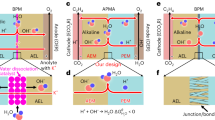
Based on the good performance, we hypothesized that Th-TF COF has two functions: increasing the concentration of K⁺ and facilitating OH− migration through its porous channels. To quantify the local K⁺ concentration at the interface of the Th-TF COF functional layer and catalyst during acidic CO₂RR, we employed in-situ operando X-ray fluorescence spectroscopy (XRFS). Supplementary Fig. 19 displays the XRF spectrum of Ar K, K Kα, and Kβ peaks across a K⁺ concentration gradient (0.5–3 M). A linear baseline correlating the K⁺/Ar peak ratio to bulk K⁺ concentration enabled the concentration of K⁺ within the reactive catalyst area to be calculated. Dynamic in-situ XRFS measurements (Fig. 4a) revealed the evolution of solvated K⁺ content on the Th-TF COF/Cu₃N electrode under varying current densities. Th-TF COF exhibits ~2.4× higher K⁺ content versus Sustainion at open-circuit potential (OCP, Fig. 4b), suggesting that its cation-adsorption sites promote K⁺ diffusion from the AEM to the catalyst. Under an applied electric field, Th-TF COF/Cu₃N shows stronger K⁺ enrichment than Sus/Cu3N, particularly at elevated current densities, confirming its role in stabilizing near-surface K⁺ during CO₂RR.

a XRF spectrum on Th-TF COF/Cu3N electrode showing the solvated K emissions at different current densities. b The local content of K of Th-TF COF/Cu3N and bare Cu3N electrodes. c In-situ Raman spectra during electrosynthesis of C2H4 over Th-TF COF and Sustainion-coated Cu3N catalysts at different current densities. Electrolyte is 0.5 M K2SO4/H2SO4 (pH~3). d In-situ ATR-SEIRS spectra recorded during acidic CO2R on Th-TF COF/Cu3N catalyst from −0.3 to −1.7 V versus Ag/AgCl. Contour map of in-situ ATR-Fourier transform infrared spectra on the Th-TF COF/Cu3N (e) and bare Cu3N catalysts (f) recorded from 1100 to 1750 cm−1. No iR correction was applied. Source data for Fig. 4 are provided as a Source Data file.
In-situ Raman spectroscopy was used to track the presence of OH− in Th-TF COF. As shown in Fig. 4c, the band located at ~ 530 cm-1 is associated with the adsorbed OH− on Cu-based catalysts26, while the red shift of the Cu–OH band on Sus/Cu3N is due to the interaction with the polymer. Comparing the OH− signal in the Th-TF COF/Cu₃N electrode versus the Sus/Cu3N electrode as a function of current density in a 0.5 M K2SO4/H2SO4 (pH~3), it can be seen that only a weak OH− adsorption peak is identified in the Th-TF COF/Cu₃N electrode. This suggests that the Th-TF COF layer promotes the migration of locally generated OH− outward and thereby maintains a low OH− local environment near the catalyst surface. Another evidence for the low local pH can be judged from the adsorption configuration of CO on the Th-TF COF/Cu₃N electrode. Previous studies revealed that CO is adsorbed in the atop configuration (COatop) on Cu sites in a low pH environment, and converts to the bridge configuration (CObridge) as the pH increases27. Supplementary Fig. 20 shows that only the COatop at 2044-2076 cm−1 is visible between 50 mA cm−2 to 250 mA cm−2 on the Th-TF COF/Cu₃N electrode, evidencing the low pH local environment in the COF layer. In contrast, at a higher pH environment, the CObridge (at 1819–1830 cm-1) is dominant, which was observed for the ionomer-coated electrode. In-situ Raman spectra in the 900–1200 cm−¹ region reveal that HCO₃− ( ~ 1001–1006 cm⁻¹) and CO₃²− ( ~ 1063–1069 cm−¹) bands increase in intensity with increasing current density on Sus/Cu₃N, whereas these signals are largely absent on Th-TF COF/Cu₃N, which indicate suppressed carbonate accumulation and a less alkaline interfacial environment (Supplementary Fig. 21).
In-situ attenuated total reflectance-Fourier transform infrared (ATR-TIR) spectroscopy was employed to investigate the intermediates formed during the C2H4 electrosynthesis process. Beyond intermediate species, the spectra also provide insight into CO adsorption. Supplementary Fig. 22 shows that the Th-TF COF/Cu₃N electrode exhibits a pronounced *COatop band (2040–2070 cm−¹), whereas bare Cu₃N predominantly shows *CObridge adsorption (1815–1830 cm−¹), consistent with CO configurations on Cu catalysts under acidic conditions. Regarding C-C coupling intermediates, *OCCOH emerges as a key species for C₂H₄ formation. As depicted in Fig. 4d, e, a characteristic peak corresponding to *OCCOH appears at 1238 and 1631 cm−1 on Th-TF COF/Cu₃N catalyst17. The intensity of the *OCCOH peak increases with the application of voltage. In contrast, the peak of *OCCOH intensity appears weak on the bare Cu3N catalyst at −1.1 V and gradually diminishes by -1.7 V. The peak area of *OCCOH (AOCCOH, TH-TF COF/Cu3N/AOCCOH, Cu3N) in Th-TF COF/Cu₃N is ~1.3 times greater compared to Cu3N from −1.1 V to −1.7 V (Supplementary Fig. 23). This indicates the Th-TF COF on the Cu3N catalyst allows *OCCOH to be stabilized and facilitates C2H4 production. Besides, the peaks of *CHO associated at 1057 cm−1 and *OC2H5 identified at 1130 cm−1 increase with potential, which are recognized as intermediates of C2H5OH28,29, further confirming enhanced C2 selectivity on Th-TF COF/Cu₃N catalyst.
Molecular dynamics (MD) modeling
To gain insights on the regulation capabilities of Th-TF COF for ions (K⁺, OH−) and CO₂ molecules, MD simulations were performed (details in Supporting Information). Three models were compared: the bare Cu catalyst (Model I), Cu coated with a Sustainion ionomer (Sus/Cu, Model II), and Cu coated with Th-TF COF (Th-TF COF/Cu, Model III), as illustrated in Supplementary Fig. 24 and described in Supplementary Data 1–4.
Ion and CO₂ microenvironment
Under concentration gradients and electric fields, K⁺ migrated toward the catalyst surface while OH− diffused into the bulk solution. The local [K⁺]/[OH−] ratio, derived from ion density calculations, reflects the catalytic microenvironment influencing the C₂H₄ conversion pathway in acidic CO₂RR. Simulations (Fig. 5a, Supplementary Figs. 25, 26) revealed a low [K⁺]/[OH−] ratio of 0.46 near the bare Cu surface (<1 nm). Introducing the Sustainion ionomer (Model II) enhanced outward OH− transport via anion-exchange nanochannels, quadrupling the [K⁺]/[OH−] ratio relative to bare Cu. Replacing ionomer with Th-TF COF (Model III) further increased the ratio to 3.2, highlighting its superior ability to promote OH− migration and restrict K⁺ flux. Local CO₂ concentrations ([CO₂]) also differed significantly across models. As shown in Fig. 5b, Supplementary Fig. 27, Th-TF COF/Cu maintained higher [CO₂] near the catalyst compared to bare Cu and Sus/Cu. This may arise from CO₂ adsorption by the hydrazone linkages (C = N − NH) in the Th-TF COF backbone, enriching CO₂ at the catalyst interface.

a Ratio of K⁺ to OH− concentration and (b) distribution of CO₂ molecules at different distances along the catalyst surface under an applied electric field for Cu, Sus/Cu, and Th-TF COF/Cu models. c Self-diffusion coefficients of K⁺, OH−, and CO₂ derived from mean square displacement (MSD) analysis. d Structure of Th-TF COF and its protonation process under acidic conditions. Two-dimensional density contour maps of protonated Th-TF COF under simulated acidic CO₂RR conditions, revealing reactive sites (e) and ionic migration pathways (f) within the channels. Detailed MD simulation parameters and procedures are described in the Methods and Supporting Information. Source data for Fig. 5a–c and Fig. 5e, f are provided as a Source Data file.
Diffusion dynamics via mean square displacement (MSD)
MSD calculations (Fig. 5c, Supplementary Fig. 28) demonstrate that Th-TF COF/Cu accelerates OH− diffusion (1.5 × 10−5 cm2 s−1) to the anion-exchange membrane (AEM), yielding a diffusion coefficient ~2.5× higher than bare Cu (0.6 × 10−5 cm2 s−1) and Sus/Cu (0.7 × 10−5 cm2 s−1). Conversely, K⁺ diffusion in Th-TF COF/Cu was slower (0.59 × 10−5 cm2 s−1) than in bare Cu (0.83 × 10−5 cm2 s−1) and Sus/Cu (0.78 × 10−5 cm2 s−1), confirming K⁺ confinement and the formation of a K⁺-rich microenvironment. Critically, CO₂ transport from the gas diffusion layer (GDL) to the catalyst/electrolyte interface was most efficient in Th-TF COF/Cu, with a diffusion coefficient (DCO₂) of 1.9 × 10−5 cm2 s−1, surpassing values for bare Cu (DCO₂ = 1.3 × 10−5 cm2 s−1) and Sus/Cu (DCO₂ = 1.4 × 10−5 cm2 s−1).
MD simulations demonstrate that Th-TF COF regulates K⁺/OH− fluxes by selectively restricting K⁺ diffusion while enhancing OH− transport and CO₂ mass transfer. These findings align with in situ characterization data, corroborating Th-TF COF’s dual role in ion/molecule regulation for acidic CO₂RR.
Ion transport mechanisms in Th-TF COF channels
To further investigate ion interactions with the Th-TF COF framework, we simulated dynamic bidirectional flow through its nanochannels. As shown in Fig. 5d, under acidic conditions, the C = N − NH and C–N groups on the pristine Th-TF COF skeleton undergo protonation, forming positively charged sites (-NH₂⁺), hereafter termed “protonated Th-TF COF.” We hypothesized that these charged groups facilitate selective ion migration. To test this, we modeled anion/cation transport pathways through pristine and protonated Th-TF COF (models showed in Supplementary Fig. 29) under electric field and concentration gradients.
OH− Migration Dynamics
In protonated Th-TF COF (Fig. 5e, f), axial OH− migration is favored due to (i) shorter hopping distances and (ii) wider transport channels enhanced by adjacently aligned H atoms from protonated sites along the axial pathway. Electrogenerated OH− at the catalyst surface migrates through COF channels via hydrogen-bond interactions with protonated groups (Eq. 2), either reacting with H⁺ at the AEM interface to form H₂O (Eq. 3) or diffusing toward the anode. The MSD results confirm that protonated functional groups significantly enhance OH− mobility: protonated Th-TF COF/Cu exhibits a diffusion coefficient of 1.5 × 10−5 cm2 s−1, 1.8× higher than the non-protonated model (0.8 × 10−5 cm2 s−1) (Supplementary Figs. 30, 31). Driven by electric field, the electrogenerated OH− near the catalyst surface migrates through the COF channels via H–bond interaction, and partially encounters with the H+ and forming the H2O at the AEM interface or traverses to the anode side (Eqs. (1)–(3)).
In contrast, K⁺ migration is governed by interactions with keto groups (C = O) on the COF backbone. These groups act as preferential binding sites for K⁺, promoting ion enrichment near the catalyst surface while restricting bulk diffusion (Supplementary Fig. 32). This selective retention creates a K⁺-rich microenvironment, critical for stabilizing reaction intermediates in acidic CO₂RR.
Discussion
We have demonstrated the use of Th-TF COF as a cathode modifier in an acid-fed AEM MEA system to enhance CO2-to-C2H4 and energy conversion efficiency. Th-TF COF serves as a multifaceted ion transport regulator in acidic CO₂RR, optimizing the catalytic microenvironment through dual functionalities: (1) enriching K⁺ near the catalyst surface and (2) accelerating OH− migration away from the reaction interface. Experimental and computational evidence demonstrates that its porous framework, functionalized with cation-adsorbing keto groups and protonated hydrazone linkages, selectively confines K⁺ while facilitating OH− diffusion via hydrogen-bond-mediated pathways. Concurrently, the COF enhances CO₂ mass transfer through adsorption, enriching reactant availability. These synergistic effects promote the stabilization of critical intermediates (e.g., *OCCOH) and favor C₂ product formation. Our studies show that careful design of functionalities in porous materials, such as COF, allows cation and anion transport dynamics to be decoupled, offering a strategy for ion-regulating frameworks in electrocatalytic systems.
Methods
Synthesis of Cu3N catalyst
The synthesis method of Cu3N has been reported previously17. Notably, 4 g of copper acetate (Cu(COOH)2) powder was ground by ball milling (BM) for 2 h at the speed of 400 reps before synthesis. In a typical synthesis of Cu3N, 200 mg of BM-Cu (COOH)2 and 2 g of urea were placed in two different porcelain boats and put into a tube furnace with urea at the upstream side. Then, the furnace was heated to 250 °C at a heating rate of 2 °C/min and maintained at 250 °C for 1 h with Ar gas flowing at 30 sccm. The resulting dark brown powder product was washed with ethanol/water and dried overnight at 60 °C to get Cu3N. All chemicals were purchased from Sigma-Aldrich and used as received unless otherwise specified.
Synthesis of Th-TF COF
Synthesis of ThzOPr
The synthesis method of ThzOPr has been reported previously30. To a mixture of methyl 2-hydroxy-4-iodobenzoate (2.78 g, 10 mmol), K2CO3 (5.6 g, 40 mmol), and KI (100 mg, 0.6 mmol) in acetone (150 mL) was added with 1-propylbromide (2 mL, 22 mmol) dropwisely. The mixture was refluxed with stirring under N2 atmosphere for 2 days and hot filtered through a Celite bed. The filtrate was evaporated and purified via flash chromatography (hexane/ethyl acetate = 5:1) to give a colorless oil. The oil was then added to a mixture of bis(pinacolato)diboron (2.67 g, 10.5 mmol), potassium acetate (2.94 g, 30 mmol), PdCl2(PPh3)2 (105 mg, 0.15 mmol) in 1,4-dioxane (50 mL) under N2 atmosphere. The mixture was heated at 100 °C for 18 h. The mixture was diluted with ethyl acetate and water. The organic layer was separated, dried over Na2SO4 and evaporated via vacuum. The residue was purified by flash chromatography (hexane/ethyl=5:1) to give a colorless oil (2.59 g, 81%). Then to a mixture of 1,3,5-tribromobenzene (315 mg, 1 mmol), K2CO3 (912 mg, 6.6 mmol), and Pd(PPh3)4 (116 mg, 0.1 mmol) in 1,4-dioxane/H2O (18 mL, 5:1), methyl 2-propoxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate (1 g, 3.3 mmol) was added under N2. The mixture was refluxed for 2 days. After the reaction was cooled to room temperature, the mixture was added with ethyl acetate and water. The organic layer was separated and dried over Na2SO4. The solvent was removed by vacuum, and the residue was purified by flash chromatography (hexane:EA = 10:1 then CH2Cl2) to give a light brown solid. The solid was suspended in ethanol (10 mL) and added with hydrazine monohydrate (1.5 mL). The mixture was refluxed for 1 day. The precipitate was filtered, washed with ethanol and dried to give a white solid (445 mg, 68%).
Synthesis of TFPBr
To a mixture of ethanolamine (726 μL, 12 mmol) in 28 mL dry DCM was added with NEt3 (2.51 mL). The mixture was cooled down to 0 °C and di-tert-butyl decarbonate (2.62 g, 12 mmol) in DCM (7 mL) was added dropwise and stirred at room temperature overnight, the product denoted as A. To a mixture of 2,4,6-Tribromophenol (992 mg, 3 mmol), Triphenylphosphine (1.53 g, 6 mmol), and 1,1′-(Azodicarbonyl)dipiperidine (1.514 g, 6 mmol) was added into dry THF (8 mL). The mixture was cooled down to 0 °C, and a solution of A (580 mg) was added dropwise to dry THF (2 mL). The mixture was then warmed to room temperature and heated at 60 °C for 2 days. The filtrate was evaporated and purified via flash chromatography (hexane/ethyl acetate = 50:1) to give a colorless oil. The oil (237 mg) was then added to a mixture of 4-Formylphenylboronic acid (247 mg, 1.65 mmol), Tetrakis(triphenylphosphine) palladium (0) (17.3 mg, 0.015 mmol), and K2CO3 (829 mg, 6 mmol) into THF (6 mL). The mixture was degassed and heated at 80 °C under an Ar atmosphere. The residue was purified by flash chromatography (hexane/ethyl = 10:1) to give a colorless oil.
Synthesis of the Th-TF COF
To a 10 mL Schlenk tube (15 mm × 80 mm) was added with 1,3,5-triformylbenzene (0.45 ml) and 1,4-Dioxane (0.05 ml). The mixture was sonicated for 5 mins, added with 6 M acetic acid (100 μL), flash frozen at 77 K, and degassed under freeze-pump-thaw for three cycles. The tube was then sealed and heated at 120 °C in an oven for three days. The solid obtained was exchanged with THF (5 mL) for 5 times and dried under vacuum to afford corresponding COF.
Charaterization
Atomic-resolution annular dark-field scanning transmission electron microscopy (ADF-STEM) was performed using an aberration-corrected JEOL JEM-ARM200F transmission electron microscope, operated at 200 kV. Energy-dispersive X-ray spectroscopy (EDS) mapping was carried out on a Thermo Fisher Spectra 300 microscope, also operated at 200 kV. The X-ray diffraction (XRD) patterns were collected on a Bruker D-8 instrument (Cu Kα radiation, λ = 0.154056 nm) at room temperature. X-ray photoelectron spectroscopy (XPS) was collected using Al Kα radiation, hv = 1486.6 eV, on a Thermo Fisher Scientific ESCALAB Xi+ instrument.
Electrode preparation
10 mL isopropanol was used as a solvent to disperse both Cu3N catalyst ( ~ 1.0 mg cm−2) and Sustainion (100 uL). COF-modified Cu3N electrodes (CNCP) were prepared by spray-coating COF nanoparticles onto the pristine Cu3N gas diffusion electrodes (GDEs). To facilitate uniform spray coating, COF nanoparticles were dispersed in 15 mL isopropanol and sonicated for at least 1 h. Unless otherwise stated, the nominal loading of COF nanoparticles on the carbon paper was set at approximately 0.5 mg cm−2. Finally, 200 µL of Sustainion XA-9 ionomer dissolved in ethanol (5 wt%) was spray-coated onto the electrode surface. The effect of interfacial K concentration on acidic CO2RR was studied by coating Cu3N catalyst with different concentrations of potassium ions. 10 mL aqueous solution containing different molar concentrations (0–0.3 M) of K2SO4 were spray-coated onto Cu3N GDEs.
MEA-cell assembly
For MEA electrolyzers, cathode and anode flow fields were used with the active areas of 1, 25, 36 and 49 cm2, corresponding to the 1 × 1, 5 × 5, 6 × 6 and 7 × 7 cm2 electrode windows, respectively. IrOx-Ti mesh was used as the anode, an anion exchange membrane (Pipeion), and COF-modified Cu3N GDE as the cathode electrode. The cell was operated under ambient temperature and pressure. Dry CO2(g) was supplied from the back side of the cathode, and 0.5 M K2SO4/H2SO4 was circulated in the anode part. The scale-up MEA cell stability test in an acidic system is assembled by using 7 × 7 cm2 electrode windows at a constant current of 10 A. The total cathode area was 49 cm2, and the flow rate of the CO2 inlet was 100 sccm.
Electrochemical measurement
All electrochemical tests were performed using an electrochemical workstation (Autolab PGSTAT302N) connected to a current booster (Metrohm Autolab,10 A). The catholyte of pH ~1.0, 2.0, and 4.0 were prepared by introducing a 0.5 M K2SO4 into specific amount of sulfuric acid was used as anolyte. For neutral electrolytes preparation (pH ~ 6.3–6.8), a 0.5 M K2SO4 solution was used directly. The CO2RR performance was tested in MEA-cell assemblies under galvanostatic mode. The current densities reported are based on the geometric surface areas. No iR correction was applied to the electrochemical data presented in this study.
CO2R product analysis
The gas products were collected from the gas outlet of the MEA cell, which were injected into a gas chromatograph for gas quantification. The gas chromatograph was equipped with a thermal conductivity detector for the detection of H2 and CO signals and a flame ionization detector for the detection of CH4 and C2H4 signals. The gas chromatograph was composed of packed columns of molecular sieves (5 Å) and Carboxen-1000 and employed Argon (99.999%) as the carrier gas. and liquid products from CO2RR were measured by high-performance liquid chromatography (YL9100) and headspace GC (YL Instruments). The FE was calculated using the equations:
where z is the number of electrons transferred, F is the Faraday’s constant (96,485 C mol−1), v is the gas flow rate at the outlet of the gas chamber (l min−1), r is the concentration of detected gas product in parts per million, j is the total current (A), Vm is the unit molar volume of gas (24.5 L mol−1), nproduct is the total moles of product derived from headspace GC analysis, and Q is the total charge (C).
The CO2 SPCE towards each product was determined using the following equation at 25 °C, 1 atm:
where jproduct is the partial current (A) of a specific CO2RR product, n is the electron transfer for the formation of each product molecule, and Vm = 24.5 L mol−1.
The full-cell energy efficiency for each product was calculated as follows:
Where \({E}_{{product}}^{0}\) is the thermodynamic potential for the formation of a specific CO2RR product, FEproduct is the calculated FE of the product and Ecell is the full-cell voltage without Ohmic loss correction evaluated in the MEA cells.
Data availability
All data are available from the authors upon reasonable request. Source data are provided with this paper.
References
Costentin, C., Robert, M. & Saveant, J. M. Catalysis of the electrochemical reduction of carbon dioxide. Chem. Soc. Rev. 42, 2423–2436 (2013).
Andreoli, E. CO2-to-ethylene electroreduction gets a boost. Nat. Catal. 4, 8–9 (2021).
Endrodi, B. et al. Operando cathode activation with alkali metal cations for high current density operation of water-fed zero-gap carbon dioxide electrolyzers. Nat. Energy 6, 439–448 (2021).
Yang, K. et al. Cation-driven increases of CO2 utilization in a bipolar membrane electrode assembly for CO2 electrolysis. ACS Energy Lett. 6, 4291–4298 (2021).
Fan, M. et al. Single-site decorated copper enables energy- and carbon-efficient CO2 methanation in acidic conditions. Nat. Commun. 14, 3314 (2023).
Xu, Y. et al. Self-cleaning CO2 reduction systems: unsteady electrochemical forcing enables stability. ACS Energy Lett. 6, 809–815 (2021).
Huang, J. E. et al. CO2 electrolysis to multicarbon products in strong acid. Science 372, 1074–1078 (2021).
She, X. et al. Pure-water-fed, electrocatalytic CO2 reduction to ethylene beyond 1000 h stability at 10 A. Nat. Energy 9, 81–91 (2024).
Xie, Y. et al. High carbon utilization in CO2 reduction to multi-carbon products in acidic media. Nat. Catal. 5, 564–570 (2022).
Pan, B. et al. Close to 90% single-pass conversion efficiency for CO2 electroreduction in an acid-fed membrane electrode assembly. ACS Energy Lett. 7, 4224–4231 (2022).
Zhao, Y. et al. Conversion of CO2 to multicarbon products in strong acid by controlling the catalyst microenvironment. Nat. Synth. 2, 403–412 (2023).
Fan, M. et al. Cationic-group-functionalized electrocatalysts enable stable acidic CO2 electrolysis. Nat. Catal. 6, 763–772 (2023).
Cao, Y. et al. Surface hydroxide promotes CO2 electrolysis to ethylene in acidic conditions. Nat. Commun. 14, 2387 (2023).
Ge, L. et al. Electrochemical CO2 reduction in membrane-electrode assemblies. Chem 8, 663–692 (2022).
Qin, H. G. et al. Surface-immobilized cross-linked cationic polyelectrolyte enables CO2 reduction with a metal cation-free acidic electrolyte. Nat. Commun. 14, 5640 (2023).
Li, H. et al. Tailoring acidic microenvironments for carbon-efficient CO2 electrolysis over a Ni–N–C catalyst in a membrane electrode assembly electrolyzer. Energy Environ. Sci. 16, 1502–1510 (2023).
Zheng, M. et al. Electrocatalytic CO2-to-C2+ with ampere-level current on heteroatom-engineered copper via tuning *CO intermediate coverage. J. Am. Chem. Soc. 144, 14936–14944 (2022).
Wang, H. et al. Cu3N nanoparticles with both (100) and (111) facets for enhancing the selectivity and activity of CO2 electroreduction to ethylene. New J. Chem. 46, 12523–12529 (2022).
Yin, Z. et al. Cu3N nanocubes for selective electrochemical reduction of CO2 to ethylene. Nano Lett. 19, 8658–8663 (2019).
Nie, W., Heim, G. P., Watkins, N. B., Agapie, T. & Peters, J. C. Organic additive-derived films on Cu electrodes promote electrochemical CO2 reduction to C2+ products under strongly acidic conditions. Angew. Chem. Int. Ed. 62, e202216102 (2023).
Watkins, N. B., Wu, Y., Nie, W., Peters, J. C. & Agapie, T. In situ deposited polyaromatic layer generates a robust copper catalyst for selective electrochemical CO2 reduction at variable pH. ACS Energy Lett. 8, 189–195 (2022).
Sun, M., Cheng, J. & Yamauchi, M. Gas diffusion enhanced electrode with ultrathin superhydrophobic macropore structure for acidic CO2 electroreduction. Nat. Commun. 15, 491 (2024).
Zhang, J. et al. Accelerating electrochemical CO2 reduction to multi-carbon products via asymmetric intermediate binding at confined nanointerfaces. Nat. Commun. 14, 1298 (2023).
Chen, Y. et al. Efficient multicarbon formation in acidic CO2 reduction via tandem electrocatalysis. Nat. Nanotechnol. 19, 311–318 (2024).
Feng, J. et al. CO2 electrolysis to multi-carbon products in strong acid at ampere-current levels on La-Cu spheres with channels. Nat. Commun. 15, 4821 (2024).
Ozden, A. et al. Energy- and carbon-efficient CO2/CO electrolysis to multicarbon products via asymmetric ion migration–adsorption. Nat. Energy 8, 179–190 (2023).
Chang, X., Zhao, Y. & Xu, B. pH dependence of Cu surface speciation in the electrochemical CO reduction reaction. ACS Catal. 10, 13737–13747 (2020).
Wang, S. et al. Manipulating C–C coupling pathway in electrochemical CO2 reduction for selective ethylene and ethanol production over a single-atom alloy catalyst. Nat. Commun. 15, 10247 (2024).
Peng, C. et al. Surface Co-modification of halide anions and potassium cations promotes high-rate CO2-to-ethanol electrosynthesis. Adv. Mater. 34, e2204476 (2022).
Li, X. et al. Rapid, scalable construction of highly crystalline acylhydrazone two-dimensional covalent organic frameworks via dipole-induced antiparallel stacking. J. Am. Chem. Soc. 142, 4932–4943 (2020).
Acknowledgments
K.P.L. acknowledges funding support from Singapore-International Synchrotron Access Program (SG-ISAP) as well as funding support from Center for Hydrogen Innovations CHI-P2022-01 and Singapore’s Ministry of Education Tier 1 Grant A8002669-00-00. Q.H.Y. acknowledges funding support from the National Natural Science Foundation of China (Nos. 52432005).
Author information
Authors and Affiliations
Contributions
D.C. conceived the research, synthesized the materials, and conducted catalytic measurements under the supervision of K.P.L. and Q.H.Y. The COF monomer designed were conducted by Y.Y.; STEM-HAADF and HRTEM were performed by X.H.; SEM was performed by Z.K.; FTIR measurements were performed by S.H.; Large current flow cell operation was performed with the assistance from J.L. and Q.H.; XAFS was performed by S.X.; The draft was written by D.C., revised by K.P.L. All authors discussed and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kun Qi and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, D., Liu, J., Yuan, Y. et al. Electrocatalytic CO2 reduction to ethylene in an acid-fed membrane electrode assembly at 10 A. Nat Commun 16, 10783 (2025). https://doi.org/10.1038/s41467-025-65831-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65831-8
