
Abstract
Mutations in the GJB2 gene cause DFNB1, the most common hereditary hearing loss. GJB2 is expressed by cochlear epithelial cells and fibrocytes, but not by sensory hair cells or neurons. Attempts to treat DFNB1 mouse models with gene therapy have not substantially restored function, as inappropriate expression in hair cells and neurons might compromise their electrical activity. Here, we use ATAC-seq to identify candidate gene regulatory elements (GREs) that can drive cell-type-specific expression of GJB2. HA-tagged GJB2, delivered to a conditional knockout mouse with AAV vectors carrying GREs, is expressed by the appropriate cells, prevents degeneration, and rescues hearing by only 10–20 dB. In a Gjb2 partial knockdown model, a vector lacking HA prevents degeneration and completely restores hearing. In cynomolgus monkey cochleas, human GJB2.HA delivered with similar vectors is located in the appropriate cell types and causes little or no compromise of hearing sensitivity. Together, these findings suggest that GRE-mediated expression of GJB2 can prevent hearing loss in DFNB1 patients.
Similar content being viewed by others

Introduction
Hearing loss is among the most common sensory disorders1,2, affecting 2–3 in 1000 newborn children. Although more than 180 genes have been causally linked to hearing loss3, a single form of recessive, non-syndromic hearing loss, DFNB1, is responsible for up to 50% of congenital deafness worldwide outside of sub-Saharan Africa4,5. In the United States alone, about 3500 children are born each year with mutations in both alleles of the causative gene, GJB24,5,6,7. Many are born with profound hearing loss, but two-thirds have some residual hearing at birth and the majority of those lose hearing over the next few years8, suggesting that a developmental window exists for therapeutic intervention.
GJB2 encodes the gap junction protein GJB2/connexin26. In the cochlea, GJB2 is expressed in two cell groups: an epithelial system comprising supporting cells of the organ of Corti, epithelial cells of the inner and outer sulcus, and interdental cells; and a connective tissue system comprising fibrocytes of the lateral wall and suprastrial zone, and basal cells of the stria vascularis9,10,11. GJB2 is absent from the sensory hair cells and the postsynaptic spiral ganglion neurons.
There are several hypotheses for why GJB2 is critical for the cochlea. In one, the K+ that enters hair cells through mechanotransduction channels and leaves through basal K+ channels is shuttled away from the organ of Corti by the epithelial system9. Failure to remove K+ from the extracellular medium would tend to depolarize hair cells and allow toxic Ca2+ influx through voltage-gated calcium channels. Another is that GJB2 in fibrocytes and strial cells is important for transporting K+ into endolymph and producing the endocochlear potential9. A third hypothesis suggests a required developmental role, perhaps involving glucose transport, as mice lacking GJB2 in the inner ear have profound loss of hair cells and supporting cells by P3012,13,14,15. If Gjb2 is deleted after P6, the phenotype is much milder16; however, there remains a long-term requirement for GJB2, as hair-cell loss occurs after months even with deletion as late as P1417. A fourth hypothesis involves ATP- and connexin-dependent intracellular Ca2+ signaling in development18. Regardless of etiology, these studies point to a critical role for GJB2 in the early postnatal period in mice, with a sustained but less critical long-term function.
Adeno-associated virus (AAV) mediated gene addition to neonatal Gjb2 conditional knockout mice might be expected to rescue this pathology. Yet this approach has thus far failed to substantially rescue function12,19,20. There may be various reasons for this failure—whether the capsid transduces all the appropriate cells, whether regulatory elements prevent expression in inappropriate cells, whether the vector dose is adequate, or whether the vector is delivered soon enough to prevent cochlear degeneration—but each study has used different combinations of these elements and their relative importance for functional rescue has not been systematically studied.
Here we describe a successful gene replacement approach for DFNB1-related hearing loss employing ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) identification of gene regulatory elements (GREs) to limit off-target expression of GJB2, which fully restores hearing sensitivity in a Gjb2 mutant mouse model. Furthermore, we demonstrate that a vector employing GREs effectively targets GJB2-expressing cells in a non-human primate (NHP; Macaca fascicularis) model without inducing toxicity. This study demonstrates significant rescue in a Gjb2 mouse model of DFNB1 deafness while also showing safety and tolerability in an NHP model.
Results
Gene regulatory elements restrict viral GJB2 expression
We previously found that several capsids, including AAV9-PHP.B and AAV-S, efficiently transduce the cells in neonatal mouse cochlea that natively express Gjb2, based on expression of a GFP marker gene driven by a ubiquitous promoter21,22,23. To determine whether we can also express Gjb2 in those cells, we packaged the coding sequence for mouse GJB2 (mmGJB2) in the AAV9-PHP.B vector under the control of the chicken β-actin (CBA) promoter (AAV-CBA-mmGJB2; Fig. 1d) and injected the vector into the cochleas of neonatal (P0 or P1) mice through the round window membrane (RWM). Ubiquitous expression of mmGJB2 was often lethal. Mice injected with AAV-CBA-mmGJB2 exhibited severe shaking and almost all animals died by P5. To determine whether the lethality was a consequence of GJB2 activity, we constructed a similar vector in which the GJB2 sequence contained the common null mutation 35delG, which leads to a frameshift and stop (AAV-CBA-mmGJB2null). Mice injected with this vector survived until at least P30, the last time point evaluated, suggesting that lethality is a consequence of inappropriate gap junction coupling. AAV vectors injected into the mouse cochlea at early postnatal ages can spread to the brain24,25,26,27. We reasoned that nonspecific expression of GJB2 in neurons, driven by the CBA promoter, could cause neurons to become electrically coupled to each other and to neighboring glial cells. The high conductance could short-circuit neuronal post-synaptic potentials; if expressed in inhibitory neurons, GJB2 could increase brain excitability and cause lethality.

a Overview of the experimental design. ATAC-seq Assay for Transposase-Accessible Chromatin with Sequencing, AAV Adeno-associated virus, ABR Auditory Brainstem Response, DPOAE Distortion Product Otoacoustic Emissions. b Identification of potential GREs with ATAC-seq. Ten cochleas from neonatal mice age P2 (n = 3), P5 (n = 3), P8 (n = 4) were harvested and processed for ATAC-seq. Peaks representing open chromatin are shown for a ~300 kb region of mouse genome near the Gjb2 locus. Peaks for a neuronal sample are shown at the top; sequence conservation among 42 mammalian genomes is plotted at the bottom. c ATAC-seq peaks at the Gjb2 gene and elements chosen for cell-specific vectors. The human or mouse coding sequence (green) was preceded by a conserved region that includes the human equivalent of mouse exon 1 (violet; GRE1) and followed by the human non-coding segment of exon 2 (violet; GRE2). d Schematics of the AAV vectors used in the study. All vectors are single stranded. ITR inverted terminal repeat, CBA hybrid CMV enhancer/chicken β-actin promoter, HA hemagglutinin tag, GRE1 gene regulatory element 1, GRE2 gene regulatory element 2, WPRE woodchuck hepatitis virus post-transcriptional regulatory element, CDS Coding DNA Sequence, BGH bovine growth hormone polyadenylation signal.
Indeed, the AAV9-PHP.B capsid was selected for its ability to transduce brain cells in neonatal mouse28. Similarly, in the cochlea, nonspecific expression of GJB2 protein in hair cells could electrically couple them to neighboring supporting cells and attenuate receptor potentials, abolishing the auditory brainstem response (ABR). When we used the promiscuous CBA promoter to drive expression of eGFP (AAV-CBA-eGFP; Fig. 1d) and injected it into the inner ears of P1 mice, we observed high expression of eGFP by P6 in cells of the sensory epithelium, including the inner and outer hair cells (Fig. 2a; Table 1).

a Expression of a reporter driven by the ubiquitous CBA promoter. An AAV9-PHP.B vector (AAV-CBA-eGFP) expressing eGFP (green) under a CBA promoter efficiently transduced inner hair cells (IHCs) and outer hair cells (OHCs), supporting cells, and other cells in mouse cochlea (n = 5). IHCs and OHCs were identified by fluorescent phalloidin (magenta). b Cells transduced with an AAV vector expressing the eGFP marker gene under the control of GREs (AAV-GRE-eGFP). Notably, no eGFP was observed in hair cells when expression was controlled by the GREs (n = 5). c, d Cells transduced with AAV vectors expressing either mmGJB2.HA (n = 3) (c) or hsGJB2.HA (n = 3) (d) under the control of human GREs. PV (parvalbumin) labels hair cells and spiral ganglion neurons. BODIPY dye labels lipid membranes. Scale bars: 10 μm (a, b), 30 µm (c, d).
We therefore sought to limit virus-mediated expression to those cells that normally express Gjb2, using specific GREs. To identify candidate GREs that control Gjb2 expression in the mouse cochlea, we dissected whole cochleas from neonatal mice (age P2, P5 and P8), purified dissociated nuclei, and employed the Assay for Transposase-Accessible Chromatin with Sequencing (ATAC-seq)29,30 to reveal regions of open chromatin (Fig. 1a), following established protocols31,32. ATAC-seq revealed several dozen such regions, both proximal and distal to the Gjb2 gene (Fig. 1b). To restrict expression to the cochlear fibrocytes and epithelial cells that express Gjb2, we avoided regions that also appeared in a neuronal sample; to select regions more likely to control gene expression, we focused on those conserved among mammalian genomes (Fig. 1b).
We chose two regions containing likely GREs (Fig. 1c, Supplementary Table 1). GRE1 encompassed the noncoding exon 1 and ~200 bp upstream, which includes a putative promoter33; GRE 2 constituted the 3’ non-coding segment of exon 2. In order to create a vector that would be more clinically relevant, we used human sequences equivalent to these mouse GREs. The human or mouse GJB2 coding sequence, with or without a C-terminal hemagglutinin epitope (HA) tag, was flanked by the two GREs.
We then constructed AAV vectors that encoded eGFP driven by our selected GREs (Fig. 1d) and injected them into neonatal mouse cochleas. We found that this vector (AAV-GRE-eGFP) resulted in a restricted pattern of eGFP expression that more closely matched endogenous expression of Gjb2 (Figs. 2b and 3a, c). Notably, there was no expression in hair cells (Fig. 2b). Next, we injected mouse cochleas at P1 with vectors encoding either mmGJB2.HA or hsGJB2.HA, adding an HA tag to the C-terminus to track virally expressed GJB2. We analyzed expression in cochleas at P30 using anti-HA staining (Fig. 2c, d). Consistent with normal GJB2 localization, HA-tag expression was detected predominantly in the spiral limbus, inner and outer sulcus epithelia, supporting cells, basal and intermediate cells of the stria vascularis, and fibrocytes of the spiral ligament (Fig. 2c). GJB2 was not observed in inner hair cells (IHCs) or outer hair cells (OHCs). Immunolabeling appeared concentrated in disc-like puncta at cell membranes, indicating that GJB2 was expressed in the appropriate cells and properly trafficked to the cell membrane to form gap junctions (Fig. 2c, d, Table 1). A similar expression pattern was observed for both mmGJB2.HA and hsGJB2.HA.

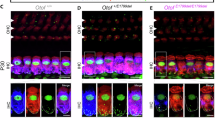
a Distribution of GJB2 (magenta) in Gjb2Pfl/Pfl, Cre− control mouse cochlea (left, n = 4) and Gjb2Pfl/Pfl, Cre+ cochlea (right, n = 5) at P6. In Gjb2Pfl/Pfl, Cre− mice, GJB2 was observed in Deiters’ cells, Hensen’s cells, Claudius cells, pillar cells, border cells, the epithelium of the inner and outer sulcus and the root cells of spiral prominence; in fibrocytes in the lateral wall and spiral limbus; and in basal and intermediate cells in the stria vascularis. Hair cells, labeled with an antibody to parvalbumin (PV; yellow) did not express Gjb2 in control cochleas. In Gjb2Pfl/Pfl, Cre+ conditional knockout mice at P6, anti-GJB2 labeling detected no signal in hair cells or epithelial cells in the organ of Corti. b At P6, scanning electron microscopy of hair cells showed normal bundle morphology in both Gjb2Pfl/Pfl, Cre+ conditional knockout mice (n = 5) and Gjb2Pfl/Pfl, Cre− controls (n = 4). c. Distribution at P30 of GJB2 in Gjb2Pfl/Pfl, Cre− (left, n = 3) and Gjb2Pfl/Pfl, Cre+ (right, n = 3) mouse cochleas. Conditional knockout mice exhibited degeneration of the organ of Corti by P30. d, e Exogenous (vector-delivered) mmGJB2.HA (n = 3) (d) or hsGJB2 (n = 3) (e) in P30 Gjb2Pfl/Pfl, Cre+ conditional knockout mouse cochlea. Hair cells did not express GJB2. It appeared in the epithelium of the inner and outer sulcus and the root cells of the spiral prominence, fibrocytes type I, II and V of the lateral wall and fibrocytes of the spiral limbus and strial cells, and these cells survived to at least P30. Hair cells, missing at P30 in untreated Gjb2Pfl/Pfl, Cre+ mice, were present in the vector-treated cochlea. LW lateral wall, SL spiral limbus, SGNs spiral ganglion neurons. Scale bars: 30 μm (a, c–e), 2 μm (b).
Hair cells and supporting cells degenerate in conditional knockout mice, resulting in profound deafness
To understand the phenotype of mice lacking GJB2 and to evaluate gene therapies in a model of DFNB1, we used mouse lines with little or no expression of the Gjb2 gene. While humans with two null alleles of GJB2 are viable, mice with constitutive deletion of both alleles are embryonic lethal34. To restrict deletion of Gjb2 to a limited number of tissues, including cochlea, we first used a conditional knockout mouse model that deleted the entire coding sequence (Gjb2tm1Ugds)35; referred to here as Gjb2Pfl, and crossed it to Sox10-Cre mice12,36; referred to here as Cre+. Mice in which Cre recombinase deleted Gjb2 (Gjb2Pfl/Pfl, Cre+) were compared to normal controls without Cre (Gjb2Pfl/Pfl, Cre−) (Fig. 3a–c).
We first characterized normal Gjb2 expression in the cochleas of control mice by immunolabeling cryosections from neonatal (P6) and adult (P30) mice with antibodies to GJB2 and to markers for hair cells and spiral ganglion neurons (SGNs) (Fig. 3). Consistent with previous reports12,36, cochleas at age P6 from Gjb2Pfl/Pfl, Cre− control mice showed prominent Gjb2 expression in the inner and outer sulcus epithelia, supporting cells, the basal and intermediate cells of the stria vascularis, and fibrocytes of spiral ligament and spiral limbus (Fig. 3a). GJB2 was not detected in IHCs or OHCs. By P30, expression levels had increased in these regions (Fig. 3c, Table 1) consistent with earlier findings12,37.
We then evaluated expression in Gjb2Pfl/Pfl, Cre+ knockout mice. At P6, there was no anti-GJB2 labeling in the organ of Corti, or more generally in the epithelial gap junction network that extends from the spiral limbus through the organ of Corti to the spiral prominence (Fig. 3a), validating the GJB2 antibody and confirming Cre-mediated knockout of GJB2 in these cells. Some anti-GJB2 signal was nevertheless present in the fibrocytes of the lateral wall and spiral limbus, suggesting that these cells do not strongly express Sox10 and do not efficiently delete Gjb2, allowing some Gjb2 expression. At P6, despite the absence of a GJB2 network in the organ of Corti, anti-parvalbumin staining showed normal hair cell morphology (Fig. 3a). Furthermore, no ultrastructural abnormalities in IHC and OHC bundle morphology were detected with scanning electron microscopy at P6 (Fig. 3b). By P30, however, Gjb2Pfl/Pfl, Cre+ mice had developed degeneration of both hair cells and supporting cells of the organ of Corti, leading to a flat sensory epithelium (Fig. 3c, Supplementary Fig. 1).
We measured auditory thresholds by ABR and distortion product otoacoustic emissions (DPOAE) recording in both the Gjb2Pfl/Pfl, Cre+ mutants and Gjb2Pfl/Pfl, Cre− control littermates at P30. The Gjb2Pfl/Pfl, Cre+ mice showed profound deafness across all frequencies tested, with ABR thresholds of 120 dB or higher. Gjb2Pfl/Pfl, Cre− animals showed thresholds consistent with wild-type hearing (Supplementary Fig. 2a, b).
Viral delivery of GJB2 with GREs rescues cochlear morphology and hearing in knockout mice
We then injected an AAV9-PHP.B vector encoding mmGJB2.HA under GRE control of expression (AAV-GRE-mmGJB2.HA; Fig. 1d) into cochleas of Gjb2Pfl/Pfl, Cre+ conditional knockout mice at P1 (dose 5 × 1010 vector genomes [vg]). As with expression of mmGJB2.HA in wild-type cochleas (Fig. 2c), the HA-tagged GJB2 was detected in many cells, including Deiters’ cells, Hensen’s cells, Claudius cells and border cells; fibrocytes type I, II and V of the lateral wall; and fibrocytes of the spiral limbus (Fig. 3d, Table 1)—congruent with the expression of eGFP (Fig. 2b) under the same regulatory elements. Importantly, no GJB2.HA signal was detected in IHCs and OHCs (parvalbumin label, yellow). We observed that restricted expression of GJB2 using AAV-GRE-mmGJB2.HA prevented the degeneration of the organ of Corti typically seen in Gjb2Pfl/Pfl, Cre+ mice at P30. However, these mice still failed to develop the tunnel of the organ of Corti. Cells of the GJB2 network—comprising the epithelium of the inner and outer sulcus and the root cells of the spiral prominence, fibrocytes of the spiral limbus and strial cells—were preserved (Fig. 3d). Hair cells, missing at P30 in untreated Gjb2Pfl/Pfl, Cre+ mice, were present in the vector-treated cochlea (Fig. 3d). Despite the preservation of cochlear structure, expression of GJB2.HA using GREs only slightly mitigated the hearing loss in Gjb2Pfl/Pfl, Cre+ mice (Supplementary Fig. 2a, b): ABR recordings at P30 demonstrated a 10–15 dB SPL improvement. However, no improvement in DPOAE was detected (Supplementary Fig. 2b).
Next, we injected Gjb2Pfl/Pfl, Cre+ mice at P1 with AAV-GRE-hsGJB2 (dose 5×1010 vg), a vector expressing human GJB2 without the HA tag. Thirty days later, the treated Gjb2Pfl/Pfl, Cre+ mice displayed a fully restored GJB2 network (Fig. 3e), matching the levels observed in Gjb2Pfl/Pfl, Cre− control animals, with no signs of histological abnormalities. Additionally, the tunnel of the organ of Corti was rescued (Fig. 3e). When we assessed hearing function in these mice, we observed a significant improvement in ABR thresholds, with an approximate gain of ~50 dB SPL (Supplementary Fig. 2a). However, no improvement in DPOAE was observed (Supplementary Fig. 2b) suggesting failure to rescue OHC function despite a significant improvement in ABR thresholds. When control mice Gjb2Pfl/Pfl, Cre− were injected with AAV-GRE-hsGJB2, their ABR and DPOAE thresholds were similar to those of untreated control mice (Supplementary Fig. 2c, d), indicating lack of vector toxicity.
AAV-mediated GJB2 expression preserves cochlear structure in a milder mouse model of DFNB1
Although incorporation of GREs produced rescue of hearing greater than that previously reported, it did not restore ABR thresholds to wild-type levels. We speculated that GJB2 has a critical developmental role as early as P018, and that vector delivered at P1 might not produce GJB2 protein until P2 or P3. We therefore sought a milder model of DFNB1, in which GJB2 protein expression is not entirely abolished, and turned to a mouse line in which the coding sequence of the upstream Gjb6 gene was replaced with a Neo selection cassette and β-galactosidase reporter38. These substitutions in Gjb6 have the effect of reducing GJB2 protein levels39,40,41, presumably due to disruption of nearby Gjb2 regulatory sequences, and the mice are referred to here as Gjb2Gjb6–.
Immunofluorescent labeling with antibodies for GJB6 in the normal control Gjb2Gjb6+ cochlea revealed that, similar to Gjb2, Gjb6 is widely expressed in the epithelial and connective tissue cells of the cochlea (Fig. 4a; Table 1). The GJB6 signal was detected in Deiters’, Hensen’s, and Claudius cells, as well as in inner and outer pillar cells, in border cells, and in the epithelium of both the inner and outer sulcus. As expected, no GJB6 signal was observed in IHCs or OHCs. Examination of the lateral wall showed that GJB6 expression was high in type I and V fibrocytes, moderate in type II fibrocytes, and absent in type III and IV fibrocytes. In the stria vascularis, GJB6 puncta were found in the basal cells. Additionally, the fibrocytes of the spiral limbus contained GJB6 puncta.

a Representative confocal microscopy images for GJB6 in Gjb2Gjb6+ control cochleas (top, n = 4) and Gjb2Gjb6– mutants (bottom, n = 5). Anti-GJB6 signal was detected in fibrocytes, Deiters’, Hensen’s, and Claudius cells, pillar cells and in both the inner and outer sulcus. No GJB6 immunoreactivity was observed in inner or outer hair cells. Gjb2Gjb6– cochleas showed normal gross morphology of the organ of Corti, hair cells, but no GJB6 was detected (bottom). b Representative images for comparison of GJB2 expression in Gjb2Gjb6– (n = 5) and Gjb2Gjb6+ (n = 4) mice at P30. In Gjb2Gjb6– mice, GJB2 was largely absent but some persisted in the lateral wall. c Representative confocal microscopy images of AAV-GRE-hsGJB2-treated (7.0 × 10¹⁰ vg/ear) Gjb2Gjb6– mice (n = 6) show complete restoration of GJB2 expression to levels seen in Gjb2Gjb6+ controls. d Scanning electron micrographs of apical, middle, and basal regions of the cochleas from Gjb2Gjb6+ (n = 6) and Gjb2Gjb6– (n = 6) mice. In untreated Gjb2Gjb6– knockout mice at P30, a tonotopic gradient of hair bundle loss was observed from base to apex, with hair bundles completely absent in the basal turn and only a few remaining in the middle and apical regions. e Gjb2Gjb6– mice (n = 7) injected with AAV-GRE-hsGJB2 (7.0 × 10¹⁰ vg/ear) display a robust restoration of hair-bundle morphology across all cochlear turns. Scale bars: 20 μm (a–c), 5 μm (d), 2.5 μm (e).
Immunofluorescence analysis of Gjb2Gjb6– cochleas consistently demonstrated complete loss of GJB6 immunoreactivity (Fig. 4a). However, the mutant mice exhibited normal overall gross morphology of the organ of Corti, with normal presence of hair cells.
We observed a significant decrease in Gjb2 expression in Gjb2Gjb6– mice compared to the Gjb2 expression in control Gjb2Gjb6+ mice, particularly in Deiters’, Hensen’s, Claudius cells, and pillar cells, as well as in the epithelium of both the inner and outer sulcus (Fig. 4b, Table 1). However, Gjb2 expression levels in Gjb2Gjb6– mice were similar to normal controls in the fibrocytes of the lateral wall and spiral limbus.
Since there were no observed differences in HA localization following the injection of AAV-GRE-mmGJB2.HA or AAV-GRE-hsGJB2.HA in wild-type control mice (Fig. 2c, d), indicating that mouse and human GJB2 exhibit similar localization and expression patterns in the mouse cochlea, we proceeded to inject Gjb2Gjb6– mice at P1 with AAV-GRE-hsGJB2.HA via the RWM. Thirty days post-injection, we analyzed cochlear morphology and hsGJB2.HA expression (Fig. 5a). Despite strong HA labeling in cells that normally express GJB2, we observed several morphological abnormalities: a reduction in scala media volume, thickening of the tectorial membrane, enlargement of inner sulcus cells attached to the tectorial membrane, and a decrease or absence of the internal spiral sulcus (Fig. 5b). Additionally, there was a reduction or absence of the tunnel of Corti compared to untreated Gjb2Gjb6– and Gjb2Gjb6+ mice. While IHCs appeared largely normal, OHCs were lost. This finding suggests that, despite strong expression and proper trafficking to the cell membrane, the GJB2 carrying the HA tag may not function properly.

a Immunolabeling of HA-tagged GJB2 protein 30 days after injection shows strong expression in cochlear cells that normally express GJB2 (n = 3). b Representative light microscopy images of Gjb2Gjb6– mice injected with hsGJB2.HA (top, n = 3), Gjb2Gjb6– injected with hsGJB2 (middle, n = 3) and Gjb2Gjb6+ control cochleas (bottom, n = 3). Despite robust expression of hsGJB2.HA seen in panel (a), cochleas from mice in the top panel (b) exhibit multiple structural abnormalities, including reduced scala media volume, thickening of the tectorial membrane, enlargement of inner sulcus cells, absence or reduction of the tunnel of Corti, and loss of OHCs. These pathological changes were not observed in Gjb2Gjb6– mice injected with hsGJB2 (middle) or in Gjb2Gjb6+ controls (bottom). Scale bars: 150 μm.
Next, we injected Gjb2Gjb6– mice at P1 with AAV-GRE-hsGJB2, a vector encoding hsGJB2 without the HA tag. Thirty days after injection, the treated Gjb2Gjb6– mice receiving AAV-GRE-hsGJB2 showed a GJB2 network completely restored to levels observed in Gjb2Gjb6– control animals, and showed no histological abnormalities (Figs. 4c and 5b, Table 1). Apparently, HA-tagged GJB2 disrupts normal GJB2 function in this mouse model.
Scanning electron microscopy of the apical, middle, and basal turns of the cochlea revealed well-organized hair bundles in both IHCs and OHCs of Gjb2Gjb6+ control mice (Fig. 4d). In untreated Gjb2Gjb6– knockout mice at P30, however, we observed a tonotopic gradient of hair bundle loss from the base to the apex, with hair bundles completely gone in the basal turn and only a few remaining in the middle and apical regions (Fig. 4d). In contrast, Gjb2Gjb6– knockout mice injected with AAV-GRE-hsGJB2 exhibited a robust preservation of hair-bundle morphology across all cochlear turns (Fig. 4e).
AAV-GRE-hsGJB2 rescues hearing in Gjb2 Gjb6– mice
We also assessed hearing function in untreated Gjb2Gjb6– and Gjb2Gjb6+ mice, as well as in animals treated with AAV-GRE-hsGJB2.HA or AAV-GRE-hsGJB2, measured with ABR recording in response to broadband clicks and tone bursts, and with DPOAEs. While Gjb2Gjb6+ mice exhibited normal hearing, Gjb2Gjb6– mice were profoundly deaf by P30 (Fig. 6a). Next, we injected Gjb2Gjb6– mice with AAV-GRE-hsGJB2.HA. As expected from previous findings with the HA tag, these mice showed no improvement in hearing, as tested by ABR or DPOAE (Fig. 6a). In contrast, when control mice were injected with AAV-GRE-hsGJB2.HA, their ABR results were similar to those of untreated control mice, and DPOAE levels were only slightly elevated (Fig. 6b). These findings suggest that GJB2.HA is not toxic but is functionally inactive.

a At P30, untreated Gjb2Gjb6+ control mice (blue, n = 12) displayed normal hearing, while untreated Gjb2Gjb6– mice (black, n = 9) were profoundly deaf. Hearing function was also evaluated in Gjb2Gjb6– mice treated with AAV-GRE-hsGJB2.HA (n = 18). Auditory brainstem responses (ABRs) were measured in response to broadband clicks and tone bursts (left), along with distortion product otoacoustic emissions (DPOAEs) (right). Gjb2Gjb6– mice injected with AAV-GRE-hsGJB2.HA showed no improvement in hearing. b Control mice injected with AAV-GRE-hsGJB2.HA displayed ABR responses comparable to untreated control mice (n = 6). However, their DPOAE levels were elevated. c At P30, Gjb2Gjb6– mice treated with a high dose (1.4 × 10¹¹ vg, n = 7), medium dose (7.0 × 10¹⁰ vg, n = 16), or low dose (3.0 × 10¹⁰ vg, n = 21) of AAV-GRE-hsGJB2 demonstrated full preservation of click-evoked and tone-burst ABR responses, matching the levels observed in untreated control mice. d The amplitude of ABR wave 1 (measured from P1 to N1) evoked by a 16 kHz tone in treated mice at P30. e Average ABR waveforms from control untreated mice and Gjb2Gjb6– mice treated with high, medium, and low doses. f Hearing recovery persisted through P90 in mice treated with high (n = 5) and medium doses (n = 7), maintaining ABR responses equivalent to those of normal-hearing controls (n = 6). Mice injected with a low dose (n = 10) exhibited elevated thresholds at P60. g The amplitude of ABR wave 1 in treated mice with high (n = 5), medium (n = 7), and low (n = 9) doses at P60 and P90. h DPOAE measurements at P30 in Gjb2Gjb6– mice treated at P1 with AAV-GRE-hsGJB2 demonstrated robust rescue with all doses of AAV-GRE-hsGJB2. This rescue persisted at P60 and P90 with DPOAE levels comparable to those of normal hearing mice for high and medium doses. All data are presented as mean values ± SEM. Source data are provided as a Source Data file.
Next, we evaluated hearing in mice injected with AAV-GRE-hsGJB2 at high (1.4 × 1011 vg), medium (7.0 × 1010 vg) and low (3.0 × 1010 vg) doses. At P30, Gjb2Gjb6– mice treated with either dose demonstrated complete rescue of click-evoked and tone burst ABR responses, to levels comparable to untreated control mice (Fig. 6c).
We analyzed the average ABR waveform responses to click stimuli at 80 dB SPL in untreated control mice and Gjb2Gjb6– mice treated with high, medium, or low doses of AAV-GRE-hsGJB2. We found that in treated Gjb2Gjb6– mice, ABR waveforms appeared comparable to those of control Gjb2Gjb6+ mice (Fig. 6e). Additionally, we analyzed the amplitude of ABR wave 1 at 16 kHz at P30. The wave 1 amplitudes in mice injected with all doses showed no difference from normal control mice (Fig. 6d). The ABR rescue remained at P60 and P90 (Fig. 6f), with hearing in animals injected with high and medium doses comparable to that of normal hearing mice. However, mice injected with a low dose exhibited an elevation of the threshold at P60 and P90. High amplitudes were preserved up to P90 in mice expressing hsGJB2 (Fig. 6g). At P30, DPOAE measurements demonstrated robust rescue in mice treated with all three doses of AAV-GRE-hsGJB2. This rescue persisted at P60 and P90 with DPOAE levels comparable to those in normal hearing mice in high and medium doses; levels were somewhat decreased with a low dose (Fig. 6h).
Expression of GJB2 with GREs in NHP cochlea is similar to that in mouse
To assess GRE-mediated viral delivery of GJB2 to the NHP cochlea, we first characterized the normal GJB2 expression. We labeled frozen sections of vehicle-injected NHP cochlea with anti-GJB2 antibodies, and co-labeled supporting and hair cells with antibodies to SOX2 and MYO7A, respectively (Fig. 7c). As expected, multiplex labeling of GJB2 showed that protein is generally present in the epithelial and connective tissue cells. GJB2 signal was detected in Deiters’, Hensen’s, and Claudius cells, inner and outer pillar cells, and border cells (Table 1). Importantly, there was no GJB2 signal in IHCs or OHCs. A well-developed GJB2 network was also seen in the epithelium of the inner and outer sulcus and the root cells of spiral prominence. Analysis of the lateral wall revealed that GJB2 was highly expressed in type I, II and V fibrocytes but was not present in type III and IV fibrocytes. In the stria vascularis, many GJB2 puncta were noted in the basal and intermediate cells but were not seen in the marginal cells. The spiral limbus fibrocytes contained many GJB2 puncta, and many were present in the cells lining the scala vestibuli floor. We almost never saw GJB2 connecting interdental cells of the spiral limbus. The epithelium of Reissner’s membrane appeared devoid of GJB2. There was no GJB2 expression in the modiolar region of the cochlea. However, we detected a very weak and diffuse signal in SGNs, which we attribute to the autofluorescence of dense SGN cytoplasm. Comparison of all cochlear turns showed no gradient in GJB2 expression from apex to base in the cells that express it.

a Overview of the experimental design. ABR: Auditory Brainstem Response. b Robust eGFP expression was observed in a non-human primate (NHP) cochlea injected with AAV-CBA-eGFP. eGFP signal (green) was detected in hair cells, Deiters’ cells, Hensen’s cells, Claudius cells, pillar cells, border cells, the epithelium of the inner and outer sulcus and the root cells of the spiral prominence, in fibrocytes type I-V in the lateral wall and in the cells of the stria vascularis, in spiral limbus fibrocytes and in interdental cells. DAPI labeled nuclei (blue). c Distribution of endogenous GJB2 (yellow) in an NHP cochlea. GJB2 signal (magenta) was detected in the following supporting cells: Deiters’ cells (DCs), Hensen’s cells, Claudius cells, pillar cells (PCs), and border cells. The GJB2 network was seen in the epithelium of the inner and outer sulcus and the root cells of spiral prominence (SP), in fibrocytes type I, II and V in the lateral wall, and in basal and intermediate cells in the stria vascularis (SV). The spiral limbus fibrocytes also contained many GJB2 puncta. Neither outer hair cells (OHCs) nor inner hair cells (IHCs) (red) expressed GJB2. DAPI labeled nuclei (blue). d Exogenous (vector-delivered) GJB2.HA expression in an NHP cochlea. The HA tag was detected in SOX2-positive cells (magenta), such as in Deiters’ cells, Hensen’s cells, Claudius cells and border cells. Little or no HA labeling was seen in pillar cells. IHCs and OHCs (yellow) did not express GJB2.HA. The GJB2.HA network was seen in the epithelium of the inner and outer sulcus and the root cells of SP, in fibrocytes type I, II and V of the lateral wall and fibrocytes of the spiral limbus. The strial cells expressed exogenous GJB2.HA at a significantly low level. DAPI labeled nuclei (blue). Scale bars: 500 µm.
GRE-driven viral GJB2 expression closely resembles endogenous expression in NHPs
While functional rescue can be tested most easily in mice, larger animals such as NHPs provide a more relevant model for transgene delivery to the human inner ear. We previously reported a successful surgical approach for delivering AAV vectors to the inner ears of cynomolgus monkeys21,22,23 and showed efficient transgene expression of a GFP reporter in a NHP cochlea following injection of the AAV9 capsid variant PHP.B via the RWM21,22,23. That vector used a ubiquitous CBA promoter, and we observed expression of the GFP reporter in many cell types, including hair cells. Following our results in mouse, we expected more specific expression of GJB2 in NHP cochlea using GRE-driven viral vectors.
To directly compare cell-specific expression between CBA and GRE-driven constructs, we injected cochleas of three juvenile cynomolgus monkeys (Macaca fascicularis). The study design is indicated in Supplementary Table 2 and Fig. 7a. The first animal received a vector encoding an eGFP marker under control of the CBA promoter by RMW injection in one ear (AAV-CBA-eGFP, 5.8 × 1011 vg, Fig. 1b). For the two other animals, three ears were injected with a vector encoding hsGJB2.HA under the control of the GREs (AAV-GRE-hsGJB2.HA; 5.0 × 1011 vg; Fig. 1d); the fourth ear was injected with formulation buffer (Fig. 1c). Five weeks post-injection, animals were tested with ABR recording, then euthanized. Cochleas were extracted from the temporal bones of animals, further trimmed, decalcified and embedded in optimal cutting temperature (OCT) compound, cryosectioned and immunostained (Fig. 7a).
Consistent with our results in mouse and earlier NHP studies21,22, the AAV-CBA-eGFP vector produced wide-ranging expression throughout the NHP organ of Corti, as revealed by anti-eGFP immunostaining of cochlea sections (Fig. 7b, Table 1). Almost all cell types of the cochlea were transduced. In particular, we observed a very high level of eGFP expression in the organ of Corti, outer and inner sulcus cells, spiral limbus, lateral wall, and Reissner’s membrane. We analyzed higher-magnification images of the organ of Corti. In cochleas that received AAV-CBA-eGFP, complete transduction was observed in hair cells, Deiters’ cells, inner phalangeal cells, pillar cells, Hensen’s cells, and Claudius cells. Additionally, the epithelial cells of the inner sulcus and fibrocytes of the basilar membrane showed high transduction. Fibrocytes of the spiral ligament in the lateral wall were significantly transduced. Notably, high-magnification images of the modiolus region revealed high expression of eGFP in satellite glial cells, Schwann cells, and SGNs.
In order to assess AAV-GRE-hsGJB2.HA expression in NHP cochleas, we first evaluated the specificity of the anti-HA antibody on frozen sections of vehicle-injected and AAV-GRE-hsGJB2.HA-injected NHP cochleas. We used antibodies to SOX2 and MYO7A to label supporting cells and hair cells, respectively. In the negative control vehicle-injected cochlea, as expected, we saw anti-SOX2 and MYO7A labeling but no HA signal (Fig. 8a). However, broad anti-HA labeling was detected in AAV-GRE-GJB2.HA-injected NHP cochleas (Fig. 8b). A weak and diffuse signal was detected in SGNs in both vehicle control and vector-injected cochleas, suggesting that it represents SGN autofluorescence.

a Vehicle-injected cochlea. Anti-SOX2 labeled supporting cells (magenta), anti-MYO7A labeled hair cells (yellow) and DAPI labeled nuclei (blue). b AAV-GRE-hsGJB2.HA-injected cochlea. Robust anti-HA labeling (red) was detected in the same locations as endogenous GJB2, and analysis of cochlear turns showed no gradient in GJB2.HA expression between apex, middle and base. Scale bars: 1 mm.
Next, we investigated the virus-mediated hsGJB2.HA expression in cryosections collected from cochleas injected with AAV-GRE-hsGJB2.HA (Fig. 7d). Because injected cochleas have both endogenous and vector-delivered GJB2, we identified vector-delivered GJB2 with an antibody to the HA tag. With GJB2 expression driven by the GJB2 GREs, IHCs and OHCs did not express hsGJB2.HA, as evidenced by lack of anti-HA labeling (Fig. 7d). Similar to the normal expression of GJB2 (Fig. 7c), the anti-HA signal was detected in SOX2-positive cells, such as Deiters’ cells, Hensen’s cells, Claudius cells and border cells, with intensity similar to that of native GJB2 (Table 1). However, few or no HA-tagged puncta were seen in inner or outer pillar cells. GJB2.HA was also seen in the inner and outer sulcus epithelia and the root cells of spiral prominence. The spiral limbus fibrocytes and the cells lining the scala vestibuli floor expressed GJB2.HA in a pattern similar to that of native GJB2. We detected weak or no HA signal in the spiral limbus interdental cells. Analysis of the lateral wall showed that, like native GJB2, exogenous GJB2.HA was highly expressed in type I, II and V fibrocytes and was not present in type III and IV fibrocytes. As expected, there was also no expression of GJB2.HA in the marginal cells or the epithelium of Reissner’s membrane. There was no GJB2.HA expression in the modiolar region of the cochlea. As with endogenous GJB2, analysis of cochlear turns showed no gradient in GJB2.HA expression between apex, middle and base (Fig. 8b). There were some differences between virus-delivered and endogenous GJB2: the stria vascularis and pillar cells expressed exogenous GJB2.HA at a lower level than the endogenous GJB2.
AAV-GRE-hsGJB2.HA is safe in NHP cochleas
We performed histological analysis on H&E-stained frozen tissue sections from injected cochleas to evaluate pathology (Fig. 9a). Tissue morphology and architecture appeared normal in both AAV-treated and vehicle control groups, with no significant abnormalities observed. Minor changes in the basal turn, likely related to surgery and vector injection, were noted in the left cochlea of NHP#2. To further evaluate immune response activation in the cochlea, we performed staining for ionized calcium-binding protein (IBA1) in frozen tissue sections from injected and control cochleas (Fig. 9b). IBA1 is primarily expressed in microglia cells, which in the cochlea are macrophages. Macrophages are abundant in the cochlea; however, insults such as AAV capsids or surgery can cause an inflammatory response, which includes activation of tissue-resident macrophages as characterized by changes in macrophage morphology, number, and distribution. We observed normal tissue morphology and architecture in all cochleas, with no notable abnormalities. (Fig. 9b). Sensory hair cells and supporting cells appeared normal. We observed no difference in macrophage infiltration in treated cochleas compared to vehicle-injected cochlea, confirming that NHPs injected with AAV-GRE-hsGJB2.HA showed minimal immune response to the vector.

a Hematoxylin and eosin-stained sections of non-human primate (NHP) cochlea injected with AAV-GRE-hsGJB2.HA (5.0 × 1011 vg). Tissue morphology and architecture appeared normal in both AAV-treated and vehicle control groups, with no significant abnormalities observed. b No immune infiltration was seen in the inner ear compared to vehicle-injected cochlea, confirmed by labeling of ionized calcium-binding protein (IBA1) (yellow). No abnormalities were seen in hair cells (red). DAPI labeled nuclei (blue). SGNs: spiral ganglion neurons. Scale bars: 500 µm.
To assess safety physiologically, we also tested the hearing before and after injection with either AAV-GRE-hsGJB2.HA or vehicle (Supplementary Fig. 3). We compared pre-injection thresholds to thresholds at 5 weeks after injection to assess changes that may be due to the vector. In the vehicle-injected cochlea (NHP#1, right ear), no significant difference was seen in the click and the pure tone ABR thresholds after injection. In two ears injected with 5.0 × 1011 vg (NHP#1 left ear and NHP#2 right ear), we saw no significant change in sensitivity in either click or tone ABRs. However, in one ear injected with 5.0 × 1011 vg (NHP#2 left ear), hearing threshold was slightly elevated by 15–25 dB nHL at all frequencies. This may be a result of early postoperative changes in the middle ear after mastoid surgery. Because two of three ears showed no elevation of threshold, we believe the injection of vector does not reproducibly cause a loss of sensitivity.
Discussion
Studies on gene therapy for deafness typically use AAV serotypes that transduce many cell types in the inner ear, including cell types that do not express the gene being delivered, and they often use strong ubiquitous promoters to drive gene expression. This approach has successfully restored hearing and balance in several other mouse models of human deafness, without documented toxicity21,23,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56. In the case of GJB2, however, we found that overexpression and misexpression led to toxicity ranging from increased hearing thresholds to lethality17,19,20, suggesting a need for more specific vector design.
One way to restrict expression is by choosing an AAV serotype that targets a cell type of interest. In the case of GJB2, there are no reported AAV serotypes that mimic its expression pattern. Moreover, GJB2 is expressed in both epithelial cells and fibrocytes of the cochlea, and it is unlikely that one AAV capsid would transduce those two cell types and no others. Instead, we chose to target expression by choosing a capsid, AAV9-PHP.B, that transduces all the cells of interest, and to limit gene expression by harnessing cis-acting gene regulatory sequences. To identify candidate GREs governing Gjb2 expression in the mouse cochlea, we performed ATAC-seq on neonatal cochleas at P2, P5, and P8, identifying multiple regions of open chromatin near the mouse Gjb2 gene. We prioritized GREs by avoiding neuronal-associated ATAC-seq peaks and focusing on elements conserved across mammalian genomes. For this study we identified a region preceding and including exon 1, and a region corresponding to the 3’ UTR of exon 2. We chose human equivalents of these regions in our vectors. Although our ATAC-seq data identified putative mouse GREs, we selected the human equivalents for testing in our vectors in order to align with translational goals and to assess their potential cross-species utility.
We first tested the selected GREs in a vector driving expression of eGFP. In injected cochleas at P6, eGFP expression overlapped normal Gjb2 expression and was eliminated from the cochlear hair cells. Next, we replaced the eGFP coding sequence with that for mmGJB2.HA (AAV-GRE-mmGJB2.HA) or hsGJB2.HA (AAV-GRE-hsGJB2.HA). Neonatal wild-type cochleas injected with mmGJB2.HA or hsGJB2.HA vectors exhibited similar HA-tag expression at P30, consistent with normal GJB2 localization and proper membrane trafficking, confirming that the GREs effectively restricted expression. Importantly, the human GREs effectively restricted expression in the mouse cochlea, yielding a spatial pattern that closely mirrored endogenous GJB2 expression in cochlear supporting cells and fibrocytes. This finding suggests that key regulatory features of these sequences are sufficiently conserved to direct appropriate expression even in the murine context, supporting their potential application in preclinical models.
In the Gjb2Pfl/Pfl, Cre+ model, restricted expression of GJB2.HA successfully preserved cochlear architecture, including hair cells and the GJB2-expressing cellular network, but resulted in only a slight improvement of hearing. In contrast, delivery of untagged human GJB2 restored both normal cochlear structure and substantially improved hearing thresholds by ~50 dB SPL at P30, without evidence of vector-related toxicity. These findings suggest that while GJB2.HA is well tolerated and capable of supporting the survival of cochlear cells, the HA tag interferes with other, critical aspects of gap junction formation or function. In this model, GJB6 is still present and may partially compensate. These results highlight the importance of preserving native protein structure for therapeutic efficacy; they suggest that GJB2.HA is not toxic but is not fully functional.
ABR thresholds did not reach wild-type levels, however, perhaps because GJB2 protein is needed for development before the vector injection at P1. To address this possibility, we used a milder DFNB1 model, Gjb2Gjb6–, in which GJB2 protein levels are reduced but not entirely abolished due to putative disruptions of Gjb2 regulatory sequences39,40,41 and ABR thresholds are severely but not profoundly elevated. Immunofluorescence analysis of untreated Gjb2Gjb6– cochleas showed no GJB6 immunoreactivity, but cochleas retained normal organ of Corti morphology with intact hair cells.
Injection of AAV-GRE-hsGJB2.HA into Gjb2Gjb6– mice resulted in strong HA-tag expression in cells that normally express GJB2, but structural abnormalities were observed, including scala media volume reduction, tectorial membrane thickening, and OHC loss, with the phenotype being more severe than in uninjected mutant mice. No such changes were observed in Gjb2Gjb6+ control mice after AAV-GRE-hsGJB2.HA injection, which displayed normal ABR and morphology.
These findings indicate that GJB6 is not redundant with GJB2 but plays a complementary role in maintaining auditory function. Although Gjb2Gjb6– mice retain some hearing and cochlear structure despite reduced GJB2 and absent GJB6, their elevated thresholds confirm that a critical level of GJB2 is required for normal auditory physiology. The severe phenotype observed when overexpressing GJB2.HA in GJB6 knockout mice suggests that, in the absence of GJB6, a precise balance of connexin proteins is essential, and that even abundant but functionally impaired GJB2 (due to the HA tag) can exacerbate dysfunction. Thus, GJB6 appears to support gap junction formation and cochlear integrity together with GJB2, reflecting a non-redundant but complementary role for passing ions and other molecules.
We injected Gjb2Gjb6– mutant mice at P1 with AAV-GRE-hsGJB2, in which GJB2 lacked the HA tag. Thirty days later, immunofluorescence analysis showed complete restoration of the GJB2 network, and showed no histological abnormalities. Hearing assessments revealed that either high, medium or low doses of the vector led to complete rescue of ABR responses at P30, with the effects persisting to at least P90 at high and medium doses. GREs apparently restrict expression to only the cells that express Gjb2, GJB2 traffics to the cell membrane to form gap junctions, and only untagged GJB2 results in fully functional gap junctions. These results highlight the importance of preserving native GJB2 structure for therapeutic efficacy and demonstrate that appropriately regulated expression of untagged GJB2 can fully restore auditory function in this model.
While testing functional rescue is more straightforward in mice, larger animals like NHPs serve as a more appropriate model for transgene delivery to the human inner ear. Here, we also report expression of GJB2.HA under cis-acting GJB2 regulatory sequences in an NHP inner ear. With AAV-mediated GJB2.HA expression in NHP cochleas, we detected vector-delivered GJB2 in SOX2-positive cells, including Deiters’ cells, Hensen’s cells, and fibrocytes, but not in IHCs or OHCs. The expression pattern of exogenous GJB2.HA was similar to that of native GJB2 in most cochlear regions (Table 1).
The differing efficacy of vectors in severe (Gjb2Pfl/Pfl, Sox10-Cre) and milder (Gjb2Gjb6–) mouse alleles, and the differing effect of the HA tag on ability to rescue of different aspects of the phenotype, support the growing appreciation that GJB2 may be serving different functions in different cells at different times in development (reviewed in ref. 18). It clearly has a role in early morphogenesis, being essential for the opening of the tunnel of Corti at ~P6 in mice. In supporting cells of the organ of Corti, GJB2 would tend to carry away the K+ that enters hair cells through mechanotransduction channels and then exits their basolateral membranes. It would thus prevent the elevation of extracellular [K+] that could depolarize hair cells and lead to excessive and perhaps lethal Ca2+ influx through their presynaptic Ca2+ channels. Later, GJB2 in the stria vascularis apparently participates in K+ pumping into the scala media, as the endocochlear potential does not form in its absence. Yet other functions, such as that mediated by GJB2 in the interdental cells, remain opaque. Some of these functions—but not all—are fulfilled by low levels of GJB2, as in the Gjb2Gjb6– mouse. Some of these functions—but not all—are fulfilled by GJB2 bearing an HA tag. Thus a more complete understanding of GJB2 functions in different cells, and the control of GJB2 expression by different regulatory elements, will be essential for restoration of hearing for the most severe DFNB1 mutations.
For both the mouse and the NHP, we chose human cis-regulatory sequences. Their efficacy in preserving morphology and hearing sensitivity, and lack of toxicity in both the control mice and NHPs, suggest that we have captured at least some conserved sequences required to mediate appropriate GJB2 expression in the human inner ear. Further work is needed to determine the minimum sequence required to provide a therapeutic effect without toxicity. ATAC-seq also revealed many candidate GREs located up to several hundred kilobases from the Gjb2 gene. Addition to the vector of one or another of these distal GREs may enhance expression and may enable good preservation of hearing even in the most profoundly deaf mouse models.
Taken together, our data show the importance of targeting a therapeutic vector to the correct cells. In this study, we regulated gene expression by employing native cis-acting regulatory sequences, rather than cell-type-specific AAV capsids. We demonstrate substantial hearing rescue in a pre-clinical model of human DFNB1 hearing loss and show that in wild-type NHPs, GREs effectively restrict GJB2 expression to the correct cells, confirming both their precision and safety. This should be an important step towards developing a gene therapy for human GJB2-related hearing loss.
Methods
Study approval
All mouse studies were conducted in compliance with ethical regulations according to protocol IS00003633 approved by the Institutional Animal Care and Use Committee (IACUC) at Harvard Medical School and were performed according to the NIH guidelines.
All NHP studies were performed at Biomere Medical Research Models (Worcester, MA) under Testing Facility Study No. NUT22-01, which was reviewed and approved by the facility’s IACUC. The facility is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International (AAALAC) and registered with the United States Department of Agriculture (USDA). All procedures complied with the Animal Welfare Act regulations (9 CFR), the Public Health Service Policy on Humane Care and Use of Laboratory Animals, and the Guide for the Care and Use of Laboratory Animals.
Mouse models (Mus musculus)
Gjb2 conditional knockout mice (Gjb2tm1Ugds)35, referred to here as Gjb2Pfl/Pfl, have loxP sites flanking the coding sequence in exon 2. Gjb2Pfl/Pfl mice were crossed to Sox10-Cre mice (Tg(Sox10-cre)1Wdr/J, Jackson Laboratory #025807)57, which have a BAC transgene encoding Cre recombinase under the Sox10 promoter, referred to here as Cre+. Gjb2Pfl/Pfl, Cre+ or Gjb2Pfl/Pfl, Cre− mice were maintained on a C57BL/6J background. Gjb6- constitutive knockout mice (Gjb6tm1Kwi; European Mouse Mutant Archive #EM:00323)38 have the Gjb6 coding region replaced by a Neo cassette and β-galactosidase reporter with a nuclear localization sequence; these substitutions have the effect of reducing GJB2 protein levels39,40,41, so mice are referred to here as Gjb2Gjb6–. These mice were maintained on mixed C57BL/6J–CBA genetic backgrounds. Animals of both sexes and ages ranging from P0 to P90 were used in experiments. The mice were housed at the animal facility of the Harvard Medical School in a 12-h-light/12-h-dark cyclic environment. The temperature ranged from 73 °F to 76 °F, and the humidity from 30% to 40%. Both sexes were used in each experiment, and no differences in genotype distribution between males and females were observed.
AAV vector production and construction
AAV backbone plasmids as initially reported21,31 were used for cloning. Human GREs and coding sequences were synthesized and inserted into AAV backbones using HiFi DNA Assembly Master Mix (NEB, Cat. #E2621). In parallel, mouse coding sequences were amplified from genomic DNA using Q5 Hot Start High-Fidelity DNA Polymerase (NEB, Cat. #M0493) according to the manufacturer’s protocol, and then inserted into AAV backbones with HiFi DNA Assembly Master Mix. Depending on the construct, candidate GREs identified through ATAC-seq were placed upstream or downstream of the transgene, or the CBA promoter was used directly. All vectors also contained a woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) and a bovine growth hormone polyadenylation signal (BGH).
For all experiments, the capsid was AAV9-PHP.B28. AAV packaging was performed by the Boston Children’s Hospital Viral Core (Boston, MA, USA) or by PackGene Biotech (Houston, TX, USA). All plasmids were sequence-verified prior to packaging. Viral titers were determined by qPCR specific for the inverted terminal repeat of the virus.
ATAC-seq library preparation and sequencing
Neonatal (P2, n = 3; P5, n = 3; P8, n = 4) CD1 mice were sacrificed and cochleas were dissected bilaterally. All tissue from each animal was combined and dounced 10 times in 5 ml of HB buffer (0.25 M sucrose, 25 mM KCl, 5 mM MgCl2, 20 mM Tricine-KOH pH 7.8, 1 mM DTT, 0.15 mM spermine, 0.5 mM spermidine, DTT 1 mM [Sigma], one-half Roche proteinase inhibitor tablet) in a 7-ml Dounce homogenizer with tight pestle, then supplemented with IGEPAL CA-630 to final concentration of 0.3%, and finally dounced an additional five times with the tight pestle. Nuclei were filtered through a 40-μm strainer and gently combined 1:1 with 50% iodixanol. Nuclei were then underlaid with a gradient of 1 ml 40% iodixanol and 1 ml 30% iodixanol and centrifuged at 10,000 × g for 19 min using the no brake setting. Purified nuclei (0.8 ml) were then collected from the 30/40% interface. A 20-μl aliquot was taken for visual inspection and cell counting, with typical yield of ~100,000–200,000 cochlear nuclei per animal.
ATAC-seq (assay for transposase accessible chromatin with sequencing) was performed using the OMNI-ATAC protocol58 immediately following the nuclear isolation steps described above. Nuclei (50,000) were pelleted at 500 × g for 10 min at 4 °C in a fixed angle centrifuge and resuspended in 50 μl transposition buffer (prepared from Illumina kit components TD buffer and 100 nM transposase, as well as 0.01% digitonin, 0.1% Tween-20) via careful pipetting. Transposition occurred at 37 °C in a thermomixer at 1000 rpm for 30 min before resuspension in 250 μl of DNA binding buffer (Zymo DNA Clean and Concentrator Kit) and reaction cleanup per manufacturer’s instructions, with final elution volume of 21 μl in elution buffer. Samples were amplified following NEBNext High-Fidelity 2X PCR Master Mix manufacturer protocols (NEB) in a total reaction volume of 50 μl (1.25 μM each of i5 and i7 sequencing primers) at 72 °C for 5 min, 98 °C for 30 s, (98 °C for 10 s, 63 °C for 30 s, and 72 °C for 1 min ×10 cycles. PCR samples were re-purified following the Zymo manufacturer protocol and eluted into 21 μl of elution buffer. Size selection was performed via agarose gel (150–1000 bp) followed by Qiagen MinElute gel extraction according to the manufacturer’s instructions, with two serial 10 μl elution steps. Sequencing (75-bp paired-end) was performed on an Illumina NextSeq500.
ATAC-seq: sequencing, alignment, and genome browser track generation
All ATAC-seq libraries were sequenced on the Illumina NextSeq 500 platform, generating 75-bp single-end reads at a minimum depth of 20 million reads per sample. Raw reads were quality trimmed with Trimmomatic (v0.33)59 using the following command: java -jar trimmomatic-0.33.jar SE -threads 1 -phred33 [FASTQ_FILE] ILLUMINACLIP:[ADAPTER_FILE]:2:30:10 LEADING:5 TRAILING:5 SLIDINGWINDOW:4:20 MINLEN:45. Nextera adapters were removed during trimming. Filtered reads were aligned to the mouse genome (GRCm38/mm10, December 2011) with Subread (v1.4.6-p3)60 using default parameters. PCR duplicates were removed using samtools rmdup. For visualization, BEDtools was used to convert BAM files to BED format (bedtools bamtobed), and mm10 blacklisted regions61 were excluded with command: bedops –not-element-of 1 [BLACKLIST_BED]. Filtered BED files were downsampled to 20 million reads, converted to coverageBED format with BEDtools genomecov, and finally transformed to bigWIG files with UCSC bedGraphToBigWig for genome browser visualization.
ATAC-seq peak calling and quantification
Putative cochlear GREs (regions of ATAC-seq enrichment) were identified using established methods32. Briefly, peaks were determined using MACS2 (v 2.1.0; parameters -p 1e-5–nolambda – keep-dup all – slocal 10000)29. Peaks from individual sample replicates were intersected to find only reproducible regions of enrichment and blacklist regions were removed according to ENCODE guidelines62. To identify sites with enriched ATAC-seq signal (peaks), we applied the IDR pipeline using the MACS2 peak calling algorithm63 with the following parameters: –nomodel –extsize 200 –keep-dup all. An IDR threshold of 0.01 was used for self-consistency, true replicate, and pooled-consistency analyses. The ‘optThresh’ cutoff was then used to obtain a final set of high-confidence, reproducible ATAC-seq peaks for each sample.
To produce a final list of reference coordinates of all genomic regions that were accessible in at least one mouse cochlear ATAC-seq sample, the MACS2-called peaks for each experimental replicate were unioned using the bedops—everything command. Bedtools merge was then used to combine any peaks that overlapped in this unioned bed file; in this way, any region that was significantly called a peak in at least one ATAC-seq dataset was incorporated in the final aggregated peak list. The featureCounts package was then used to obtain ATAC-seq read counts for each of these accessible putative CREs for downstream analyses64, including comparison to previously published mouse neuronal ATAC-seq data65 to identify putative cochlear-restricted sites.
AAV round window membrane injection in neonatal mice
RWM injections were performed under a stereomicroscope (Nikon SMZ1500). P0-P1 pups were anesthetized using cryoanesthesia and kept on an ice pack during the procedure21,23,45. A small incision was made beneath the external earbud, which was then extended to allow for separation of the soft tissues and exposure of the bulla. The round window niche was identified visually. Using a Nanoliter 2000 Injector (World Precision Instruments), 1 µl of the viral vector solution was injected through a micropipette needle. The incision was closed with a 7-0 Vicryl surgical suture, and standard postoperative care was provided following the injection.
Mouse cochlear histology and imaging
In experiments where transduction efficiency of AAV-eGFP was detected via intrinsic eGFP fluorescence, organ of Corti explants were dissected at P6 in L-15 medium and fixed with 4% formaldehyde in Hank’s balanced salt solution (HBSS) for 1 h, washed three times with HBSS, and then blocked and permeabilized with 10% donkey serum (Jackson ImmunoResearch, #017-000-121) with 0.3% Triton X-100 for 1 h at room temperature. Rabbit anti-GJB2 antibody (1:100, Fisher, #71-0500) was used to label GJB2. Antibody was diluted in 10% donkey serum and incubated overnight at room temperature followed by several washes in HBSS. Next, samples were incubated in blocking solution for 30 min and incubated overnight at room temperature with a donkey anti-rabbit secondary antibody conjugated to Alexa Fluor 594 (1:500, Invitrogen, #A21207) in blocking solution. To label hair bundle actin, we used phalloidin conjugated to Alexa Fluor 405 (1:20; Life Technologies).
In experiments assessed with cryosections, mouse cochleas were dissected in L-15 medium, immediately fixed with 4% formaldehyde in HBSS for 1 h at room temperature, then washed with HBSS and transferred to fresh 10% EDTA for 1-3 days. Next, samples were cryoprotected and embedded in OCT compound and stored at −80 °C prior to sectioning. Cryosections were generated using a Leica CM 3050 S cryostat at 20 μm step size.
For immunofluorescence labeling, the following primary antibodies and secondary antibodies were used: rabbit anti-HA C29F4 (1:200, Cell Signaling, #3724), rabbit anti-GJB2 (1:50, Fisher, #71-0500), Guinea pig anti-parvalbumin (1:200, Synaptic System, #195 004), rabbit anti-GJB6 (1:200, Fisher, #71-2200), rabbit anti-MYO7A (1:200, Proteus Biosciences, #25-6790), donkey anti-rabbit IgG secondary antibody conjugated to Alexa Fluor 594 (1:200, Invitrogen, #A21207), donkey anti-rabbit IgG conjugated to Alexa Fluor 488 (1:200, Invitrogen, #A32790), and donkey anti-guinea pig Alexa Fluor 647 (1:200, Invitrogen, #A21450). We used phalloidin conjugated to Alexa Fluor 405 (1:20; Life Technologies) to label hair-bundle actin (1:20), DAPI to label nuclei (1:500, Invitrogen), and BODIPY (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene; Invitrogen, D3835) to label membranes (1:1500). Samples were blocked for 1 h at room temperature. Antibodies were diluted in 10% donkey serum and incubated with samples overnight at room temperature, followed by several rinses in HBSS. Next, samples were incubated in a blocking solution for 30 min and incubated overnight at room temperature with a secondary antibody in the blocking solution. Tissues were mounted on a Colorfrost glass slide (Thermo Fisher Scientific) using Prolong Gold Antifade mounting medium (Thermo Fisher Scientific). Imaging was performed with a Nikon Ti2 inverted spinning disk confocal using a Plan Fluor 40×/1.3 oil objective, Plan Apo λ 60×/1.4 oil objective, or Plan Apo λ 100×/1.45 oil objective.
Scanning electron microscopy
Scanning electron microscopy in neonatal and adult mice was performed using an established method66. Neonatal organ of Corti explants were dissected at P6 in L-15 medium and fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) supplemented with 2 mM CaCl2 for 1–2 h at room temperature. The samples were rinsed three times in sodium cacodylate buffer, 0.1 M, pH 7.4 for 10 min, and then briefly rinsed once in distilled water, dehydrated in an ascending series of ethanol concentrations, and critical-point dried from liquid CO2 (Tousimis Autosamdri 815).
For adult mice, cochleas were rapidly extracted from the temporal bones and placed in Leibovitz’s L-15 solution. Under a stereomicroscope, a small hole was created at the cochlear apex using a 27 G needle. Immediately after dissection, the cochleas were fixed in 2.5% glutaraldehyde in the same buffer with 2 mM CaCl₂ for an hour at room temperature. They were then rinsed in 0.1 M cacodylate buffer (pH 7.2) and subsequently in distilled water. Using a 27 G needle, the cochlear bone was carefully removed, allowing for the microdissection of the organ of Corti and the extraction of the tectorial membrane to expose the sensory epithelium. The samples were then incubated in a saturated aqueous solution of 1% osmium tetroxide for 1 h in the dark, followed by a 10-min water rinse and postfixation in 1% tannic acid aqueous solution for another hour in the dark. Finally, the samples were rinsed, dehydrated, and subjected to critical point drying.
Samples were mounted on aluminum stubs with carbon conductive tabs and were sputter-coated (EMS 300 T dual-head sputter coater or EMS 150 T S Metal Sputter Coater) with platinum to 5 nm and observed in a field-emission scanning electron microscope (Hitachi S-4700).
Mouse ABR and DPOAE testing
ABRs and DPOAEs were recorded using an established protocol45,67, utilizing an acoustic system developed by Massachusetts Eye and Ear, Boston, MA, USA. Briefly, adult mice were administered anesthesia and maintained on a 37 °C heating pad throughout the recording session. Tone-pip stimuli (5 ms duration and 0.5 ms rise-fall time) were delivered at frequencies ranging from 4 kHz to 32 kHz. Sound levels were increased in 5-dB increments from 20 to 120 dB sound pressure level (SPL). ABR Peak Analysis software (version 1.1.1.9, Massachusetts Eye and Ear, Boston, MA, USA) was used to measure ABR thresholds and peak amplitudes. DPOAEs were recorded for primary tones with a frequency ratio of f2/f1 = 1.2, with L1 = L2 + 10 dB. The f2 frequency ranged from 5.6 kHz to 32 kHz in half-octave increments. Primary tone levels were adjusted in 5-dB increments, from 10 to 120 dB SPL for f2.
NHP study design
One female and two male juvenile cynomolgus monkeys (Macaca fascicularis; 2–4 years old, weighing 2.6–3.2 kg) were used in this study. Primate work was performed at Biomere Medical Research Models (Worcester, MA, USA) according to Biomere’s animal use guidelines and approved procedures. Each animal was considered acclimated to the environment at the time of the study. NHPs were healthy and without a history of ear inflammation, ear surgery, signs of balance disorders, or other risk factors for dysfunction of the inner or middle ear. Animals received transmastoid/trans-RWM injections of 10 μl of AAV-GRE-hsGJB2.HA vector with doses of 5.0 × 1011 vg (n = 3 ears) and 20 μl AAV-CBA-eGFP of 5.8 × 1011 vg (n = 1 ear). One animal was injected in one ear with 10 μl formulation buffer control (see Supplementary Table 2). A hearing test was performed before, and 5 weeks after unilateral injection in NHP #1 and NHP#2. Five weeks post-injection, animals were euthanized. Cochleas were extracted from the temporal bones of animals for histological analysis.
NHP surgical procedures
Animals were sedated with ketamine (10 mg/kg) and pre-medicated with atropine (0.04 mg/kg) following overnight food deprivation22. Prior to surgery, dexamethasone (1 mg/kg), buprenorphine (0.03 mg/kg), sustained-release buprenorphine (0.2 mg/kg, SC), and cefazolin (20 mg/kg) were administered. During the surgery, anesthesia was induced with isoflurane (1–4%) and maintained with oxygen (0.5–4 L) and isoflurane (~1–5%). Animals received warmed intravenous fluids, corneal protection, and continuous monitoring of vital signs. The surgical site was prepared aseptically and local anesthesia was provided with bupivacaine. All viral inner-ear administrations were performed via the facial recess approach, with round-window injection22,23. Under direct microscopic visualization, a microinjection pump delivered 20 μl or 10 μl at 2 μl/min using a Hamilton syringe and 29 G stainless steel needle. After injection, the surgical site was flushed with warm saline and closed with tension and absorbable sutures. The skin was cleaned with saline and a topical antibiotic ointment was applied. The wounds were monitored for proper healing for 2 weeks. Postoperatively, animals received bupivacaine for incision analgesia, dexamethasone for up to 5 days, and cefazolin on days 0 and 3, and were monitored daily for pain, recovery, and wound healing for 2 weeks.
NHP cochlear processing, histology, and imaging
NHPs were euthanized at 35 days after vector injection and were perfused with heparinized saline followed by 4% formaldehyde. Collected temporal bones were postfixed for another 48 h in 4% formaldehyde. Then, cochleas were dissected from the temporal bones under a microscope22,23 and decalcified in EDTA. Next, samples were cryoprotected by incubating in gradient concentrations of sucrose and were embedded in OCT compound and stored at −80 °C prior to sectioning. Cryosections were generated using a Leica CM 3050 S cryostat at 30-μm step size.
For immunofluorescence labeling, the following primary antibodies and secondary antibodies were used: mouse anti-MYO7A (1:50; Santa Cruz, #sc-74516), rabbit anti-HA C29F4 (1:200, Cell Signaling, #3724), rabbit anti-GJB2 (1:50, Fisher, #71-0500), goat anti-SOX2 (1:50, R&D Systems, #AF2018), rabbit anti-IBA1 (1:200, Wako Chemicals, #019-19741), donkey anti-rabbit IgG secondary antibody conjugated to Alexa Fluor 594 (1:200, Invitrogen, #A21207), donkey anti-goat IgG conjugated to Alexa Fluor 647 (1:200, Invitrogen, #A21447), donkey anti-mouse IgG conjugated to Alexa Fluor 488 (1:200, Invitrogen, #A32766), donkey anti-mouse IgG conjugated to Alexa Fluor 594 (1:200, Invitrogen, #A21135), and donkey anti-rabbit IgG conjugated to Alexa Fluor 488 (1:200, Invitrogen, #A32790). Samples were blocked for 1 h at room temperature. Antibodies were diluted in 10% donkey serum and incubated with samples overnight at room temperature, followed by several rinses in HBSS. Next, samples were incubated in a blocking solution for 30 min and incubated overnight at room temperature with a secondary antibody in the blocking solution. DAPI labeled cell nuclei. Tissues were mounted on Colorfrost glass slides (Thermo Fisher Scientific) using Prolong Gold Antifade mounting medium (Thermo Fisher Scientific). Imaging was performed with a Nikon Ti2 inverted spinning disk confocal using a Plan Fluor 40×/1.3 oil objective and Plan Apo λ 60×/1.4 oil objective.
ABR testing in NHPs
Testing was performed using an established protocol23. A hearing test was performed before, and 5 weeks after the unilateral injection. The animals were anesthetized with a mixture of ketamine and atropine and were masked with isoflurane. During the procedure, an animal was maintained on isoflurane anesthesia. ABRs were acquired using a clinical-grade auditory evoked potential system (GSI Audera v2.7; Grayson-Stadler, Eden Prairie, MN, USA). ABRs were recorded using three subdermal needle electrodes: the reference electrode was positioned on the midline of the scalp between the ears, the recording electrode was placed just posterior to the pinna near the slit, and the ground electrode was situated on the lower back near the tail. The click and tone-pip stimuli were presented at alternate polarity with a tubal insert phone (TIP 50 GSI generic; Grayson-Stadler, Eden Prairie, MN, USA). The acoustic click was presented at a repetition rate of 11.1 Hz, and waveforms were averaged across 2004 stimulus repetitions. Clicks were a condensation stimulus of 100-μs duration. Sound levels were incremented in 5-dB steps, from ~15 dB nHL up to 54.5 dB nHL. Tone frequencies of 500, 1000, 2000 and 4000 Hz were used. The tone pips were presented at a repetition rate of 27 Hz and waveforms were averaged across 2016 stimulus repetitions. Sound levels were incremented in 5-dB steps, from ~10 dB nHL up to 59.5 dB nHL. GSI Audera v2.7 software (Grayson-Stadler, Eden Prairie, MN, USA) was used to determine the ABR thresholds.
Statistics and reproducibility
All experiments, except for the ones noted below, were performed in at least three independent replicates using independent biological samples. Only samples displaying notable dissection-related damage were excluded from the analysis. No other data were omitted.
In NHP experiments (Figs. 7–9), an initial cohort of three juvenile cynomolgus monkeys was used, as described in the study design (Supplementary Table 2). Across these animals, a total of five ears were injected (three with AAV-GRE-hsGJB2.HA, one with AAV-CBA-eGFP, and one with formulation buffer control). For each cochlea, at least five slides from different regions were analyzed independently. Histological analyses consistently yielded similar results across animals and slides, confirming reproducibility of the findings.
All figures were assembled using Fiji (version 1.54), Adobe Illustrator 2024 (version 28.7.8), Adobe Illustrator 2025 (version 29.7.1), and Adobe Firefly (web-based; no version number available). ABR data were collected and sorted using ABR Peak Analysis (version 1.3.0) and Microsoft Excel for Microsoft 365 (version 2507). GraphPad Prism (version 10.5.0) was used to generate graphs and perform statistical analyses. SnapGene (version 5.1) was used for plasmid cloning and sequence analysis. Microsoft Word for Microsoft 365 (version 2507) was used to prepare the manuscript. The results are shown as mean ± SEM as indicated in the figure legends. Randomization was used whenever possible.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw ATAC-seq data generated in this study have been deposited in the GEO repository database under the accession code GSE308613. All other data generated or analyzed during this study are included in this article and its supplementary information files. Source data are provided with this paper.
References
Sheffield, A. M. & Smith, R. J. H. The epidemiology of deafness. Cold Spring Harb. Perspect. Med. 9, a033258 (2019).
Lydia, M. H. et al. Hearing loss prevalence and years lived with disability, 1990-2019: findings from the Global Burden of Disease Study 2019. Lancet 397, 996–1009 (2021).
Walls, W. D., Azaiez, H. & Smith, R. J. H. Hereditary Hearing Loss Homepage.Hereditary Hearing Loss Homepage. https://hereditaryhearingloss.org (accessed Feb 19, 2025).
Zelante, L. et al. Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum. Mol. Genet. 6, 1605–1609 (1997).
Kelsell, D. P. et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 387, 80–83 (1997).
Azaiez, H. et al. Genomic Landscape and Mutational Signatures of Deafness-Associated Genes. Am. J. Hum. Genet. 103, 484–497 (2018).
Azaiez, H. et al. GJB2: the spectrum of deafness-causing allele variants and their phenotype. Hum. Mutat. 24, 305–311 (2004).
Kenna, M. A. et al. Audiologic phenotype and progression in GJB2 (Connexin 26) hearing loss. Arch. Otolaryngol. Head. Neck Surg. 136, 81–87 (2010).
Kikuchi, T., Kimura, R. S., Paul, D. L. & Adams, J. C. Gap junctions in the rat cochlea: immunohistochemical and ultrastructural analysis. Anat. Embryol. 191, 101–118 (1995).
Forge, A. et al. Gap junctions and connexin expression in the inner ear. Novartis Found. Symp. 219, 151–136 (1999).
Forge, A. et al. Gap junctions in the inner ear: comparison of distribution patterns in different vertebrates and assessement of connexin composition in mammals. J. Comp. Neurol. 467, 207–231 (2003).
Crispino, G. et al. BAAV mediated GJB2 gene transfer restores gap junction coupling in cochlear organotypic cultures from deaf Cx26Sox10Cre mice. PLoS One 6, e23279 (2011).
Wang, Y. et al. Targeted connexin26 ablation arrests postnatal development of the organ of Corti. Biochem. Biophys. Res. Commun. 385, 33–37 (2009).
Sun, Y. et al. Connexin30 null and conditional connexin26 null mice display distinct pattern and time course of cellular degeneration in the cochlea. J. Comp. Neurol. 516, 569–579 (2009).
Johnson, S. L. et al. Connexin-mediated signaling in nonsensory cells is crucial for the development of sensory inner hair cells in the mouse cochlea. J. Neurosci. 37, 258–268 (2017).
Chang, Q., Tang, W., Kim, Y. & Lin, X. Timed conditional null of connexin26 in mice reveals temporary requirements of connexin26 in key cochlear developmental events before the onset of hearing. Neurobiol. Dis. 73, 418–427 (2015).
Guo, J. et al. GJB2 gene therapy and conditional deletion reveal developmental stage-dependent effects on inner ear structure and function. Mol. Ther. Methods Clin. Dev. 23, 319–333 (2021).
Mammano, F. Inner ear connexin channels: roles in development and maintenance of cochlear function. Cold Spring Harb. Perspect. Med. 9, a033233 (2019).
Iizuka, T. et al. Perinatal Gjb2 gene transfer rescues hearing in a mouse model of hereditary deafness. Hum. Mol. Genet. 24, 3651–3661 (2015).
Yu, Q. et al. Virally expressed connexin26 restores gap junction function in the cochlea of conditional Gjb2 knockout mice. Gene Ther. 21, 71–80 (2014).
György, B. et al. Gene transfer with AAV9-PHP.B rescues hearing in a mouse model of usher syndrome 3A and transduces hair cells in a non-human primate. Mol. Ther. Methods Clin. Dev. 13, 1–13 (2019).
Ivanchenko, M. V. et al. Preclinical testing of AAV9-PHP.B for transgene expression in the non-human primate cochlea. Hear Res. 394, 107930 (2020).
Ivanchenko, M. V. et al. AAV-S: a versatile capsid variant for transduction of mouse and primate inner ear. Mol. Ther. Methods Clin. Dev. 21, 382–398 (2021).
Talaei, S., Schnee, M. E., Aaron, K. A. & Ricci, A. J. Dye tracking following posterior semicircular canal or round window membrane injections suggests a role for the cochlea aqueduct in modulating distribution. Front. Cell Neurosci. 13, 471 (2019).
Zhao, Y., Zhang, L., Wang, D., Chen, B. & Shu, Y. Approaches and vectors for efficient cochlear gene transfer in adult mouse models. Biomolecules 13, 38 (2022).
Landegger, L. D. et al. A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat. Biotechnol. 35, 280–284 (2017).
Lalwani, A. K., Walsh, B. J., Reilly, P. G., Muzyczka, N. & Mhatre, A. N. Development of in vivo gene therapy for hearing disorders: introduction of adeno-associated virus into the cochlea of the guinea pig. Gene Ther. 3, 588–592 (1996).
Deverman, B. E. et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 34, 204–209 (2016).
Buenrostro, J. D., Wu, B., Chang, H. Y. & Greenleaf, W. J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 109, 21 29 21–21 29 29 (2015).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Hrvatin, S. et al. A scalable platform for the development of cell-type-specific viral drivers. Elife 8, e48089 (2019).
Nagy, M. A. et al. Cis-regulatory elements driving motor neuron-selective viral payload expression within the mammalian spinal cord. Proc. Natl. Acad. Sci. USA 121, e2418024121 (2024).
Tu, Z. J. & Kiang, D. T. Mapping and characterization of the basal promoter of the human connexin26 gene. Biochim. Biophys. Acta. 1443, 169–181 (1998).
Gabriel, H. D. et al. Transplacental uptake of glucose is decreased in embryonic lethal connexin26-deficient mice. J. Cell Biol. 140, 1453–1461 (1998).
Cohen-Salmon, M. et al. Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death. Curr. Biol. 12, 1106–1111 (2002).
Takada, Y. et al. Connexin 26 null mice exhibit spiral ganglion degeneration that can be blocked by BDNF gene therapy. Hear Res 309, 124–135 (2014).
Lautermann, J., Frank, H. G., Jahnke, K., Traub, O. & Winterhager, E. Developmental expression patterns of connexin26 and −30 in the rat cochlea. Dev. Genet. 25, 306–311 (1999).
Teubner, B. et al. Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum. Mol. Genet. 12, 13–21 (2003).
Ortolano, S. et al. Coordinated control of connexin 26 and connexin 30 at the regulatory and functional level in the inner ear. Proc. Natl. Acad. Sci. USA 105, 18776–18781 (2008).
Lynn, B. D., Tress, O., May, D., Willecke, K. & Nagy, J. I. Ablation of connexin30 in transgenic mice alters expression patterns of connexin26 and connexin32 in glial cells and leptomeninges. Eur. J. Neurosci. 34, 1783–1793 (2011).
Boulay, A. C. et al. Hearing is normal without connexin30. J. Neurosci. 33, 430–434 (2013).
Oestreicher, D., Picher, M. M., Rankovic, V., Moser, T. & Pangrsic, T. Cabp2-gene therapy restores inner hair cell calcium currents and improves hearing in a DFNB93 mouse model. Front. Mol. Neurosci. 14, 689415 (2021).
Wu, X. et al. Gene therapy via canalostomy approach preserves auditory and vestibular functions in a mouse model of Jervell and Lange-Nielsen syndrome type 2. Nat. Commun. 12, 697 (2021).
Gyorgy, B. et al. Rescue of hearing by gene delivery to inner-ear hair cells using exosome-associated AAV. Mol. Ther. 25, 379–391 (2017).
Ivanchenko, M. V. et al. Mini-PCDH15 gene therapy rescues hearing in a mouse model of Usher syndrome type 1F. Nat. Commun. 14, 2400 (2023).
Ivanchenko, M. V. et al. PCDH15 dual-AAV gene therapy for deafness and blindness in Usher syndrome type 1F models. J. Clin. Invest. 134, e177700 (2024).
Akil, O. et al. Dual AAV-mediated gene therapy restores hearing in a DFNB9 mouse model. Proc. Natl. Acad. Sci. USA 116, 4496–4501 (2019).
Lu, Y. C. et al. Gene therapy with a synthetic adeno-associated viral vector improves audiovestibular phenotypes in Pjvk-mutant mice. JCI Insight 7, e152941 (2022).
Zhao, X., Liu, H., Liu, H., Cai, R. & Wu, H. Gene therapy restores auditory functions in an adult Vglut3 knockout mouse model. Hum. Gene Ther. 33, 729–739 (2022).
Shubina-Oleinik, O. et al. Dual-vector gene therapy restores cochlear amplification and auditory sensitivity in a mouse model of DFNB16 hearing loss. Sci. Adv. 7, eabi7629 (2021).
Taiber, S. et al. Neonatal AAV gene therapy rescues hearing in a mouse model of SYNE4 deafness. EMBO Mol. Med. 13, e13259 (2021).
Nist-Lund, C. A. et al. Improved TMC1 gene therapy restores hearing and balance in mice with genetic inner ear disorders. Nat. Commun. 10, 236 (2019).
Pan, B. et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat. Biotechnol. 35, 264–272 (2017).
Dulon, D. et al. Clarin-1 gene transfer rescues auditory synaptopathy in model of Usher syndrome. J. Clin. Invest. 128, 3382–3401 (2018).
Geng, R. et al. Modeling and preventing progressive hearing loss in usher syndrome III. Sci. Rep. 7, 13480 (2017).
Isgrig, K. et al. Gene therapy restores balance and auditory functions in a mouse model of usher syndrome. Mol. Ther. 25, 780–791 (2017).
Matsuoka, T. et al. Neural crest origins of the neck and shoulder. Nature 436, 347–355 (2005).
Corces, M. R. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Liao, Y., Smyth, G. K. & Shi, W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108 (2013).
Schneider, V. A. et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864 (2017).
Landt, S. G. et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22, 1813–1831 (2012).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Stroud, H. et al. Early-life gene expression in neurons modulates lasting epigenetic states. Cell 171, 1151–1164 e1116 (2017).
Ivanchenko, M. V., Indzhykulian, A. A. & Corey, D. P. Electron microscopy techniques for investigating structure and composition of hair-cell stereociliary bundles. Front. Cell Dev. Biol. 9, 744248 (2021).
Wu, X. et al. PKHD1L1 is a coat protein of hair-cell stereocilia and is required for normal hearing. Nat. Commun. 10, 3801 (2019).
Acknowledgements
We thank Dr. Yehoash Raphael, University of Michigan, for providing the Gjb2Pfl mice. We greatly appreciate molecular biology support and laboratory management by Bruce Derfler. We appreciate the use of the Nikon Ti2 inverted spinning disk confocal and Olympus VS200 Slide Scanner at the Harvard Medical School MicRoN Microscopy Core, use of the Hitachi S-4700 scanning electron microscope at the Harvard Medical School Electron Microscopy Facility, and use of instruments at the Harvard University Center for Nanoscale Systems. We thank Alan W. LaRochelle, Biomere (Biomedical Research Models, Inc.), for his assistance with the non-human primate study. This work was supported by a Blavatnik Biomedical Accelerator Pilot Grant, a Blavatnik Biomedical Accelerator Development Grant, a Q-FASTR Grant, and a Blavatnik Sensory Disorders Grant (from Harvard Medical School to D.P.C.); by a sponsored research agreement from WayVector, Inc. to D.P.C.; by a T32GM007748 Training Grant in Genetics fellowship to K.T.B.; by a Warren Alpert Fellowship to S.H.; and by a T32GM007753 MSTP Training Grant and a Paul & Daisy Soros Fellowship for New Americans to M.A.N.
Author information
Authors and Affiliations
Contributions
M.V.I. conceptualization, data acquisition, data analysis, data interpretation, visualization of in vivo experiments in mice and NHPs, manuscript writing; K.T.B. conceptualization, data acquisition, data analysis, data interpretation, visualization of in vivo experiments in mice, manuscript writing; K.D.K. conceptualization, data analysis, data interpretation in mice; L.M.A. ABR data acquisition, data analysis, data interpretation; M.A.N., E.C.G., S.H., M.E.G. data acquisition, data analysis, data interpretation, ATAC-seq experiments, AAV design, manuscript writing; C.W.P. data acquisition, data analysis, data interpretation of in vivo experiments in mice; S.P. data acquisition, AAV cloning; Y.L. data acquisition and neonatal injections in mice; A.L., A.W. data acquisition, visualization of in vivo experiments in mice; D.P.C. conceptualization, supervision, data interpretation, manuscript writing.
Corresponding authors
Ethics declarations
Competing interests
M.V.I. is a consultant to and holds equity in Skylark Bio. K.D.K. is an employee and holds equity in Skylark Bio. D.P.C. holds equity in Skylark Bio. These relationships are unrelated to the specific research presented in this manuscript. M.V.I., K.T.B., S.H., M.A.N., C.W.P., E.C.G., M.E.G., and D.P.C. are inventors on patent application US20230340038A1, Recombinant adeno associated virus (raav) encoding gjb2 and uses thereof. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Karen Steel and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ivanchenko, M.V., Booth, K.T.A., Karavitaki, K.D. et al. Cell-specific delivery of GJB2 restores auditory function in mouse models of DFNB1 deafness and mediates appropriate expression in NHP cochlea. Nat Commun 16, 11157 (2025). https://doi.org/10.1038/s41467-025-66110-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66110-2
