
Abstract
Single-cell RNA sequencing (scRNA-seq) provides high-resolution insights into cellular heterogeneity but remains costly, restricting its use to small cohorts that often lack comprehensive clinical data, reducing translational relevance. In contrast, bulk RNA sequencing is scalable and cost-effective but obscures critical single-cell insights. We introduce SIDISH, a neural network framework that integrates the granularity of scRNA-seq with the scalability of bulk RNA-seq. Using a variational autoencoder, deep Cox regression, and transfer learning, SIDISH identifies high-risk cell populations while enabling robust clinical predictions from large-cohort data. Its in silico perturbation module identifies therapeutic targets by simulating interventions that reduce high-risk cells associated with adverse outcomes. SIDISH also generalizes to spatial transcriptomics, identifying high-risk cells and mapping them within their native tissue microenvironment. Applied across diverse diseases, SIDISH establishes the link between cellular dynamics and clinical phenotypes, facilitating biomarker discovery and precision medicine. By unifying single-cell insights with large-scale clinical data, SIDISH advances computational tools for disease risk assessment and therapeutic prioritization, offering an integrative and scalable approach to precision medicine.
Similar content being viewed by others

Introduction
The cellular and phenotypic heterogeneity inherent to complex diseases, such as cancer, drives diverse progression patterns and variable treatment responses, complicating accurate diagnosis, prognosis, and therapeutic target identification1,2,3. This heterogeneity arises from the intricate molecular composition of tumors and their microenvironments, which remain poorly understood at a granular level4,5,6. Next-generation sequencing (NGS) technologies have advanced disease biology research, with bulk RNA sequencing (bulk RNA-seq) serving as a widely used approach for large-scale transcriptomic studies due to its cost-effectiveness and scalability7,8,9. While bulk RNA-seq has been widely used for biomarker discovery and disease subtyping10,11, it averages gene expression across all cells in a sample, masking the cellular heterogeneity essential for deciphering disease mechanisms12,13. In contrast, single-cell RNA sequencing (scRNA-seq) provides high-resolution insights into cellular diversity, enabling the identification of distinct subpopulations within tissue samples. By uncovering cell-specific gene expression profiles, scRNA-seq has transformed our understanding of disease biology, immune responses, and therapeutic vulnerabilities14. Despite its precision, the high cost of scRNA-seq–approximately $5000 for 20,000 cells as of 2025 (costpercell)–limits its application in large patient cohorts15. Furthermore, the frequent absence of corresponding clinical metadata, such as patient survival outcomes, in scRNA-seq studies restricts their translational relevance16. The dichotomy between the scalability of bulk RNA-seq and the granularity of scRNA-seq underscores the need for an integrative framework that bridges these modalities, enabling a deeper understanding of complex disease mechanisms, the discovery of actionable biomarkers, and potential therapeutic targets.
Several computational tools have been developed to bridge this gap. For instance, methods such as Scissor17 and scAB18 integrate bulk and single-cell data using approaches like Pearson correlation, sparse regression, and matrix factorization to infer clinically relevant cell subpopulations. Concurrently, DEGAS19 employs a multitask neural network20 with domain adaptation, to project both data types into a single shared latent space, enabling the transfer of clinical phenotypes to individual cells as association scores. However, these tools has notable limitations. The reliance of Scissor and scAB on Pearson correlation21 assumes linear relationships between both data types21,22. Gene expression patterns often exhibit nonlinear dynamics driven by intricate regulatory networks, which Pearson correlation cannot adequately capture. This approach is also highly sensitive to noise and outliers, both prevalent in scRNA-seq data23,24, which may compromise the accuracy of cell-phenotype associations. While DEGAS is capable of capturing non-linearities, its strategy of projecting single-cell and bulk expression data into a single shared latent space may risk diluting the distinct clinical signal present in the bulk data with the cellular resolution provided by single-cell data. A more fundamental limitation shared by these frameworks is their reliance on a single-pass integration strategy. Because these models are trained only once to establish a fixed relationship between bulk and single-cell data, they cannot adapt to the dynamic and context-dependent nature of biological systems. In contrast, an iterative refinement strategy, in which insights from bulk and single-cell analyses are continuously fed back to improve one another, offers the potential to capture more nuanced cellular dynamics and uncover clinically relevant signals that static integration may miss. Moreover, these frameworks are not designed to integrate spatial transcriptomics, which preserves tissue architecture alongside gene expression profiles, limiting their ability to capture how microenvironmental context shapes disease phenotypes. Finally, existing methods lack in silico perturbation capabilities, an essential feature for systematically evaluating transcriptomic perturbations and identifying therapeutic targets in complex diseases25. The absence of in silico perturbation reflects a broader translational gap: these existing frameworks have not been utilized for precision medicine. They fail to investigate transcriptomic differences between patients or to screen for patient-specific therapeutic strategies, thereby limiting their potential to inform personalized treatment approaches.
To address these limitations, we introduce SIDISH (Semi-supervised Iterative Deep Learning for Identifying Single-cell High-Risk Populations), a deep learning framework designed to bridge clinical outcomes with cellular resolution by shifting from single-step data integration to a dynamic, iterative learning process. At its core, SIDISH leverages a feedback loop between scRNA-seq and bulk RNA-seq data to progressively refine the identification of clinically relevant cell subpopulations associated with specific phenotypes, such as poor survival outcomes. This iterative architecture allows SIDISH to achieve a precise, disease-focused integration between the two modalities and to discover survival-critical genes that might be obscured in static methods. Furthermore, the framework is designed to be adaptable to emerging data modalities like spatial transcriptomics, allowing poor-survival-associated cells to be mapped back onto their tissue context. To bridge the translational gap of prior tools, SIDISH incorporates an in silico perturbation module that systematically simulates gene knockouts, enabling the evaluation of their impact on phenotype-associated cell subpopulations. This feature also facilitates the screening of patient-specific therapeutic responses, supporting the identification of targeted interventions tailored to individual patient profiles. By linking patient-specific transcriptomic features to clinical outcomes, SIDISH addresses key challenges in precision medicine, facilitating the discovery of actionable biomarkers and potential therapeutic strategies.
In this work, using datasets from pancreatic ductal adenocarcinoma (PDAC), breast cancer (BRCA), and lung adenocarcinoma (LUAD), we demonstrate SIDISH’s applicability and versatility in identifying survival-associated features in cancer. Specifically, we showcase the clinical and biological relevance of phenotype-associated cell subpopulations identified by SIDISH in both non-spatial and spatial contexts. In benchmarking evaluations, SIDISH outperforms previously mentioned tools such as Scissor, scAB, and DEGAS, demonstrating its effectiveness in linking cellular features to poor-survival outcomes. Additionally, we conduct an ablation study to evaluate the contributions of SIDISH’s architectural components, underscoring the significance of each feature in driving its performance. SIDISH’s in silico perturbation feature is applied across all three cancer types, leading to the identification of both FDA-approved and investigational therapeutic targets. Furthermore, SIDISH’s patient-specific analyses reveal considerable inter-patient heterogeneity in phenotype-associated cell subpopulations, therapeutic responses, and clinical outcomes. Notably, patients with higher proportions of these cells show greater enrichment of malignant markers and significantly worse survival outcomes. By integrating the high-resolution insights of scRNA-seq with the scalability of bulk RNA-seq, SIDISH provides a cost-effective and adaptable framework with significant potential for advancing precision medicine, offering various opportunities to uncover actionable biomarkers and develop targeted therapeutic strategies across diverse diseases.
Results
Method overview
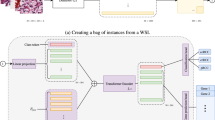
The SIDISH framework integrates bulk survival signals with scRNA-seq resolution to identify high-risk cell subpopulations associated with poor outcomes through an iterative four-phase process. For clarity, SIDISH can be summarized in three main steps: (1) learn cellular features with a variational autoencoder, (2) transfer them to a Cox survival model, and (3) iteratively refine high-risk populations. In Phase 1, a variational autoencoder (VAE), FS26,27, compresses high-dimensional scRNA-seq data into a biologically meaningful latent space (Fig. 1a), capturing cellular heterogeneity through a Zero-Inflated Negative Binomial (ZINB) reconstruction. The loss function, \({{{{\mathcal{L}}}}}_{{F}_{S}}\), incorporates a cell-by-gene weight matrix W, initialized uniformly and iteratively refined in later iterations to prioritize clinically relevant genes. In the spatial extension, a graph convolutional network (GCN) layer propagates features across a K-nearest spatial neighbor graph of cells, ensuring that the learned cellular representation reflects both transcriptomic variation and spatial context. Phase 2 focuses on survival prediction. A deep Cox regression model, FC28, is trained on bulk RNA-seq data B and patient survival outcomes (Fig. 1b). The pretrained encoder ES from Phase 1 is transferred into FC, providing survival modeling with high-resolution features that capture rare and clinically important cell states otherwise lost in bulk profiles. Crucially, these transferred weights are not static: ES is finetuned alongside survival-specific hidden layers weights, enabling the model to adapt the learned single-cell representations to the patient-level. The loss function, \({{{{\mathcal{L}}}}}_{{F}_{C}}\), includes a patient-specific weight vector v, iteratively updated to emphasize high-risk patients and strengthen the detection of survival-associated signals. In Phase 3, the trained Cox model assigns continuous risk scores Y to both patients and cells, linking cellular states with clinical outcomes (Fig. 1c). Stratification into high-risk and background groups is achieved by fitting a Weibull distribution29 to predicted risk scores. The threshold Pcut is defined as an upper-tail cutoff of the fitted distribution, ensuring statistically principled stratification while allowing dataset-specific customization. Phase 4 iteratively refines the framework to sharpen high-risk cell and gene identification (Fig. 1d). SHapley Additive exPlanations (SHAP)30,31 quantify gene contributions to high-risk classifications, and these values are passed through a transformed function σ* to generate updated weights for W. Patient risks update v through a bounded transformation, aligning survival optimization with clinical severity. Both W and v are reintegrated into the losses \({{{{\mathcal{L}}}}}_{{F}_{S}}\) and \({{{{\mathcal{L}}}}}_{{F}_{C}}\), ensuring that SIDISH progressively adapts across iterations to emphasize the most prognostically informative features. This iterative exchange continues until the predefined iteration limit τ is reached, improving robustness and sensitivity in distinguishing high-risk from background cells. SIDISH also includes an in silico perturbation module for therapeutic discovery (Fig. 1e). By simulating single and combinatorial gene knockouts within gene interaction networks, it quantifies the ability of genes to reduce high-risk cells while preserving background states. This strategy enables systematic ranking of candidate therapeutic targets32,33. Finally, Fig. 1f summarizes SIDISH’s core functionalities, including high-risk subpopulation identification in non-spatial and spatial single-cell datasets, biomarker discovery, prognosis modeling, and therapeutic target screening. Together, these capabilities allow SIDISH to bridge the resolution gap between single-cell and bulk transcriptomics, offering a unified and clinically grounded framework for risk prediction and precision oncology.

a Phase 1: Extraction of cellular heterogeneity via a variational autoencoder. A VAE FS is trained on scRNA-seq data X to capture low-dimensional representations of key cellular patterns, guided by a weighted loss \({{{{\rm{L}}}}}_{{F}_{S}}\). b Phase 2: Survival prediction using transfer learning. The encoder ES from the VAE is integrated into a deep Cox regression model, FC, which is optimized using a weighted survival loss function, \({{{{\rm{L}}}}}_{{F}_{C}}\). The model is trained on bulk RNA-seq data (b) and patient survival data to predict survival risks (Y). c Phase 3: Risk prediction and stratification using predicted survival scores. The trained FC predicts risk scores Y for cells and patients, categorizing them into high-risk or background groups based on a Weibull distribution. d Phase 4: iterative weight updates. High-risk labels are used to train a binary classifier, which is then applied to identify survival-associated genes via SHAP values. These SHAP values are transformed into weights to refine FS through \({{{{\rm{L}}}}}_{{F}_{S}}\). Simultaneously, patients' survival risks are transformed into weights to optimize FC through \({{{{\rm{L}}}}}_{{F}_{C}}\). This iterative process is repeated at each iteration. e In silico perturbation. Gene knockouts are simulated in silico to predict therapeutic targets by ranking genes based on their ability to reduce high-risk cells. f Core functionalities of SIDISH. SIDISH enables the identification of high-risk cells in both non-spatial and spatial scRNA-seq datasets, the discovery of biomarkers associated with these cells, prognosis analysis of biomarkers derived from them, screening for potential therapeutic targets, and its application in precision medicine. Created in BioRender. Jolasun, Y. (2025) https://BioRender.com/r2p5q0y.
SIDISH uncovers high-risk cell subpopulations and prognostic biomarkers in pancreatic ductal adenocarcinoma
The cellular composition of malignant tumors exhibits substantial heterogeneity, complicating the identification of cell subpopulations with clinical significance. To address this challenge, SIDISH was applied to a pancreatic ductal adenocarcinoma (PDAC) scRNA-seq dataset34, comprising 41,986 tumor cells from 24 tumor samples and 15,544 cells from 11 normal pancreatic tissues. Bulk RNA-seq data and survival information were obtained from 181 PDAC patients in the Cancer Genome Atlas (TCGA)35. Unsupervised clustering and cell type annotation revealed distinct cell types, including ductal cells, fibroblasts, T cells, acinar cells, B cells, stellate cells, endothelial cells, macrophages, and endocrine cells (Fig. 2a). By focusing exclusively on tumor cells, SIDISH prioritized tumor-specific signals to identify cellular subpopulations associated with poor survival outcomes. Given the poor survival rates in PDAC36, it serves as a suitable disease for evaluating SIDISH’s ability to identify prognostic cellular and molecular signatures.
SIDISH identified 3623 high-risk cells, representing ~8.63% of the total cell population (Fig. 2b and Supplementary Data 1). The majority of these high-risk cells (55.8%) originate from type 2 ductal cells, a cell type previously associated with aggressive tumor behavior and poor prognosis34 (Fig. 2c). High-risk cells were also identified among type 1 ductal cells (16.4%), fibroblasts (7.9%), and T cells (5.9%), underscoring the complexity of the tumor microenvironment. Notably, stromal components, including fibroblasts and stellate cells, played a significant role, reflecting their critical contribution to supporting tumor growth and progression in PDAC37,38,39,40,41. Interestingly, 5.7% of high-risk cells originated from acinar cells, a rare cell type linked to distinct PDAC subtypes characterized by particularly poor survival outcomes42,43. These findings highlight the biological relevance of high-risk cells identified by SIDISH, as these subpopulations are enriched in cell types with well-documented roles in PDAC progression and unfavorable clinical outcomes. SIDISH was applied to the original PDAC scRNA-seq dataset, which included cells from both tumor and control tissues. The identified high-risk cells were predominantly confined to tumor samples, with 98.5% originating from tumor tissues (Fig. 2d). In contrast, background cells mirrored the tumor-control composition of the original dataset, with 69.6% derived from tumor samples and 30.4% from control tissues. This enrichment of high-risk cells within tumor tissues underscores SIDISH’s ability to isolate clinically meaningful subpopulations and distinguish biologically relevant signals from random noise.

a UMAP clustering of major cell types, including two subtypes of ductal cells (ductal cell types 1 and 2), fibroblasts, T cells, acinar cells, B cells, stellate cells, endothelial cells, macrophages, and endocrine cells. b UMAP visualization of 3623 high-risk cells identified by SIDISH is shown in red, while the 38,363 background cells are depicted in gray. c Bar plot showing the distribution of high-risk cells across identified cell types. Type 2 ductal cells comprise the largest proportion of high-risk cells (55.8%), followed by type 1 ductal cells (16.4%), fibroblasts (7.9%), and T cells (5.9%). d Bar plot comparing the distribution of high-risk cells between tumor-only samples and combined tumor-control samples. e Volcano plot of differential gene expression analysis between high-risk and background cells. Upregulated genes are marked in pink, while downregulated genes are shown in blue. Adjusted P values were calculated using a two-tailed Wilcoxon rank-sum test. f Violin plots comparing the expression levels of key upregulated genes between high-risk and background cells, with the middle bar indicating the median expression for each cell subpopulation. P values comparing the expression levels between both cell groups were calculated using a two-tailed Wilcoxon rank-sum test. g Functional enrichment bar plots. The SIDISH-derived marker genes are enriched in biological processes and pathways related to PDAC progression and poor survival. These genes also show a strong association with PDAC-related disease terms. h, i Kaplan–Meier survival curves for two independent bulk validation datasets: GSE224564 (h) and GSE85916 (i). High-risk patients shown in pink show clear stratification from background patients shown in gray (P = 6.67 × 10−16 and P = 9.60 × 10−19, respectively), validating the clinical relevance of SIDISH-derived marker genes. P values were calculated using the two-tailed log-rank-sum test to compare survival curves between high-risk and background patient groups.
To further evaluate the stability of SIDISH’s high-risk cell selection, we conducted a series of simulations under varying biological and technical conditions (Supplementary Figs. 1–4). First, to determine whether SIDISH learns a distributed prognostic signal rather than relying on the most abundant malignant population, we progressively removed 50% (Supplementary Fig. 1c) and 75% (Supplementary Fig. 2c) of the primary high-risk cell type, ductal type 2. In both settings, SIDISH demonstrated adaptive recalibration, up-weighting the contribution of other poor-prognosis cell types known to affect survival in PDAC, including fibroblasts and acinar cells44,45. Second, we introduced a 10% increase in dropout noise to simulate a lower-quality scRNA-seq dataset. Despite the added technical noise, SIDISH continued to detect ductal type 2 population as the largest contributor to the high-risk cells (36.5%; Supplementary Fig. 3c), indicating that noisy measurements do not obscure the survival-related signals the model learns. Finally, to test sensitivity to clinical signals, we rebalanced the bulk cohort by increasing the proportion of censored patients. SIDISH maintained its focus on patients with poor outcomes and increased the proportion of ductal type 2 cells within the high-risk population from 55.8 to 83.5% (Supplementary Fig. 4c). This behavior is consistent with the design of the Phase 2 loss function, which iteratively up-weights patients at higher risk of death, thereby emphasizing biological and clinical information from the most adverse outcomes. Overall, these simulations indicate that SIDISH consistently identifies high-risk subpopulations across changes in cell type composition, technical quality, and case-control balance, underscoring its reliability for diverse datasets.
To characterize the molecular features of high-risk cells, we performed differential gene expression analysis between high-risk and background subpopulations, identifying 323 upregulated genes (Fig. 2e). Among the upregulated genes, several have been previously associated with aggressive tumor phenotypes46,47,48,49,50, and showed higher expression in high-risk cells compared to background cells (Fig. 2f). For example, KRT17, which is overexpressed in PDAC50, and C15orf48 have been linked to poor clinical outcomes46,47,49, while CEACAM6 has been implicated in promoting cancer cell invasion and metastasis in PDAC48. These molecular expression patterns indicate that high-risk cells are enriched for genes associated with malignant behavior. Functional enrichment analysis supported these results, revealing significant disruptions in pathways related to metabolic reprogramming, particularly lipid metabolism, alongside alterations in cell cycle regulation, both hallmark features of PDAC progression51,52,53,54 (Fig. 2g). Additionally, disease enrichment analysis further showed associations with hereditary pancreatitis, chronic pancreatitis, and pancreatic cancer subtypes, indicating that the gene signatures derived from the high-risk subpopulation are aligned with known biological and clinical characteristics of PDAC.
The clinical utility of the SIDISH-derived 323 gene signature was validated using two independent PDAC bulk datasets (GSE22456455 and GSE8591656). In both datasets, Kaplan–Meier survival analysis demonstrated that patients classified as high-risk exhibited worse survival outcomes compared to those classified as background (P = 6.67 × 10−16 and P = 9.60 × 10−19, respectively) (Fig. 2h, i). These findings highlight the reproducibility and robustness of SIDISH in identifying gene biomarkers capable of stratifying patients by survival risk. To test the generalizability of the framework, we evaluated the trained SIDISH model on an independent PDAC scRNA-seq cohort57. The model identified a high-risk subpopulation of 2588 cells that was predominantly enriched within the annotated malignant cell population (22.41%), confirming that SIDISH had learned a transferable biological signature related to poor prognosis (Supplementary Fig. 5a–c). Differential expression analysis revealed 148 upregulated genes in the identified high-risk cells, many of which were linked to extracellular matrix organization and cancer-associated pathways, processes known to drive metastasis and poor prognosis58 (Supplementary Fig. 5d). Importantly, marker genes derived from the high-risk cell subpopulation stratified patients by survival with high statistical significance, not only in the original TCGA-PDAC training cohort (P = 4.38 × 10−29) but also across the two independent bulk validation datasets, GSE224564 (P = 4.51 × 10−18) and GSE85916 (P = 4.67 × 10−17) (Supplementary Fig. 5e–g).
By integrating scRNA-seq and bulk RNA-seq data, SIDISH identifies high-risk cell subpopulations and gene biomarkers associated with poor survival in PDAC. These results, validated across independent single-cell and bulk datasets, demonstrate that SIDISH can recover clinically relevant molecular signatures and provide a framework for advancing precision medicine.
SIDISH unveils high-risk cells and disease markers in breast cancer
To demonstrate the robustness and generalizability of SIDISH, we further applied SIDISH to a breast cancer (BRCA) scRNA-seq dataset59 from three BRCA subtype tumors: 11 estrogen receptor-positive (ER+), five human epidermal growth factor receptor 2-positive (HER2+), and ten triple-negative breast cancer (TNBC). Given the distinct clinical and molecular differences among these subtypes60, we focused exclusively on TNBC cells to eliminate potential confounding factors and to ensure that the downstream results reflected the unique biological and clinical features of this aggressive subtype. The TNBC dataset included 42,512 cells, which were paired with bulk RNA-seq data from 1194 TCGA-BRCA patients with survival data. Using SIDISH-derived cellular embeddings, we clustered the TNBC scRNA-seq data and annotated the resulting clusters based on pre-existing cell type labels provided in the dataset59. The clusters included cancer epithelial cells, cancer-associated fibroblasts (CAFs), perivascular-like (PVL) cells, normal epithelial cells, myeloid cells, T cells, B cells, plasmablasts, and endothelial cells (Fig. 3a). SIDISH identified 3789 high-risk cells, representing 8.91% of the total TNBC cell population (Fig. 3b and Supplementary Data 2). Among these, cancer epithelial cells dominated (65.4%), followed by CAFs (16.4%) and PVL cells (8.6%) (Fig. 3c). This dominance is consistent with the established role of cancer epithelial cells in BRCA progression, particularly their involvement in epithelial-to-mesenchymal transition (EMT)–a process critical for metastasis and therapy resistance61,62. The identification of PVL cells is also noteworthy, as these cells are implicated in immune evasion mechanisms63. Together, these findings underscore the biological and clinical significance of high-risk cells in advancing BRCA pathogenesis.

a UMAP clustering of major cell types in the TNBC scRNA-seq dataset, including cancer epithelial cells, cancer-associated fibroblasts (CAFs), perivascular-like (PVL) cells, normal epithelial cells, myeloid cells, T cells, B cells, plasmablasts, and endothelial cells. b UMAP visualization of 3789 high-risk cells identified by SIDISH is shown in red, while the 38,723 background cells are depicted in gray. c Bar plot depicting the distribution of high-risk cells across various cell types. Cancer epithelial cells account for the majority (65.4%) of high-risk cells, followed by CAFs (16.4%) and PVL cells (8.6%). d SIDISH was applied to HER2+ and TNBC datasets, revealing a higher prevalence of high-risk cells in TNBC. e Heatmap of differential gene expression analysis between high-risk and background cells. High-risk cells show significantly higher expression of marker genes (P = 9.25 × 10−9). P value highlighting the differences in expression levels between high-risk and background cell subpopulations was calculated using a one-sided Mann–Whitney U-test. f Functional enrichment analysis, including GO terms, pathways, and disease terms, highlights terms related to BRCA progression. g Kaplan–Meier survival curve for the TCGA breast cancer cohort (TCGA-BRCA). High-risk patients shown in pink show clear stratification from background patients shown in gray (P = 7.44 × 10−19). h, i Kaplan–Meier survival curves for two independent bulk validation datasets: Caldas 2007 cohort (h) and Chin 2006 cohort (i). Both plots confirm significantly worse survival outcomes for high-risk patients (P = 2.19 × 10−10 and P = 1.51 × 10−12, respectively). P values were calculated using the two-tailed log-rank-sum test to compare survival curves between high-risk and background patient groups in all three cohorts.
To assess SIDISH’s specificity, we evaluated its performance on TNBC versus HER2+ subtypes within the same scRNA-seq dataset (Fig. 3d). Consistent with TNBC’s more aggressive nature, SIDISH identified a higher proportion of high-risk cells in TNBC compared to HER2+ tumors64. For example, in Supplementary Fig. 6c, only 0.7% of high-risk cells in HER2+ tumors originated from cancer epithelial cells, compared to 65.4% in TNBC. In Supplementary Fig. 6f, the survival analysis of HER2+-specific marker genes further confirmed their limited prognostic relevance (P = 4.40 × 10−13) in comparison to TNBC-specific markers (P = 7.44 × 10−19). These findings illustrate SIDISH’s ability to integrate clinical and transcriptomic information, effectively distinguishing between BRCA subtypes and their associated outcomes.
To further elucidate the molecular mechanisms of high-risk cells, we conducted differential gene expression analysis, identifying 265 upregulated genes (Fig. 3e). Enrichment analysis highlighted critical pathways such as extracellular matrix (ECM) organization and angiogenesis, which are essential for tumor progression and metastasis (Fig. 3f)65,66,67. Disease enrichment terms, including “malignant neoplasm of the breast" and “mammary carcinoma, human," further validated the relevance of these pathways to BRCA pathogenesis. Kaplan–Meier survival analyses across the TCGA-BRCA dataset and two independent bulk cohorts–Chin 200668 and Caldas 200769–validated the prognostic significance of SIDISH-derived marker genes. In the TCGA dataset (Fig. 3g), patients with high expression of these marker genes exhibited significantly worse survival outcomes compared to those with low expression (P = 7.44 × 10−19). This trend was replicated in the Caldas 2007 dataset (Fig. 3h), where higher marker gene expression correlated with worse survival outcomes (P = 2.19 × 10−10). Similarly, the Chin 2006 dataset (Fig. 3i) corroborated these findings (P = 1.51 × 10−12), validating the clinical significance of SIDISH-derived markers.
SIDISH effectively identified high-risk cell subpopulations and clinically relevant biomarkers in BRCA. The framework prioritized biologically significant pathways, derived robust prognostic markers, and validated these findings across independent datasets, demonstrating its utility for biomarker discovery and therapeutic development in BRCA.
SIDISH uncovers high-risk cell subpopulation associated with poor survival in lung adenocarcinoma
Lung adenocarcinoma (LUAD), a leading cause of cancer-related deaths worldwide, is characterized by substantial cellular and clinical heterogeneity, posing significant challenges for understanding its progression and improving patient outcomes70,71.
To address this complexity, we applied SIDISH to a scRNA-seq dataset consisting of 4102 tumor cells71. This dataset, which lacks predefined cell type labels and exclusively contains tumor cells, provided a unique opportunity to evaluate SIDISH’s ability to identify high-risk cells within a homogeneous malignant population. SIDISH categorized these cells into ten distinct clusters, potentially representing rare and biologically unique subpopulations (Fig. 4a). By integrating bulk RNA-seq data from 572 LUAD patients in TCGA35, SIDISH identified 168 high-risk cells that were strongly associated with significantly worse survival outcomes (Fig. 4b and Supplementary Data 3).

a UMAP clustering reveals cellular diversity within the tumor microenvironment, grouping cells into distinct clusters (0–9). b UMAP visualization of 4102 cells from the LUAD scRNA-seq dataset. High-risk cells identified by SIDISH are shown in red (168 cells), while background cells are depicted in gray (3934 cells). c Bar plot quantifying the distribution of high-risk cells across identified clusters. Most high-risk cells are concentrated in clusters 3 (52.4%), 2 (22.6%), and 7 (22.6%). d Volcano plot of differential gene expression analysis between high-risk and background cells. Upregulated genes are marked in pink, while downregulated genes are shown in blue. Adjusted P values were calculated using a two-tailed Wilcoxon rank-sum test. e Violin plots comparing the expression levels of key upregulated genes between high-risk and background cells, with the middle bar indicating the median expression for each subpopulation. Genes such as LDHA, ENO1, BNIP3, NDUFA4L2, VEGFA, KIT, CA9, and WFDC2. P values comparing the expression levels of the key upregulated genes between high-risk and background cells were calculated using a one-sided Wilcoxon test. f Functional enrichment analysis, including GO terms, pathways, and disease terms, highlights terms relevant to LUAD poor survival. g, h Kaplan–Meier survival curves for the GSE157009 (g) and GSE37745 (h) independent validation datasets show significantly worse survival outcomes for patients with a higher expression level of signature genes identified by SIDISH (P = 5.40 × 10−19 and P = 1.89 × 10−21, respectively). High-risk patients in pink exhibit clear stratification from background patients in gray. P values were calculated using the two-tailed log-rank-sum test to compare survival curves between high-risk and background patient groups in both cohorts.
These high-risk cells were predominantly enriched in clusters 3, 2, and 7, underscoring the biological and clinical relevance of these subpopulations (Fig. 4c). These findings highlight SIDISH’s effectiveness in prioritizing clinically significant subpopulations, even in the absence of predefined cell type validation, and its potential to reveal insights into the molecular drivers of poor prognosis in LUAD.
Differential gene expression analysis between high-risk and background cells identified 212 upregulated genes, shedding light on molecular pathways associated with poor-survival outcomes (Fig. 4d). Among these, ENO1, LDHA, and CA9 emerged as key contributors to tumor progression. Specifically, ENO1 enhances tumor invasion and metastasis by driving glycolysis under both aerobic and anaerobic conditions72. LDHA facilitates anaerobic glycolysis in hypoxic environments, while CA9 regulates pH in the tumor microenvironment, promoting cell survival and metastasis70,73. These results underscore the biological and clinical importance of SIDISH-identified high-risk cells in LUAD progression (Fig. 4e). Functional enrichment analysis of the 212 upregulated genes confirmed their involvement in key pathways and processes, including responses to hypoxia and reduced oxygen levels, hallmarks of tumor progression and metastasis74,75 (Fig. 4f). These results align with established mechanisms of LUAD progression, further validating the clinical significance of the identified high-risk subpopulations.
To assess the prognostic relevance of these marker genes, we evaluated their clinical utility using two independent LUAD bulk datasets (GSE15700976 and GSE3774577). Kaplan–Meier survival analyses stratified patients into high-risk and background groups based on marker gene expression levels. In the GSE157009 dataset (Fig. 4g), patients with high marker gene expression exhibited significantly worse overall survival compared to those with lower expression levels (P = 5.40 × 10−19). Similar trends were observed in the GSE37745 dataset (P = 1.89 × 10−21) (Fig. 4h). These findings highlight the robust prognostic value of SIDISH-derived markers in LUAD and their potential to guide biomarker discovery and therapeutic development.
SIDISH identifies tumor-enriched high-risk cell subpopulations in spatial transcriptomics
To further evaluate the robustness and generalizability of SIDISH, we applied the framework to spatial transcriptomics (ST) data, which capture gene expression alongside spatial organization but often present challenges such as reduced resolution or limited transcript coverage.
This extension enables the investigation of spatially localized high-risk subpopulations and is particularly valuable in PDAC, where tumor-stroma interactions critically shape progression and therapeutic resistance78,79,80.
To evaluate this extension, we applied SIDISH to a high-resolution 10x Xenium PDAC ST dataset comprising 190,965 cells from a complex tumor ecosystem (Fig. 5a)81. The analysis identified 41,323 high-risk cells distributed across the spatial architecture of the PDAC tissue. A high degree of concordance was observed between the high-risk population and annotated tumor cells, as visualized in the UMAP embedding where the two populations largely overlapped (Fig. 5b). Quantitatively, 67.5% of high-risk cells were tumor cells, representing the largest contributor, followed by acinar cells (15.7%) and metaplastic cells (7.7%) (Fig. 5c). The inclusion of these additional cell types, which are implicated in pancreatic carcinogenesis, suggests that the high-risk phenotype may involve a broader set of cells within the tumor microenvironment (TME)82,83. The precision of this alignment was further quantified, showing a 74.5% overlap between high-risk and tumor cell categories, with McNemar’s test84 demonstrating a highly significant association (P = 3.54 × 10−146) (Fig. 5d), thereby supporting SIDISH’s accuracy in identifying cellular drivers of poor prognosis within their spatial context.

a Spatial atlas of annotated cell types across the pancreatic tumor section, including fibroblasts, B cells, CXCL9/10 cells, T cells, mast cells, lymphatic endothelial cells, macrophages, endothelial cells, endocrine cells, tumor cells, metaplastic cells, smooth muscle cells, acinar cells, and ductal cells. b UMAP visualization showing strong concordance between high-risk cells (41,323) and tumor cells, with high-risk tumor cells (27,887) in red, high-risk non-tumor cells in cyan, and background cells in gray. c Bar plot showing the proportion of high-risk cells by cell type, with tumor cells representing 67.5% of all high-risk cells, followed by acinar cells (15.7%) and metaplastic cells (7.7%), indicating strong tumor enrichment. d Confusion matrix quantifying overlap between high-risk and tumor cells, revealing a 74.5% concordance (McNemar’s test P = 3.54 × 10−146). P values were calculated using McNemar’s test to evaluate the association between tumor and high-risk cell labels. e Functional enrichment bar plots. The SIDISH-derived marker genes are enriched in biological processes and pathways related to PDAC progression and poor survival. f Kaplan–Meier survival curves for the independent bulk validation dataset (GSE224564). High-risk patients, shown in pink, exhibit clear stratification from background patients, shown in gray (P = 2.42 × 10−11), validating the clinical relevance of SIDISH-derived marker genes. P values were calculated using the two-tailed log-rank-sum test to compare survival curves between high-risk and background patient groups.
To further characterize these high-risk cells, we performed functional enrichment analysis on their upregulated marker genes. The analysis revealed significant enrichment of pathways central to PDAC biology, including “regulation of programmed cell death” and “focal adhesion signaling” (Fig. 5e). These pathways are associated with tumor progression, enabling cells to evade apoptosis and enhance invasion and extracellular matrix remodeling55,82,83,85,86. The enrichment of these pathways further showcases the clinical relevance of the high-risk cells identified by SIDISH.
The clinical relevance of the spatially-derived marker genes was assessed in an independent bulk PDAC dataset (GSE224564). Kaplan–Meier survival analysis showed that higher expression of the high-risk marker genes correlated with worse survival (P = 2.42 × 10−11) (Fig. 5f). This result indicates that biomarkers identified in the spatial context are also prognostic and that spatially-informed biomarkers may be predictive in non-spatial datasets.
SIDISH outperforms established methods in identifying high-risk cells and predicting survival outcomes
To comprehensively evaluate SIDISH’s performance in identifying clinically relevant cell subpopulations associated with poor survival and prognostic biomarkers, we benchmarked it against three established tools: Scissor, scAB, and DEGAS17,18,19. The evaluation utilized scRNA-seq and bulk RNA-seq datasets from LUAD, BRCA, and PDAC, focusing on SIDISH’s ability to identify high-risk cells and their associated disease-relevant biomarkers. Performance was assessed across several key metrics, including patient stratification using Kaplan–Meier survival analysis, prediction accuracy measured by the concordance index (C-Index) to evaluate alignment between predicted and observed survival rankings, and enrichment of high-risk cells for known disease marker gene sets evaluated by gene set expression scores. High-risk cells identified in the scRNA-seq datasets by SIDISH, Scissor, scAB, and DEGAS were first subjected to differential gene expression analysis. The resulting marker genes were then evaluated for their ability to stratify patients in the bulk TCGA cohorts using Kaplan–Meier survival analysis (Supplementary Data 4–6). Results, including UMAP visualizations of identified high-risk cells, volcano plots of upregulated genes, and Kaplan–Meier survival plots, are shown in Supplementary Figs. 7 (LUAD), 8 (BRCA), 9 (PDAC). The resulting distributions of the negative log-transformed P values from the two-tailed log-rank-sum tests are shown in Fig. 6a–c. Across all datasets, SIDISH consistently demonstrated better stratification performance, achieving the highest significance in survival analyses. For example, in the TCGA-LUAD bulk dataset, SIDISH achieved significantly stronger patient stratification compared to other methods (Fig. 6a). Similar results were observed in the TCGA-BRCA and TCGA-PDAC bulk datasets, where markers identified by SIDISH stratified patients more effectively based on survival than those from other benchmarked methods (Fig. 6b, c). These findings demonstrate that high-risk cells identified by SIDISH capture clinically relevant information strongly associated with poor survival than other tools.

This benchmarking analysis demonstrates SIDISH's effectiveness in identifying clinically relevant subpopulations and prognostic biomarkers compared to Scissor, DEGAS, and scAB. a–c Prognostic power of SIDISH-derived marker genes evaluated by Kaplan–Meier survival analysis in TCGA bulk cohorts for LUAD (a), BRCA (b), and PDAC (c). The y-axis represents the negative log-transformed P value from the two-tailed log-rank-sum test, where higher values indicate stronger patient stratification. Each score is based on N = 10 independent runs per method using different random seeds (technical replicates). Competing methods, including Scissor, DEGAS, and scAB, show lower performance. P values for differences in −log(P) between SIDISH and competing methods were calculated using a one-sided Mann–Whitney U-test. d–f Predictive accuracy of patient survival using SIDISH-derived signatures assessed with the concordance Index (C-Index) in the same TCGA cohorts. Each score is based on N = 10 independent runs per method using different random seeds (technical replicates). P values for differences in C-Index scores between SIDISH and competing methods were calculated using a one-sided Mann–Whitney U-test. g–i Biological relevance of SIDISH-identified high-risk cells evaluated by gene set scores for known marker genes in LUAD (g), BRCA (h), and PDAC (i) scRNA-seq datasets. Gene set scores were calculated in a single scoring pass using Scanpy’s score_genes function, with marker genes obtained from databases such as cBioPortal and OncoKB. Sample sizes (N) for LUAD were: SIDISH (168), Scissor (201), DEGAS (1181), and scAB (753). For BRCA, the sample sizes were: SIDISH (3789), Scissor (4613), DEGAS (7128), and scAB (11,422). For PDAC, the sample sizes were: SIDISH (3623), Scissor (4605), DEGAS (14,382), and scAB (6252). P values for differences in gene set scores between SIDISH and competing methods were calculated using a one-sided Mann–Whitney U-test. Boxes indicate the interquartile range (IQR; 25th–75th percentile), with the line inside each box representing the median. Whiskers extend to the 5th–95th percentiles. P values from Mann–Whitney U-tests: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; n.s. not significant. Exact P values are provided in the Source Data File.
Kaplan–Meier analysis provides a robust framework for evaluating survival stratification; however, the C-Index offers a complementary and quantitative measure of predictive accuracy by assessing the concordance between predicted and observed survival rankings in patients. To rigorously assess the predictive power of SIDISH-derived genes compared to other tools, we employed Cox survival analysis and used the C-index as the evaluation metric. In the TCGA-LUAD bulk dataset, SIDISH achieved the highest C-index values, demonstrating performance comparable to scAB (Fig. 6d). Conversely, across the TCGA-BRCA and TCGA-PDAC bulk datasets, SIDISH consistently outperformed Scissor, DEGAS, and scAB, exhibiting higher predictive accuracy and enhanced clinical relevance (Fig. 6e, f). These results highlight SIDISH’s ability to not only identify high-risk cells but also uncover gene signatures with strong prognostic value.
To further validate the clinical relevance of high-risk cells identified by each method, we analyzed the expression levels of known cancer marker gene sets. Gene sets from cBioPortal87 and OncoKB88 were utilized to calculate expression scores using Scanpy’s gene activity scoring function89. High-risk cells identified by SIDISH consistently exhibited higher expression levels of these known cancer biomarkers compared to those identified by Scissor, DEGAS, and scAB across all three scRNA-seq cancer datasets, as shown in LUAD (Fig. 6g), BRCA (Fig. 6h), and PDAC (Fig. 6i). Enrichment analysis further revealed that SIDISH-derived markers were strongly associated with key biological processes, pathways, and disease terms linked to poorsurvival outcomes. For instance, in the LUAD scRNA-seq dataset, SIDISH-derived markers were enriched in pathways associated with hypoxia response, metabolic reprogramming, and cell proliferation, which are known drivers of LUAD progression and poor survival (Supplementary Fig. 10a). Similar patterns were observed in the BRCA (Supplementary Fig. 11) and the PDAC scRNA-seq datasets (Supplementary Fig. 12). In contrast, markers identified by Scissor, DEGAS, and scAB showed weaker associations with these critical processes, further underscoring SIDISH’s ability to uncover actionable insights and clinically meaningful phenotype-related subpopulations.
Ablation study confirms data integration and iterative learning are critical for SIDISH’s performance
To quantify the contributions of its core architectural components, we conducted an ablation study of the SIDISH framework. This analysis was designed to isolate the impact of three key features: the integration of single-cell and bulk data, the injection of clinical information to guide single-cell embedding, and the dynamic iterative learning process. We compared the full SIDISH model against ablated versions, including a bulk only analysis, models without transfer learning or finetuning, and a simple variational autoencoder (VAE) trained without clinical guidance. Patient stratification performance was assessed using Kaplan–Meier survival analysis two-tailed log-rank-sum test P values, while the concordance Index (C-Index) was used to measure predictive power for patient survival. Additionally, a suite of metrics, including adjusted Rand index (ARI), normalized mutual information (NMI), completeness score, Fowlkes–Mallows index (FMI), and Silhouette score, to evaluate the quality of cell embeddings (Fig. 7). The results indicate that the fully integrated, iterative framework outperforms all ablated versions, confirming that each component is essential for biomarker discovery and clinical risk identification.

This analysis evaluates the contribution of core architectural components of SIDISH, including single-cell and bulk data integration, clinical signal injection, and iterative optimization. a–c Kaplan–Meier survival curves in TCGA-LUAD (a), TCGA-BRCA (b), and TCGA-PDAC (c) cohorts, comparing stratification based on biomarkers identified by SIDISH (top row) versus a bulk-only analysis (bottom row). SIDISH consistently yields more significant survival separation between high-risk (pink) and background (gray) patients. P values were calculated using the two-tailed log-rank-sum test. d–e Predictive accuracy of biomarkers, assessed by the concordance index (C-Index) in TCGA-BRCA (d) and TCGA-PDAC (e). SIDISH outperforms bulk-only derived biomarkers. Each score is based on N = 50 independent runs using different random seeds (technical replicates). Each boxes indicate the interquartile range (IQR, 25th–75th percentile), with the line inside each box representing the median. Whiskers extend to the 5th–95th percentiles. P value was calculated using a one-sided Mann–Whitney U-test. f–j Impact of clinical guidance on single-cell embeddings, comparing SIDISH (green) against simple VAE without survival guidance (brown). Clustering accuracy was evaluated using adjusted Rand index (ARI; f), normalized mutual information (NMI; g), completeness score (h), Fowlkes–Mallows index (FMI; i), and Silhouette score (j). Each box plot is based on N = 100 independent runs with different random seeds (technical replicates). Each boxes indicate the interquartile range (IQR, 25th–75th percentile), with the line inside each box representing the median. Whiskers extend to the 5th–95th percentiles. P value was calculated using a one-sided Mann–Whitney U-test. k Iterative optimization improves prognostic power in BRCA scRNA-seq data, with higher negative log-transformed P values (−log(P)) across training cycles. l, m Iterative refinement of embeddings further enhances clustering, with progressive improvements in NMI (l) and ARI (m) over four iterations. Each box plot is based on N = 100 independent runs across iterations. Each boxes indicate the interquartile range (IQR, 25th–75th percentile), with the line inside each box representing the median. Whiskers extend to the 5th–95th percentiles. P value was calculated using a one-sided Mann–Whitney U-test. P values from one-sided Mann–Whitney U-tests: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; n.s. not significant. Exact P values are provided in the Source Data File.
First, we tested the hypothesis that integrating the cellular resolution of scRNA-seq with the clinical context of bulk RNA-seq yields more powerful prognostic signatures than either modality alone. We compared biomarkers derived from the full SIDISH framework against those identified from a bulk only analysis. Across all three cancer cohorts, SIDISH-derived markers achieved patient stratification that was substantially more significant than bulk-derived markers (Fig. 7a–c). In the TCGA-LUAD dataset, for instance, SIDISH markers yielded a P value of 2.45 × 10−18, compared to 3.41 × 10−3 from the bulk-only approach. This enhancement was consistent for BRCA (P = 7.45 × 10−19 vs. 3.38 × 10−5) and PDAC (P = 4.06 × 10−18 vs. 1.45 × 10−4). The predictive accuracy of SIDISH was further confirmed by higher C-Index scores in both the BRCA and PDAC datasets, underscoring that the integration of single-cell biological insights is important for identifying prognostic biomarkers obscured in bulk-level analyses (Fig. 7d, e). To dissect the mechanism of this integration, we evaluated the specific roles of transfer learning and finetuning. We first compared the full SIDISH model to a version where the VAE encoder weights were not transferred to initialize the survival model. The absence of transfer learning reduced prognostic performance, with the P value in the TCGA-PDAC cohort weakening from 4.06 × 10−18 to 3.53 × 10−9 (Supplementary Fig. 13). This was corroborated by a decrease in the C-Index across all datasets, indicating a loss of predictive power (Supplementary Fig. 14). Next, we assessed a variant where the encoder weights were transferred but kept frozen (i.e., without finetuning). This also resulted in a decrease in performance, with the stratification P value in the TCGA-PDAC cohort falling to 9.56 × 10−7 (Supplementary Fig. 15) and a corresponding reduction in C-Index (Supplementary Fig. 16). These results confirm that both the initial transfer of learned single-cell representations and their subsequent finetuning with clinical data are essential processes for the model’s performance.
Furthermore, we compared SIDISH’s clinically integrated approach with a standard single-cell analysis in which marker genes were derived from annotated cell types without survival context, since none of the scRNA-seq datasets used included the necessary matched sample-level survival metadata. This comparison evaluates whether the marker genes enriched in the high-risk cell subpopulations identified by SIDISH are more informative for patient survival than marker genes derived from conventional cell type annotations. While markers from some cell types, such as ductal cell type 2 in the PDAC dataset, showed prognostic ability (P = 9.23 × 10−8), none approached the statistical significance of the markers derived from SIDISH’s high-risk population (P = 4.06 × 10−18) (Supplementary Fig. 17a). These findings highlight that incorporating clinical outcome information into single-cell analyses yields far more clinically meaningful results than analyses performed without such context. In particular, SIDISH’s ability to identify poor survival-associated subpopulations that cut across fixed cell type labels produces more relevant prognostic signals than approaches restricted to predefined annotations.
We next examined whether the clinical signals injected during training enhance the biological fidelity of cell embeddings. We compared the clustering performance of SIDISH’s clinically informed embeddings against those from a simple VAE trained without survival data (Fig. 7f–j). SIDISH’s embeddings demonstrated an improved ability to resolve ground-truth cell types, achieving a 35.18% higher ARI score, a 2.12% higher NMI, an 8.22% higher completeness score, and an 8.23% higher FMI score than the simple VAE in the BRCA dataset. Similar gains were observed in the PDAC dataset, with an 11.78% increase in ARI, 3.31% in NMI, 4.79% in completeness, and 6.74% in FMI. Although the simple VAE obtained a marginally higher Silhouette score–a measure of cluster density–its weaker performance on metrics assessing cell type specificity indicates that its embeddings do not fully capture key biological variation. These results indicate that injecting clinical information from bulk data during SIDISH’s iterative training helps shape the latent space in ways that improve biologically meaningful clustering. Notably, SIDISH embeddings outperformed state-of-the-art tools for both scRNA-seq and spatial transcriptomics analysis. As shown in Supplementary Fig. 18, clinically informed embeddings generated by SIDISH achieved higher accuracy than SCVI90, Scanpy89, scGEN91, and scGPT92, as well as spatial methods (Supplementary Fig. 19) such as SpaGCN93, SEDR94, and stLearn95. Together, these findings further establish the added value of embedding clinical information into single-cell for a more informative analysis.
Finally, we assessed the impact of SIDISH’s iterative learning framework, a key feature that distinguishes it from static, single-pass models. In the BRCA dataset, we observed that both the clinical relevance of the identified biomarkers and the quality of the cell embeddings improved progressively with each iteration (Fig. 7). The prognostic power of the marker genes, measured by the negative log-transformed P value from Kaplan–Meier analysis, increased with each cycle, indicating a refinement of the prognostic signal (Fig. 7k). Concurrently, the quality of the cell embeddings improved, with the ARI increasing by 6.67% and the NMI by 6.13% from the first to the final iteration (Fig. 7l, m). A one-sided Mann–Whitney U-test confirmed that these improvements in P values, ARI, and NMI scores were statistically significant in later iterations compared to earlier ones. These findings show that the iterative feedback loop is a robust mechanism, enabling SIDISH to dynamically learn and uncover increasingly relevant biological and clinical patterns over time.
SIDISH enables in silico gene perturbation to identify therapeutic targets and drugs
A defining feature of SIDISH is its in silico perturbation capability, which simulates gene knockouts to evaluate their impact on high-risk cells. Following perturbation, SIDISH predicts new risk scores and reclassifies cells, identifying genes that shift cells from high-risk to background states, thereby revealing potential therapeutic candidates. In the LUAD scRNA-seq dataset, gene perturbation analyses identified several potential therapeutic targets, including AKT1, VEGFA, MAP2K1, and CDK1, which achieved among the highest perturbation scores (Fig. 8a and Supplementary Data 7). Perturbing these genes significantly reduced the high-risk cell subpopulation. Notably, VEGFA and MAP2K1 are targeted by FDA-approved therapies, such as Bevacizumab96 and Trametinib97, respectively. Similarly, AKT1 and CDK1 inhibitors, including Ipatasertib98 and Dinaciclib99, are under active clinical investigation, demonstrating the translational potential of this feature of SIDISH and its derived targets. Kaplan–Meier survival plot validated the clinical relevance of top-ranking perturbed genes, revealing significantly worse survival outcomes for patients with high expression of them (P = 1.77 × 10−57), further underscoring SIDISH’s ability to detect potential therapeutic targets (Fig. 8b). Gene Ontology (GO) analysis further demonstrated that perturbing these signature genes regulated critical biological processes, including apoptosis, protein phosphorylation, and cell proliferation (Fig. 8c). These processes are essential to cancer progression and therapeutic resistance, suggesting that targeting these genes may help modulate biological processes central to aggressive malignancies, such as LUAD100,101,102.

a Bar plot ranking top therapeutic targets in LUAD scRNA-seq dataset based on perturbation scores calculated by SIDISH. Genes such as AKT1, VEGFA, MAP2K1, and CDK1 exhibit the highest perturbation scores, with FDA-approved drugs highlighted in red, such as Bevacizumab and Trametinib. AKT inhibitors such as Ipatasertib and CDK inhibitors like Dinaciclib, highlighted in purple, are under clinical investigation for LUAD treatment. b Kaplan–Meier survival analysis of TCGA-LUAD patients demonstrates significantly worse survival outcomes for high-risk patients, characterized by higher expression of the top perturbed genes in pink, compared to background patients with lower expression levels in gray (P = 1.77 × 10−57). P values were calculated using the two-tailed log-rank-sum test. c Gene Ontology (GO) analysis reveals biological processes impacted by gene perturbation. d Heatmaps of single-gene (single gene knockout) and combinatorial-gene (dual-gene knockout) knockouts display perturbation scores for top perturbed genes. e UMAP visualizations show the effects of perturbing CDK1 and MAP2K1, as well as gene combinations CDK1 and BTG2, and MAP2K1 and FN1, demonstrating a higher reduction of high-risk cell subpopulations when perturbing a combination of two genes compared to single-gene perturbation. The color legend indicates: gray for unchanged background cells (background to background), blue for high-risk cells transitioned to background cells (high-risk to background), purple for background cells transitioned to high-risk cells (background to high-risk), and red for persistent high-risk cells (high-risk to high-risk). f PDAC spatial map (10x Xenium) showing high-risk cells before perturbation.g PDAC spatial map after EGFR perturbation, showing a reduction of high-risk cell subpopulation. h Top perturbed genes in the spatial dataset highlight clinically actionable targets, including EGFR (Erlotinib), CXCR4 (BL8040/Motixafortide), CDK1 (Dinaciclib), and MDM2 (Brigimadlin). i Kaplan–Meier survival analysis of TCGA-LUAD patients shows significantly worse survival outcomes for high-risk patients, characterized by higher expression of the top perturbed genes in the spatial dataset, compared to background patients with lower expression levels (gray) (P = 1.55 × 10−22). P values were calculated using the two-tailed log-rank-sum test to compare survival curves between high-risk and background patient groups.
To investigate synergistic therapeutic opportunities, SIDISH performed combinatorial perturbation analyses by simultaneously knocking out gene pairs (Fig. 8d). Dual perturbations often produced stronger effects than single-gene knockouts. For instance, co-perturbation of AKT1 and CCND1 resulted in a more pronounced reduction in high-risk cells, consistent with their roles in the PI3K/AKT signaling pathway and cell cycle regulation103,104. UMAP visualizations provided further insights into cellular state transitions, demonstrating substantial shifts in high-risk cells to background states following gene perturbations (Fig. 8e). For example, individual perturbation of CDK1 significantly reduced the number of high-risk cells, resulting in a perturbation score of 77.87, emphasizing its role in oncogenic signaling and tumor progression105,106. Similarly, targeting MAP2K1 achieved a perturbation score of 79.76, highlighting its essential role in sustaining malignant phenotypes107,108. Interestingly, combinatorial targeting of CDK1 and BTG2, as well as MAP2K1 and FN1, resulted in a perturbation score of 85.11 and 85.71, respectively, underscoring the potential of combination therapies. The UMAP visualization categorized cell transitions into four states: background to background in gray, high-risk to background in blue, background to high-risk in purple, and high-risk to high-risk in red. Predominantly blue regions across perturbations reflect the effectiveness of targeting these genes in reprogramming high-risk cells, while minimal purple regions underscore the specificity of SIDISH predictions in preventing malignant transformation. Persistent red regions indicate resistant subpopulations, suggesting the need for complementary therapeutic strategies.
SIDISH demonstrated its versatility beyond LUAD by effectively identifying therapeutic targets in other cancer types. In PDAC, the top therapeutic targets identified based on predicted gene perturbation scores included SPARC and PLK1. These genes are strongly associated with Nab-paclitaxel (Abraxane)109,110, an FDA-approved drug, and Volasertib111, which is currently undergoing clinical trials (Supplementary Fig. 20 and Supplementary Data 8). Similarly, when applied to BRCA scRNA-seq, key targets such as KDR, IGF1R, and NOTCH1 were identified. These genes are targeted by drugs under active investigation, including Apatinib (KDR)112,113, Atezolizumab (IGF1R)114, and NADI-351 (NOTCH1)115 (Supplementary Fig. 21 and Supplementary Data 9).
To further assess the adaptability of the framework, we applied the in silico perturbation module in a spatial context using the PDAC spatial dataset. Perturbation of EGFR resulted in a marked reduction of high-risk cells (Fig. 8f, g). Among the top-ranked perturbed genes (Fig. 8h), several were clinically actionable, including EGFR, the target of the FDA-approved PDAC therapy Erlotinib116, as well as CXCR4, CDK1, and MDM2, which are targets of inhibitors (Motixafortide, Dinaciclib, and Brigimadlin) currently in clinical trials117,118,119. Importantly, Kaplan–Meier survival analysis confirmed the clinical relevance of these targets, showing that patients with high expression of the top perturbed genes had significantly worse survival outcomes compared to those with lower expression (P = 1.55 × 10−22; Fig. 8i). Together, these results validate the robustness and translational potential of SIDISH’s perturbation feature in identifying actionable therapeutic targets across multiple cancer types and data modalities.
SIDISH enables patient-specific high-risk cell analysis and therapeutic insights for precision medicine
Precision medicine requires detailed patient-level biological information to enable the development of targeted therapies. Using the SIDISH framework, we performed high-resolution analyses of individual patients and identified substantial heterogeneity in high-risk cell populations across multiple cancers, underscoring the influence of unique tumor microenvironments on therapeutic vulnerabilities. In the PDAC dataset of 24 patients, high-risk cells were predominantly derived from ductal cell types 1 and 2, with contributions from acinar cells, fibroblasts, and B cells. However, the proportions of these cell types varied markedly across patients, reflecting distinct tumor susceptibilities. For instance, patients with a predominance of ductal high-risk cells may exhibit epithelial vulnerabilities120, whereas those with enriched B cells suggest potential for immune-modulating therapies121 (Fig. 9a). To further assess whether the observed inter-patient variability in high-risk cell composition reflects poor survival-related biological signals rather than patient-specific noise, we examined the expression of each patient’s identified marker genes across the high-risk populations of other individuals. As shown in Supplementary Fig. 22, the marker genes from one patient were not confined to that individual but were consistently expressed across multiple others. This indicates that SIDISH captures a poor survival phenotype rather than patient-specific artifacts, and reinforces the robustness of the framework in identifying clinically meaningful molecular patterns. In the BRCA dataset of ten patients, high-risk cells were largely composed of cancer epithelial cells, although significant variability was observed in patients CID3946, CID4523, and CID44041, where T cells and myeloid cells substantially contributed to the high-risk landscape (Fig. 9b). These findings underscore the cellular diversity between tumors and highlight the necessity of patient-specific therapeutic strategies122,123.

a, b Composition of high-risk cells across patients in the PDAC (24 patients) and BRCA TNBC (ten patients) scRNA-seq datasets, highlighting variability in cell type contributions. c Heatmap comparing the marker gene expression profiles of the top-ranking PDAC patient (T20) and the lowest-ranking patient (T3). d Box plots showing gene set scores for known PDAC markers in patients T20 and T3. Gene set scores were calculated for each cell in a single scoring pass. Sample sizes were T20 (N = 482 cells) and T3 (N = 1317 cells). Boxes indicate the interquartile range (IQR; 25th–75th percentile), with the line inside each box representing the median. Whiskers extend to the 5th–95th percentiles. P value was calculated using a one-sided Mann–Whitney U-test. e, f Kaplan–Meier survival curves stratifying TCGA-PDAC patients based on disease markers derived from the top-ranking patient T20 (e) and the lowest-ranking patient T3 (f). P values were calculated using the two-tailed log-rank-sum test to compare survival curves between high-risk and background patient groups. g Heatmap comparing the marker gene expression profiles between the top-ranking patient (CID3946) and the lowest-ranking patient (CID4523). h Box plots showing gene set scores for known BRCA markers in patient CID3946 and CID4523. Sample sizes were CID3946 (N = 774 cells) and CID4523 (N = 1754 cells). Boxes indicate the interquartile range (IQR; 25th–75th percentile), with the line inside each box representing the median. Whiskers extend to the 5th–95th percentiles. P value was calculated using a one-sided Mann–Whitney U-test. i, j Kaplan–Meier survival curves stratifying TCGA-BRCA patients based on disease markers derived from the top-ranking patient CID3946 (i) and the lowest-ranking patient CID4523 (j). P values were calculated using the two-tailed log-rank-sum test to compare survival curves between high-risk and background patient groups. k Heatmap displaying single-gene perturbation scores for PDAC patients. l UMAP visualizations illustrating the effects of SPARC perturbation in PDAC for patients T3 (top), T8 (middle), and T20 (bottom). m Heatmap of single-gene perturbation scores for BRCA patients. n UMAP visualizations showing the impact of CTLA4 perturbation in BRCA for CID4495 (top), CID3946 (middle), and CID4523 (bottom). o Heatmap of combinatorial in silico perturbation scores in PDAC patients. p UMAP visualizations for PDAC patients showing effects of combinatorial perturbation of SPARC and SLC12A2 in T3 (top), T8 (middle), and T20 (bottom). q Heatmap of combinatorial perturbation scores in BRCA patients. r UMAP visualizations for BRCA patients showing combinatorial perturbation effects of CTLA4 and IL6 in CID4495 (top), CID3946 (middle), and CID4523 (bottom). The color legend in the UMAPs indicates: gray for unchanged background cells (background to background), blue for high-risk cells transitioned to background cells (high-risk to background), purple for background cells transitioned to high-risk cells (background to high-risk), and red for persistent high-risk cells (high-risk to high-risk). ST represents the perturbation score. Significance thresholds: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; n.s. not significant. Exact P values are provided in the Source Data File.
To establish the clinical relevance of high-risk cells, we analyzed patients with the highest and lowest proportions of these subpopulations. In the PDAC scRNA-seq dataset, patient T20 exhibited the highest proportion of high-risk cells (31.53%), while T3 had the lowest (1.52%) (Supplementary Fig. 23). Differential gene expression analysis revealed significant transcriptomic differences between these two patients, with marker genes from T20 showing markedly higher expression levels than in T3 (Fig. 9c). Furthermore, high-risk cells from patient T20 were significantly more enriched in known PDAC marker genes derived from cBioPortal87 and OncoKB88 compared to cells from patient T3 (P < 0.0001; Fig. 9d). Importantly, Kaplan–Meier survival analyses of TCGA-PDAC bulk RNA-seq data demonstrated that marker genes derived from T20 stratified TCGA-PDAC patients into groups with significantly worse survival outcomes compared to those stratified by T3-derived marker genes (P = 1.78 × 10−10 vs. P = 5.48 × 10−2; Fig. 9e, f). Similar results were observed in the BRCA scRNA-seq dataset. Patient CID3946 exhibited the highest proportion of high-risk cells (15.5%), while CID4523 had the lowest (2.39%) (Supplementary Fig. 24). Differential gene expression analysis revealed that marker genes derived from CID3946 were poorly expressed in CID4523 (Fig. 9g). High-risk cells from patient CID3946 exhibited significantly higher expression of known BRCA biomarkers, also derived from cBioPortal87 and OncoKB88, compared to CID4523 (P < 0.0001; Fig. 9h). Kaplan–Meier survival analysis further demonstrated that CID3946 signature genes stratified TCGA-BRCA patients into groups with significantly worse survival outcomes compared to CID4523 signature genes (P = 1.64 × 10−21 vs. P = 1.31 × 10−13; Fig. 9i, j). These findings underscore that patients with higher proportions of high-risk cells exhibit increased expression of malignant markers, which are strongly associated with worse survival outcomes. This demonstrates that high-risk cells identified by SIDISH serve as robust indicators of poor survival, even at the individual patient level, supporting its potential for advancing precision oncology.
SIDISH’s ability to model patient-specific therapeutic responses was evaluated using in silico perturbation analyses. In the PDAC scRNA-seq dataset, perturbation of SPARC, a gene involved in tumor progression124 and a carrier for the FDA-approved drug Abraxane125, revealed striking inter-patient variability (Fig. 9k and Supplementary Fig. 25). For patient T3, SPARC perturbation caused a modest reduction in high-risk cells, whereas T8 and T20 exhibited substantial transitions of high-risk cells to background cells, indicating therapeutic potential (Fig. 9l and Supplementary Fig. 26). Similarly, in the BRCA scRNA-seq dataset, perturbation of CTLA4, an immune checkpoint regulator126, demonstrated various patient-level effects. CID3946 and CID4523 exhibited near-complete transitions of high-risk cells to background cells, while CID4495 showed only modest reductions (Fig. 9m, n and Supplementary Figs. 27, 28). These results emphasize the necessity of personalized therapeutic strategies tailored to the unique molecular landscapes of individual patients.
Given this observed variability, we evaluated whether the perturbation effect in each patient is influenced by their cell type heterogeneity. Using Spearman’s rank correlation22, we assessed the relationship between the abundance of each specific cell type and perturbation scores (ST) after SPARC knockout in PDAC patients (Supplementary Fig. 29). Correlations were generally weak and often statistically insignificant across most cell types. For example, the most enriched cell type in high-risk populations, ductal cell type 2, displayed negligible correlation (ρ = −0.32, P = 0.12). To further test whether global heterogeneity contributes to these effects, we compared patient-level perturbation scores with mean cLISI scores, a measure of inter-patient cell state diversity127. As shown in Supplementary Fig. 30, no significant correlation was observed (ρ = 0.072, P = 0.74). Together, these results indicate that reductions in high-risk cells after perturbation reflect the intrinsic biological impact of the knockout rather than baseline differences in cell type composition, reinforcing that SIDISH captures clinically relevant and patient-specific perturbation effects.
Combination therapies are increasingly recognized as a cornerstone of effective cancer treatment due to their ability to simultaneously target multiple molecular pathways128. This approach disrupts the complex mechanisms driving tumor progression and mitigates resistance development, a common limitation of single-drug therapies129. In this context, SIDISH simulated patient-specific combinatorial-gene perturbations to identify more efficacious therapeutic options tailored to individual tumor profiles. In PDAC, dual perturbation of SPARC and SLC12A2 resulted in substantial reductions in high-risk cells in patient T3, while patients T8 and T20 exhibited limited responses (Fig. 9o, p and Supplementary Fig. 31). Similarly, in BRCA, co-targeting CTLA4 and IL6 significantly reduced high-risk cells in patients CID4495 and CID3946, whereas CID4523 remained largely unresponsive (Fig. 9q, r and Supplementary Fig. 32). These results underscore SIDISH’s ability to screen for patient-specific combinatorial therapeutic strategies, effectively addressing inter-patient heterogeneity and advancing the field of precision oncology.
Discussion
SIDISH represents a robust advancement for precision medicine by providing a deep iterative neural framework to integrate scRNA-seq, bulk RNA-seq, and spatial transcriptomics data. Using a semi-supervised, iterative deep-learning approach, SIDISH bridges cellular resolution with clinical breadth, enabling the precise identification of high-risk cell subpopulations and clinically relevant biomarkers. This framework addresses limitations of existing approaches by iteratively refining predictions and leveraging nonlinear relationships between gene expression and patient survival outcomes. Applied to multiple cancer types, including PDAC, BRCA, and LUAD, and extended to spatial transcriptomics, SIDISH has demonstrated its capacity to uncover actionable insights that can drive improved diagnostic and therapeutic strategies.
SIDISH introduces a suite of methodological and translational components that collectively establish it as a framework capable of integrating single-cell, bulk, and spatial transcriptomics data. These contributions are summarized below. First, SIDISH overcomes the linearity and noise-sensitivity of existing association methods. Prior frameworks, such as Scissor and scAB, employ Pearson correlation or sparse regression to connect single-cell transcriptional states with bulk-level clinical outcomes. These linear methods are inherently sensitive to noise and incapable of capturing the nonlinear dependencies that characterize gene regulation and disease progression. While DEGAS attempted to address nonlinearity by employing multitask neural networks, its reliance on a single shared latent space compresses bulk and single-cell data into one representation, reducing either clinical breadth or cellular resolution. In contrast, SIDISH employs a transfer learning paradigm that preserves the full heterogeneity of single-cell data while anchoring predictions to bulk RNA-seq and survival outcomes. This design allows SIDISH to simultaneously retain single-cell granularity and patient-level clinical relevance, enabling the discovery of high-risk subpopulations and survival-associated genes that competing methods may fail to detect. Second, SIDISH introduces an iterative refinement strategy that dynamically strengthens biological and clinical signals. Existing methods operate in a single pass, deriving associations between cells and outcomes without re-evaluating the robustness of their predictions. SIDISH uniquely implements a cyclical learning framework, in which cell embeddings, patient stratification, and biomarkers are updated across multiple iterations. With each cycle, survival-associated signals are sharpened, leading to progressively improved concordance indices and more stable marker discovery. This iterative learning acts as an internal self-correction mechanism, filtering out noise and enhancing reproducibility. In benchmarking analyses, iterative refinement improved both embedding quality and prognostic significance, demonstrating its central role in SIDISH’s overall performance. Third, SIDISH extends phenotype-associated population discovery into spatial transcriptomics. Although several recent methods (e.g., SpaGCN, stLearn, SEDR, and BayesSpace130) can cluster cells or spots spatially, none are designed to directly identify clinically relevant, outcome-associated subpopulations in a spatial context. SIDISH fills this gap by projecting high-risk subpopulations into tissue architecture, revealing that ~67.5% of these cells correspond to malignant tumor cell types while maintaining spatial fidelity. This enables the detection of poor survival phenotypes at the single-cell level and facilitates their visualization and interpretation within the tumor microenvironment. Fourth, SIDISH incorporates an in silico perturbation module for therapeutic discovery. Beyond identifying high-risk subpopulations, SIDISH simulates the knockout of individual genes or combinations thereof to predict their effects on cellular risk states. This feature systematically uncovers potential therapeutic targets by quantifying shifts in high-risk cells toward background states. Importantly, SIDISH recovered well-established targets such as VEGFA (Bevacizumab) and MAP2K1 (Trametinib), validating its predictive accuracy. It also identified additional candidates such as SPARC, PLK1, CDK1, and BTG2, several of which align with drugs in active clinical trials. Combinatorial perturbation analyses revealed synergistic effects. For instance, dual targeting of MAP2K1 and FN1 achieved stronger reductions in high-risk subpopulations than either alone. This module, therefore, extends SIDISH from an analytic framework into a prioritization engine for translational interventions. Fifth, SIDISH advances precision oncology by linking high-risk subpopulations to patient survival at scale. In multiple cohorts (PDAC, BRCA, and LUAD), patients with higher proportions of SIDISH-defined high-risk cells exhibited consistently worse outcomes. These populations thus serve as robust biomarkers of prognosis, offering a new dimension of patient stratification beyond bulk expression signatures or predefined cell types. The framework also validated its generalizability across cancers, as survival associations derived from one dataset remained predictive in independent cohorts. By enabling risk assessment at both cellular and patient levels, SIDISH demonstrates how integrated modeling can directly inform prognosis and patient management. Sixth, SIDISH enables individualized therapeutic predictions by modeling inter-patient variability in perturbation responses. Cancer heterogeneity often limits the efficacy of one-size-fits-all treatments, underscoring the need for tools that predict patient-specific vulnerabilities. SIDISH addresses this challenge by simulating perturbations within each patient’s high-risk subpopulation, revealing striking variability in therapeutic responses. For example, perturbation of SPARC nearly eliminated high-risk cells in some PDAC patients but produced only modest effects in others, while in BRCA, perturbation of CTLA4 demonstrated differential efficacy across individuals. These results highlight the potential of SIDISH to support personalized treatment design, by matching therapies to the molecular patterns of each patient’s tumor microenvironment.
While SIDISH offers significant advancements, it also faces limitations that warrant future investigation. Its computational requirements may limit accessibility for users without high-performance computing resources, particularly in large-scale studies involving millions of cells or thousands of bulk RNA-seq samples. Optimizing SIDISH for scalability could address these challenges. Additionally, integrating other omics modalities, such as ATAC-seq, proteomics, and imaging, would expand its ability to capture broader biological insights and further enhance its translational applications. Finally, exploring SIDISH’s utility beyond oncology, in diseases such as autoimmune disorders or infectious diseases, would broaden its impact on precision medicine.
SIDISH provides an integrated framework that combines high-resolution scRNA-seq data with the clinical context of bulk RNA-seq and spatial transcriptomics. By iteratively refining survival associated features and incorporating an in silico perturbation module, SIDISH links cellular-level heterogeneity to patient-level outcomes and enables systematic evaluation of therapeutic targets. Beyond data integration, the framework supports the prioritization of single-gene and combinatorial perturbation strategies while accounting for inter-patient variability in tumor susceptibility and treatment response. Through unified modeling of biomarker discovery, therapeutic target prioritization, and patient-specific insights, SIDISH provides a comprehensive and adaptable approach for precision medicine across multiple cancer types and data modalities.
Methods
Data processing and preparation
Raw single-cell RNA sequencing (scRNA-seq) count data were processed using the Scanpy Python library (v1.9.1)89. An initial quality control filter was applied to the raw count matrix, in which cells with fewer than 200 detected genes and genes expressed in fewer than three cells were excluded. The filtered matrix was normalized to 10,000 total counts per cell and log-transformed using the log1p function. Highly variable genes (HVGs) were identified from the normalized data using the highly_variable_genes function with default parameters. The original, unnormalized count matrix, filtered to retain only the selected HVGs, was used as the final input for the SIDISH model, as its VAE component requires raw counts for the Zero-Inflated Negative Binomial (ZINB) likelihood. Additionally, to correct for inter-individual variability in multi-sample datasets, batch correction using the Harmony algorithm (v0.1.7)127 and the Combat algorithm were131 were incorporated into the data processing pipeline. However, no batch correction was applied to any of the datasets used in this study.
Spatial transcriptomics (ST) data were processed following the same quality control strategy as scRNA-seq data. Quality control filters were applied to remove cells with fewer than 200 detected genes and genes expressed in fewer than three cells. Spatial coordinates were retained to enable both model training and downstream visualization of high-risk and background cell populations across tissue sections. Due to the lower gene coverage typical of targeted spatial platforms, all genes captured in the raw count matrix were retained.
Bulk RNA-seq data for The Cancer Genome Atlas (TCGA) cohorts were obtained as preprocessed count matrices. To create a consistent feature space for model integration, only the intersection of genes present in both the final scRNA-seq and bulk count matrices was retained for all subsequent analyses. The same strategy was applied to spatial transcriptomics datasets to maintain consistency across all modalities. Clinical metadata, including survival time and event status, were merged with the corresponding bulk expression data. Patients with missing clinical survival information or incomplete gene expression profiles were excluded. No imputation of missing survival outcomes or gene expression values was performed.
To maintain a consistent feature space for validation, all external single-cell and bulk datasets were harmonized with the training data. The intersection of genes between the training and validation sets was retained, and any genes from the training set that were missing in a validation set were added with expression values of zero.
SIDISH model description
Phase 1: Extraction of cellular heterogeneity via a variational autoencoder
In Phase 1, SIDISH performs unsupervised extraction of cellular heterogeneity using a Variational Autoencoder (VAE). Applied to scRNA-seq datasets X, consisting of M cells and N genes, the VAE FS compresses high-dimensional gene expression data into a lower-dimensional latent space Z, capturing biologically meaningful patterns while also enabling the generation of hypothetical new samples.
The VAE consists of two main components: an encoder ES( ⋅ ; ϕ), parameterized by weights ϕ, and a decoder DS( ⋅ ; ψ), parameterized by ψ. The encoder maps each input expression profile Xi to a latent variable Zi, while the decoder reconstructs the input by generating parameters of a Zero-Inflated Negative Binomial (ZINB) distribution. Specifically, the decoder outputs the mean expression ρψ(Zi), the dispersion θψ, and the dropout probability πψ(Zi). The ZINB distribution is particularly suited to single-cell RNA-seq data, as it models both overdispersion and sparsity due to dropout events.
Training optimizes the weighted Evidence Lower Bound (ELBO):
In this loss function, M represents the total number of cells in the scRNA-seq dataset. Each cell is described by Xi, a vector representing the gene expression values for the i-th cell. The latent variable Zi is sampled from the approximate posterior distribution qϕ(Zi ∣ Xi), which is parameterized by the encoder ES. The prior distribution \({{{\mathcal{N}}}}({{{\bf{0}}}},{{{\bf{I}}}})\) is a standard multivariate Gaussian with mean 0 and identity covariance matrix I. The reconstruction term pψ (Xi ∣ ρψ (Zi), πψ (Zi), θψ) represents the ZINB likelihood of the observed gene expression vector given the decoder outputs. The weight vector Wi, representing the gene-level weights for cell i, is initially set to 1 in this phase. These weights are derived from row i of the weight matrix W (details provided in Phase 4) and assign clinical significance to each gene for cell i. This ensures that biologically and clinically relevant features are prioritized in the reconstruction loss. The encoder parameters ϕ and decoder parameters ψ are optimized jointly to balance latent space regularization with accurate data reconstruction, ensuring robust extraction of cell-level biological features. After convergence, the optimized encoder weights are denoted as \(\widehat{\phi }\). This ensures the robust extraction of meaningful representations of cellular heterogeneity at the cellular level, providing a strong foundation for the subsequent phases of SIDISH’s framework.
Spatial Transcriptomics extension
To extend SIDISH for spatial transcriptomics (ST) data, we introduced a Graph Convolutional Network (GCN) layer between the encoder and latent space of the VAE. This enables spatial relationships to be incorporated into the latent embeddings while preserving the transfer learning design (details provided in Phase 2). The spatial graph was constructed by treating each cell as a node and connecting it to its K-nearest neighbors (KNN) based on Euclidean distance in the spatial coordinates. The adjacency matrix \({{{\bf{A}}}}\in {{\mathbb{R}}}^{M\times M}\) encodes these connections, with entries defined as:
where \({{{{\mathcal{N}}}}}_{k}(i)\) denotes the set of k-nearest neighbors of cell i, and d(i, j) is the Euclidean distance between cells i and j. The adjacency matrix was symmetrically normalized as \(\widetilde{{{{\bf{A}}}}}={\widetilde{{{{\bf{D}}}}}}^{-1/2}({{{\bf{A}}}}+{{{\bf{I}}}}){\widetilde{{{{\bf{D}}}}}}^{-1/2},\) where \(\widetilde{{{{\bf{D}}}}}\) is the diagonal degree matrix of (A + I). Given an input feature matrix H(0) = ES(X; ϕ), the GCN propagation rule is defined as \({{{{\bf{H}}}}}^{(1)}=\varphi (\widetilde{{{{\bf{A}}}}}\,{{{{\bf{H}}}}}^{(0)}\,{{{{\bf{W}}}}}_{{{{\rm{GCN}}}}})\), where \({{{{\bf{W}}}}}_{{{{\rm{GCN}}}}}\) is the trainable weight matrix of the GCN, and φ( ⋅ ) is a nonlinear activation function (ReLU). The resulting matrix H(1) replaces the latent representation Z in the standard VAE, ensuring that the learned embeddings incorporate both transcriptomic variation and spatial proximity. This design allows the SIDISH framework to jointly capture gene-level, cell-level, and spatial-level information for robust survival prediction.
Phase 2: Survival prediction using transfer learning
In Phase 2, SIDISH links the cell-level information learned in Phase 1 to patient survival outcomes by training a supervised deep Cox regression model FC. This model is trained on bulk RNA-seq data B, represented as a matrix with P patients (rows) and N genes (columns), together with clinical outcomes: the vector of event times T and event indicators δ (1 for observed events, 0 for censored). The output is a set of predicted survival risk scores Y.
Unlike classical Cox models, which assume a linear relationship between gene expression and hazard, the deep Cox regression model leverages nonlinear feature transformations to capture complex dependencies among genes. A central innovation of SIDISH is its use of transfer learning: the encoder trained in Phase 1, parameterized by the learned weights \(\widehat{\phi }\), is used to initialize the feature extraction layers of the Cox network. This ensures that the model starts from a biologically informed representation of cellular heterogeneity rather than random initialization. Importantly, the transferred encoder is not frozen; instead, its parameters are finetuned jointly with additional hidden layers, allowing the model to adapt the single-cell features to patient-level outcomes.
Formally, for patient j with bulk expression profile \({{{{\bf{B}}}}}_{j}\in {{\mathbb{R}}}^{N}\), the predicted risk score is:
where \({E}_{S}(\cdot ;\,\widehat{\phi })\) denotes the transferred encoder initialized with weights from Phase 1, \({F}_{{C}_{{{{\rm{hidden}}}}}}(\cdot ;\,\omega )\) represents the additional hidden layers with parameters ω, and the full parameter set is \(\Theta=\{\widehat{\phi },\omega \}\). During training, both \(\widehat{\phi }\) and ω are optimized, enabling finetuning of the transferred encoder.
Training minimizes a weighted negative partial log-likelihood adapted from DeepSurv132:
where δj is the event indicator, ensuring that only patients with observed events contribute to the optimization. The risk set \({\mathfrak{R}}({T}_{j})\) includes all patients still at risk at the time of patient j’s event (Tj). A distinctive feature of this formulation is the incorporation of the patient-specific weight vj, derived from the weight vector v. This weight dynamically prioritizes high-risk patients during training by reducing the relative contribution of background patients to the loss. By dividing the cumulative risk term by vj, the influence of background patients is attenuated, thereby amplifying emphasis on clinically adverse outcomes.
This weighted framework enables the model to capture nonlinear relationships between gene expression and survival outcomes while aligning optimization with biologically and clinically significant high-risk patients. This design is particularly advantageous in bulk RNA-seq studies characterized by diverse patient populations and varying survival risk distributions.
Phase 3: Risk prediction and stratification using predicted survival scores
In Phase 3, we identify high-risk cell and patient populations using the survival risk scores predicted by the trained deep Cox regression model (FC). These scores are denoted as YX for cells and YB for patients. This phase transfers the learned clinical information back to the cellular level, enabling a bidirectional exchange of insights between single-cell and bulk data. Risk scores were modeled using parametric survival distributions to establish a statistical basis for stratification. While the Weibull distribution was chosen as the default due to its flexibility and widespread use in survival analysis29,133,134, we performed a goodness-of-fit evaluation to confirm its suitability compared to alternative distributions like the Gamma135,136 and Exponential137. The model fit was assessed using complementary metrics such as the Akaike Information Criterion (AIC), the Bayesian Information Criterion (BIC), the Kolmogorov–Smirnov (KS) statistic, the coefficient of determination (R2), and quantile–quantile (Q–Q) plots for direct visual assessment of alignment between empirical and theoretical quantiles. Across most cancer datasets, the Weibull distribution consistently provided the best overall fit (Supplementary Figs. 33, 34 and Supplementary Tables 1–3). To enhance flexibility and generalizability, SIDISH implements an optional data-driven procedure that compares the Weibull, Gamma, and Exponential fits during the first iteration. The optimal distribution is then selected based on the same metrics and fixed for all subsequent iterations. Using the SciPy Python library, the Weibull distribution was fitted by maximizing the log-likelihood function, yielding parameter estimates for scale (λ) and shape (u). Separate distributions were derived for cells and patients, ensuring a tailored representation of risk across the two sets of data.
Cells and patients were classified as high-risk or background using the hyperparameter Pcut, which defines an upper-tail cutoff on the fitted distribution of risk scores. Specifically, Pcut corresponds to the cumulative distribution function (CDF) threshold, retaining the top 100 × (1 − Pcut)% of scores. For example, Pcut = 0.95 selects the top 5% of cells or patients with the highest risk, analogous to a one-sided significance threshold of P < 0.05. The CDF of the Weibull distribution was computed using the estimated parameters, \(\lambda \cdot {(-{{{\rm{ln}}}}(1-{P}_{cut}))}^{1/u}\)). Classification was then performed using this labeling function:
Here, FC(Xi) denotes the predicted risk score for cell i, while \(\lambda {\left[-ln\left(1-{P}_{{{{\rm{cut}}}}}\right)\right]}^{1/u}\) defines the cutoff value derived from the fitted Weibull distribution. If a cell’s risk score exceeds this threshold, the indicator function \({\mathbb{I}}(\cdot )\) returns 1 and the cell is labeled as high-risk. Conversely, if the score is less than or equal to the threshold, the indicator returns 0, and the cell is labeled as background. This binary assignment–0 for background and 1 for high-risk–was applied identically to both cell-level and patient-level risk scores, ensuring consistent stratification across modalities.
Phase 4: Iterative weight updates
Calculation of Gene Weights for Individual Cells
In Phase 4, Shapley additive explanations (SHAP)30,31 are used to quantify the contribution of each gene to the classification of high-risk (class 1) versus background (class 0) cells. These contributions are represented by a weight matrix \({{{\bf{W}}}}\in {{\mathbb{R}}}^{M\times N}\), where M is the number of cells and N is the number of genes. Each element Wij represents the importance of gene j for cell i in the classification process. Initially, all entries of W are set to 1 and are refined iteratively during training. A binary random forest classifier from the Scikit-learn Python library138 is trained using the cell labels generated in Phase 3. The SHAP values are then computed using a trained classifier. Because SHAP values are unbounded, they are normalized using a modified sigmoid function σ*, defined as \({\sigma }^{*}(x)=\frac{1}{1+{e}^{-ax}}\). Where the hyperparameter a controls the steepness of the curve, increasing the sensitivity to smaller values. This transformation maps SHAP values to the range [0, 1], ensuring numerical stability and interpretability. Only high-risk cells (HRC) contribute to the weight updates, with background cells excluded from the refinement and always set to 0.
At iteration r, the weight matrix is updated as:
where 1 < r < τ, and τ denotes the total number of iterations. Here, W(r−1) represents the weight matrix from the previous iteration, and \(\Phi \in {{\mathbb{R}}}^{M\times N}\) is the SHAP value matrix. Each element Φij reflects the contribution of gene j for cell i. The indicator matrix \({\mathbb{I}}\) identifies elements where Φi, j > 0 and the cell i belongs to the high-risk group (HRC), ensuring that only relevant SHAP values are included in the updates. The Hadamard (element-wise) product (⊙) combines this filtered subset with the sigmoid-transformed SHAP values, σ*(Φ), ensuring biologically meaningful weight adjustments.
This iterative refinement ensures Wr dynamically encodes the evolving importance of genes across cells while prioritizing high-risk classifications. To maintain stability and prevent overemphasis on individual genes, weights are capped at a maximum value of 2 after each update.
Assigning weights to patients
A patient-specific weight vector \({{{\bf{v}}}}\in {{\mathbb{R}}}^{P}\) is constructed and iteratively refined to capture the relative survival risk of each patient based on bulk RNA-seq data. The vector is initialized as a uniform vector of ones, v(0) = 1P = [1, 1, …, 1], representing an uninformative prior with no initial distinction among patients. Over successive iterations, v is updated using model-predicted survival risks, progressively encoding patient-specific clinical relevance.
At iteration r, the update rule is defined as:
where v(r) represents the weight vector at iteration r, v(r−1) is the weight vector from the previous iteration, and σ(YB) is the sigmoid-transformed vector of predicted risk scores YB. The indicator function\({\mathbb{I}}(i\in H{R}_{P})\) identifies whether a patient belongs to the high-risk group (HRP), assigning a value of 1 for high-risk patients and 0 otherwise. The Hadamard product (⊙) ensures that only patients classified as high-risk contribute to the weight updates, while background patients are excluded from the refinement process.
This iterative update mechanism ensures that only high-risk patients contribute to the refinement of v, while background patients remain unchanged. The sigmoid transformation bounds the risk scores between 0 and 1, providing numerical stability and preserving relative risk differences. As iterations progress, v(r) increasingly emphasizes patients with clinically adverse outcomes, enabling the model to align survival prediction with biologically meaningful high-risk signals.
Integration into loss functions
Both the weight matrix W and the weight vector v are incorporated into the loss functions of Phase 1 and Phase 2 during subsequent iterations. In Phase 1, W modifies the reconstruction term of \({L}_{{F}_{S}}\) to prioritize genes and cells with greater survival relevance. In Phase 2, v modifies the loss function \({L}_{{F}_{C}}\), emphasizing patients with higher risk. This iterative integration enables the framework to progressively refine its representation of high-risk contributions at both the cellular and patient levels, aligning model optimization with evolving biological insights.
SIDISH’s iterative process
A defining characteristic of SIDISH is its iterative learning process, which enables a continuous exchange of information between single-cell and bulk data, progressively enhancing its ability to identify high-risk cells. Each iteration begins with the VAE (FS), initialized with a weight matrix W set to ones, and the deep Cox regression model (FC), initialized with a uniform patient weight vector v. This provides an unbiased starting point for learning. At every iteration, cell representations learned by FS are transferred to FC, where they are used to predict patient-specific survival risks. In return, feedback from FC, in the form of updated patient and gene weights from Phase 4, refines the reconstruction process in FS, forming an adaptive feedback loop. The updated weights are incorporated into both W and v, ensuring that each cycle builds upon prior biological and clinical insights. Importantly, FS retains and further optimizes its learned representations across iterations, while FC is reinitialized at the start of each cycle but retains the updated weight vector v to preserve patient-level risk information. This bidirectional information exchange continues until convergence, defined either by stabilization of the gene weight matrix W (change less than 5% between iterations) or by reaching a predefined maximum number of iterations. This approach balances computational efficiency with robust performance.
In silico perturbation strategy
Single in silico gene knockout
SIDISH incorporates an in silico gene perturbation strategy to simulate knockouts, enabling a systematic evaluation of the functional importance of genes in maintaining high-risk cellular states. This is achieved by modifying the single-cell gene expression matrix X using a curated Gene Interaction Network that integrates high-confidence interactions from the HIPPIE139 and STRINGDB140 databases. Interaction strengths are filtered using thresholds of 800 (HIPPIE) and 0.8 (STRINGDB) to retain biologically relevant connections. The expression of the target gene and its directly interacting neighbors is set to zero, resulting in a perturbed matrix \({{{{\bf{X}}}}}^{{\prime} }\).
SIDISH then re-evaluates cell-level risk post-perturbation, producing updated labels using the function RS(Xi), which classifies each cell as high-risk (HR) or background (BG). A perturbation score (ST) is computed to quantify the net reduction in high-risk cells: This relabeling allows for a direct comparison of cell states between the original (X) and perturbed (\({{{{\bf{X}}}}}^{{\prime} }\)) matrices, thereby facilitating the calculation of the perturbation score (ST):
where \({\sum }_{i=1}^{N}RS({{{{\bf{X}}}}}_{i}^{{\prime} })\) denotes the total number of high-risk cells after perturbation. The perturbation score quantifies the net effect of the knockout, measuring the percentage of high-risk cells that transition to the background state. A positive ST indicates a reduction in high-risk cells, suggesting that the gene is essential for maintaining high-risk states. Each cell’s contribution to the score depends on its state transition. A cell transitioning from HR to BG contributes +1, reflecting a loss of high-risk status. Conversely, a transition from BG to HR contributes −1, indicating a gain of high-risk status. Cells that remain in their original state contribute 0. This systematic approach provides insights into gene function and identifies potential therapeutic targets for experimental validation.
In spatial transcriptomics (ST), perturbations are further informed by spatial proximity. Cells are embedded in a spatial graph based on their coordinates, and perturbation effects propagate to neighboring cells through this spatial connectivity. As a result, this enables SIDISH to capture both intrinsic molecular alterations and microenvironmental effects.
Combinatorial in silico gene knockout
The combinatorial gene perturbation strategy extends the single gene analysis by evaluating the combined effects of two genes. To reduce computational complexity, we selected the top 30 marker genes from the single gene perturbation results. Each pair of genes was perturbed sequentially, as the order of perturbation influences the outcome. The perturbation score was calculated for each pair using the same formula as in the single gene knockout. This enables detection of synergistic interactions between genes, providing deeper insights into their roles in maintaining high-risk cellular states.
Alternative continuous perturbation score
In addition to the default binary classification approach, SIDISH also supports an alternative method for quantifying perturbation effects based on continuous risk scores predicted by the deep Cox regression model. This score (\({S}_{T}^{{{{\rm{continuous}}}}}\)) is defined as the average difference in continuous risk scores across all cells before and after a gene perturbation:
where Yi and \({Y}_{i}^{{\prime} }\) represent the continuous risk scores of cell i before and after perturbation, respectively. While this formulation provides a more nuanced quantification by capturing subtle shifts in predicted risk across cells, it was less effective in identifying clinically validated and relevant therapeutic targets (Supplementary Fig. 35), in comparison to the binary approach. Therefore, the binary strategy was used as the default for primary analyses.
Therapeutic target statistical significance calculation
To evaluate the statistical significance of the absolute change in the number of high-risk cells before and after perturbation, we construct a contingency table comparing the distribution of high-risk and background cells. For each gene perturbed, we apply Fisher’s exact test141 to assess whether perturbation induces a statistically significant shift, specifically a decrease in the number of high-risk cells accompanied by a corresponding increase in background cells.
Fisher’s exact test calculates the exact probability of observing the given 2 × 2 contingency table (high-risk vs. background before and after perturbation), making it well-suited for single-cell data where correlations between cells may be present. This exact inference approach ensures more conservative and reliable results by avoiding assumptions of independence or large-sample approximations.
The resulting P value for each gene quantifies the significance of the observed changes, ensuring that our perturbation-based prioritization of therapeutic targets is statistically rigorous and biologically meaningful.
SIDISH architectural design
SIDISH integrates a variational autoencoder (VAE) and a deep Cox regression model for survival analysis, both implemented in PyTorch. Below is an overview of the architecture, with complete details available in the GitHub repository (refer to the Code availability section).
The VAE encoder consists of sequential linear layers with ReLU activations that compress high-dimensional input data into a lower-dimensional latent space. Dropout and batch normalization were incorporated to prevent overfitting and stabilize training, while a standard Gaussian prior on the latent space and weight decay provided additional regularization. The decoder mirrors the encoder structure in reverse to reconstruct the input data. The VAE architecture was adapted from SCVI and tailored to the specific requirements of SIDISH. For instance, in the BRCA dataset, the VAE was trained for 225 epochs in the first iteration with a latent dimension of 16 and hidden layers of size 512, 256, and 64. For subsequent finetuning iterations, training was shortened to 20 epochs to avoid overfitting. Optimization was performed with the Adam optimizer at a learning rate of 1 × 10−4.
For the deep Cox regression model, transfer learning was applied by initializing the model with encoder weights (ES) learned during VAE training. Rather than serving only as a feature extractor, the encoder was integrated and finetuned within the Cox model. The architecture included an additional hidden layer of 192 units with Tanh activations and dropout for regularization, followed by a final linear layer to produce survival risk predictions. This component was trained on the bulk dataset (1194 patients) using the Adam optimizer with a learning rate of 1 × 10−5.
Training the SIDISH model involved iterative optimization of both the VAE and the deep Cox regression components. Initially, the VAE was pretrained on scRNA-seq data to learn meaningful latent representations. The encoder weights from this phase were then transferred to the deep Cox model to predict patient survival. The model iteratively identified high-risk and background subpopulations for both cells and patients using the trained Cox model, enabling the calculation of the weight matrix (W) and weight vector (v) for subsequent optimization steps in phases 1 and 2. The training time and memory usage of SIDISH on the BRCA dataset are presented in the Supplementary Table 4.
SIDISH also incorporates two key hyperparameters: a and Pcut, which influence sensitivity and thresholding during training. The hyperparameter a, shown in Supplementary Fig. 36a, b, adjusts the steepness of the modified sigmoid function σ*, which is used in Phase 4 to update weights based on SHAP values. While larger a values improve ARI and NMI metrics, they come at the expense of reduced C-Index performance, highlighting a trade-off between clustering accuracy and predictive survival performance. The hyperparameter Pcut defines the risk stratification threshold by specifying an upper-tail cutoff on the cumulative distribution function (CDF) of the fitted Weibull distribution over predicted survival scores. Alternative parametric distributions, such as the Gamma or Exponential, can also be used. While Pcut governs the selection of high-risk cells and patients, the discovery of signature biomarkers is performed on the selected high-risk cells during downstream differential expression analysis. To ensure robustness in both high-risk cell identification and biomarker discovery, we systematically evaluated SIDISH’s sensitivity to Pcut by varying it over a broad range (0.60–0.99) and assessing the stability of the resulting high-risk subpopulations and corresponding DEG signatures. As shown in Supplementary Fig. 36c, increasing values of Pcut result in smaller high-risk populations, enabling tunable refinement of clinically relevant subgroups. To quantify the stability of marker gene sets across different thresholds, we employed the Overlap Coefficient (Szymkiewicz–Simpson index)44,142, which measures the extent to which one gene set is contained within another. Formally, for two gene sets A and B, the coefficient is defined as \(\,{{{\rm{Overlap}}}}\,(A,B)=\frac{| A\cap B| }{\min (| A|,| B| )}\), where values near 1 indicate strong containment of the smaller set within the larger, reflecting high signature stability.
As illustrated in Supplementary Fig. 43, DEG signatures derived from high-risk populations formed stable plateaus at higher Pcut thresholds across all datasets. In LUAD, stability was observed between 0.80 and 0.95, supporting our choice of 0.95. For BRCA and PDAC, stabilization occurred between 0.85 and 0.90, aligning with our selected operating threshold of 0.90. These results demonstrate that our selected Pcut values are statistically principled and empirically validated, and that SIDISH reliably identifies robust high-risk subpopulations and associated biomarkers for clinical stratification and downstream therapeutic analyses.
This architectural design enables SIDISH to seamlessly integrate single-cell and survival data, identifying clinically relevant high-risk subpopulations while balancing clustering performance, survival prediction accuracy, and computational efficiency through the careful adjustment of hyperparameters.
Simulation studies for robustness analysis
A series of simulation experiments was carried out on the PDAC dataset to examine the robustness of SIDISH under varying conditions. Each simulation modified the input data in a specific way, after which the standard SIDISH pipeline was rerun to assess the stability of high-risk cell identification.
Varying cell type proportions
To determine whether SIDISH depends on the most abundant malignant cell type, we randomly reduced the ductal type 2 population in the PDAC scRNA-seq dataset. Cells annotated as ductal type 2 were randomly subsampled to achieve reductions of 50 and 75%, thereby generating two modified datasets. These datasets were then re-analyzed with SIDISH, and the resulting composition of high-risk cells was compared with that obtained from the unmodified dataset.
Technical noise simulation
To mimic lower-quality single-cell sequencing data, additional dropout events were introduced into the PDAC count matrix. In this procedure, 10% of the nonzero entries were randomly masked and set to zero, increasing sparsity without altering overall cell and gene structure. The noisy dataset was subsequently re-analyzed with SIDISH to evaluate the effect of technical degradation on the detection of high-risk subpopulations.
Case-control imbalance simulation
To investigate the effect of survival cohort composition, the PDAC bulk dataset was rebalanced by randomly duplicating samples annotated as alive, thereby increasing their proportion relative to deceased patients. This modification attenuated the survival signal available to the Cox regression model. SIDISH was then retrained on the altered bulk data, and the distribution of high-risk cells was examined.
Kaplan–Meier survival analysis
To evaluate the clinical relevance of biomarkers identified from the SIDISH-derived high-risk cell subpopulation, we performed Kaplan–Meier survival analysis143. These biomarkers were used to stratify patients in the bulk RNA-seq dataset into high-risk and background groups based on their expression levels of the identified signature genes. For each patient, only the expression data of the marker genes were retained. A simple Cox regression model was trained on the subsetted bulk dataset, which included survival information, to derive a coefficient vector for the marker genes. Using this coefficient vector, a risk score for each patient was calculated as \({\sum }_{j=1}^{N}({{{{\bf{B}}}}}_{j}\times \,{{{\rm{coefficient\; of}}}}\,{{{{\bf{B}}}}}_{j})\). Patients were then stratified into high-risk and background groups based on their risk scores, using the median score as the threshold. Kaplan–Meier survival curves were generated using the KaplanMeierFitter function from the lifelines Python package144 to illustrate survival differences between the two groups. Statistical significance was assessed using a two-tailed log-rank-sum test, which calculated the P value to confirm the distinction in survival outcomes between patients with high versus low expression of SIDISH-derived signature genes.
Precision medicine analysis
To assess SIDISH’s precision medicine capabilities, we performed patient-specific analyses by partitioning the scRNA-seq dataset into individual patient subsets. This approach allowed us to investigate inter-patient variability in high-risk cell populations and their therapeutic susceptibilities. The analysis comprised three key components: patient-level high-risk cell characterization, patient-specific single-gene perturbation analysis, and combinatorial perturbation assessment.
Patient-level high-risk cell analysis
SIDISH identified significant inter-patient variability in the composition and proportion of high-risk cell populations. For each patient, we quantified the distribution of cell types within their high-risk cell population. The proportion of high-risk cells for an individual patient was calculated as the ratio of high-risk cells to the total number of cells from the same patient. Patients were ranked in descending order based on their high-risk cell proportion to distinguish those with the highest and lowest prevalence of high-risk cells. To highlight the transcriptomic difference between the top-ranking patients and the lowest-ranking patients, we performed differential gene expression analysis based on high-risk cell proportion using SCANPY89. More specifically, we performed differential gene expression analysis between the set of cells of the top/lowest-ranking patient and the rest of the patients. We used the Wilcoxon rank-sum test to calculate the P value and the log2 fold-change (log2FC) for each gene. Using the Benjamin–Hochberg correction145 on the P values, we obtained the adjusted P values for each gene. Genes with an adjusted P value below 0.05 and a log2FC above a specific threshold were considered significant. Additionally, we examined the expression of known disease marker genes curated from cBioPortal87 and OncoKB88 to determine whether patients with a higher proportion of high-risk cells exhibited stronger expression of these disease-specific markers, particularly in cancer. Using Scanpy’s gene activity scoring function89, we computed gene expression scores for individual high-risk cells in each patient. Finally, we performed Kaplan–Meier survival analysis (see previous section) to evaluate whether signature genes from the top- and lowest-ranking patients stratified survival outcomes, further validating the clinical relevance of high-risk cells.
Patient-specific single-gene perturbation
We performed in silico single-gene knockouts to evaluate the impact of individual genes on high-risk cell populations for each patient. The perturbation effect was quantified using the perturbation score (ST), which measures the reduction in the proportion of high-risk cells following a gene knockout. To contextualize patient-specific results, we compared the top perturbation genes identified from individual patient analyses with those identified in the single-gene perturbation analysis of the complete dataset. This comparison emphasized the differences between generalized therapeutic strategies and tailored interventions.
Patient-specific combinatorial perturbation
To investigate synergistic therapeutic strategies, we extended the analysis to include in silico dual-gene knockouts for each patient. Combinatorial perturbations were performed to identify synergistic or additive effects in reducing high-risk cell populations. Similar to single-gene perturbations, we compared the top-ranked combinatorial perturbations from patient-specific analyses with those derived from the complete dataset. This framework demonstrated that combinatory therapeutic strategies tailored to individual patients outperformed generalized approaches, addressing patient-specific tumor heterogeneity and vulnerabilities.
SIDISH disease biomarker identification
Differential gene expression analysis
Differential gene expression analysis was performed to identify genes distinguishing SIDISH-labeled high-risk and background cell subpopulations. At the cell level, we applied the Wilcoxon rank-sum test to normalized single-cell expression matrices, followed by multiple-testing correction using the Benjamini–Hochberg procedure to control the false discovery rate (FDR). In addition to statistical significance, we applied log2FC thresholds as biologically motivated filters to prioritize genes with effect sizes likely to be meaningful in heterogeneous cancer datasets. Importantly, log2FC thresholds are not statistical measures but user-defined filters of biological significance; therefore, we adapted them in a dataset-specific manner rather than using a fixed universal cutoff, consistent with established practice in single-cell transcriptomics146,147,148.
Considering that the Wilcoxon rank-sum test does not account for the nested structure of cells within individuals, we compared this approach to pseudobulking. In the PDAC dataset, we aggregated counts at the patient level and performed differential expression analysis with DESeq2, which explicitly models inter-individual variability. We then compared the results to our original cell-level Wilcoxon-based analysis. As shown in Supplementary Fig. 37, pseudobulk-derived markers exhibited weaker clinical relevance than cell-level markers, with reduced stratification power (Supplementary Fig. 37a) and a lower Concordance Index (Supplementary Fig. 37b). These findings indicate that while pseudobulking mitigates potential inflation of false discoveries, it also reduces sensitivity to signals from rare populations or subtle gradients of cell states that are critical for identifying high-risk subpopulations. Supplementary Fig. 38 shows that cells from different patients are well-mixed within clusters, indicating that strong patient-specific batch effects are not the dominant source of variation in these datasets.
To verify that our downstream conclusions were not dependent on dataset-adaptive cutoffs, and that SIDISH’s improved performance is robust to threshold choice, we performed benchmarking under a standardized cutoff (log2FC = 1 and adjusted P < 0.05) across all cancer datasets and methods (Scissor, scAB, DEGAS). As shown in Supplementary Fig. 39, SIDISH consistently outperformed the benchmarked tools.
Weight-matrix analysis
In addition to the differential gene expression analysis approach, SIDISH also supports an alternative method for identifying marker genes using its learned weight matrix. The weight matrix in this context is generated after the final training iteration of SIDISH, where gene-level attribution scores are computed for each high-risk cell using SHAP-based importance values and then transformed through the modified sigmoid function σ*, controlled by the hyperparameter a. While DEGs are identified by comparing average expression differences between high-risk and background cells, the weight matrix serves a complementary role: it directly reflects what the SIDISH framework has learned from integrating clinical and molecular information. Marker genes were derived from the weight matrix by ranking genes according to their mean weight score and selecting the top genes based on a percentile threshold, using the SciPy Python library.
As shown in Supplementary Figs. 40–42, weight-matrix derived markers exhibited strong enrichment for disease-relevant pathways, and the overlap with DEG-based markers was statistically significant across PDAC, BRCA, and LUAD scRNA-seq datasets (P = 1.10 × 10−15, P = 1.10 × 10−18, and P = 2.41 × 10−12; respectively) as assessed by a hypergeometric test. In PDAC, for instance, 55 of the top 100 genes were shared between the two methods (Supplementary Fig. 40), confirming substantial agreement. At the same time, both approaches recovered distinct sets of genes. Importantly, the unique genes identified by the weight matrix were not noise but carried prognostic relevance. Kaplan–Meier survival analyses showed that DEG-based and weight-matrix–based markers were both strongly prognostic, with comparable P values across datasets. Moreover, even when restricting to genes uniquely identified by the weight matrix, significant patient stratification was observed in PDAC (P = 5.76 × 10−11; Supplementary Fig. 40g), BRCA (P = 1.03 × 10−15; Supplementary Fig. 41d), and LUAD (P = 5.76 × 10−11; Supplementary Fig. 42g).
The high concordance between differentially expressed genes and weight-matrix-derived markers highlights SIDISH’s ability to learn disease-relevant gene patterns. Genes with higher weights are consistently linked to poor prognosis. By jointly leveraging bulk and single-cell RNA-seq data, SIDISH provides a biologically meaningful representation of high-risk subpopulations, offering valuable insights for disease characterization and therapeutic target identification.
Enrichment analysis
To investigate the biological significance of the identified marker genes, we performed Gene Ontology (GO), Reactome, and Disease Ontology enrichment analyses. Marker genes were identified through Scanpy’s differential expression analysis and subsequently analyzed using ToppGene Suite (https://toppgene.cchmc.org/)149, a widely used tool for functional enrichment and pathway analysis. Enrichment results were assessed using Benjamini–Hochberg adjusted P values to correct for multiple comparisons, ensuring statistical robustness. Significant pathways and biological processes were prioritized based on their adjusted P values, allowing for the identification of key functional associations relevant to the marker genes. This approach provided deeper insights into the biological roles and potential disease relevance of the identified markers.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
We evaluated SIDISH using publicly available datasets from three cancer types. The BRCA scRNA-seq dataset from ref. 59 is available through the Gene Expression Omnibus under accession number GSE176078. The single-cell PDAC dataset from ref. 34 can be accessed via the Genome Sequence Archive under project number PRJCA001063. The PDAC scRNA-seq spatial transcriptomics dataset can be accessed via the 10x Genomics dataset webpage. Additionally, the LUAD from ref. 71 was downloaded from the ArrayExpress with accession numbers E-MTAB-6149 and E-MTAB-6653; for comparison with Scissor17, we utilized the same subset of 4102 cancer cells as the original authors. Bulk RNA-seq data and corresponding survival information used to train SIDISH were sourced from The Cancer Genome Atlas (TCGA)35 and downloaded through the UCSC Xena platform150. Two independent bulk BRCA validation datasets with survival information, from Chin et al.68 and Teschendorff et al.69 were also downloaded through the UCSC Xena platform. Independent bulk PDAC datasets with survival information were retrieved from GEO under accession numbers GSE224564 and GSE85916. The independent PDAC scRNA-seq from ref. 57 dataset was also retrieved from GEO under accession number GSE242230. Independent LUAD validation bulk datasets, including sample survival information, were obtained through GEO under accession numbers GSE157009 and GSE37745. The example datasets associated with this study are available on Zenodo at 10.5281/zenodo.17419767151. Source data are provided with this paper.
Code availability
Code for the models and results reproduction is publicly available on GitHub: https://github.com/mcgilldinglab/SIDISH. The specific version of the code associated with this publication is archived in Zenodo and is accessible via https://doi.org/10.5281/zenodo.17420265152.
References
Sun, Y., Wu, Y. & Liao, B. Phenotypic heterogeneity in human genetic diseases: ultrasensitivity-mediated threshold effects as a unifying molecular mechanism. J. Biomed. Sci. 30, 58 (2023).
Dagogo-Jack, I. & Shaw, A. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 15, 81–94 (2018).
Ramón y Cajal, S., Sesé, M. & Capdevila, C. Clinical implications of intratumor heterogeneity: challenges and opportunities. J. Mol. Med. 98, 161–177 (2020).
Dentro, S. C. Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes. Cell 184, 2239–2254.e39 (2021).
Marusyk, A. Intratumor heterogeneity: The rosetta stone of therapy resistance. Cancer Cell 37, 471–484 (2020).
Jacquemin, V. et al. Dynamic cancer cell heterogeneity: diagnostic and therapeutic implications. Cancers 14, 280 (2022).
Wang, Z., Gerstein, M. & Snyder, M. Rna-seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63 (2009).
Momeni, K., Ghorbian, S. & Ahmadpour, E. Unraveling the complexity: understanding the deconvolutions of rna-seq data. Transl. Med. Commun. 8, 21 (2023).
Maden, S. K., Kwon, S.-H. & Huuki-Myers, L. A. Challenges and opportunities to computationally deconvolve heterogeneous tissue with varying cell sizes using single-cell rna-sequencing datasets. Genome Biol. 24, 288 (2023).
Kuksin, M. et al. Applications of single-cell and bulk rna sequencing in onco-immunology. Eur. J. Cancer 149, 193–210 (2021).
Li, X. & Wang, C. From bulk, single-cell to spatial rna sequencing. Int. J. Oral. Sci. 13, 36 (2021).
Haque, A., Engel, J. & Teichmann, S. A. A practical guide to single-cell rna-sequencing for biomedical research and clinical applications. Genome Med. 9, 75 (2017).
Little, P., Liu, S. & Zhabotynsky, V. A computational method for cell type-specific expression quantitative trait loci mapping using bulk rna-seq data. Nat. Commun. 14, 3030 (2023).
Lei, Y., Tang, R. & Xu, J. Applications of single-cell sequencing in cancer research: progress and perspectives. J. Hematol. Oncol. 14, 91 (2021).
Wang, J., Fonseca, G. & Ding, J. scsemiprofiler: advancing large-scale single-cell studies through semi-profiling with deep generative models and active learning. Nat. Commun. 15, 5989 (2024).
Wang, Y., Fan, J. L. & Melms, J. C. Multimodal single-cell and whole-genome sequencing of small, frozen clinical specimens. Nat. Genet. 55, 19–25 (2023).
Sun, D., Guan, X. & Moran, A. E. Identifying phenotype-associated subpopulations by integrating bulk and single-cell sequencing data. Nat. Biotechnol. 40, 527–538 (2022).
Zhang, Q., Jin, S. & Zou, X. scAB detects multiresolution cell states with clinical significance by integrating single-cell genomics and bulk sequencing data. Nucleic Acids Res. 50, 12112–12130 (2022).
Johnson, T. S., Yu, C.-Y. & Huang, Z. Diagnostic evidence gauge of single cells (degas): a flexible deep transfer learning framework for prioritizing cells in relation to disease. Genome Med. 14, 11 (2022).
LeCun, Y., Bengio, Y. & Hinton, G. Deep learning. Nature 521, 436–444 (2015).
Rodgers, J. L. & Nicewander, W. A. Thirteen ways to look at the correlation coefficient. Am. Statistician 42, 59–66 (1988).
Schober, P., Boer, C. & Schwarte, L. A. Correlation coefficients: appropriate use and interpretation. Anesthesia Analgesia 126, 1763–1768 (2018).
Pernet, C. R., Wilcox, R. R. & Rousselet, G. A. Robust correlation analyses: false positive and power validation using a new open source matlab toolbox. Front. Psychol. https://www.frontiersin.org/journals/psychology/articles/10.3389/fpsyg.2012.00606 (2013).
Smirnov, P., Smith, I. & Safikhani, Z. Evaluation of statistical approaches for association testing in noisy drug screening data. BMC Bioinformatics 23, 188 (2022).
Ji, Y. Machine learning for perturbational single-cell omics. Cell Syst. 12, 522–537 (2021).
Kingma, D. P. & Welling, M. Auto-encoding variational bayes. In Conference proceedings: papers accepted to the International Conference on Learning Representations ICLR, 2014. arXiv: http://arxiv.org/abs/1312.6114 (2014).
Kingma, D. P. & Welling, M. An introduction to variational autoencoders. Found. Trends Mach. Learn. 12, 307–392 (2019).
Cox, D. R. Regression models and life-tables. J. R. Stat. Soc. Ser. B Methodol. 34, 187–220 (1972).
Weibull, W. A statistical distribution function of wide applicability. J. Appl. Mech. 18, 293–297 (2021).
Lundberg, S. M. & Lee, S.-I. A unified approach to interpreting model predictions. In 31st Conference on Neural Information Processing Systems (NIPS) (eds Guyon, I. et al.) (Curran Associates, Inc., 2017).
Shapley, L. S. A Value for N-Person Games (RAND Corporation, 1952).
Pun, F. W. et al. Ai-powered therapeutic target discovery. Trends Pharmacol. Sci. 44, 561–572 (2023).
López-Cortés, A., Cabrera-Andrade, A., Echeverría-Garcés, G. et al. Unraveling druggable cancer-driving proteins and targeted drugs using artificial intelligence and multi-omics analyses. Sci. Rep. 14, 19359 (2024).
Peng, J., Sun, B., Chen, C. et al. Single-cell rna-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 29, 725–738 (2019).
The Cancer Genome Atlas Research Network, Weinstein, J. C. E. et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 45, 1113–1120 (2013).
Perrotta, G. et al. Trends in incidence and survival of early-stage pancreatic ductal adenocarcinoma in the united states. J. Clin. Oncol. 42, 633–633 (2024).
Marzoq, A., Mustafa, S., Heidrich, L. et al. Impact of the secretome of activated pancreatic stellate cells on growth and differentiation of pancreatic tumour cells. Sci. Rep. 9, 5303 (2019).
Allam, A. et al. Pancreatic stellate cells in pancreatic cancer: in focus. Pancreatology 17, 514–522 (2017).
Saúde-Conde, R. et al. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma or a metaphor for heterogeneity: from single-cell analysis to whole-body imaging. Biomedicines https://www.mdpi.com/2227-9059/12/3/591 (2024).
Zhang, T., Ren, Y., Yang, P. et al. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. Cell Death Dis. 13, 897 (2022).
Garcia, P., Scales, M., Allen, B. & Pasca di Magliano, M. Pancreatic fibroblast heterogeneity: from development to cancer. Cells 9, 2464 (2020).
Calimano-Ramirez, L. et al. Pancreatic acinar cell carcinoma: a comprehensive review. World J. Gastroenterol. 28, 5827–5844 (2022).
Ikezawa, K. et al. Comprehensive review of pancreatic acinar cell carcinoma: epidemiology, diagnosis, molecular features and treatment. Jpn. J. Clin. Oncol. 54, 271–281 (2023).
Franco-Pereira, A. M., Nakas, C. T., Reiser, B. & Pardo, M. C. Inference on the overlap coefficient: the binormal approach and alternatives. Stat. Methods Med. Res. 30, 2672–2684 (2021).
Sun, Q., Zhang, B. & Hu, Q. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics 8, 5072–5087 (2018).
Xu, C., Wang, S. & Sun, Y. The role of krt7 in metastasis and prognosis of pancreatic cancer. Cancer Cell Int. 24, 321 (2024).
Li, Y., Su, Z., Wei, B. & Liang, Z. Krt7 overexpression is associated with poor prognosis and immune cell infiltration in patients with pancreatic adenocarcinoma. Int. J. Gen. Med. 14, 2677–2694 (2021).
Pandey, R., Zhou, M., Islam, S. et al. Carcinoembryonic antigen cell adhesion molecule 6 (ceacam6) in pancreatic ductal adenocarcinoma (pda): an integrative analysis of a novel therapeutic target. Sci. Rep. 9, 18347 (2019).
Li, C. et al. The prognostic and immune significance of c15orf48 in pan-cancer and its relationship with proliferation and apoptosis of thyroid carcinoma. Front. Immunol. https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2023.1131870 (2023).
Delgado-Coka, L., Horowitz, M., Torrente-Goncalves, M. et al. Keratin 17 modulates the immune topography of pancreatic cancer. J. Transl. Med. 22, 443 (2024).
Vishwa, R., BharathwajChetty, B., Girisa, S. et al. Lipid metabolism and its implications in tumor cell plasticity and drug resistance: what we learned thus far?. Cancer Metastasis Rev. 43, 293–319 (2024).
Shu, M. L., Yang, W. T., Li, H. M. et al. Circ_0124346 facilitates cell proliferation of pancreatic adenocarcinoma cells by regulating lipid metabolism via mir-223-3p/acsl3 axis. Discov. Oncol. 15, 670 (2024).
Li, F., Si, W., Xia, L. et al. Positive feedback regulation between glycolysis and histone lactylation drives oncogenesis in pancreatic ductal adenocarcinoma. Mol. Cancer 23, 90 (2024).
Matthews, H., Bertoli, C. & de Bruin, R. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 23, 74–88 (2022).
Ohara, Y. Serpinb3-myc axis induces the basal-like/squamous subtype and enhances disease progression in pancreatic cancer. Cell Rep. 42, 113434 (2023).
Puleo, F. et al. Stratification of pancreatic ductal adenocarcinomas based on tumor and microenvironment features. Gastroenterology 155, 1999–2013.e3 (2018).
Storrs, E. P. et al. High-dimensional deconstruction of pancreatic cancer identifies tumor microenvironmental and developmental stemness features that predict survival. npj Precis. Oncol. 7, 105 (2023).
Ferrara, B. et al. The extracellular matrix in pancreatic cancer: description of a complex network and promising therapeutic options. Cancers 13, 4442 (2021).
Wu, S., Al-Eryani, G., Roden, D. et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 53, 1334–1347 (2021).
Fragomeni, S., Sciallis, A. & Jeruss, J. Molecular subtypes and local-regional control of breast cancer. Surg. Oncol. Clin. North Am. 27, 95–120 (2018).
Singh, D. & Siddique, H. Epithelial-to-mesenchymal transition in cancer progression: unraveling the immunosuppressive module driving therapy resistance. Cancer Metastasis Rev. 43, 155–173 (2024).
Ji, S., Yu, H., Zhou, D. et al. Cancer stem cell-derived chi3l1 activates the maf/ctla4 signaling pathway to promote immune escape in triple-negative breast cancer. J. Transl. Med. 21, 721 (2023).
Wu, S. et al. Stromal cell diversity associated with immune evasion in human triple-negative breast cancer. EMBO J. 39, e104063 (2020).
Obidiro, O., Battogtokh, G. & Akala, E. Triple negative breast cancer treatment options and limitations: future outlook. Pharmaceutics 15, 1796 (2023).
Yuan, Z., Li, Y., Zhang, S. et al. Extracellular matrix remodeling in tumor progression and immune escape: from mechanisms to treatments. Mol. Cancer 22, 48 (2023).
Jiang, X., Wang, J., Deng, X. et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 39, 204 (2020).
Chen, R., Zhang, R., Ke, F. et al. Mechanisms of breast cancer metastasis: the role of extracellular matrix. Mol. Cell. Biochem. https://doi.org/10.1007/s11010-024-05175-x (2024).
Chin, K. et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 10, 529–541 (2006).
Teschendorff, A., Miremadi, A., Pinder, S. et al. An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome Biol. 8, R157 (2007).
Yue, S., Liu, H., Su, H. et al. m6a-regulated tumor glycolysis: new advances in epigenetics and metabolism. Mol. Cancer 22, 137 (2023).
Lambrechts, D., Wauters, E., Boeckx, B. et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 24, 1277–1289 (2018).
Huang, C., Sun, Y., Lv, L. & Ping, Y. Eno1 and cancer. Mol. Ther. Oncolytics 24, 288–298 (2022).
Yang, Y., Chong, Y., Chen, M. et al. Targeting lactate dehydrogenase a improves radiotherapy efficacy in non-small cell lung cancer: from bedside to bench. J. Transl. Med. 19, 170 (2021).
Chen, Y., Liu, M., Niu, Y. & Wang, Y. Romance of the three kingdoms in hypoxia: Hifs, epigenetic regulators, and chromatin reprogramming. Cancer Lett. 495, 211–223 (2020).
Ortmann, B. M. Hypoxia-inducible factor in cancer: from pathway regulation to therapeutic opportunity. BMJ Oncol. 3, e000154 (2024).
Bueno, R., Richards, W. G., Harpole, D. H. & Ballman, K. V. Multi-institutional prospective validation of prognostic mrna signatures in early stage squamous lung cancer (alliance). J. Thorac. Oncol. 15, 1748–1757 (2020).
Botling, J., Edlund, K., Lohr, M. & Hellwig, B. Biomarker discovery in non-small cell lung cancer: integrating gene expression profiling, meta-analysis, and tissue microarray validation. Clin. Cancer Res. 19, 194–204 (2013).
Hosein, A. N., Brekken, R. A. & Maitra, A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 17, 487–505 (2020).
Ren, B. et al. Tumor microenvironment participates in metastasis of pancreatic cancer. Mol. Cancer 17, 108 (2018).
Götze, J. et al. Tumor-stroma interaction in pdac as a new approach for liquid biopsy and its potential clinical implications. Front. Cell Dev. Biol. 10, 918795 (2022).
10x Genomics. Pancreatic cancer with xenium human multi-tissue and cancer panel—in situ gene expression dataset (2023).
Neesse, A., Algül, H. & Tuveson, D. A. Stromal biology and therapy in pancreatic cancer: a changing paradigm. Gut 64, 1476–1484 (2015).
Hamacher, R., Schmid, R. M., Saur, D. & Schneider, G. Apoptotic pathways in pancreatic ductal adenocarcinoma. Mol. Cancer 7, 64 (2008).
Pembury Smith, M. Q. R. & Ruxton, G. D. Effective use of the mcnemar test. Behav. Ecol. Sociobiol. 74, 133 (2020).
Carragher, N. O. & Frame, M. C. Focal adhesion and actin dynamics: a place where kinases and proteases meet to promote invasion. Trends Cell Biol. 14, 241–249 (2004).
Kanteti, R., Batra, S. K., Lennon, F. E. & Salgia, R. Fak and paxillin, two potential targets in pancreatic cancer. Oncotarget 7, 31586–31601 (2016).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci. Signal. 6, pl1–pl1 (2013).
Chakravarty, D. et al. Oncokb: a precision oncology knowledge base. JCO Precis. Oncol. https://ascopubs.org/doi/pdf/10.1200/PO.17.00011 (2017).
Wolf, F., Angerer, P. & Theis, F. Scanpy: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Lopez, R., Regier, J., Cole, M. et al. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
Lotfollahi, M., Wolf, F. A. & Theis, F. J. scgen predicts single-cell perturbation responses. Nat. Methods 16, 715–721 (2019).
Cui, H., Wang, C., Maan, H. et al. scgpt: toward building a foundation model for single-cell multi-omics using generative ai. Nat. Methods 21, 1470–1480 (2024).
Hu, J. et al. SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods 18, 1342–1351 (2021).
Xu, H., Fu, H., Long, Y. et al. Unsupervised spatially embedded deep representation of spatial transcriptomics. Genome Med. 16, 12 (2024).
Pham, D., Tan, X., Balderson, B. et al. Robust mapping of spatiotemporal trajectories and cell–cell interactions in healthy and diseased tissues. Nat. Commun. 14, 7739 (2023).
Institute, N. C. Bevacizumab (n.d.). https://www.cancer.gov/about-cancer/treatment/drugs/bevacizumab Accessed: December 12 2024.
Institute, N. C. Trametinib (n.d.) https://www.cancer.gov/about-cancer/treatment/drugs/trametinib. Accessed: December 12 2024.
Institute, N. C. Clinical trial id: Nci-2021-08861 (n.d.) https://www.cancer.gov/research/participate/clinical-trials-search/v?id=NCI-2021-08861&r=1. Accessed: December 12 2024.
Danilov, A., Hu, S., Orr, B. et al. Dinaciclib induces anaphase catastrophe in lung cancer cells via inhibition of cyclin-dependent kinases 1 and 2. Mol. Cancer Ther. 15, 2758–2766 (2016).
Mohammad, R. M. et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 35, S78–S103 (2015).
Lu, C., Xiong, M., Luo, Y. et al. Genome-wide transcriptional analysis of apoptosis-related genes and pathways regulated by h2ax in lung cancer a549 cells. Apoptosis 18, 1039–1047 (2013).
Galassi, C., Chan, T. A., Vitale, I. & Galluzzi, L. The hallmarks of cancer immune evasion. Cancer Cell 42, 1825–1863 (2024).
Duggal, S., Jailkhani, N., Midha, M. et al. Defining the akt1 interactome and its role in regulating the cell cycle. Sci. Rep. 8, 1303 (2018).
Chen, K., Jiao, X., Ashton, A. et al. The membrane-associated form of cyclin d1 enhances cellular invasion. Oncogenesis 9, 83 (2020).
Elebiyo, T. C. et al. Reassessing vascular endothelial growth factor (vegf) in anti-angiogenic cancer therapy. Cancer Treat. Res. Commun. 32, 100620 (2022).
Sofi, S., Mehraj, U., Qayoom, H. et al. Targeting cyclin-dependent kinase 1 (cdk1) in cancer: molecular docking and dynamic simulations of potential cdk1 inhibitors. Med. Oncol. 39, 133 (2022).
Mizuno, S., Ikegami, M., Koyama, T. et al. High-throughput functional evaluation of map2k1 variants in cancer. Mol. Cancer Ther. 22, 227–239 (2023).
Olejarz, W., Kubiak-Tomaszewska, G., Chrzanowska, A. & Lorenc, T. Exosomes in angiogenesis and anti-angiogenic therapy in cancers. Int. J. Mol. Sci. 21, 5840 (2020).
Al-Hajeili, M., Azmi, A. & Choi, M. Nab-paclitaxel: potential for the treatment of advanced pancreatic cancer. Oncotargets Ther. 7, 187–192 (2014).
Su, S. et al. Abraxane approved for metastatic pancreatic cancer. Cancer Discov. 3, OF3 (2013).
Su, S., Chhabra, G., Singh, C., Ndiaye, M. & Ahmad, N. Plk1 inhibition-based combination therapies for cancer management. Transl. Oncol. 16, 101332 (2022).
Hu, X., Cao, J., Hu, W. et al. Multicenter phase ii study of apatinib in non-triple-negative metastatic breast cancer. BMC Cancer 14, 820 (2014).
Lin, Y., Wu, Z., Zhang, J. et al. Apatinib for metastatic breast cancer in non-clinical trial setting: Satisfying efficacy regardless of previous anti-angiogenic treatment. Tumor Biol. https://doi.org/10.1177/1010428317711033 (2017).
Rugo, H., Trédan, O., Ro, J. et al. A randomized phase ii trial of ridaforolimus, dalotuzumab, and exemestane compared with ridaforolimus and exemestane in patients with advanced breast cancer. Breast Cancer Res. Treat. 165, 601–609 (2017).
Alvarez-Trotta, A., Guerrant, W., Astudillo, L. et al. Pharmacological disruption of the notch1 transcriptional complex inhibits tumor growth by selectively targeting cancer stem cells. Cancer Res. 81, 3347–3357 (2021).
Faller, B. A. et al. Treatment of pancreatic cancer with epidermal growth factor receptor-targeted therapy. Clin. Colorectal Cancer 8, 419–428 (2009).
Bockorny, B. et al. Bl-8040, a cxcr4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the combat trial. Nat. Med. 26, 878–885 (2020).
Wijnen, R. et al. Cyclin dependent kinase-1 (cdk-1) inhibition as a novel therapeutic strategy against pancreatic ductal adenocarcinoma (pdac). Cancers 13, 4389 (2021).
Yoo, C. et al. Brightline-2: a phase iia/iib trial of brigimadlin (bi 907828) in advanced biliary tract cancer, pancreatic ductal adenocarcinoma or other solid tumors. Ann. Oncol. 35, 1069–1077 (2024).
Bulle, A. & Lim, K. Beyond just a tight fortress: contribution of stroma to epithelial-mesenchymal transition in pancreatic cancer. Signal Transduct. Target. Ther. 5, 249 (2020).
Ju, Y., Xu, D., Liao, M. et al. Barriers and opportunities in pancreatic cancer immunotherapy. npj Precis. Oncol. 8, 199 (2024).
Marusyk, A., Janiszewska, M. & Polyak, K. Intratumor heterogeneity: the rosetta stone of therapy resistance. Cancer Cell 37, 471–484 (2020).
Baghban, R., Roshangar, L., Jahanban-Esfahlan, R. et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 18, 59 (2020).
Yoshida, S., Asanoma, K., Yagi, H. et al. Fibronectin mediates activation of stromal fibroblasts by sparc in endometrial cancer cells. BMC Cancer 21, 156 (2021).
Vaz, J., Ansari, D., Sasor, A. & Andersson, R. Sparc: A potential prognostic and therapeutic target in pancreatic cancer. Pancreas 44, 1024–1035 (2015).
Zhang, H., Dai, Z., Wu, W. et al. Regulatory mechanisms of immune checkpoints pd-l1 and ctla-4 in cancer. J. Exp. Clin. Cancer Res. 40, 184 (2021).
Korsunsky, I., Millard, N. & Fan, J. Fast, sensitive and accurate integration of single-cell data with harmony. Nat. Methods 16, 1289–1296 (2019).
Bayat Mokhtari, R., Homayouni, T., Baluch, N. et al. Combination therapy in combating cancer. Oncotarget 8, 38022–38043 (2017).
Plana, D., Palmer, A. & Sorger, P. Independent drug action in combination therapy: Implications for precision oncology. Cancer Discov. 12, 606–624 (2022).
Zhao, E., Stone, M. R. & Ren, X. Spatial transcriptomics at subspot resolution with bayesspace. Nat. Biotechnol. 39, 1375–1384 (2021).
Zhang, Y., Parmigiani, G. & Johnson, W. E. Combat-seq: batch effect adjustment for rna-seq count data. NAR Genom. Bioinform. 2, lqaa078 (2020).
Katzman, J., Shaham, U., Cloninger, A. et al. Deepsurv: personalized treatment recommender system using a cox proportional hazards deep neural network. BMC Med. Res. Methodol. 18, 24 (2018).
Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019).
Carroll, K. On the use and utility of the weibull model in the analysis of survival data. Control. Clin. Trials 24, 682–701 (2003).
Bowman, K. O., Shenton, L. R. & Karlof, C. Estimation problems associated with the three parameter gamma distribution. Commun. Stat. Theory Methods 24, 1355–1376 (1995).
Matheson, M., Muñoz, A. & Cox, C. Describing the flexibility of the generalized gamma and related distributions. Commun. Stat. Theory Methods 4, 15 (2017).
Lovric, M. The Concise Encyclopedia of Statistics (Springer, New York, 2008).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Alanis-Lobato, G., Andrade-Navarro, M. A. & Schaefer, M. H. Hippie v2.0: enhancing meaningfulness and reliability of protein–protein interaction networks. Nucleic Acids Res. 45, D408–D414 (2016).
Szklarczyk, D. et al. The string database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646 (2022).
Kim, H. Y. Statistical notes for clinical researchers: chi-squared test and fisher’s exact test. Restor. Dent. Endod. 42, 152–155 (2017).
Wang, D. & Tian, L. Parametric methods for confidence interval estimation of overlap coefficients. Comput. Stat. Data Anal. 106, 12–26 (2017).
Kartsonaki, C. Survival analysis. Diagnostic Histopathol. 22, 263–270 (2016).
Davidson-Pilon, C. lifelines: survival analysis in python. J. Open Source Softw. 4, 1317 (2019).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300 (1995).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 15, 550 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edger: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Schurch, N. J. et al. How many biological replicates are needed in an rna-seq experiment and which differential expression tool should you use?. RNA 22, 839–851 (2016).
Chen, J., Xu, H., Aronow, B. & Jegga, A. Improved human disease candidate gene prioritization using mouse phenotype. BMC Bioinformatics 8, 392 (2007).
Goldman, M., Craft, B., Hastie, M. et al. Visualizing and interpreting cancer genomics data via the xena platform. Nat. Biotechnol. 38, 675–678 (2020).
Jolasun, Y. et al. Sidish integrates single-cell and bulk transcriptomics to identify high-risk cells and guide precision therapeutics through in silico perturbation. https://doi.org/10.5281/zenodo.17419767 (2025).
Jolasun, Y. et al. Sidish integrates single-cell and bulk transcriptomics to identify high-risk cells and guide precision therapeutics through in silico perturbationhttps://doi.org/10.5281/zenodo.17420265 (2025).
Acknowledgements
This work is supported by grants from the Canadian Institutes of Health Research (CIHR) [PJT-180505 to J.D]; the Funds de recherche du Québec - Santé (FRQS) [295298 to J.D., 295299 to J.D., 366764 to J.D.]; the Natural Sciences and Engineering Research Council of Canada (NSERC) [RGPIN2022-04399 to J.D.]; and the Meakins-Christie Chair in Respiratory Research [to J.D.]. This research was enabled in part by support provided by Calcul Québec (calculquebec.ca) and the Digital Research Alliance of Canada (alliancecan.ca). Schematics were generated with Biorender.
Author information
Authors and Affiliations
Contributions
J.D. conceived the study. Y.J. and J.D. jointly designed the methodology, with Y.J. implementing it. Y.J. was responsible for data collection. J.D. supervised the production of results and conceived the in silico perturbation feature. Y.Z. contributed to the design and analysis of the in silico perturbation feature. Y.J. implemented the in silico perturbation methodology and conducted the analysis. D.E. reviewed the biological significance of the results. All authors (Y.J., K.S., J.D., Y.Z., J.W., D.H.E., and G.J.F.) contributed to the writing and revision of the manuscript. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jolasun, Y., Song, K., Zheng, Y. et al. SIDISH integrates single-cell and bulk transcriptomics to identify high-risk cells and guide precision therapeutics through in silico perturbation. Nat Commun 16, 11271 (2025). https://doi.org/10.1038/s41467-025-66162-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66162-4
