
Abstract
Quantum interference between reaction pathways plays a crucial role in understanding the microscopic mechanisms of chemical reactions. The hydrogen exchange reaction, involving a well-characterized conical intersection, exhibits rich dynamics arising from interference between multiple reactive pathways. Here, we report high-resolution scattering measurements at a collision energy of 2.38 eV, which reveal pronounced sideways angular oscillations in the HD product distribution. Together with exact quantum theoretical calculations, these results provide unambiguous evidence of geometric phase-induced quantum interference in the H + D2 reaction. Detailed analysis reveals that the observed sideways scattering oscillations, hallmarks of quantum interference, are strongly influenced by transient D–D bond elongation in D2 reactants. This behavior, in contrast to the reaction with HD, highlights a pronounced isotopic effect and reflects the unique dynamic competition along the roaming insertion pathway. Collectively, these findings underscore the critical role of pathway interference and provide new mechanistic insights into how the isotopic effect modulated the geometric phase in an elementary chemical reaction.
Similar content being viewed by others


Introduction
Understanding chemical reaction pathways, the detailed routes through which reactants transform into products, is fundamental to the study of chemistry1,2,3,4,5. Investigating these pathways not only clarifies the final products of a reaction, but also provides essential insights into the underlying mechanisms that govern chemical transformations. Such insights enable the potential control of reaction rates, product yields, and branching ratios6,7,8,9,10, and are particularly vital in fields like atmospheric chemistry, interstellar chemistry, combustion science, and chemical engineering2,3,4,5,6,7,8,9,10.
In a molecular reaction system involving a conical intersection (CI), when nuclear motion traces a closed loop around a CI, the accompanying change in the electronic wave function must be compensated by a phase change in the nuclear wave function to maintain the single-valuedness of the total wave function. This topological effect, known as the geometric phase (GP), plays a central role in quantum reaction dynamics11,12,13,14. According to topological theory, adiabatic and non-adiabatic wave functions can be recombined to extract two types of distinct reaction pathways, corresponding to odd and even numbers of crossings around the CI15.
The H + H2 → H2 + H reaction and its isotopic variants, featuring a D3h-symmetric CI at a total energy of 2.75 eV, have served as benchmark systems for studying the GP effect and associated quantum interference phenomena12,16,17. Notably, in the H3 reaction, pathways that loop around the CI more than once are effectively nonexistent, simplifying the dynamics to two fundamental pathways: path 1 (with 0 CI crossings, i.e., direct pathway) and path 2 (with 1 CI crossing, i.e., looping pathway). When two such distinct pathways, each represented by a different wave function, lead to the same final state, quantum interference arises. This interference could manifest as measurable oscillatory patterns in the reaction observables18,19,20,21,22,23. These distinctive interference patterns in reactive scattering offer fundamental insights into the quantum dynamics of elementary chemical reactions2,3,4,5,6,7,8,9,10,24,25.
Over the past few years, by combining high-resolution reactive scattering experiments with highly accurate theoretical calculations, researchers have been able to directly capture key information about the two fundamental reaction pathways in hydrogen exchange reactions. In particular, the reaction H + HD → H2 + D has been well studied, providing compelling evidence for these quantum effects16,21,22,26. Notably, the GP effect has been experimentally observed in the H + HD → H2 + D reaction21,22,26, where it demonstrably modulates the reaction dynamics via pathway interference. The experimental identification of quantum interference between the direct abstraction pathway and the roaming insertion pathway represents a significant breakthrough in understanding reaction mechanisms at the quantum level.
While the GP effect has been well identified in H + HD reactions16,20,21,27, experimental studies on the isotopic variant H + D2 system, despite extensive investigation at collision energies up to 3.26 eV28,29,30,31,32, have so far been unable to discover any GP signatures. This is particularly surprising given that the H + D2 reaction yields only a single product channel (HD + D). The challenge lies in disentangling the complex quantum-mechanical behavior of the system, particularly the subtle interference patterns encoded in the nuclear wave functions. Therefore, a detailed quantum-level investigation of the reaction pathways in this system is indispensable.
In this work, we probe the quantum dynamics of the H + D2 reaction by performing high-resolution scattering experiments on the H + o-D2 → HD + D reaction at a collision energy of 2.38 eV. The measurements reveal clear angular oscillatory structures in the quantum state specific differential cross sections (DCSs) for HD products, with pronounced sideways scattering as a characteristic feature. By combining these observations with accurate quantum theoretical calculations, we unambiguously identify the GP effect in the H + D2 reaction. Further analysis shows that the interaction between the incoming H atom and the D2 molecule elongates the D-D bond, promoting product formation via Path 2, and demonstrates the role of the potential barrier height in modulating the GP effect. In contrast to the H + HD reaction, the results reflect that the isotopic effect plays a key role in relative contributions of Path 2 in the two systems. These findings provide the definitive evidence for the GP effect in the H + D2 reaction, highlighting the crucial role of the isotopic effect in modulating reactive quantum interference and the GP effect. Our results offer a new platform for understanding fundamental quantum effects in chemical reactivity and underscore the importance of coherent pathway analysis in elementary chemical reactions.
Results
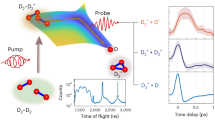
A rotational state-resolved experimental image was obtained using the crossed molecular beams method combined with the velocity map ion imaging technique. The D-atom products were probed using a near-threshold laser ionization scheme. (for details, see Methods). The H-atom beam was generated by photolyzing HI at 213 nm (5th harmonic of an Nd:YAG laser, Continuum Powerlite DLS 9020; produced via sum frequency mixing of 1064 nm fundamental and 266 nm 4th harmonic using a BBO crystal). The faster H-atom component (22.89 km/s) was selected. The D2 beam (1030 m/s) had ~92% in (v = 0, j = 0). The H and D2 beams crossed at 160°, with the D-atom product velocity map ion image shown in Fig. 1 (collision energy 2.38 eV).

Ring 1 corresponds to the quantum states of the product HD, including (v’ = 3, j’ = 12), (v’ = 4, j’ = 8) and (v’ = 1, j’ = 18). Ring 2 corresponds to the quantum states of HD with (v’ = 2, j’ = 14), (v’ = 4, j’ = 5) and (v’ = 1, j’ = 17). “F” and “B” represent the forward and the backward scattering direction, respectively. The symbols “D2” and “H” represent the velocity vectors of the D2 molecular beam and the H-atom beam, respectively. “CM” represents the origin of the center-of-mass coordinate system. Source data are provided as a Source Data file.
In Fig. 1, the concentric rings in the image of D-atom products correspond to the ro-vibrational states of the co-product HD molecules. The red arrows in Fig. 1 indicate the specific HD product states: (v’ = 3, j’ = 12), (v’=4, j’ = 8) and (v’ = 1, j’ = 18), as well as (v’ = 2, j’ = 14), (v’ = 4, j’ = 5) and (v’ = 1, j’ = 17). Notably, the fine angular oscillations of these states in the sideways scattering direction, indicated by ring 1 and ring 2, were experimentally observed. Based on the total kinetic energy release spectrum (TKERs) and rotational state-specific DCSs of the products can be obtained (please see data analysis in Supplementary Note 1).
To better illustrate the fine angular structure of products at specific rotational states, the experimental angular distributions of these states (indicated by ring 1 and ring 2) in the angular range of 0–70° are shown alongside the theoretical results in Fig. 2a and Fig. 2b, respectively.

a, b Comparisons between experimental results and theoretical calculation results of HD angular distribution for specific rovibrational states: a HD (v’ = 3, j’ = 12), (v’ = 4, j’ = 8), and (v’ = 1, j’ = 18); b HD (v’ = 2, j’ = 14), (v’ = 4, j’ = 5), and (v’ = 1, j’ = 17). Experimental data are displayed with hollow circles (EXP). Theoretical results without incorporating the geometric phase are depicted by red lines (NGP), whereas those with the geometric phase included are represented by blue lines (GP). c Theoretical DCS comparison between HD (v’ = 3, j’ = 12) state and the combination of states: the blue line represents the theoretical DCS of the combinational state of (v’ = 3, j’ = 12), (v’ = 4, j’ = 8), and (v’ = 1, j’ = 18) with the inclusion of GP; the purple line represents the theoretical DCS of the (v’ = 3, j’ = 12) state with the inclusion of GP. d Theoretical relative DCSs of path 1 and path 2 for product HD (v’ = 3, j’ = 12).
The hollow points in Fig. 2 represent the experimental DCS results, which are compared with the outcomes of two different theoretical calculations. The light blue lines correspond to the DCSs calculated using theoretical models that incorporate the GP, while the light red lines represent theoretical results obtained without considering the GP. The experimental angular oscillatory structure and its fine details are in good agreement with the theoretical calculations that include the GP effect, clearly deviating from the results that omit the GP. As a result, the GP effect in the H + D2 reaction has been unambiguously identified. Notably, the pronounced sideways oscillatory pattern serves as a distinctive signature, confirming the presence of the GP effect in the H + D2 → HD + D reaction.
Theoretical analysis reveals that the (v’=3, j’=12) state primarily contributes to the angular distribution shown in Fig. 2a, with the observed oscillatory pattern largely arising from the fine structure of this state (see Fig. 2c). Therefore, in the following, we focus on investigating the oscillations associated with the (v’=3, j’=12) state. Using a straightforward topological approach proposed by Althorpe and coworkers12, the adiabatic and diabatic scattering amplitude results were recombined into two reaction pathways around the CI21. The scattering amplitudes for these pathways are expressed as:
As previously mentioned, pathways encircling the CI more than once are negligible. Therefore, the reconstructed paths represent a direct abstraction (path 1) and a looping path around the CI (path 2). Figure 2d illustrates the contributions of these two paths to the DCSs for the product (v’ = 3, j’ = 12). Notably, path 1 overwhelmingly dominates, producing ~100 times more products than path 2. Additionally, products from path 1 are primarily distributed in the forward-sideways direction, while those from path 2 exhibit a more uniform distribution. To evaluate the contribution of each partial wave to the scattering, Fig. 3a, c presents the QM generalized deflection functions (GDFs) together with the QCT results. The quantum GDF represents the quantum correlation between the total angular momentum, J, and the scattering angle, and can be considered as a joint quasi-probability distribution function of J and θ that includes all coherences between different partial waves. The summation of this function over all partial waves recovers the exact differential cross section33. For a more direct comparison with the trajectory analysis, the functions are presented without the usual factor of sinθ.

Topological methods were used to analyze the contributions of different paths to DCS. a–d, a and c correspond to the contribution of Path 1; b and d correspond to the contribution of Path 2. The quantum dynamics method used in (a) and (b); The quasi-classical trajectory dynamics used in (c) and (d). For path 1, back-scattering products primarily arise from head-on collisions involving low partial waves, while sideways-scattering products result from glancing collisions involving high partial waves. Yet, for path 2, low-partial-wave collisions preferentially produce forward scattering, whereas high-partial-wave collisions predominantly result in backward scattering. The angular range in this Figure begins at 30° due to the scarcity of reactive trajectories following Path 2. The most pronounced interference structures are observed in the 30°–60° range.
The collision dynamics of atom–diatom reactions can often be interpreted within the framework of the billiard ball model32. In this model, back-scattering products primarily originate from head-on collisions involving low partial waves, whereas sideways-scattering products arise from glancing collisions involving high partial waves. Figure 3a shows a scattering pattern for path 1 that is consistent with these qualitative predictions. In sharp contrast, the scattering pattern for path 2 displays an unexpected trend: products from low-partial-wave collisions exhibit forward scattering, whereas high-partial-wave collisions predominantly yield backward scattering. Moreover, Fig. 3 reveals a distinctive feature of path 2. The peaks of the DCSs shift progressively from the forward to the backward direction as the impact parameter increases, challenging conventional understanding of scattering mechanisms.
Notably, the partial waves from path 2 that contribute to the observed sideways (30°–60°) angular oscillations are concentrated in two distinct ranges: J = 0–15 and J = 16–30. To assess their contributions, we calculated the product amplitudes from path 2 separately for J = 0–15 and J = 16–40, and combined them with those from path 1 to obtain the corresponding differential cross sections (DCSs). As shown in Fig. 4, there is no significant interference between the products from path 1 and those from path 2 at higher partial waves (J > 15). This suggests that the oscillations in the sideways and forward scattering angles primarily originate from interference between path 1 and the low-J (J ≤ 15) contributions of path 2. To further support this, we have plotted the reaction probability P(J) for path 2 of the H + D2 → D + HD (v’ = 3, j’ = 12) reaction as a function of the total angular momentum J. As shown in Supplementary Fig. S5, P(J) decreases significantly for J > 15.

All partial waves were included for path 1, while different partial wave components were included for path 2. The gray line corresponds to all partial waves for path 1. The red line corresponds to all partial waves for path 1 and the partial waves with J < 15 for path 2. The blue line likely corresponds to a different set of partial waves J ≥ 15 for path 2. The green line corresponds to all partial waves for both path 1 and path 2. Source data are provided as a Source Data file.
A partial wave analysis was also performed for the dominant product channels shown in Fig. 2b, (v′ = 2, j′ = 14 and v′ = 1, j′ = 17; see Supplementary Fig. S6), with the results shown in Supplementary Figs. S7, S8. For path 2 in these states, the DCS peaks progressively shift from the forward to the backward direction as the impact parameter increases, consistent with the trend observed in Fig. 3b, d. The partial-wave decomposition further reveals that, as in the v’ = 3, j’ = 12 case, the oscillatory features in the 30°–60° range arise mainly from interference between path 1 and the low-J (J ≤ 15) component of path 2. The consistency of these features across both cases shown in Fig. 2 indicates that the unconventional roaming-insertion mechanism associated with path 2 is a general aspect of the reaction dynamics, persisting even when the HD product populates different final vibrational states.
The looping pathway (path 2) was considered to be a roaming-like pathway20,21. Roaming describes a process in which a reactant deviates from the minimum energy path, with a fragment of the molecule orbiting the remaining part until it encounters a reactive site to form products. This mechanism was previously assumed to be rare in the reaction system with minimal electronic complexity5,34,35,36,37,38. The H + D2 reaction is a system with only three electrons yields only one product channel (HD + D). The fact that a roaming insertion pathway arises even in this more constrained and electronically minimal system highlights its generality and makes its appearance here particularly remarkable.
To gain insight into the underlying dynamics, we performed a quasi-classical trajectory (QCT) study. In this study, we ran a total of 20 million trajectories, of which about 13.65% exhibited reactivity, resulting in a total reaction cross section of about 1.08 Å2. The majority of these reactive trajectories followed the direct abstraction mechanism (path 1). However, 0.42% of the reactive trajectories occurred via path 2, which involves a two-transition-state mechanism. We focused our analysis on a specific product state with vibrational and rotational quantum numbers v’ = 3 and j’ = 12. For trajectories following path 2, both QCT and quantum dynamics results revealed a pronounced angular dependence in the scattering outcomes. Trajectories with higher angular momentum, corresponding to larger partial waves, tended to produce backward scattering. In contrast, those with lower angular momentum favored forward scattering. This trend differs markedly from the dynamics observed for path 1.
In Fig. 5b, d, the green curve represents the variation of the system’s potential energy with time. For the ball-and-stick models included therein, “H” stands for a hydrogen atom, “a” stands for Da, and “b” stands for Db. The bond lengths between molecules are represented by the straight lines and curves between them. Source data are provided as a Source Data file.

Panels (a) and (b) correspond to the time evolution of bond lengths, and panels (c) and (d) correspond to the evolution of potential energy. Panels (a) and (c) correspond to low-impact-parameter trajectories, while panels (b) and (d) correspond to high-impact-parameter trajectories. In (a) and (c), different curves represent the bond length variation of different molecules. The red curve corresponds to D₂, the blue curve corresponds to H-Da, and the black curve corresponds to H-Db. In (b) and (d), the green curve represents the variation of the system’s potential energy with time. For the ball-and-stick models included therein, “H” stands for a hydrogen atom, “a” stands for Da, and “b” stands for Db. The bond lengths between molecules are represented by the straight lines and curves between them. Source data are provided as a Source Data file.
To further examine the physical origin of this behavior, we analyzed the effect of the impact parameter on path 2 trajectories. Collisions with larger impact parameters that led to backward scattering displayed features similar to the roaming insertion mechanism previously reported for the H + HD → H2 + D reaction21. In contrast, for smaller impact parameters in the H + D2 system, we observed a distinct roaming behavior characterized by a different scattering direction. Specifically, the incoming H atom circled one of the D atoms (Da), rotated by approximately 180 degrees, and then inserted into the Da-Db bond to form a transient Da-H-Db configuration. This intermediate subsequently evolved into the final product.
To clearly illustrate the roaming process in the H + D2 reaction, we analyzed the time-dependent changes in bond lengths during the reaction (Fig. 5a, c). The critical step in the roaming process, which occurs during the dynamically competing interactions at 30–45 femtoseconds, was determined. Interestingly, the initial step of this phenomenon closely resembles the inelastic collision results reported by Greaves et al. in 2008 for the H + D2 system, which involved a vibrational excitation mechanism commonly observed in neutral particles with strong mutual attractions39. In the present study, during the dynamic competing interactions, the H-Da and Da-Db bonds soften and elongate when the interatomic forces are approximately balanced. This enables the H atom to orbit around the Da atom. Simultaneously, the interaction between the H atom and Db competes with the Da–Db bond, stretching it and reducing steric hindrance to facilitate the insertion process. The H atom then inserts between the Da-Db bond and approaches this site, ultimately forming the reaction products. Notably, the roaming insertion pathway is more efficient and feasible than the direct insertion pathway. As a result, most path 2 reactions process via the roaming pathway regardless of impact parameters, leading to the unconventional scattering direction of path 2 products.
It is instructive to compare the present findings for the H + D2 reaction with the H + HD reaction. To enable a direct comparison, we performed a detailed QCT analysis for H + HD under the same initial state and collision energy as for H + D2. The comparison clarifies the subtle mechanistic differences in the roaming-like insertion pathway between the two reactions. The contributions of the two pathways to the DCSs, and the classical trajectories for the forward and backward scattering products are illustrated and presented in the new Supplementary Figs. S9–S11 of the revised Supplementary Information. A side-by-side comparison of the time evolution of bond length and potential energy along the roaming-like insertion pathway (path 2) was conducted and presented in the new Supplementary Figs. S12–S15 of the revised Supplementary Information. As mentioned above, the critical step in the roaming process, which occurs during the dynamically competing interactions at 30–45 femtoseconds, was determined. To compare the differences in the reaction mechanisms of the two isotope reactions though path 2, theoretical calculations were conducted on the product quantum states (v’ = 0, j’ = 0) and (v’ = 3, j’ = 12), systematically investigating the bond length stretching characteristics of the two reactant molecules within the 30–45 femtosecond time range (as shown in Supplementary Fig. S13). As shown in the Supplementary Fig. S13, for the product with the same ro-vibrational state, the stretching rate of the D₂ bond in the H + D2 reaction is slower than that of the HD bond in the H + HD reaction; this result indicates that the reaction rate of H + D2 via path 2 is slower than that of H + HD via path 2. It’s commonly accepted that the reaction rate strongly relies on the height of the potential barrier along the reaction pathway. The evolution of the forward and backward potential energies of the H + D2 → D + HD (v’, j’) reaction via path 2 is further analyzed. The results for the H + D2 → D + HD (v’ = 3, j’ = 12) reaction ranging from 20 to 75 fs are presented in Fig. 5b, d. The potential energy evolution along path 2 can be viewed as an effective barrier that reactants must overcome to form products. In order to explore the differences in barrier height in the isotopic reaction system and its impact on the path 2 reaction. Further comparison of the time evolution of the total potential energy for product in the (v’ = 0, j’ = 0) and (v’ = 3, j’ = 12) states between the H + HD → D + H2 and the H + D2 → D + HD reaction, as shown in Supplementary Figs. S14, 15. In Supplementary Fig. S14, for high-impact-parameter trajectories, the effective barrier along path 2 is lower than the one with low impact parameter in both systems, which explains the predominance of backward scattering and the GP effect observed for H + HD at a collision energy of 1.72 eV21. In contrast, for low-impact-parameter trajectories, the path 2 barrier in H + D2 is about 0.2 eV higher than in H + HD, leading to a sideways path 2 yield in H + D2 that is approximately half of that in H + HD in our QCT calculations as shown in Supplementary Fig. S13a, b. Furthermore, as shown in Supplementary Fig. S15, the path 2 barrier for the reaction H + D2 → D + HD (v’ = 0, j’ = 0) is ~0.36 eV higher than that for H + HD → D + H2 (v’ = 0, j’ = 0). This comparison clarifies both the common features and the subtle mechanistic differences in the roaming-like insertion pathway between the two reactions. In summary, the QCT simulations highlight the dynamical significance of the newly identified roaming-like pathway. They reveal the emergence of a roaming-like path 2 involving transient D–D bond stretching, which provides a clear explanation for why the GP effect is much more difficult to probe in H + D2 than in its isotopic variants.
In the H + DaDb reaction at collision energy 2.38 eV, dynamic competing interactions lead to the softening and extension of H-Da and Da-Db bonds, resulting in the vibrational excitation (v ≥ 3) of D2, similar to what was reported in previous work about inelastic collisions in H + D239. As shown in Fig. 6, this excitation, via the roaming pathway, results in the formation of HD products. This also gives insight into the reactive collision between the highly vibrationally excited D2 molecule, which was prepared via collisions. While this type of molecular vibrational excitation is not an actively induced process, it provides a valuable research strategy and experimental basis for studying vibrationally excited reaction dynamics.

a The H atom collides with the D2 molecule. Due to the strong interaction between the neutral particles, the D2 molecule vibrates and excites, but the H atom does not move away from D2. Instead, it participates in the reaction through a new reaction pathway created by the vibrational excitation of D2, which is why the roaming pathway is formed. For the ball-and-stick models included therein, “H” stands for a hydrogen atom, “D” stands for D atom. The bond lengths between molecules are represented by the straight lines and curves between them. b The reaction proceeds through the roaming pathway, forming HD products. The reaction coordinate diagram shows the process of the H + D2 reaction via the roaming pathway. Firstly, collisions and interactions between H atoms and D2 molecules promote the excitation of D2 molecules to create new reactive pathways, and H atoms react with D2 along this reactive pathway to form the excited state of the HD product. Source data are provided as a Source Data file.
Through high-resolution scattering measurements, we have revealed significant sideways angular oscillations in the HD product distribution of the H + D2 → HD + D reaction. Combined with accurate quantum mechanical calculations, these results provide unambiguous evidence of GP-induced quantum interference. The observed sideways scattering is strongly modulated by dynamic competition along the roaming insertion pathway, driven by transient elongation of the D–D bond. Unlike the reaction with HD, this behavior highlights a pronounced isotopic effect while reflecting the unique dynamic competition within the roaming insertion pathway. Collectively, these findings not only underscore the critical role of pathway interference but also provide novel mechanistic insights into how the isotopic effect modulates the geometric phase in an elementary chemical reaction.
Methods
Experimental setup
The H + D2 → HD + D reaction was investigated using a velocity map imaging (VMI) - crossed molecular beam (CMB) apparatus. The apparatus consists of three differentially pumped chambers: the scattering chamber, the fixed source chamber, and the rotating source chamber. In this experiment, a liquid N2-cooled Even Lavie valve was mounted in the rotating source chamber to generate the pulsed molecular beam of ortho-D2 (o-D2). The o-D2 sample was obtained by catalytic conversion of normal D2 (n-D2) at low temperatures. To catalytically convert the n-D2 into o-D2, n-D2 was frozen within a spiral copper pipe containing the catalyst, which was mounted to the second stage of a closed cycle cryocooler maintained at a temperature below 20 K. Subsequently, the catalytically converted D2 was collected with an aluminum gas cylinder to prevent back conversion. The backing pressure of the D2 molecular beam was 13 bar. The most probable velocity of the D2 molecules was 1030 m/s. The pulsed o-D2 atom beam was collimated by two skimmers (Beam Dynamics, Model 50.8, size 2 mm and Model 1, size 1.5 mm). Approximately 90% of the D2 molecules in the molecular beam are populated in the ground rovibrational state, (v, j) = (0, 0). The rotational state distribution of the D2 beam is measured using the (3 + 1) resonant enhanced multiphoton ionization (REMPI) method at around 280.22 nm via the D2 (C1Πu, v = 5 ←X1Σ+, v = 0), R (0), and R (1)) transitions.
In the study of the H + D2 → HD + D reaction at 2.38 eV, pure HI molecules are supersonically expanded by a pulsed Parker valve (Series 9, vertically mounted in the fixed source chamber) at the backing pressure of 1 bar. After the expansion, the HI molecules are dissociated by a linearly polarized laser at 213 nm (30 mJ/pulse) about 10 mm above the nozzle of the pulsed valve. The photodissociation laser at 213 nm is generated from the 5th harmonic of an Nd: YAG laser (Continuum, Powerlite DLS 9020). The 5th harmonic is realized by the sum frequency of the fundamental output (1064 nm) and the 4th harmonic (266 nm) of the Nd: YAG using a BBO crystal. At this wavelength, H + I(2P3/2) and H + I(2P1/2) dissociation channels of the HI molecule are both energetically allowed, which give rise to two sharp and separated peaks in the velocity distribution of H atom products, exhibiting either faster or slower velocities. Due to the different angular distributions of the H atom in the two channels, in this experiment, when the photolysis laser is set to be vertically polarized, the faster H atom at a speed of 22.89 km/s from the H + I(2P3/2) channel is selectively collimated to the scattering chamber using a skimmer, intersecting with D2. The crossing angle of H and D2 beams is set to be 160°, giving a collision energy of 1.72 eV. Other experimental conditions are consistent with the aforementioned.
The D-atom products generated from the H + D2 reaction was ionized using a near-threshold resonance-enhanced multiphoton ionization (REMPI) approach with a 1 + 1’ scheme, employing laser wavelengths of 121.6 nm and 364.5 nm. The D atoms were excited from n = 1 to n = 2 state by the VUV laser beam at 121.6 nm, and subsequently pumped by a UV laser beam at 364.5 nm to an energy which is just above the ionization limit, and thus ionized. The ionized species were then directed toward a microchannel plate (MCP) detector via ion optical components, while the resulting ion images were captured using a charge-coupled device (CCD) camera.
Theoretical method
We performed adiabatic quantum dynamics calculations on the BKMP2 PES and extracted state-to-state dynamical information by using the product-coordinate-based wave packet method40,41. The numerical parameters provided in the supplementary materials facilitate the convergence of DCSs for collision energies up to 2.4 eV. To further explore the underlying dynamics of H + D2 and H + HD reaction, we carried out a quasi-classical trajectory (QCT) dynamics study. All the calculations were performed on the BKMP2 PES42.
Quantum scattering calculations
The state-to-state quantum dynamics information were extracted using the product-coordinate-based time-dependent wave packet method. We prepared an initial wave function in reactant coordinates. A coordinate transformation was then carried out to transfer the wave packet from the reactant coordinates to the D + HD product coordinates. After the transformation, we propagated the wave packet for additional 800 iterations with Δt = 10 in the product coordinates. We applied a total of 161 sine basis functions (including 127 for the interaction region) in the R range of 0.1–15.0 bohr, a total of 110 potential optimized discrete variable representation basis functions (including 10 for the asymptotic region) in the r range of 0.4–14.0 bohr, and 110 rotational basis functions to converge calculation results. The range of the total angular momentum was taken as 0 ≤ J ≤ 45, and all helicity channels were included. Damping functions were employed to prevent the wave packet from reflecting back from the boundaries in Ra = 11.0 bohr and ra = 12.0 bohr.
Quasi-classical trajectory calculations
To confirm the reaction mechanism in the classical dynamics picture, quasi-classical trajectory (QCT) calculations were performed on the adiabatic BKMP2 PES. Although these simulations are unable to predict quantum effects such as the GP, they are expected to accurately capture the underlying dynamics, which are essentially semiclassical. In our calculations, the maximum impact parameter bmax is set to be 3 bohr and the time step is equal to 3 in atom unit. Similar to the calculations of H + D2, for H + HD reaction, we ran a total of 20 million trajectories, of which about 8.98% exhibited reactivity, resulting in a total reaction cross section of about 0.71 Å2. As illustrated in Supplementary Figs. S10 and S11, the position of the transition state can be identified in hyperspherical coordinates, and the number of trajectories passing through the transition state allows the assignment of Path 1 and Path 2. The majority of these reactive trajectories followed the direct abstraction mechanism (path 1). However, 0.44% of the reactive trajectories occurred via path 2. The snapshots of a representative QCT trajectory of the path 2 mechanism for forward and backward scattered HD (v’ = 3, j’ = 12) product are shown in Supplementary Figs. S3 and S4. In the trajectory, the incoming H atom approaches D2 molecule and passes the first H-Da-Db linear transition state. As the Da-Db molecular stretches, H atom inserts between D-D bond by experiencing the second Da-H-Db transition state. For comparison, the snapshots of a representative H + HD QCT trajectory of the path 2 mechanism for forward and backward scattered H2 (v’ = 3, j’ = 12) product are also given in Supplementary Figs. S10 and S11.
Data availability
Data supporting the findings of this study are available from the corresponding authors upon request. Additionally, source data are provided with this paper and can be accessed at https://doi.org/10.6084/m9.figshare.30417382.
Code availability
The code that supports the theoretical calculations in this paper is available from the corresponding authors upon request.
References
Bowman, J. M. & Suits, A. G. Roaming reactions: The third way. Phys. Today 64, 33–37 (2011).
Baiardi, A. et al. Expansive Quantum Mechanical Exploration of Chemical Reaction Paths. Acc. Chem. Res. 55, 35–43 (2022).
Dai, D. et al. Interference of Quantized Transition-State Pathways in the H + D2 → D + HD Chemical Reaction. Science 300, 1730–1734 (2003).
Continetti, R. E. The view from a transition state. Nat. Chem. 9, 931–932 (2017).
Hause, M. L., Herath, N., Zhu, R., Lin, M. C. & Suits, A. G. Roaming-mediated isomerization in the photodissociation of nitrobenzene. Nat. Chem. 3, 932–937 (2011).
Yosa Reyes, J., Nagy, T. & Meuwly, M. Competitive reaction pathways in vibrationally induced photodissociation of H2SO4. Phys. Chem. Chem. Phys. 16, 18533–18544 (2014).
Suits, A. G. & Parker, D. H. Hot molecules—off the beaten path. Science 346, 30–31 (2014).
Shi, Y.-F., Kang, P.-L., Shang, C. & Liu, Z.-P. Methanol Synthesis from CO2/CO Mixture on Cu–Zn Catalysts from Microkinetics-Guided Machine Learning Pathway Search. J. Am. Chem. Soc. 144, 13401–13414 (2022).
Ellerbrock, R., Zhao, B. & Manthe, U. Vibrational control of the reaction pathway in the H + CH3D → H2 + CD3 reaction. Sci. Adv. 8, eabm9820 (2022).
Zhang, J. et al. State to State to State Dynamics of the D + H2 → HD + H Reaction: Control of Transition-State Pathways via Reagent Orientation. Phys. Rev. Lett. 96, 093201 (2006).
Valahu, C. H. et al. Direct observation of geometric-phase interference in dynamics around a conical intersection. Nat. Chem. 15, 1503–1508 (2023).
Juanes-Marcos, J. C., Althorpe, S. C. & Wrede, E. Theoretical Study of Geometric Phase Effects in the Hydrogen-Exchange Reaction. Science 309, 1227–1230 (2005).
Martinazzo, R. & Burghardt, I. Dynamics of the Molecular Geometric Phase. Phys. Rev. Lett. 132, 243002 (2024).
Huang, J., Kendrick, B. K. & Zhang, D. H. Mechanistic Insights into Ultracold Chemical Reactions under the Control of the Geometric Phase. J. Phys. Chem. Lett. 12, 2160–2165 (2021).
Whitlow, J. et al. Quantum simulation of conical intersections using trapped ions. Nat. Chem. 15, 1509–1514 (2023).
Yuan, D. et al. Observation of the geometric phase effect in the H + HD → H2 + D reaction. Science 362, 1289–1293 (2018).
Aoiz, F. J., BaÑares, L. & Herrero, V. J. The H + H2 reactive system. Progress in the study of the dynamics of the simplest reaction. Int. Rev. Phys. Chem. 24, 119–190 (2005).
Dixon, R. N. et al. Chemical “Double Slits”: Dynamical Interference of Photodissociation Pathways in Water. Science 285, 1249–1253 (1999).
Jambrina, P. G. et al. Quantum interference between H + D2 quasi-classical reaction mechanisms. Nat. Chem. 7, 661–667 (2015).
Xie, Y. et al. Quantum interference in H + HD→H2+ D between direct abstraction and roaming insertion pathways. Science 368, 767–771 (2020).
Li, S. et al. Observation of geometric phase effect through backward angular oscillations in the H + HD → H2 + D reaction. Nat. Commun. 15, 1698 (2024).
Yarkony, D. R. Suppressing the geometric phase effect: Closely spaced seams of the conical intersection in Na3(2 2E′). J. Chem. Phys. 111, 4906–4912 (1999).
Kendrick, B. K. Geometric phase effects in chemical reaction dynamics and molecular spectra. J. Phys. Chem. A 107, 6739–6756 (2003).
Foley, C. D., Xie, C., Guo, H. & Suits, A. G. Orbiting resonances in formaldehyde reveal coupling of roaming, radical, and molecular channels. Science 374, 1122–1127 (2021).
del Mazo-Sevillano, P., Aguado, A., Jiménez, E., Suleimanov, Y. V. & Roncero, O. Quantum Roaming in the Complex-Forming Mechanism of the Reactions of OH with Formaldehyde and Methanol at Low Temperature and Zero Pressure: A Ring Polymer Molecular Dynamics Approach. J. Phys. Chem. Lett. 10, 1900–1907 (2019).
Yuan, D. et al. Observation of the geometric phase effect in the H + HD → H2 + D reaction below the conical intersection. Nat. Commun. 11, 3640 (2020).
Jankunas, J., Sneha, M., Zare, R. N., Bouakline, F. & Althorpe, S. C. Hunt for geometric phase effects in H + HD → HD (v′, j′) + H. J. Chem. Phys. 139, 144316 (2013).
Fernandez-Alonso, F. et al. Forward scattering in the H + D2→ HD + D reaction: Comparison between experiment and theoretical predictions. J. Chem. Phys. 115, 4534–4545 (2001).
Gao, H., Sneha, M., Bouakline, F., Althorpe, S. C. & Zare, R. N. Differential Cross Sections for the H + D2 → HD (v’ = 3, j’ = 4-10) + D Reaction above the Conical Intersection. J. Phys. Chem. A 119, 12036–12042 (2015).
Wrede, E. et al. High-resolution study of the H+D2 → HD+D reaction dynamics at a collision energy of 2.2 eV. Chem. Phys. Lett. 265, 129–136 (1997).
Jankunas, J. et al. Seemingly Anomalous Angular Distributions in H + D2 Reactive Scattering. Science 336, 1687–1690 (2012).
Yuan, D. et al. Imaging the State-to-State Dynamics of the H + D2 → HD + D Reaction at 1.42 eV. J. Phys. Chem. Lett. 11, 1222–1227 (2020).
Jambrina, P. G., Menéndez, M. & Aoiz, F. J. Angular momentum–scattering angle quantum correlation: a generalized deflection function. Chem. Sci. 9, 4837–4850 (2018).
Fu, Y.-L. et al. Collision-induced and complex-mediated roaming dynamics in the H + C2H4 → H2 + C2H3 reaction. Chem. Sci. 11, 2148–2154 (2020).
Fu, Y. L., Bai, Y., Han, Y. C., Fu, B. & Zhang, D. H. Double-Roaming Dynamics in the H + C2H2 → H2 + C2H Reaction: Acetylene-Facilitated Roaming and Vinylidene-Facilitated Roaming. J. Phys. Chem. Lett. 12, 4211–4217 (2021).
Townsend, D. et al. The Roaming Atom: Straying from the Reaction Path in Formaldehyde Decomposition. Science 306, 1158–1161 (2004).
Grubb, M. P. et al. No Straight Path: Roaming in Both Ground- and Excited-State Photolytic Channels of NO3 → NO + O2. Science 335, 1075–1078 (2012).
Pomerantz, A. E. et al. Reaction Products with Internal Energy beyond the Kinematic Limit Result from Trajectories Far from the Minimum Energy Path: An Example from H + HBr → H2 + Br. J. Am. Chem. Soc. 127, 16368–16369 (2005).
Greaves, S. J. et al. Vibrational excitation through tug-of-war inelastic collisions. Nature 454, 88–91 (2008).
Yuan, K. Experimental and quantum dynamical study on an asymmetric insertion reaction: state-to-state dynamics of O (1D) + HD → OH + D. Phys. Rev. Lett. 96, 103202 (2006).
Huang, J. & Zhang, D. H. An efficient way to incorporate the geometric phase in the time-dependent wave packet calculations in a diabatic representation. J. Chem. Phys. 153, 141102 (2020).
Arnold, I. et al. A refined H3 potential energy surface. J. Chem. Phys. 104, 7139–7152 (1996).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nos. 22125302(X.W.), 22288201(D.Z.), 22473020(J.H.), 92476207(J.H.), 22173097(Z.Z.), 22322305(Z.Z.), 22327801(X.W.), and 22233003(J.H.)), the Guangdong Science and Technology Program (Grant Nos. 2019ZT08L455 and 2019JC01X091) (X.Y.), the Innovation Program for Quantum Science and Technology (Grant No. 2021ZD0303304)(X.Y.), the Innovation Program for Quantum Science and Technology (Grant No. 2021ZD0303305)(D.Z.) and the Dalian Innovation Support Program (Grant No. 2021RD05)(D.Z.).
Author information
Authors and Affiliations
Contributions
X.Y., D.Z., X.W., and Z.Z conceived and supervised the research. The experiments were carried out by S.L., C.L., Z.L., Y.S., W.C., D.Y., and X.W. Data analysis and interpretation were performed by S.L., C.L., Z.L., Y.S., W.C., D.Y., X.W., and X.Y. Theoretical calculations were performed by J.H., Z.Z., B.F., and D.Z. The manuscript was written by X.W., Z.Z., S.L., and J.H., with contributions from all authors. All authors contributed to discussions about the content of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, S., Huang, J., Luo, C. et al. Sideways scattering oscillations reveal geometric phase effect and isotope effect in the H + D2 → HD + D reaction. Nat Commun 16, 11166 (2025). https://doi.org/10.1038/s41467-025-66114-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66114-y
