
Abstract
Codon usage biases play a significant role in determining gene expression levels. Codon usage was thought to primarily influence translation-dependent processes. Using an unbiased genome-wide screening to search for factors involved in codon usage effects on gene expression, we identified the CCR4-NOT complex subunit CNOT4 and many nuclear factors, including nuclear RNA exosome, PAXT complex components, and transcription factors. The CCR4-NOT complex has been shown to affect codon usage-dependent co-translational mRNA decay in yeast. Surprisingly, human CNOT4 was found to influence codon usage-dependent gene expression largely by its impact on nuclear RNA levels due to transcriptional effect. On the other hand, nuclear exosome and PAXT complex, regulate nuclear mRNA stability through the RNA quality control pathway, leading to preferential cytoplasmic accumulation of mRNAs with optimal codon usage. Overall, our results show that different nuclear mechanisms play a key role in determining nucleotide composition-dependent gene expression levels in human cells.
Similar content being viewed by others

Introduction
Codon usage bias, the preference for certain synonymous codons for almost all amino acids, is a fundamental feature of eukaryotic and prokaryotic genomes. Although synonymous codon changes were previously thought to be silent, gene codon usage has been shown to play a major role in regulating gene expression and protein biogenesis in diverse organisms1,2,3,4. In various organisms, genome-wide correlations between codon usage bias and protein levels have been observed5,6,7. As expected from its role in encoding amino acids, the effects of codon usage on gene expression were thought to be mainly due to influences on translation-dependent processes. In support of this, codon usage has been shown to control translation elongation speed and subsequently affects co-translational protein folding8,9,10,11,12,13,14. Due to the translational pausing at non-optimal codons during elongation, codon usage affects mRNA translational efficiency by causing premature translation termination15,16. The effects of codon usage on elongation kinetics can also feedback to regulate translation initiation process7,17,18.
In addition to directly acting on translation process, accumulating evidence indicates that codon usage influences gene expression by determining mRNA levels genome-wide5,6,19,20,21. Codon usage has been shown to indirectly affect translation efficiency by regulating translation-dependent mRNA decay in different organisms3,22,23,24,25,26. In yeast, for example, codon usage influences the recruitment of the CCR4-NOT complex to the ribosome via an interaction between the NOT5 subunit and the ribosomal E-site when translation elongation encounters nonoptimal codons, which can result in a ribosome with an empty A site27,28,29. The CCR4-NOT complex is the major deadenylating enzyme in eukaryotes and plays a key role in translation-dependent mRNA decay30,31. Thus, the elongation pausing triggered by non-optimal codon promotes mRNA deadenylation and decay. A cooperation between PABPC and human CCR4-NOT complex was shown to promote mRNA deadenylation and prevent premature uridylation and decay32. Reconstitution of recombinant human CCR4-NOT complex has also permitted elucidation of mechanistic insights of its regulated mRNA deadenylation33. Furthermore, the NOT complex subunits influence elongation kinetics and regulate mRNA solubility in a codon optimality-dependent manner31,34,35.
In addition to its roles in mRNA metabolism, the CCR4-NOT complex is also an important regulator of chromatin structure, transcription, nuclear RNA quality control and mRNA export in the nucleus30. Mutants of CCR4-NOT complex show impaired transcription elongation in yeast36. Several genetic and biochemical evidences support that CCR4–NOT, especially NOT4 subunit, controls H3 K4me3 by regulating the stability of Jhd2, a major histone demethylase in yeast37. Recently, the human CCR4-NOT complex has been shown to globally regulate gene expression by transcriptionally silencing retrotransposon activation38 but its nuclear role in the codon usage effect is unknown.
Although effects on translation were thought to be the main mechanism through which codon usage regulates gene expression, recent evidence suggests that codon usage also regulates gene expression by influencing translation-independent processes4. We previously demonstrated that a major effect of codon usage on expression of reporter genes in the filamentous fungus Neurospora crassa is through an influence on chromatin structure that impacts transcription efficiency5. Consistent with a translation-independent effect, codon usage or GC content (which correlates with codon usage) within gene coding regions influences mRNA levels without influencing mRNA decay in human cells39,40. Codon usage has also been shown to influence transcription and chromatin structures in Drosophila and human cells, suggesting a conserved mechanism in eukaryotes21,41. In addition, non-optimal codons can impact mRNA levels by causing premature transcription termination42. Furthermore, high GC content promotes cytoplasmic mRNA localization, and mRNA splicing enhances nuclear export of AU-rich mRNAs43,44. The nuclear RNA export pathway depends on multiple mRNA export factors such as NXF1, TREX and RBM33 that differentially recognize and facilitate export of either GC-rich or AU-rich transcripts19,45,46,47. However, it is unclear whether the preferential nuclear export of GC-rich mRNAs is mainly due to the preference at the nuclear export step or at an upstream process. Together, these results suggest that gene codon usage biases are due to selection by both translation-dependent and translation-independent processes.
The relative contributions of translation-independent nuclear effects and translation-dependent effects of codon usage on gene expression in mammalian cells remain unknown. Additionally, the mechanism responsible for the preferential export of GC-rich transcripts, which are enriched with optimal codons, is unclear. In this study, we show that codon optimality correlates genome-wide with nuclear mRNA and transcription levels in human cells, indicating a broader role for codon usage in regulating nuclear mRNA metabolism. To identify mechanisms involved in codon usage effects in gene expression, we performed a genome-wide CRISPR-Cas9 screen using a dual-codon usage reporter human cell line. This screen identified CNOT4, a RING E3 ligase providing the ubiquitination activity of the CCR4-NOT complex, and many components of the nuclear RNA quality control pathway as factors that mediate the effects of codon usage on gene expression in human cells. Our findings reveal that different nuclear, translation-independent effects of codon usage/nucleotide compositions have important impacts on gene expression levels in human cells by influencing transcription, the nuclear quality control pathway and mRNA localization.
Results
Codon usage and nuclear RNA levels correlate genome-wide in human cells
Our previous results on the nuclear role of codon usage in fungi and Drosophila cells prompted us to examine the nuclear effect of codon usage genome-wide in human cells. We previously demonstrated in Drosophila cells that the genome-wide effect of codon usage on mRNA levels can be masked by tissue-specifically expressed genes and the genome-wide correlations between gene codon usage and mRNA levels are much stronger for constitutively expressed genes than for tissue-specifically expressed genes21. Thus, we compared genome-wide Pearson correlation coefficients between mRNA levels and gene codon usage as measured by codon adaptation indices (CAI) using available total and nuclear RNA-seq (sequencing) data for human cells45,48 for all genes and for constitutively expressed genes. As expected, there was a weak positive correlation between codon usage and total RNA or nuclear RNA levels genome-wide when all genes were included in the analyses (Fig. 1a). However, for the constitutively expressed genes (Const1) (i.e., which are up- or downregulated by more than 2-fold in less than five tissues among the 53 human tissues examined)21, the positive correlations became much stronger (Fig. 1a). Importantly, the positive correlations were similar for both total and nuclear RNA-seq data. Furthermore, very similar correlation results were seen in two independent global nuclear run-on (GRO-seq) datasets (ref. 49 and (GSE70449)), which reflect genome-wide transcriptional levels (Fig. 1a). Because the nuclear effects of codon usage should be independent of translation, we also performed the analyses of these data using gene GC contents (which reflect nucleotide composition) instead of CAI and found similar positive correlations (Fig. 1b). Although CAI and GC content can reflect different aspects of nucleotide composition, CAI values positively correlated with GC contents in the human genome.

Plots showing Pearson correlation coefficients between CAI (a) or GC content (b) and abundance of RNA in total, nuclear and nascent RNA samples from human cells as indicated, calculated for either all genes or constitutive1 genes (number of genes used for analysis are indicated). Data are presented as mean ± SD. c Pearson correlation coefficients between occurrence of codon frequencies and RNA abundance for 59 synonymous codons in the total, and nuclear RNA samples from MCF7 cells and nascent RNA samples from HeLa cells (Gro Seq) calculated for constitutive1 genes are shown here. The correlation coefficients estimate the effect of codon optimality of each codon on transcript abundance in RNA samples. G/C ending, and A/T ending codons are indicated by blue, and red bars, respectively. d Pearson correlation coefficients between occurrence of codon frequencies and RNA abundance for 59 synonymous codons in nascent RNA samples from HeLa cells (Gro Seq) versus those in the total, nuclear and cytosolic RNA from MCF7 cells is plotted for constitutive1 genes. Correlation coefficients (r) between different comparisons are indicated. e Western blot result showing wild-type (wt) and codon optimized (opt) GFP protein levels in HEK293T cells transfected with indicated GFP mRNA. f Densitometric analyses of Western blot results of wt and opt-GFP protein levels as in (e) normalized with respective mRNA levels from six independent experiments. Data are presented as mean ± SD. g Western blot result showing wild-type (wt) and codon optimized (opt) GFP protein levels in HEK293T cells co-transfected with indicated GFP plasmid constructs and opt-mCherry plasmid construct for 72 h. mCherry level was used to normalize for transfection efficiency. (h) Densitometric analyses of Western blot results of wt and opt-GFP protein levels as in (g) normalized with mCherry protein levels from three independent experiments. Data are presented as mean ± SD. i Diagram showing the design of the CMV promoter driven wt-mCherry/opt-GFP dual codon usage reporter construct. CAI of both reporters are indicated. j Plot showing relative RNA levels of opt-GFP and wt-mCherry reporters in nascent and input RNA, measured by RT-qPCR using dual reporter stable cell line from three independent experiments. Nascent RNA levels were determined by nuclear run-on assays. Data are presented as mean ± SD. Source data are provided as a Source Data file.
To study the impact of codon optimality of synonymous codons on mRNA levels, we determined the Pearson correlation coefficients between individual codon frequency (except for stop, methionine, and tryptophan codons) in constitutive genes and their RNA levels using the total mRNA-seq, nuclear RNA-seq, and GRO-seq data. Individual codons were assigned into G/C ending or A/T ending codon group because human codon usage is biased for C/G ending codons. In all three sets of analyses, the codons that are positively correlated with total, nuclear and Gro-seq RNA levels are almost all G/C ending codons and those with negative correlations are almost all A/T ending codons (Fig. 1c). In addition, the codons were also assigned into optimal, intermediate, and non-optimal groups based on a previous study on codon optimality based on RNA stability in human cells23. As shown in Suppl. Fig. 1a, the codons with positive correlations with RNA levels are mostly optimal codons and the codons with negative correlations are mostly non-optimal codons. Furthermore, the correlations of individual codon with RNA levels between the Gro-seq data and those of total RNA, nuclear and cytoplasmic RNA data are highly correlative (Pearson r values > 0.9) (Fig. 1d). Together, these results suggest that codon optimality has a genome-wide nuclear effect on gene expression.
To examine if nuclear effect plays a substantial role in determining codon-usage dependent gene expression in human cells, we compared the effect of codon usage on protein expression by transfecting the wild-type and codon-optimized GFP mRNAs directly into HEK293T cells (involving mainly translation/cytoplasmic effects) with that by transfecting DNA constructs expressing the same GFP genes (involving both nuclear and cytoplasmic effects). As shown in Fig. 1e, f, the codon optimization resulted in ~6x increase of GFP protein level (normalized by mRNA levels) by mRNA transfection. In contrast, the codon optimization resulted in more than 30x increase of GFP protein level (normalized with mCherry protein levels from co-transfected mCherry DNA construct) by DNA transfection (Fig. 1g, h). These results are consistent with our previous results comparing the codon usage effects of the human Kras gene (mRNA vs DNA plasmid)41. Together, these results suggest that the nuclear effects of codon usage play a major role in mediating codon-usage dependent gene expression in human cells.
To confirm the transcriptional impact of codon usage, we created a HEK293 cell line stably transfected with a previously created dual-reporter construct that expresses the codon-optimized GFP reporter gene (opt-GFP) and the wild-type mCherry gene (wt-mCherry), which is enriched with rare codons, independently controlled by the same cytomegalovirus (CMV) promoter (Fig. 1i)50. We then used the dual reporter cell line and nuclear run-on assay to compare the transcription of the two codon usage reporter genes. As shown in Fig. 1j, we found that the opt-GFP transcription was ~5x as that of the wt-mCherry. Thus, codon usage indeed has an important impact on transcription. However, in the input nuclear RNA, we found that the opt-GFP mRNA is close to 20x of that of the wt-mCherry (Fig. 1j). Because the nuclear RNA level is affected by different nuclear effects (transcription, nuclear decay, and export), this result suggests that transcriptional regulation is not the only nuclear process determining the effect of codon usage on mRNA levels. This conclusion is consistent with previous results on nascent RNA production in fungi, Drosophila, and human cells when other reporter genes were analyzed5,21,40,41,43. Together, these results suggest that the nuclear, translation-independent effects have an important role in determining how codon usage influences gene expression in eukaryotic organisms.
Because gene GC contents correlate with codon usage bias, the observed codon usage-dependent nuclear nascent RNA effect could be due to mRNA GC content effect. To examine this, we re-analyzed the data in Fig. 1c based on the GC contents of individual codons (Supplementary Fig. 1b). Although the 100% GC and 100% AT codons all have positive or negative correlations with nascent RNA levels, respectively, the codons with positive correlations in the 66.7% and 33.3% GC codon groups are almost all optimal codons. These results suggest that the observed codon usage effects are not solely determined by GC contents. Thus, the observed codon usage biases are the selection results by both nucleotide compositions for nuclear effects and optimal translation in cytoplasm.
To explore the contribution of gene regions other than coding regions in determining their RNA levels, we compared Pearson correlations between mRNA levels and GC contents of 5’-UTR, 3’-UTR and intronic regions with the correlations with GC contents of coding regions (CDS). As seen in Supplementary Fig. 2a–d, the GC contents of intronic regions and 3’-UTRs of genes correlated positively with the total, nuclear and Gro-seq RNA levels to similar extents as with CDS. On the other hand, the GC contents of 5’-UTRs exhibited relatively modest positive correlations (Supplementary Fig. 2c). Such a difference might be due to the much higher GC contents of human 5’-UTRs than those of 3’-UTRs (60.8 ± 12.6% vs 44.4 ± 11%, respectively)51. Another plausible reason of lower correlations between RNA levels and the GC contents of 5’-UTRs might be their shorter lengths (average 210 nucleotides) than 3’-UTRs (average length of 1027.7 nucleotides) and CDS52. Further, CAI/GC/GC3 contents of CDS show a much higher correlation with the GC contents of 3’-UTR and intronic regions than those of 5’-UTR regions51 (Supplementary Fig. 2e). These results suggest that GC contents of all the regions of genes function collectively in impacting their RNA levels genome-wide.
A genome-wide CRISPR-Cas9 screen identified many nuclear factors involved in codon usage effect on gene expression
To identify factors involved in regulating the codon usage effect on human gene expression, we created a HEK293 cell line stably transfected with a dual-reporter construct that expresses the codon-optimized mCherry reporter gene (opt-mCherry) and the wild-type GFP gene (wtGFP), which is enriched with rare codons, independently controlled by CMV promoter (Fig. 2a)50. The use of the dual reporter cell line enables simultaneous measurements of both reporters as well as their transcripts levels and can avoid the differences in the efficiencies of different experimental treatments when the reporters are expressed individually. As expected, the cell line exhibited much higher mCherry fluorescence levels compared to the levels of GFP fluorescence, indicating a strong codon usage effect on gene expression. We then carried out an unbiased genome-wide CRISPR-Cas9 screen in the opt-mCherry and wtGFP dual reporter cell line by sorting cell population with highest GFP/mCherry fluorescence ratio (0.5% of all cells) and sequencing for enriched single-guide RNAs (sgRNAs) (Fig. 2b). This screen allowed the identification of potential factors, when depleted in cells, resulting in reduced codon usage effect on reporter expression.

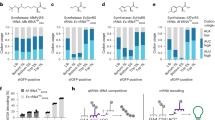
a Diagram showing the design of the CMV promoter driven wtGFP/opt-mCherry dual codon usage reporter construct. CAI of both reporters are indicated. The fluorescence measurements of GFP (green) and mCherry (purple) reporters in the wtGFP/opt-mCherry stable cells, as assessed by FACS, are shown below. b Design of CRISPR–Cas9 screening is depicted here. CRISPR-Cas9 screen was performed using the wtGFP/opt-mCherry dual reporter cell line, cell population with highest GFP/mCherry fluorescence ratio (0.5% of all cells) was sorted by FACS and was used for sequencing to identify enriched sgRNAs. c Model-based Analysis of Genome-wide CRISPR/Cas9 Knockout (MAGeCK) was used to identify genes targeted by enriched sgRNAs in sorted populations as described78. MAGeCK analysis of screening results identified many nuclear factors potentially involved in regulation of codon-usage mediated gene expression (highlighted in black). Genes related to nuclear RNA exosome and CNOT4 are highlighted in red. d Validation of screening results by the fluorescence measurements of GFP and mCherry reporters in the wtGFP/opt-mCherry stable cells treated with control siRNA (green) or siCNOT4 (purple) for 72 h, as assessed by FACS. e Western blot results showing wtGFP and opt-mCherry protein levels in the wtGFP/opt-mCherry stable cells treated with control siRNA or siCNOT4 for 72 h. Actin protein levels are used as loading controls. f Densitometric analyses of Western blot results of wtGFP and opt-mCherry protein levels as in (e) from four independent experiments. Data are presented as mean ± SD, unpaired two-tailed Student’s t test. g FACS analysis of wtGFP/opt-mCherry stable cells treated with indicated lentiviral sgRNA and selected with puromycin for 9 days. Non targeting control sgRNA treated cells are shown in green while knockout cells are shown in purple in dot plots. h Western blot result showing wtGFP and opt-mCherry protein levels in the wtGFP/opt-mCherry stable cells following sgRNA treatment as indicated. i Densitometric analyses of Western blot results of wtGFP and opt-mCherry protein levels as in (h) from six independent experiments. Data are presented as mean ± SD, one-way ANOVA with Dunnett’s multiple comparisons test. Source data are provided as a Source Data file.
The top hits of the screen include CNOT4, a subunit of the CCR4-NOT complex, in addition to many factors localized to the nucleus (Fig. 2c). These factors include the multiple RNA exosome complex components EXOSC2, 4, 7 and 8; the catalytic subunit of nuclear RNA exosome DIS3; components of the nuclear PAXT complex, which binds the poly-A tails of nascent mRNAs and targets them to the nuclear exosome for decay53, including the zinc-finger protein ZFC3H1, the nuclear poly-A binding protein PABPN1, and the nuclear CAP binding protein NCBP2 (also known as CBP20); transcription factors TAF6, 7, 10; and other proteins known to be enriched in the nucleus. Consistent with a previous study on the role of codon usage-dependent mRNA splicing and export43, several nuclear factors involved in splicing (DBR1, ZCRB1, and HNRNPL) were also identified as top hits in the screen.
Because the CCR4-NOT complex was previously shown to regulate translation-dependent non-optimal codons-mediated mRNA decay and mRNA solubility in yeast27,31,35, the identification of CNOT4 suggest that it also plays a role in regulating the codon usage-dependent effects on gene expression in human cells. When the wtGFP/opt-mCherry dual reporter cells were depleted of CNOT4 using a small interfering RNA (siRNA) targeting CNOT4, there was a significant increase in wtGFP fluorescence levels but not opt-mCherry (Fig. 2d). In addition, there was a specific increase of wtGFP protein level as compared to opt-mCherry protein level (Fig. 2e, f).
To confirm the codon usage-dependent effect of CNOT4 depletion, we also examined the opt-GFP/wt-mCherry as well as opt-GFP/opt-mCherry dual-reporter cells to determine if the observed codon usage effect is reporter-specific. As shown in Supplementary Fig. 3a–f, there was a specific increase of wt-mCherry/opt-GFP protein ratio similar to the wtGFP/opt-mCherry protein ratio observed after CNOT4 depletion, while no significant change was observed in opt-GFP/opt-mCherry protein ratio in the opt-GFP/opt-mCherry dual-reporter cells after CNOT4 depletion. These results indicate that the effects observed are codon usage-specific and are independent of reporter genes used.
We also examined the effect of CNOT4 depletion in wtGFP and opt-GFP single reporter cells in which the reporter is either with or without an intron43 (Supplementary Fig. 3g). As expected, the CNOT4 depletion resulted in a significant increase in wtGFP protein levels in both absence and presence of an intron (Supplementary Fig. 3h-k), while no significant changes were observed in opt-GFP protein levels. These results indicate that the role of CNOT4 on the codon usage-dependent effect on reporter expression is independent of the presence of an intron.
The identification of DIS3 and components of the PAXT complex suggested that the nuclear exosome and the nuclear RNA quality control are involved in mediating the codon usage effect. PAXT complex directs polyadenylated RNA substrates to the nuclear exosome core for decay53. To confirm their role, we separately depleted DIS3, ZFC3H1, and PABPN1 in the wtGFP/opt-mCherry dual reporter cells by using gene-specific sgRNAs. As shown in Fig. 2g–i, depletion of each of these proteins led to a significant and specific increase of wtGFP/opt-mCherry protein ratios. On the other hand, there was no significant increase in the opt-GFP/opt-mCherry protein ratio in the opt-GFP/opt-mCherry dual-reporter cells after siRNA depletion of ZFC3H1 or PABPN1 (Supplementary Fig. 4a–c), confirming the codon usage-dependent effect of PAXT complex components. Examination of the effects of ZFC3H1 and PABPN1 depletions in intron-containing or intron-lacking wtGFP and opt-GFP single reporter cells also revealed a significant increase in wtGFP protein levels both in the presence or absence of an intron (Supplementary Fig. 4d–g) with no significant changes in opt-GFP protein levels. These results further confirmed the involvement of these factors in mediating codon usage effects on gene expression.
CNOT4 impacts codon usage-dependent gene expression largely due to its nuclear effect on mRNA levels
To determine whether the codon usage effect of CNOT4 is mainly mediated by the translation-dependent role of the CCR4-NOT complex, we transfected 5’ capped and 3’ polyadenylated mCherry mRNA that was either codon-optimized or not into HEK293 cells by electroporation. The transfection of the mRNA instead of plasmid DNA eliminated potential transcription-dependent nuclear effects that can influence cytoplasmic mRNA levels. Surprisingly, depletion of CNOT4 did not significantly impact the codon usage effect on mCherry protein levels (Fig. 3a, b), suggesting that the translation-dependent effect on translation efficiency or mRNA decay is not the main mechanism that determines the codon usage effect of CNOT4 for the reporter transgenes.

a Western blot result showing wild-type (WT) and optimal (Opt) codons enriched mCherry protein levels in HEK293 cells first treated with siControl or siCNOT4 for 72 h and then transfected with indicated mCherry mRNA for 8 h. Western blot results confirmed depletion of CNOT4 protein after siRNA mediated knockdown (middle panel). Tubulin protein levels are used as loading controls. b Densitometric analyses of Western blot results of WT and Opt-mCherry protein levels as in (a) normalized with Tubulin levels from three independent experiments are shown in left panel. Relative WT and Opt-mCherry mRNA levels measured in transfected cells harvested after 8 h as determined by RT-qPCR from three independent experiments are shown in right panel. Data are presented as mean ± SD, one-way ANOVA with Sidak’s multiple comparisons test. c Representative Western blot results showing indicated protein levels in total, cytosolic and nuclear protein fractions of HEK293 cells. This experiment was repeated two times independently with similar results. d Representative immunofluorescence imaging results in HEK293 cells treated with siControl or siCNOT4 for 72 h using a rabbit polyclonal anti-CNOT4 antibody. This experiment was repeated three times independently with similar results. The scale bar corresponds to 5 µm. e Quantification of the ratio of mRNA levels of GFP and mCherry reporters in total, cytosolic and nuclear RNA fractions isolated from the wtGFP/opt-mCherry stable cells treated with indicated siRNA for 72 h and with cycloheximide (CHX) as indicated for 24 h as measured by RT-qPCR. Data are presented as mean ± SD, n = 4 independent experiments for samples with no CHX treatment, n = 6 independent experiments for samples with CHX treatment, one-way ANOVA with Sidak’s multiple comparisons test. f Quantification of the ratio of cytoplasmic to nuclear levels (left panel) or nuclear to cytoplasmic levels (right panel) of GAPDH mRNA and GAPDH-IN1 (intron1 containing) pre-mRNA in RNA samples from Fig. 3e as measured by RT-qPCR to determine cellular fractionation efficiency. Data are presented as mean ± SD, n = 4 independent experiments. g Quantification of the nuclear RNA levels of wtGFP and opt-GFP reporters in the presence or absence of in-frame stop codons as measured by RT-qPCR of RNA samples isolated from stably transfected single reporter cells treated with indicated siRNA for 72 h. Data are presented as mean ± SD, n = 3 independent experiments, one-way ANOVA with Sidak’s multiple comparisons test. h Nuclear run-on assay results showing relative nascent opt-mCherry and wtGFP RNA levels (normalized with 18S RNA levels) as measured by RT-qPCR using the wtGFP/opt-mCherry stable cells. The cells were subjected to indicated siRNA mediated knockdown for 72 h. Data are presented as mean ± SD, n = 3 independent experiments, one-way ANOVA with Sidak’s multiple comparisons test. Source data are provided as a Source Data file.
The NOT proteins were originally identified as global transcriptional regulators and are known to participate in various steps of transcriptional process in yeast30,31,54. To evaluate whether CNOT4 has a function in the nucleus of human cells, we examined its cellular localization in HEK293 cells. Western blot analysis showed that various CNOT4 isoforms were highly enriched in the nuclear fraction and their cytoplasmic levels were low (Fig. 3c), suggesting that the CNOT4 mainly acts in the nucleus. Immunofluorescence assay also confirmed the nuclear enrichment of CNOT4 in HEK293 cells although some signals were also seen in the cytoplasm (Fig. 3d). The immunofluorescence signals were reduced substantially in the cells depleted of CNOT4, confirming the specificity of the fluorescence signal for CNOT4. Consistent with these results, it was previously shown that CNOT1 and CNOT3, two other subunits of the CCR4-NOT complex, were also present in the nuclei of human and mouse cells55,56.
The nuclear enrichment of CNOT4 prompted us to determine whether the codon usage effect we observed is largely caused by the nuclear effect of CNOT4. We compared the ratios of wtGFP mRNA to opt-mCherry mRNA in the total, cytoplasmic, and nuclear RNA preparations of the dual reporter cells. The opt-mCherry mRNA was detected at significantly higher levels than the wtGFP mRNA in total RNA samples of the control cells (Fig. 3e), indicating that the codon usage effect is largely due to changes in mRNA levels. CNOT4 depletion not only resulted in a marked increase in the wtGFP/opt-mCherry mRNA ratios in the total and cytoplasmic RNA samples but also in nuclear RNA (Fig. 3e). As expected, opt-GFP/opt-mCherry mRNA ratio was not increased when tested using the opt-GFP/opt-mCherry dual-reporter cells after CNOT4 depletion (Suppl. Fig. 5a). Importantly, a marked increase in the wtGFP/opt-mCherry mRNA ratios was observed in the total RNA in the CNOT4 depleted cells compared to control cells even in the presence of a translation inhibitor, cycloheximide, suggesting that this effect of CNOT4 on the reporter mRNA levels is largely translation independent (Fig. 3e).
Relative GAPDH mRNA and nascent intron-containing GAPDH pre-mRNA levels in the cytosolic and nuclear RNA fractions demonstrated the purity of our cellular fractionation preparations (Fig. 3f). To exclude the possibility of contamination of mRNA attached to ER membrane in nuclear fractions, we examined the levels of Calnexin, an ER marker, in total, cytoplasmic and nuclear fractions. As seen in Suppl. Fig. 5b, Calnexin was present in total and cytoplasmic fractions but not in the nuclear fraction. In addition, tubulin was found in the total and cytoplasmic fractions but not in nuclear fraction. On the other hand, the nuclear marker Lamin was absent from the cytoplasmic fraction. These results further confirmed the purity of our cytoplasmic and nuclear fractionations. These results suggest that CNOT4 regulates codon usage effect on gene expression by preferentially suppressing the nuclear levels of mRNAs enriched in non-optimal codons. We also examined the effect of another CCR4-NOT complex component, CNOT3, on the nuclear RNA levels of wtGFP and opt-mCherry by its depletion. As seen in Suppl. Fig. 5c-e, CNOT3 depletion by siRNA also resulted in a significant increase in wtGFP/opt-mCherry RNA ratio in total and nuclear RNA. These results support the involvement of CCR4-NOT complex in the nuclear effect of codon usage on gene expression.
To further validate the translation-independent role of CNOT4 on codon usage effect, we examined the effects of CNOT4 depletion on the expression of wtGFP and opt-GFP (enriched in optimal codons) RNAs in the presence or absence of in-frame stop codons immediate downstream of the start codon (Suppl. Fig. 5f). Single reporter stably transfected cells were generated. As expected, CNOT4 depletion in these cells (with/without premature stop codons) resulted in a higher and significant increase in nuclear wtGFP RNA levels as compared to the opt-GFP reporter RNA levels (Fig. 3g). These results further suggest the translation-independent role of CNOT4 in mediating the codon usage effect on nuclear RNA levels.
We next explored the impact of the nucleotide composition of 3’-UTR on the effect of CNOT4 depletion on wtGFP and opt-GFP nuclear expression levels by changing its 3’-UTR (385 nt) GC content from 42% to 71% (high GC) (Suppl. Fig. 5g). The high GC in the 3’-UTR should counter the low GC content effect of wtGFP, resulting in impaired codon usage/GC content-dependent effects on the nuclear mRNA levels caused by CNOT4 depletion. As expected, in contrast to the original reporter cells, the preferential increase of wtGFP mRNA level in the nucleus due to CNOT4 depletion was not observed in these new reporters (Suppl. Fig. 5h), indicating that the high 3’-UTR GC impaired the differential codon usage/GC content-dependent effect caused by CNOT4 depletion. Thus, the GC content of 3’-UTR also contributes to the nuclear effect of codon usage (GC composition).
We also examined the effect of CNOT4 depletion on mRNA levels of intron-containing wtGFP and opt-GFP reporters as compared to controls. The functionality of the intron was confirmed by the dramatic decrease of the intron-containing mRNA in the cytosolic RNA compared to nuclear RNA. As seen in Supplemental Fig. 5i (left panel), the total wtGFP mRNA levels of the intron-containing reporter exhibited a significant increase in CNOT4 depleted cells compared to control cells, but no significant increase was observed in opt-GFP mRNA levels from intron-containing reporter cells. In addition, the preferential increase of wtGFP mRNA levels upon CNOT depletion was also observed in cytosolic as well as nuclear RNA fractions (Suppl. Fig. 5h, right panel), suggesting that its increase in cytosolic RNA was not mainly due to increased nuclear export due to the presence of the intron. These results further support the nuclear function of CNOT4 on gene expression in a codon usage-dependent manner.
We next examined if CNOT4 affects the transcription of reporter genes in a codon-usage dependent manner by nuclear run-on assays. As shown in Fig. 3h, CNOT4 depletion indeed resulted in a significant increase of wtGFP transcription while a much smaller increase was seen at the transcription levels of opt-mCherry as compared to control cells, suggesting a role of CNOT4 in mediating codon usage-dependent transcriptional regulation. This result is consistent with the known role of CCR4-NOT complex as a transcriptional regulator30. On the other hand, the depletion of the PAXT component ZFC3H1 did not affect the transcription of wtGFP as compared to control cells (Fig. 3h). Thus, CNOT4 and PAXT complex have different roles in the nuclear effects of codon usage.
Depletion of CNOT4 impairs genome-wide codon usage effects on transcription
We next examined the global effects of CNOT4 depletion on RNA levels genome-wide by sequencing RNA from total and nuclear fractions of HEK293 cells treated with an siRNA specific for CNOT4. Analyses of the total RNA-seq data revealed that CNOT4 depletion resulted in a significant reduction of correlation between GC content or CAI and mRNA levels for all genes and constitutively expressed gene groups (constitutive 1: up- or downregulated by >2-fold in no more than five human tissues; constitutive 2: up- or downregulated by >3-fold in no more than seven tissues)21, (Fig. 4a and Supplementary Fig. 6a). We then performed nuclear run-on sequencing (BrU-IP and input RNA sequencing) to investigate the effect of CNOT4 depletion on genome-wide transcription. As shown in Fig. 4b and Supplementary Fig. 6b, CNOT4 depletion substantially decreased the correlation between the gene GC contents/CAI and their nascent RNA abundance (measured as BrU-IP/input counts ratio) genome-wide as compared to control cells. In addition, the fold changes of total mRNA levels in CNOT4 depleted cells as compared to control treated cells showed clear negative correlations for both GC contents and CAI of genes in total RNA (Fig. 4c, and Supplementary Fig. 6c). Similarly, the fold changes of nascent RNA abundance in CNOT4 depleted cells as compared to control treated cells also showed negative correlations for both GC contents and CAI of different genes (Fig. 4d). These results suggest that CNOT4 is important for the transcriptional effect of codon usage genome-wide.

Plots showing Pearson correlation coefficients between GC content (nucleotide composition effect) and abundance of RNA in total RNA sequencing samples (a) and nascent RNA samples (b) as determined by nuclear run-on sequencing from HEK293 cells treated with either siControl or siCNOT4 as indicated for 72 h, calculated for all genes or constitutive genes. Data are presented as mean ± SD, n = 2 independent experiments. Scatter plots showing Pearson correlation coefficients between Log2 fold change in total RNA (c) and nascent RNA (d) levels (in CNOT4 depleted cells as compared to control treated cells) vs their GC content (left) or CAI (right) of constitutive1 genes. Pearson R and P (two-tailed) values are indicated. Dots, genes. (e) Gene ontology (GO) enrichment analysis of the downregulated genes in nuclear RNA of CNOT4 depleted cells compared to control cells was done using ClusterProfiler package in R82. Source data are provided as a Source Data file.
Upregulated mRNAs are preferentially genes with non-optimal codon usage profiles, which is consistent with its role in transcription by suppressing genes enriched with non-optimal codons. Furthermore, depletion of CNOT4 resulted in downregulation of nuclear mRNAs enriched for genes involved in chromatin regulation (Fig. 4e), which is consistent with the known role of the CCR4-NOT complex in transcriptional regulation. This conclusion is also consistent with our previous studies that demonstrated the roles of chromatin regulators in codon usage-dependent transcriptional effects in other organisms5,41,57.
The nuclear, but not cytosolic, RNA exosome and the nuclear PAXT complex mediate the codon usage effect on gene expression
The multi-subunit RNA exosome is located in both nucleus and cytosol of eukaryotic cells58 (Fig. 5a). In the nucleus, its primary function is in RNA quality control as the RNA exosome processes and degrades a variety of aberrant noncoding and pre-mRNA transcripts. In the cytoplasm, the complex is involved in mRNA turnover and quality control. The CCR4-NOT complex is known to mediate mRNA decay in a translation- and codon usage-dependent manner by monitoring the speed of translation elongation and causing mRNA deadenylation and subsequent decay22,23,27,29. DIS3L is the catalytic subunit of the cytosolic exosome59. Its co-factor is the superkiller (Ski) complex, which consists of SKIV2L, the tetratricopeptide repeat-containing protein TTC37 (also known as Ski3), and two copies of the WD40-repeat protein Ski860. In the cytosol, HBS1L (also known as Ski7) connects the Ski complex to the RNA exosome for co-translational mRNA quality control61.

a A schematic showing components of nuclear and cytosolic RNA exosome and their RNA targeting complexes. b FACS analysis of wtGFP/opt-mCherry stable cells following siRNA mediated knockdown of indicated genes. Control siRNA treated cells are shown in green while knockdown cells depleted of different factors are shown in purple in dot plots. c Western blot result showing wtGFP and opt-mCherry protein levels in wtGFP/opt-mCherry stable cells following siRNA mediated knockdown of genes as indicated. Actin protein levels are used as loading controls. d Densitometric analyses of Western blot results of wtGFP and opt-mCherry protein levels as in (c) from three independent experiments. e Quantification of the ratio of mRNA levels of GFP and mCherry in total RNA isolated from wtGFP/opt-mCherry stable cells treated with indicated siRNA for 72 h as measured by RT-qPCR. f Quantification of SNHG19 (left) and SNHG10 (right) RNA levels in total RNA isolated as in (e). Data in (d–f) are presented as mean ± SD, n = 3 independent experiments, one-way ANOVA with Dunnett’s multiple comparisons test. Source data are provided as a Source Data file.
To identify the exosome and adaptor complex that regulate codon usage-dependent expression of our reporter genes, we used siRNA to silence expression of different components of these complexes. siRNA-mediated depletions of PAXT complex components ZFC3H1 and PABPN1 significantly increased the expression of wtGFP but not opt-mCherry in the wtGFP/opt-mCherry dual reporter cell line, whereas depletion of DIS3L, HBS1L, or TTC37 did not significantly affect the expression of reporter proteins despite of the accumulation of previously identified cytoplasmic exosome substrates62 (Fig. 5b–d and Supplementary Fig. 7a, b). We also investigated the codon usage dependent effect on the expression of our reporter genes at the transcripts level and found a significant increase in wtGFP/opt-mCherry mRNA ratio in the cells depleted of ZFC3H1 and PABPN1, but not in HBS1L and TTC37 depleted cells (Fig. 5e). In addition, we examined the levels of previously identified PAXT complex substrates, SNHG19 and SNHG1053, and found that depletions of ZFC3H1 and PABPN1 result in an accumulation of SNHG19 and SNHG10 RNA, while HBS1L and TTC37 depleted cells did not show any effect on these substrates (Fig. 5f), confirming that PAXT and Ski complexes are functionally distinct.
Unlike the expected significant increase observed in wtGFP/opt-mCherry mRNA ratio, the opt-GFP/opt-mCherry mRNA ratio was not increased in the opt-GFP/opt-mCherry dual-reporter cells after ZFC3H1 and PABPN1 depletions despite a similar increase in SNHG19 levels was observed (Supplementary Fig. 7c, d). Overall, these results suggest that the nuclear RNA exosome and the PAXT complex but not the cytosolic RNA exosome components mediate the observed codon usage effect on reporter gene expression.
We also examined the effects of ZFC3H1 and PABPN1 depletions on the mRNA levels of intron-lacking or intron-containing wtGFP and opt-GFP reporters as compared to control treatment using single reporter cells. As shown in Supplementary Fig. 7e, the wtGFP mRNA level increased significantly higher than the opt-GFP mRNA levels irrespective of the presence of introns, indicating the effect is independent of splicing process.
Two RNA-binding proteins, RBM26 and RBM27, were recently identified as components of the nuclear PAXT complex; loss of either protein results in an accumulation of some RNA substrates of the PAXT complex63. RBM26 and RBM27 co-depletions resulted in a dramatic accumulation of the PAXT substrate SNHG19 RNA but did not alter the expression of reporter proteins in a codon usage-dependent fashion (Supplementary Fig. 7b), suggesting that the PAXT complex mediates codon usage-dependent nuclear mRNA decay independently of RBM26 and RBM27. Thus, the RBM26 and RBM27-associated PAXT may specifically target aberrant noncoding and pre-mRNA transcripts but not mature mRNAs of non-optimal codon usage.
PAXT complex affects codon usage-dependent nuclear mRNA decay
ZFC3H1 acts as a nuclear retention factor for polyadenylated RNA substrates by competing with the RNA export factors64. Similarly, PABPN1 prevents nuclear export of unspliced RNA65. We therefore investigated if these PAXT components affect the relative reporter mRNA levels in a codon usage-dependent fashion in the total, cytoplasmic, and nuclear RNA preparations. As shown in Fig. 6a, siRNA-mediated silencing of ZFC3H1 and PABPN1 resulted in an increase in the ratio of wtGFP/opt-mCherry mRNA levels in both nuclear and cytoplasmic RNA fractions. The specific presence of Tubulin and Lamin in the cytoplasmic and nuclear preparations respectively, indicated the success of our fractionation procedure (Fig. 6b). As expected, we observed an increase in wtGFP amounts in total protein and cytosolic protein fractions in ZFC3H1 and PABPN1 depleted cells as compared to control treatment, whereas the levels of opt-mCherry protein remained unchanged (Fig. 6b). The RNA fractionation efficiency was confirmed by the enrichment of GAPDH mRNAs in cytosolic fraction and of intron-containing GAPDH pre-mRNAs in the nuclear RNA fractions (Supplementary Fig. 8a, b). The efficiency of ZFC3H1 and PABPN1 depletion was confirmed by the accumulation of SNHG19 RNA (Supplementary Fig. 8c).

a Quantification of the ratio of mRNA levels of GFP and mCherry reporters in total, cytosolic and nuclear RNA fractions isolated from wtGFP/opt-mCherry stable cells treated with indicated siRNAs for 72 h as measured by RT-qPCR. Data are presented as mean ± SD, n = 3 independent experiments, one-way ANOVA with Sidak’s multiple comparisons test. b Western blot result showing wtGFP and opt-mCherry protein levels in total, cytosolic and nuclear protein fractions isolated from wtGFP/opt-mCherry stable cells as in (a). Lamin and Tubulin protein levels are used as controls for subcellular fractionation efficiency. c Plots showing relative cytoplasmic/nuclear ratios of opt-mCherry and wtGFP mRNA isolated from wtGFP/opt-mCherry stable cells treated with indicated siRNAs for 72 h as measured by RT-qPCR. Data are presented as mean ± SD, n = 3 independent experiments, one-way ANOVA with Dunnett’s T3 multiple comparisons test. d Quantification of relative mRNA levels of GFP and mCherry reporters in cytoplasmic and nuclear RNA fractions isolated from wtGFP/opt-mCherry stable cells subjected to siRNA mediated knockdown for 72 h followed by actinomycin D treatment for indicated time intervals. RNA levels were normalized with GAPDH mRNA levels in respective samples, as measured by RT-qPCR. Data are presented as aligned replicates, n = 3 independent experiments, two-way ANOVA was performed with Dunnett’s multiple comparisons test. P values are shown for the comparison between siControl and siZFC3H1 samples for the indicated time points (e) Plots showing relative RNA levels of SNHG19, wtGFP and opt-mCherry in input nuclear RNA (left) and PABPN1-associated RNA after immunoprecipitation from wtGFP/opt-mCherry stable cells subjected to indicated siRNA mediated knockdown for 72 h (right), as measured by RT-qPCR. Data are presented as mean ± SD, n = 3 independent experiments, one-way ANOVA with Sidak’s multiple comparisons test. Source data are provided as a Source Data file.
We also measured the cytosolic/nuclear mRNA ratios for wtGFP and opt-mCherry in ZFC3H1 and PABPN1 depleted cells compared to control treatment. As seen in Fig. 6c, cytosolic/nuclear mRNA ratio of wtGFP is lower than the ratio of opt-mCherry in control cells, suggesting that non-optimal codons containing transcripts are poorly exported to cytosol compared to optimal codons-containing transcripts. The depletion of PAXT components results in an increase in the cytoplasmic/nuclear mRNA ratio of wtGFP, while the ratio for opt-mCherry did not change significantly. Thus, loss of PAXT activity results in an increased level of wtGFP mRNA in cytoplasm, suggesting that the PAXT complex may act to prevent the export of mRNAs enriched with non-optimal codons in the nucleus. In addition, ZFC3H1 and PABPN1 depletions also led to a significant increase in cytosolic/nuclear mRNA ratios for wtGFP irrespective of the absence or presence of an intron in the reporter genes, while those for opt-GFP remained largely unchanged (Supplementary Fig. 8d, e).
Although codon optimality is known to affect translation-dependent mRNA decay, whether it affects mRNA decay or clearance in the nucleus is unknown. Thus, we examined the decay rates of wtGFP and opt-mCherry mRNAs in the nucleus after the addition of transcription inhibitor actinomycin D. In the presence of the transcription inhibitor, the wtGFP mRNA was cleared from the nucleus faster than the opt-mCherry mRNA (Fig. 6d, comparing top and bottom panels). Because mRNA export is known to be affected by codon usage and GC-rich mRNAs are preferentially exported43,45, this result suggests that the fast clearance of wtGFP mRNA may be due to its rapid decay in the nucleus rather than its preferential export. Although ZFC3H1 depletion did not affect the opt-mCherry mRNA clearance rate from the nucleus, it significantly slowed wtGFP mRNA clearance (Fig. 6d, right panels), suggesting that PAXT complex preferentially mediates the clearance of mRNAs enriched with non-optimal codons. Thus, in addition to removing aberrant transcripts in the nucleus, the nuclear RNA quality control machinery also preferentially targets mRNAs with non-optimal codon usage for decay through the nuclear exosome. In the cytoplasmic fractions, depletion of ZFC3H1 also resulted in an increase in wtGFP but not opt-mCherry transcripts levels (Fig. 6d, left panels), suggesting that wtGFP mRNA stabilization in nucleus may be coupled with an increased export to cytoplasm.
We also examined whether CNOT4 is involved in mRNA decay in the human cell nucleus. CNOT4 depletion did not result in a significant change of wtGFP transcript decay in the nucleus, while an increase in wtGFP transcript levels was observed in cytoplasmic RNA after 2 hrs of actinomycin D treatment (Fig. 6d, lower panels). This latter result is consistent with a role for the CCR4-NOT complex in promoting the decay of mRNA enriched with non-optimal codons in cytoplasm27,29,31. Unlike ZFC3H1 depletion, CNOT4 depletion did not increase SNHG19 RNA accumulation (Supplementary Fig. 8f), indicating that CNOT4 does not act through the PAXT complex. Thus, CNOT4 and PAXT play different roles in the nuclear codon usage effects: CNOT4 mainly suppresses transcription of genes enriched with non-optimal codons, while PAXT promotes the nuclear decay of RNA enriched with non-optimal codons. Together, these combined effects result in preferential cytoplasmic accumulation of mRNAs enriched with optimal codons.
PAXT complex preferentially associates with mRNA enriched with non-optimal codons
We next examined if PAXT complex interacts with RNA in codon-optimality dependent manner by performing PABPN1 immunoprecipitation in control and ZFC3H1 depleted reporter cells. As shown in Fig. 6e, PABPN1 preferentially associated with wtGFP RNA than opt-mCherry RNA in the control cells. Note that the PCR amplification efficiencies for these reporter RNAs were very similar. Association of PABPN1 with its known RNA substrate, SNHG19, was used as a positive control. These results suggest that PAXT preferentially binds to nuclear mRNAs enriched with non-optimal codons. Importantly, the association of PABPN1 with wtGFP and SNHG19 RNA was markedly reduced by ZFC3H1 depletion despite similar PABPN1 immunoprecipitation levels in both control and ZFC3H1 depleted cells (Fig. 6e and Supplementary Fig. 8g).
We also investigated the association of PABPN1 with wtGFP and opt-GFP mRNAs by performing PABPN1 immunoprecipitation in single reporter cells. Similar to wtGFP mRNA in dual reporter cells, wtGFP mRNA from single reporter cells was found to associate more than opt-GFP mRNA with PABPN1 (Supplementary Fig. 8h). SNHG19, was used as a positive control for PABPN1 binding. The association between wtGFP mRNA and PABPN1 is present both in the presence or absence of ZFC3H1, but reduces significantly when ZFC3H1 was depleted, suggesting a cooperative and independent roles of PABPN1 and ZFC3H1 in targeting non-optimal codons enriched mRNA. These results indicate an important role of these components in the recognition of non-optimal codons enriched nuclear transcripts by the PAXT complex.
PAXT components affect global codon usage-dependent mRNA cellular localization
To determine the genome-wide effects of PAXT components on gene expression, we depleted ZFC3H1 and PABPN1 from HEK293 cells using siRNA and then sequenced RNAs from total, cytoplasmic, and nuclear fractions. To determine the mRNA cellular distribution, we calculated the ratio between cytoplasmic and nuclear mRNA levels for each gene. For constitutively expressed gene groups, there are positive correlations between mRNA location and CAI/GC contents (Fig. 7a, b), indicating that optimal codon usage promotes preferential cytoplasmic distribution of mRNAs with optimal codon usage. Depletion of either ZFC3H1 or PABPN1 resulted in a dramatic decrease in the positive correlation between gene CAI/GC content and ratios of cytoplasmic/nuclear RNA levels for constitutively expressed genes (Fig. 7a, b), indicating a key role for the PAXT components in the regulation of codon usage-dependent mRNA localization. When the analyses were performed for total RNA levels instead of the cytoplasmic/nuclear RNA ratios, depletion of ZFC3H1 or PABPN1, only resulted in a slight decrease in the Pearson R correlation between CAI and RNA levels of the constitutive genes as compared to control cells (Supplementary Fig. 9a), suggesting that the effect of PAXT complex on codon-usage dependent gene expression are mainly mediated by influencing the mRNA localization in the cells.

a Plot showing Pearson correlation coefficients between CAI (to evaluate the codon-usage mediated effects) and cytoplasmic/nuclear ratio of mRNA (a measure of cytoplasmic localization of RNA) in HEK293 cells treated with indicated siRNA for 72 h followed by cellular fractionation and RNA sequencing, calculated for constitutive genes as indicated. b Plot showing Pearson correlation coefficients between GC content (a measure of nucleotide composition) and cytoplasmic/nuclear ratio of mRNA in ZFC3H1 and PABPN1 knockdown cells, calculated for constitutive genes as indicated. Pearson correlation coefficients between occurrence of codon frequencies and cytoplasmic/nuclear ratio of mRNA for 59 synonymous codons in RNA samples from the control cells and the ZFC3H1 and PABPN1 knockdown cells for constitutive1 (c) or all genes (d) to estimate the effect of optimality of each codon on transcript abundance. G/C ending, and A/T ending codons are indicated by blue, and red bars, respectively. Number of genes (n) used in analyses in different groups are shown in parenthesis. e A model showing the roles of CCR4-NOT complex and PAXT complex in determining nuclear and cytosolic RNA levels of AT-rich and GC-rich genes in a codon-optimality/nucleotide composition-dependent manner. Following export to the cytosol, mRNA translation efficiency is further subjected to codon usage control. Source data are provided as a Source Data file.
To determine the effect of individual codons on mRNA localization, Pearson correlations between gene codon frequencies and cytoplasmic/nuclear ratios were determined. When only constitutive genes were used in the analysis, codons that were positively correlated with mRNA cytoplasmic/nuclear ratios are all G/C ending codons whereas codons with negative correlations were almost all A/T ending codons (Fig. 7c). A similar observation was also observed when synonymous codons were assigned as optimal codons and non-optimal as previously described for mRNA stability in human cells23 (Supplementary Fig. 9b). Most of the exceptions in the latter analysis are codons that end in G/C nucleotide (and are not the rarest synonymous codons). These results further confirm the genome-wide codon usage effect on mRNA localization. It is important to note that a similar codon optimality effect is also seen when all expressed genes were included in the analysis (Fig. 7d and Supplementary Fig. 9c), indicating that the codon usage effect on mRNA localization is global and not limited to constitutively expressed genes.
When either ZFC3H1 or PABPN1 was depleted, the codon usage-dependent correlations for both G/C ending/optimal and A/U ending/non-optimal codons were markedly affected for almost all codons (Fig. 7c, d and Supplementary Fig. 9b, c, red or blue data points), indicating global impairment of the codon-usage-dependent mRNA localization in the absence of these PAXT components. It should be noted that the analyses using constitutive genes (Fig. 7c and Supplementary Fig. 9b) and all genes (Fig. 7d and Supplementary Fig. 9c) resulted in similar observations, indicating that the effect of PAXT on codon usage-dependent mRNA localization is broad and not specific for constitutive genes.
Discussion
Codon usage was previously thought to mediate its effect on gene expression mainly through translation-dependent processes such as translation elongation, initiation, premature termination, and translation elongation-dependent mRNA decay4,29,31. In this study, we showed that translation-independent nuclear effects of codon usage also play a major role in determining gene expression levels by affecting different nuclear gene regulatory processes, including transcription and nuclear RNA quality control. Our results suggest that the evolution of codon usage biases is due to selection on both translation-related processes in the cytoplasm and nuclear translation-independent processes. For the latter, the open-reading frame sequences are not read as codons but nucleotide compositions that resemble codon usage biases. The nucleotide compositions are recognized by the nuclear machineries in forms of DNA/RNA elements to activate or suppressing transcription or nascent RNA stability/export.
Based on our results, we propose that the CCR4-NOT complex preferentially represses the transcription of AT-rich genes. Newly transcribed transcripts are further subjected to quality control by PAXT complex in nucleus, which marks GC-rich transcripts for nuclear export but leads to nuclear retention and subsequent degradation of AT-rich transcripts. Following export to the cytosol, mRNA translation efficiency is further regulated by codon usage due to its effects on translation initiation, elongation and translation-dependent mRNA decay, resulting in the preferential expression of proteins encoded by mRNAs enriched for optimal codons (Fig. 7e).
By examining the relationship between codon usage and nuclear RNA/transcription levels, we showed that codon optimality correlates genome-wide with nuclear mRNA and transcription levels in human cells, suggesting a broader role for codon usage/nucleotide composition in regulating nuclear mRNA levels. Individual genome-wide codon optimality for total mRNAs and nuclear nascent mRNAs is highly similar to each other, indicating that selection on transcription also results in codon usage/GC compositions similar to those selected by translation-dependent processes. In addition to the coding regions, the nucleotide compositions of other regions (UTRs and introns) can also contribute to their RNA levels. However, a low correlation of GC contents of 5’UTRs of genes with their RNA levels was observed which might be due to their short lengths compared to those of their CDS and 3’UTRs52.
To identify factors involved in regulating codon usage effects on gene expression, we performed a non-biased genome-wide CRISPR-Cas9 screen in a dual codon usage reporter cell line. The identified factors include the CCR4-NOT subunit CNOT4 and many factors involved in nuclear RNA quality control pathway. The CCR4-NOT complex has been shown to mediate the codon usage effect on gene expression in yeast by affecting translation-dependent mRNA decay and mRNA solubility27,29,31. Here we evaluated the role of this complex in human cells. Depletion of CNOT4 from HEK293 cells indeed impaired the codon usage-dependent expression of our reporter genes, but it did not significantly affect the codon usage-dependent translation efficiency of mRNA reporters. Instead, we found that CNOT4 is highly enriched in nucleus and its depletion significantly affected the nascent and steady state nuclear levels and transcription of the reporter genes in a codon usage-dependent manner, indicating a role of CNOT4 in regulating transcription in a codon usage-dependent manner. Importantly, the effect of CNOT4 depletion on reporter mRNA level was maintained when translation was blocked. Although the CCR4-NOT complex functions in mRNA decay and translation in the cytoplasm, it was first identified by genetic selections for transcriptional regulators in yeast66,67, and its nuclear functions in chromatin structure, transcription initiation and elongation, nuclear RNA quality control, and export have been previously described30.
We previously showed that codon usage influences chromatin structure, and therefore transcription, in fungi, Drosophila, and human cells5,21,41. Consistent with a role for CNOT4 in transcription, we found that depletion of CNOT4 affects mRNA levels genome-wide by preferentially upregulating mRNAs enriched with non-optimal codons. The mechanism by which CNOT4 impacts gene transcription in a codon usage-dependent manner is not known. It is possible such an effect is caused by its impact on transcription of genes encoding for chromatin regulators. Consistent with this notion, we found that CNOT4 depletion results in significant downregulation of many chromatin regulators. Furthermore, we previously showed that some chromatin regulators are involved in determining the codon usage effect on gene expression in Neurospora57.
In human cells, repressive action of the CCR4-NOT complex in de novo transcription of MHC II genes was previously shown68, likely due to a direct or indirect effect of its association with chromatin. In addition, the recruitment of CCR4-NOT complex subunits to hormone-inducible genes was previously shown to cause transcriptional repression in human cells69, suggesting that the chromatin association of CCR4-NOT complex influences transcription initiation. Moreover, such transcriptional repression could be partially relieved by the addition of the histone deacetylases inhibitor, suggesting the involvement of chromatin remodeling in this repression process. Effects of CNOT-mediated transcriptional repression involving transcription factors such as estrogen receptor α, c-Myb, or E26-related gene were also previously described68. Furthermore, CCR4-NOT was recently found to suppress gene expression of many gene by regulating the expression of the transcriptional repressor, KRAB-Zinc-Finger-protein (KZNFs)38. Although how CNOT4 acts to regulate gene transcription in a codon usage-dependent manner in human cells is unclear, we propose that CCR4-NOT complex preferentially suppresses transcription of genes enriched with non-optimal codons due to its direct chromatin recruitment or its indirect regulation on chromatin regulators or transcription repressor.
In addition to CNOT4, most of the top hits identified in our genome-wide screen are nuclear factors including multiple nuclear exosome components and subunits of its nuclear substrate adaptor, PAXT complex. Although the cytoplasmic exosome was expected to contribute to the codon usage effect by mediating translation-dependent mRNA decay, depletion of the nuclear exosome catalytic subunit DIS3, but not the cytoplasmic exosome catalytic subunit and its co-factors, significantly affected the codon usage-dependent expression of the reporter genes, indicating that the nuclear exosome has a larger impact on the regulation of genes with non-optimal codon usage profiles than does the cytoplasmic exosome.
ZFC3H1 and PABPN1 are PAXT complex subunits that associate with polyadenylated RNA substrates and target them to the nuclear exosome for decay53. In human cells, depletion of either ZFC3H1 or PABPN1 significantly increased expression levels of the reporter genes with non-optimal codon usage. In addition, depletion of ZFC3H1 preferentially stabilized the mRNA with non-optimal codon usage with a concomitant rise in its cytoplasmic level. In this study, we showed that PABPN1 subunit of PAXT complex preferentially binds mRNA enriched for non-optimal codons and ZFC3H1 is important for the recognition mRNA targets by PAXT. An increase in the cytosolic/nuclear mRNA ratio of non-optimal codons enriched reporters as opposed to optimal codons enriched reporters also support this notion. Furthermore, cytoplasmic and nuclear RNA sequencing of cells that were depleted with ZFC3H1 or PABPN1 revealed that PAXT is important for the global codon usage-dependent mRNA localization due to their roles in nuclear RNA quality control process. Interestingly, ZFC3H1 was previously shown to functionally compete with AlyREF, which is involved in recruitment of transcription/export (TREX) machinery to mRNA64. TREX machinery has been shown to export primarily GC-rich mRNAs45. It is possible that the specificity of TREX for GC-rich mRNAs is contributed by the exclusion of ZFC3H1 mRNA substrates from the TREX machinery.
Altogether, these results suggest that PAXT preferentially targets nascent mRNAs enriched with non-optimal codon usage for decay by the nuclear exosome. Identification of multiple nuclear exosome components in the screen also indicates a major contribution of this pathway in shaping codon-usage mediated transcripts levels in human cells. It was previously shown that codon usage and GC content of mRNA affect nuclear RNA export43,44,45,46,70. Thus, different nuclear processes, including transcription, nuclear RNA quality control and mRNA export, are regulated by codon usage/nucleotide compositions, which preferentially promotes the expression and nuclear export of G/C-rich mRNAs with optimal codon usage and their subsequent translation in the cytoplasm. This conclusion is consistent with the recent hypotheses which proposed that high GC-content mRNAs, which are those with optimal codon usage, is a key mRNA feature that separates “wanted” transcripts from “unwanted” transcripts44,47,71,72. The latter are mostly spurious or mis-spliced transcripts and RNAs transcribed from transposable elements or viruses. This hypothesis further proposes that in humans, a species with a small effective population size and long generation time, selection by translation is not prominent, instead much of the selection on human codon usage suppressed the production of “unwanted” transcripts through various nuclear effects to promote GC-rich transcripts and suppress AU/CG-rich ones for translation71. Although mRNAs of most genes with A/U-rich nucleotide composition or non-optimal codon usage are not “unwanted” mRNAs, their nucleotide composition-dependent regulation by transcription, nuclear RNA quality control and export pathways act to suppress their cytoplasmic levels to ensure their encoded proteins are not overexpressed to interfere with normal cellular functions.
Together, our results indicate that CNOT4, ZFC3H1 and PABPN1 differentially contribute to the nuclear codon usage effects on transcription, RNA quality control, and the preferential export of mRNAs with optimal codon usage. Several transcriptional regulators, such as TAFs and CCDC12, were also among the top hits identified in our CRISPR-Cas9 screen; however, it should be noted that the gene disruption-based CRISPR-Cas9 screen will likely miss many genes that encode factors involved in transcription and chromatin regulation due to their essential roles in normal cell growth and survival. Overall, these results demonstrate that the nuclear, translation-independent effects of codon usage have a large impact on gene expression levels in human cells.
Methods
Cell culture, vector construction, transfection, and stable cell generation
HEK293T and HEK293 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Sigma, catalog #P4333). The cells were maintained in a humidified incubator at 37 °C with 5% CO2.
The dual reporter constructs, CMV-mCherrycom-CMV-GFPrare, CMV-mCherrycom-CMV-GFPcom, and CMV-mCherryrare-CMV-GFPcom50, were a generous gift from Christopher M. Counter, Duke University. For the construction of pCMV-wtGFP, pCMV-opt-GFP and pCMV-opt-mCherry single reporter plasmids, PCR amplification was performed to generate fragments of GFPrare, GFPcom, and mCherrycom, using dual reporter constructs mentioned above as templates. For the construction of in-frame stop codons containing wtGFP and opt-GFP single reporter plasmids, PCR amplifications were performed as above using forward primers containing in-frame stop codon sequences. Gel-purified DNA fragments were digested with BamHI, HindIII, EcoRI and NotI and subcloned into pCMV-Tag-2B vector backbone at corresponding sites. For the construction of intron containing pCMV-wtGFP and pCMV-opt-GFP single reporter plasmids, a chimeric intron43 (133 nt, GC- 44%) DNA fragment was synthesized by Eurofins. The synthetic DNA was digested with SacI and NotI and subcloned into wtGFP and opt-GFP single reporter plasmids at corresponding sites within their 5’UTRs. For the construction of pCMV-wtGFP and pCMV-opt-GFP single reporter plasmids containing high GC content in their 3’UTRs, a 377 nt DNA fragment (73.4% GC, based on human HCN2 3’UTR sequence) was synthesized by Genscript. The synthetic DNA containing plasmid was digested with HindIII and MfeI and subcloned into wtGFP and opt-GFP single reporter plasmids at corresponding sites within their 3’UTRs. Primer sequences used for subcloning are provided in Supplementary Data 1.
For transfection, polyethyleneimine transfection reagent, PEI (Polysciences, catalog #24765) was used according to the manufacturer’s instructions. Briefly, cells were seeded in wells of culture plates and allowed to adhere overnight. The transfection mixture was prepared by diluting the transfection reagents in Opti-MEM and adding DNA to the mixture. The mixtures were incubated at room temperature for 20 minutes and then added to the cells. After incubation, the transfection medium was replaced with fresh growth medium, and cells were allowed to recover before further analysis. The dual reporter constructs CMV-mCherrycom-CMV-GFPrare, CMV-mCherrycom-CMV-GFPcom, and CMV-mCherryrare-CMV-GFPcom were transfected into HEK293 cells followed by selection using G418 at 500 µg/mL concentration to generate cells that stably express wtGFP and opt-mCherry or opt-GFP and wt-mCherry. The stably transfected cells of CMV-mCherrycom-CMV-GFPrare, and CMV-mCherryrare-CMV-GFPcom were sorted for dual fluorescence using FACS and diluted to generate clonal cell lines. The single reporter constructs pCMV-wtGFP, or pCMV-opt-GFP with or without stop codons, were transfected into HEK293 cells as above, followed by selection using G418 (500 µg/mL) to generate cells that stably express wtGFP and opt-GFP with/without premature stop codons. The single reporter intron containing constructs pCMV-wtGFP (+Intron), pCMV-opt-GFP(+Intron) and 3’UTR high GC variants of pCMV-wtGFP and pCMV-opt-GFP were transfected into HEK293 cells as above, followed by selection using G418 (500 µg/mL) to generate cells that stably express wtGFP and opt-GFP with respective variations.
Co-transfection of single reporter plasmids was done as follows: pCMV-wtGFP or pCMV-opt-GFP was mixed with 1/10 amount of pCMV-opt-mCherry to normalize for transfection efficiency and diluted in Opti-MEM for transfection of HEK293T cells. The transfection mixture was prepared by diluting PEI in Opti-MEM and adding diluted DNA to the mixture. Transfections were performed as above and cells were harvested after 72 hours for western blot analyses.
Generation of lentivirus and knockout cells
The lentiCRISPR_v2 vectors encoding Cas9, sgRNAs, and a puromycin-resistance gene and packaging plasmids were co-transfected into HEK293T cells at approximately 70% confluency in a 10-cm dish, and the medium was changed 24 h later. After 48 hours of incubation, the cell culture supernatant was transferred to a 15-mL centrifuge tube and centrifuged at 3000×g for 10 minutes. The supernatant was then filtered through a 0.45-micron syringe filter and collected into a new sterile tube. The viral solution was further concentrated using the Lenti-X™ Concentrator (Takara, catalog #631231) according to the manufacturer’s instructions followed by snap freezing in liquid nitrogen and storage at −80 °C for long-term use.
Lentiviral sgRNA mediated knockout of genes was performed as described previously73. HEK293 or HEK293T cells were split into media containing puromycin at a concentration of 1 µg/mL at 48 hours post-transduction. For experiments using DIS3-, ZFC3H1-, and PABPN1-knockout cells, selection in puromycin was done for 6–9 days before harvesting the cells for analysis. sgRNA sequences are provided in Supplementary Data 1.
siRNA and mRNA transfections
For siRNA-mediated knockdown, cells were transfected with 10 nM siRNA (Millipore Sigma) using Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer’s instructions. The details of siRNA sequences are provided in Supplementary Data 1. For double knockdowns, 5 nM of each siRNA was used for a total mix of 10 nM. Cells were harvested at 72 h post-transfection for all assays. The mRNA templates were prepared using in vitro transcription as previously described16. 1 µg mRNA/sample was electroporated into cells using Gene Pulser Xcell electroporation system (Biorad) according to the manufacturer’s instructions for mammalian cells, and plated cells were harvested after 8 h.
Immunofluorescence assay
HEK293 cells were transfected with 10 nM siRNA (Millipore Sigma) using Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer’s instruction. Cells were washed three times with phosphate-buffered saline (PBS) and fixed with 4% PFA for 20 min at 20 °C at 72 h post-transfection. Immunofluorescence assays were performed as previously described74. The cells were analyzed using a Zeiss LSM 880 confocal microscope. The sources of antibodies are listed in Supplementary Data 1.
Flow cytometry analysis
Cells stably transfected with reporter constructs were harvested and resuspended in PBS supplemented with 2% bovine serum albumin, 5 mM EDTA, and 0.05% sodium azide. Cells were filtered through a 35-micron filter and analyzed with a BD LSRFortessa cell analyzer. Data analysis was performed using FlowJo version 10.10.0. FSC-A/SSC-A gating of starting population was used to select live cells, FSC-A/FSC-H gating was used to select single cells, Double positive cells with fluorescence levels above 100 were used for analysis.
Western blot analysis
The protein concentration of samples was determined by Bradford assay, and 10–80 µg of total, cytosolic, or nuclear protein extracts were separated by SDS-PAGE, transferred onto a PVDF membrane (Millipore), and detected using Pierce ECL western blotting substrate (Thermo Scientific, catalog #32106). The intensities of the bands were quantified using Fiji Image J 1.54 d software. The sources of antibodies are listed in Supplementary Data 1.
Reverse transcription and quantitative real-time PCR (RT-qPCR) assays
For analysis of gene expression and RNA subcellular localization, extracted RNA was treated with Turbo DNase (Ambion) and extracted using acidic phenol:chloroform (Ambion) prior to reverse transcription using a High-Capacity cDNA Reverse Transcription kit (Applied Biosystems) with random primers or HiScript III RT Supermix (Vazyme) according to the manufacturer’s protocol. Real-time PCR was performed as described previously5. Primers used for RT-qPCR are listed in Supplementary Data 1. For differential expression analysis, expression was normalized to 18S rRNA or GAPDH mRNA. Fractionation quality was validated by primers targeting the nuclear GAPDH-IN pre-mRNA and the cytoplasmic GAPDH mRNA.
Cellular fractionation
Cells were washed with PBS and trypsinised briefly. After stopping the reaction with DMEM, cells were collected by spinning at 400 × g for 5 min. The cell pellets were resuspended in ice-cold PBS, and subcellular fractionation was performed as described previously75. Cytosolic and nuclear protein and RNA extractions were performed as previously described75.
Translation inhibition by cycloheximide
Cells were treated with siRNAs targeting CNOT4 or with a control siRNA as described above. At 48 hours post-transfection, growth medium was replaced and cycloheximide (Sigma, catalog #C7698) dissolved in ethanol (10 mg/mL) was added to a final concentration of 100 µg/mL. Cells were harvested after 24 hours and RT-qPCR analyses were performed as above.
Transcription inhibition by actinomycin D
Cells were treated with siRNAs targeting ZFC3H1 or CNOT4 or with a control siRNA in 10-cm plates as described above. At 72 h post-transfection, growth medium was replaced and actinomycin D (Sigma, catalog #A9415) dissolved in dimethyl sulfoxide was added to a final concentration of 8 µg/mL. Cells were harvested at 0 h (non-treated cells), after 1 h, and after 2 h of actinomycin D treatment. Cellular fractionations and RT-qPCR analyses were performed as above.
Nuclear run-on assay
Cells were washed with cold PBS and collected by centrifugation at 500×g for 5 min at 4 °C. Cells were lysed in 0.5 mL ice cold lysis buffer (10 mM Tris–HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40, and 10% Glycerol) supplemented with 200 U/mL Superase-in (Thermo Fisher) for 5 min on ice. Nuclei were isolated by centrifugation at 500×g for 5 min at 4 °C and were resuspended in 50 µL freezing buffer (50 mM Tris–HCl, pH 7.4, 5 mM MgCl2, 40% glycerol and 0.1 mM EDTA) by pipetting. Transcription was performed by adding 50 µL transcriptional buffer (20 mM Tris–HCl, pH 8.0, 5 mM MgCl2, 300 mM KCl, 2 mM DTT, 500 µM ATP, 500 µM CTP, 500 µM GTP, 500 µM BrUTP (Sigma, B7166), 1% Sarkosyl and 200 U/mL Superase-in). After incubation at 30 °C for 30 min (with shaking after every 5 min), 900 µL TRIzol was added to each reaction to stop transcription. RNA was isolated and resuspended in 100 µL IP buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.05% NP-40, 1 mM EDTA, and 200 U/mL Superase-in), after removing an aliquot of 1/10 volume as input control. Anti-BrU antibody at 2 µg/IP (Santa Cruz Biotechnology, IIB5: sc-32323) and EZview Red Protein G beads (Sigma, E3403) were incubated with RNA for 3 hours at 4 °C, followed by washing beads with IP buffer three times and isolation of RNA using TRIzol. Levels of newly transcribed RNA were measured by RT-qPCR.
RNA immunoprecipitation (IP)
Cells were washed with cold PBS and collected by centrifugation at 500 × g for 5 min at 4 °C. Cells were lysed in 0.5 mL ice cold lysis buffer (10 mM Tris–HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40, and 10% Glycerol) supplemented with 200 U/mL Superase-in and protease inhibitors by pipetting and incubated for 5 min on ice. Nuclei were isolated by centrifugation at 500 × g for 5 min at 4 °C and were resuspended in IP lysis buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.5% Sarkosyl, 1 mM EDTA, and 10% Glycerol) supplemented with 200 U/mL Superase-in and protease inhibitors by pipetting and kept on ice for 10 min. Cleared lysate was obtained by centrifugation at 16,000 × g for 10 min at 4 °C repeated twice. An aliquot of 1/10 volume was taken from cleared lysate as input control, and RNA was extracted using TRIzol. Anti-mouse IgG or Anti-PABPN1 monoclonal antibodies were added to cleared lysates at 5 µg/IP concentration (Proteintech, catalog # 66807-1) and EZview Red Protein G beads were incubated with lysates overnight at 4 °C, followed by washing beads with IP lysis buffer three times and isolation of RNA using TRIzol. Input and IP protein aliquots were subjected to western blot analysis using PABPN1 antibodies. Input RNA and IP-RNA were treated with DNase using Turbo DNA-free kit (Invitrogen, catalog # AM1907) following manufacturer’s protocols and levels of co-immunoprecipitated RNAs were measured by RT-qPCR.
Genome-wide CRISPR-Cas9 screen
The Brunello lentiCRISPR_v2 library (Addgene, catalog #73179)76 was used for genome-wide CRISPR-Cas9 screening as described previously77. Two biological replicates were performed. Dual reporter-expressing HEK293 were spun down, resuspended in fresh media with 8 µg/mL polybrene (EMD Millipore), and mixed with the lentiviral library at a multiplicity of infection of ~0.3. Beginning 48 h after transduction, cells were selected in 1 µg/mL puromycin and grown for 10 more days before sorting. At least 2 × 107 cells were transduced for each screen, corresponding to ~300× or greater coverage. For cell sorting, cells were washed with PBS and resuspended in PBS supplemented with 3% FBS at 1.4 × 107 cells/ml. The 0.5% of cells with the highest GFP to mCherry fluorescence ratio were sorted until 3 × 105 cells were obtained per replicate. Cells were pelleted and frozen at −80 °C prior to genomic DNA extraction. At least 5 × 107 unsorted cells per replicate were also collected and frozen. Genomic DNA from unsorted cells and sorted cells was extracted using DNeasy Blood & Tissue Kit (Qiagen, catalog #69504) according to manufacturer’s protocol.
For sequencing library preparation, two sequential rounds of PCR were performed using Herculase II Fusion DNA polymerase (Agilent) as described previously77. The pooled PCR reaction mixture was used for the second PCR (9 to 11 cycles) with primers containing barcodes and adaptors for Illumina sequencing. Agencourt AMPure XP beads (Beckman Coulter Life Sciences) were used to purify amplicons. Library sequencing was performed on an Illumina NextSeq500 High Output sequencer to obtain 76-nucleotide single-end reads. Primer sequences are provided in Supplementary Data 1. Approximately 400,000,000 sequencing reads were obtained for each replicate and Model-based Analysis of Genome-wide CRISPR/Cas9 Knockout (MAGeCK) was used to identify genes targeted by enriched sgRNAs in sorted populations as described78.
RNA-seq and data analysis
Knockdown efficiency was confirmed by FACS analysis. For total RNA-seq of siCNOT4- and siControl-treated cells, total RNA (from two biological replicates) was isolated using TRIzol reagent (Ambion). The RNA samples were then treated with Turbo DNase (Ambion) and extracted using acidic phenol:chloroform (Ambion). A poly-A RNA library was prepared using TruSeq Stranded mRNA Library Prep Kit (Illumina) following the manufacturer’s instructions, and sequencing was performed on the Illumina NextSeq 2000 sequencer to obtain 100-nucleotide single-end reads. Between 25,000,000 and 35,000,000 RNA-seq reads were mapped to the human genome (hg19 assembly). Fastq files were quality-checked using fastqc (v0.11.2). Reads from each sample were mapped to the reference genome using STAR (v2.5.3a)79. Read counts were generated using featureCounts80, and differential expression analysis was performed using edgeR81.
For the RNA-seq analysis of total, cytosolic, and nuclear RNA, cellular fractionation and RNA extractions were performed as above. Ribosomal RNA depletion and library preparation of total, cytosolic, and nuclear RNA were done using QIAseq FastSelect rRNA HMR (Qiagen) and UltraII Directional RNA Library Prep (NEB) kits following manufacturer’s protocols. The libraries were sequenced on Illumina NovaSeq X plus to obtain 60,000,000 (30,000,000 each strand) 150-nucleotide paired-end reads. FastQC (version v0.11.8) was used to check the quality of raw reads. Trimmomatic (version v0.38) was applied to remove adaptors and trim low-quality bases with default settings. RNA-seq reads were aligned to the human genome (hg38 assembly, GRCh38) with STAR Aligner version 2.7.1a, and Picard tools (version 2.20.4) were applied to mark duplicates. StringTie version 2.0.4 was used to assemble the RNA-seq alignments into potential transcripts, and featureCounts (version 1.6.0)/HTSeq was used to count mapped reads for genomic features such as genes, exons, promoter, gene bodies, genomic bins, and chromosomal locations. Differential expression was computed using DESeq2 (version 1.14.1), and the Gene Ontology analysis was done using ClusterProfiler package in R82.
GC contents of introns, 5’-UTR and 3’-UTRs of human genes (hg38 assembly, GRCh38) were calculated using Bedtools. GC contents of longest sequence of 5’-UTR and 3’-UTR for each gene were considered for further analysis. GC contents of individual introns of each gene were considered separately for further analysis. For previously published total and nuclear RNA-seq data analyses, RNA-seq datasets were used from the study, GSE139151 (siNT total, cytosolic, and nuc samples in triplicates)45. For GRO-seq data analyses, two different datasets from previously published studies were used49: Dataset 1: duplicate samples (DMSO_GRO-seq) of GSE136024. Dataset 2: duplicate samples (LP-1_DMSO) of GSE70449.
Nuclear run-on RNA-seq and data analysis
HEK293 cells (2 × 106 cells) were plated in 10 cm plates, followed by treatment with siRNA targeting CNOT4 or with a control siRNA the next day. At 72 h post-transfection, cells were harvested by trypsinization and nuclear run-on assays were performed. Two biological replicates were performed. Input and BrU-IP RNA were treated with DNase using Turbo DNA-free kit following manufacturer’s protocols. Ribosomal RNA depletion and library preparation were done using KAPA Hyper RNA with Riboerase HMR kit following manufacturer’s protocols. The libraries were sequenced on Illumina NovaSeq X plus to obtain 60,000,000 (30,000,000 each strand) 150-nucleotide paired-end reads. FastQC (version v0.11.8) was used to check the quality of raw reads. Trimmomatic (version v0.38) was applied to remove adaptors and trim low-quality bases with default settings. RNA-seq reads were aligned to the human genome (hg38 assembly, GRCh38) with STAR Aligner version 2.7.1a, and Picard tools (version 2.20.4) were applied to mark duplicates. StringTie version 2.0.4 was used to assemble the RNA-seq alignments into potential transcripts, and featureCounts (version 1.6.0)/HTSeq was used to count mapped reads for genomic features such as genes, exons, promoter, gene bodies, genomic bins, and chromosomal locations. Differential expression was computed using DESeq2 (version 1.14.1), and the Gene Ontology analysis was done using ClusterProfiler package in R82. Nascent RNA abundance was calculated as the ratio of BrU IP/ Input RNA TPM of respective samples and the genes with a mean ratio >1 in control samples were considered for further analysis. Gene-level fold changes were calculated as Log2((BrU IP/Input) siCNOT4/ (BrU IP/Input) siControl).
Statistical Analysis
All statistical analysis was performed using GraphPad Prism version 10.0.0.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All high-throughput RNA sequencing data generated during this study have been deposited in Gene Expression Omnibus under accession numbers GSE261504, GSE261505, and GSE294397. All other data are available from the corresponding author upon request. Source Data are provided with this paper. All materials and cell lines are available through University Texas Southwestern Medical Center from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Plotkin, J. B. & Kudla, G. Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 12, 32–42 (2011).
Chaney, J. L. & Clark, P. L. Roles for synonymous codon usage in protein biogenesis. Annu. Rev. Biophys. 44, 143–166 (2015).
Hanson, G. & Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 19, 20–30 (2018).
Liu, Y., Yang, Q. & Zhao, F. Synonymous but not silent: the codon usage code for gene expression and protein folding. Annu. Rev. Biochem. 90, 375–401 (2021).
Zhou, Z. et al. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. USA 113, E6117–E6125 (2016).
Jeacock, L., Faria, J. & Horn, D. Codon usage bias controls mRNA and protein abundance in trypanosomatids. Elife 7, e32496 (2018).
Lyu, X., Yang, Q., Zhao, F. & Liu, Y. Codon usage and protein length-dependent feedback from translation elongation regulates translation initiation and elongation speed. Nucleic Acids Res. 49, 9404–9423 (2021).
Yu, C. H. et al. Codon usage influences the local rate of translation elongation to regulate co-translational protein folding. Mol. Cell 59, 744–754 (2015).
Zhou, M. et al. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature 495, 111–115 (2013).
Komar, A. A., Lesnik, T. & Reiss, C. Synonymous codon substitutions affect ribosome traffic and protein folding during in vitro translation. FEBS Lett. 462, 387–391 (1999).
Zhang, G., Hubalewska, M. & Ignatova, Z. Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat. Struct. Mol. Biol. 16, 274–280 (2009).
Sander, I. M., Chaney, J. L. & Clark, P. L. Expanding Anfinsen’s principle: contributions of synonymous codon selection to rational protein design. J. Am. Chem. Soc. 136, 858–861 (2014).
Fu, J. et al. Codon usage affects the structure and function of the Drosophila circadian clock protein PERIOD. Genes Dev. 30, 1761–1775 (2016).
Zhao, F., Yu, C. H. & Liu, Y. Codon usage regulates protein structure and function by affecting translation elongation speed in Drosophila cells. Nucleic Acids Res. 45, 8484–8492 (2017).
Man, O. & Pilpel, Y. Differential translation efficiency of orthologous genes is involved in phenotypic divergence of yeast species. Nat. Genet. 39, 415–421 (2007).
Yang, Q. et al. eRF1 mediates codon usage effects on mRNA translation efficiency through premature termination at rare codons. Nucleic Acids Res. 47, 9243–9258 (2019).
Chu, D. et al. Translation elongation can control translation initiation on eukaryotic mRNAs. Embo J. 33, 21–34 (2014).
Barrington, C. L. et al. Synonymous codon usage regulates translation initiation. Cell Rep. 42, 113413 (2023).
Coghlan, A. & Wolfe, K. H. Relationship of codon bias to mRNA concentration and protein length in Saccharomyces cerevisiae. Yeast 16, 1131–1145 (2000).
Harigaya, Y. & Parker, R. Analysis of the association between codon optimality and mRNA stability in Schizosaccharomyces pombe. BMC Genom. 17, 895 (2016).
Yang, Q., Lyu, X., Zhao, F. & Liu, Y. Effects of codon usage on gene expression are promoter context dependent. Nucleic Acids Res. 49, 818–831 (2021).
Presnyak, V. et al. Codon optimality is a major determinant of mRNA stability. Cell 160, 1111–1124 (2015).
Wu, Q. et al. Translation affects mRNA stability in a codon-dependent manner in human cells. Elife 8, e45396 (2019).
Narula, A., Ellis, J., Taliaferro, J. M. & Rissland, O. S. Coding regions affect mRNA stability in human cells. RNA 25, 1751–1764 (2019).
Forrest, M. E. et al. Codon and amino acid content are associated with mRNA stability in mammalian cells. PLoS ONE 15, e0228730 (2020).
Hia, F. et al. Codon bias confers stability to human mRNAs. EMBO Rep. 20, e48220 (2019).
Buschauer, R. et al. The Ccr4-Not complex monitors the translating ribosome for codon optimality. Science 368, eaay6912 (2020).
Veltri, A. J. et al. Distinct elongation stalls during translation are linked with distinct pathways for mRNA degradation. Elife 11, e76038 (2022).
Bae, H. & Coller, J. Codon optimality-mediated mRNA degradation: linking translational elongation to mRNA stability. Mol. Cell 82, 1467–1476 (2022).
Collart, M. A. The Ccr4-Not complex is a key regulator of eukaryotic gene expression. Wiley Interdiscip. Rev. RNA 7, 438–454 (2016).
Collart, M. A., Audebert, L. & Bushell, M. Roles of the CCR4-Not complex in translation and dynamics of co-translation events. Wiley Interdiscip. Rev. RNA 15, e1827 (2023).
Yi, H. et al. PABP Cooperates with the CCR4-NOT complex to promote mRNA deadenylation and block precocious decay. Mol. Cell 70, 1081–1088 e5 (2018).
Raisch, T. et al. Reconstitution of recombinant human CCR4-NOT reveals molecular insights into regulated deadenylation. Nat. Commun. 10, 3173 (2019).
Allen, G. E. et al. Not4 and Not5 modulate translation elongation by Rps7A ubiquitination, Rli1 moonlighting, and condensates that exclude eIF5A. Cell Rep. 36, 109633 (2021).
Allen, G. et al. Not1 and Not4 inversely determine mRNA solubility that sets the dynamics of co-translational events. Genome Biol. 24, 30 (2023).
Reese, J. C. The control of elongation by the yeast Ccr4-not complex. Biochim. Biophys. Acta 1829, 127–133 (2013).
Mersman, D. P., Du, H. N., Fingerman, I. M., South, P. F. & Briggs, S. D. Polyubiquitination of the demethylase Jhd2 controls histone methylation and gene expression. Genes Dev. 23, 951–962 (2009).
Kulkarni, S. et al. Human CCR4-NOT globally regulates gene expression and is a novel silencer of retrotransposon activation. bioRxiv (2024).
Kudla, G., Lipinski, L., Caffin, F., Helwak, A. & Zylicz, M. High guanine and cytosine content increases mRNA levels in mammalian cells. PLoS Biol. 4, e180 (2006).
Newman, Z. R., Young, J. M., Ingolia, N. T. & Barton, G. M. Differences in codon bias and GC content contribute to the balanced expression of TLR7 and TLR9. Proc. Natl. Acad. Sci. USA 113, E1362–E1371 (2016).
Fu, J., Dang, Y., Counter, C. & Liu, Y. Codon usage regulates human KRAS expression at both transcriptional and translational levels. J. Biol. Chem. 293, 17929–17940 (2018).
Zhou, Z., Dang, Y., Zhou, M., Yuan, H. & Liu, Y. Codon usage biases co-evolve with transcription termination machinery to suppress premature cleavage and polyadenylation. Elife 7, e33569 (2018).
Mordstein, C. et al. Codon usage and splicing jointly influence mRNA localization. Cell Syst. 10, 351–362 e8 (2020).
Palazzo, A. F. & Kang, Y. M. GC-content biases in protein-coding genes act as an “mRNA identity” feature for nuclear export. Bioessays 43, e2000197 (2021).
Zuckerman, B., Ron, M., Mikl, M., Segal, E. & Ulitsky, I. Gene architecture and sequence composition underpin selective dependency of nuclear export of long RNAs on NXF1 and the TREX complex. Mol. Cell 79, 251–267 e6 (2020).
Thomas, A. et al. RBM33 directs the nuclear export of transcripts containing GC-rich elements. Genes Dev. 36, 550–565 (2022).
Palazzo, A. F., Qiu, Y. & Kang, Y. M. mRNA nuclear export: how mRNA identity features distinguish functional RNAs from junk transcripts. RNA Biol. 21, 1–12 (2024).
Sharp, P. M. & Li, W. H. The codon Adaptation Index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281–1295 (1987).
Bahat, A., Lahav, O., Plotnikov, A., Leshkowitz, D. & Dikstein, R. Targeting Spt5-Pol II by small-molecule inhibitors uncouples distinct activities and reveals additional regulatory roles. Mol. Cell 76, 617–631 e4 (2019).
Peterson, J., Li, S., Kaltenbrun, E., Erdogan, O. & Counter, C. M. Expression of transgenes enriched in rare codons is enhanced by the MAPK pathway. Sci. Rep. 10, 22166 (2020).
Pesole, G., Liuni, S., Grillo, G. & Saccone, C. Structural and compositional features of untranslated regions of eukaryotic mRNAs. Gene 205, 95–102 (1997).
Pesole, G. et al. Structural and functional features of eukaryotic mRNA untranslated regions. Gene 276, 73–81 (2001).
Meola, N. et al. Identification of a nuclear exosome decay pathway for processed transcripts. Mol. Cell 64, 520–533 (2016).
Collart, M. A., Panasenko, O. O. & Nikolaev, S. I. The Not3/5 subunit of the Ccr4-Not complex: a central regulator of gene expression that integrates signals between the cytoplasm and the nucleus in eukaryotic cells. Cell Signal. 25, 743–751 (2013).
Cejas, P. et al. Transcriptional regulator CNOT3 defines an aggressive colorectal cancer subtype. Cancer Res. 77, 766–779 (2017).
Sarkar, M. et al. CNOT3 interacts with the Aurora B and MAPK/ERK kinases to promote survival of differentiating mesendodermal progenitor cells. Mol. Biol. Cell 32, ar40 (2021).
Zhao, F. et al. Genome-wide role of codon usage on transcription and identification of potential regulators. Proc. Natl. Acad. Sci. USA 118, e2022590118 (2021).
Chlebowski, A., Lubas, M., Jensen, T. H. & Dziembowski, A. RNA decay machines: the exosome. Biochim. Biophys. Acta 1829, 552–560 (2013).
Tomecki, R. et al. The human core exosome interacts with differentially localized processive RNases: hDIS3 and hDIS3L. EMBO J. 29, 2342–2357 (2010).
Weick, E. M. & Lima, C. D. RNA helicases are hubs that orchestrate exosome-dependent 3’-5’ decay. Curr. Opin. Struct. Biol. 67, 86–94 (2021).
Kogel, A., Keidel, A., Bonneau, F., Schafer, I. B. & Conti, E. The human SKI complex regulates channeling of ribosome-bound RNA to the exosome via an intrinsic gatekeeping mechanism. Mol. Cell 82, 756–769 e8 (2022).
Kalisiak, K. et al. A short splicing isoform of HBS1L links the cytoplasmic exosome and SKI complexes in humans. Nucleic Acids Res. 45, 2068–2080 (2017).
Silla, T. et al. The human ZC3H3 and RBM26/27 proteins are critical for PAXT-mediated nuclear RNA decay. Nucleic Acids Res. 48, 2518–2530 (2020).
Silla, T., Karadoulama, E., Makosa, D., Lubas, M. & Jensen, T. H. The RNA exosome adaptor ZFC3H1 functionally competes with nuclear export activity to retain target transcripts. Cell Rep. 23, 2199–2210 (2018).
Kwiatek, L., Landry-Voyer, A. M., Latour, M., Yague-Sanz, C. & Bachand, F. PABPN1 prevents the nuclear export of an unspliced RNA with a constitutive transport element and controls human gene expression via intron retention. RNA 29, 644–662 (2023).
Collart, M. A. Global control of gene expression in yeast by the Ccr4-Not complex. Gene 313, 1–16 (2003).
Collart, M. A. & Panasenko, O. O. The Ccr4-not complex. Gene 492, 42–53 (2012).
Rodriguez-Gil, A. et al. The CCR4-NOT complex contributes to repression of major histocompatibility complex class II transcription. Sci. Rep. 7, 3547 (2017).
Winkler, G. S., Mulder, K. W., Bardwell, V. J., Kalkhoven, E. & Timmers, H. T. Human Ccr4-Not complex is a ligand-dependent repressor of nuclear receptor-mediated transcription. EMBO J. 25, 3089–3099 (2006).
Bhat, P. et al. Influenza virus mRNAs encode determinants for nuclear export via the cellular TREX-2 complex. Nat. Commun. 14, 2304 (2023).
Radrizzani, S., Kudla, G., Izsvak, Z. & Hurst, L. D. Selection on synonymous sites: the unwanted transcript hypothesis. Nat. Rev. Genet. 25, 431–448 (2024).
Palazzo, A. F. & Akef, A. Nuclear export as a key arbiter of “mRNA identity” in eukaryotes. Biochim. Biophys. Acta 1819, 566–577 (2012).
Manjunath, H. et al. Suppression of ribosomal pausing by eIF5A is necessary to maintain the fidelity of start codon selection. Cell Rep. 29, 3134–3146 e6 (2019).
Xie, P. et al. Mammalian circadian clock proteins form dynamic interacting microbodies distinct from phase separation. Proc. Natl. Acad. Sci. USA 120, e2318274120 (2023).
Gagnon, K. T., Li, L., Janowski, B. A. & Corey, D. R. Analysis of nuclear RNA interference in human cells by subcellular fractionation and Argonaute loading. Nat. Protoc. 9, 2045–2060 (2014).
Doench, J. G. et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191 (2016).
Golden, R. J. et al. An Argonaute phosphorylation cycle promotes microRNA-mediated silencing. Nature 542, 197–202 (2017).
Li, W. et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Acknowledgements
We thank Drs. Jaeil Han and Joshua Mendell for help in library screening; Vanessa Schmid in the McDermott Center Next Generation Sequencing Core and Admera Health for help in high throughput sequencing and data analysis; Drs. Angela Mobley and Alyssa Guzman in the University of Texas Southwestern Flow Cytometry Core for assistance with cell sorting and Drs. Xueliang Lyu and Fangzhou Zhao for help in RNA sequencing analysis. We thank members of our laboratory for assistance and discussion. This work was supported by grants from National Institutes of Health (R35 GM118118) and the Welch Foundation (I-1560) to Y.L.
Author information
Authors and Affiliations
Contributions
R.G. and Y.L. conceptualized the study, analyzed and interpreted data and wrote the manuscript, which was revised and approved by all authors. R.G. and P.X. help designed and generated cell lines used in this study, R.G. J.D., and H.L. performed cellular experiments, R.G. performed all bioinformatic analyses, and P.X. performed the immunofluorescent experiment.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Garg, R., Xie, P., Duan, J. et al. Nuclear effects play an important role in determining codon usage-dependent human gene expression. Nat Commun 16, 10865 (2025). https://doi.org/10.1038/s41467-025-65907-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65907-5
