
Abstract
Cell adhesion molecules (CAMs) are pivotal in establishing and maintaining synaptic connectivity. Emerging evidence indicates that some secreted factors within the synaptic cleft, including C1q-like proteins (C1qls), play a crucial role in bridging pre- and post-synapses by connecting the bilateral CAMs. However, the mechanisms of those secreted factors in synapse assembly remain incomplete. Here, we explore C1ql-mediated synaptic connectivity, focusing on the assembly of C1ql1 and its postsynaptic receptor brain-specific angiogenesis inhibitor 3 (BAI3, also called ADGRB3). Our biochemical, structural, and computational analyses reveal that the trimeric globular C1q (gC1q) domain of C1ql1 undergoes a calcium-modulated domain-swapping event to form a hexamer. Cryo-EM study manifests the stabilizing role of calcium ions on the C1ql1_gC1q hexamer in complex with the extended CUB domain of BAI3. Using the gC1q hexamer, full-length C1ql1 further assembles into linear clusters, possibly providing a scaffold to accumulate BAI3 receptors on the plasma membrane. Our cellular and in vivo studies support a role for the gC1q-mediated dynamic assembly of C1ql1 in receptor accumulation and synapse maintenance. Collectively, our findings provide a plausible mechanism of secreted factor-mediated synaptic connectivity, driven by the calcium-modulated assembly of C1qls and their interactions with CAMs.
Similar content being viewed by others


Introduction
Synapses are specialized functional units designed for neural signal transmission from presynaptic to postsynaptic cells. Cell adhesion molecules (CAMs) have been established as essential players in synapse organization1,2,3. During neuronal development, synaptic CAMs bridge the pre- and postsynaptic compartments across the synaptic cleft and facilitate synapse formation and/or maturation4,5,6,7. Intriguingly, some secreted factors, such as cerebellins8,9,10,11 and C1q-like proteins (C1qls)11,12,13,14,15, have been recently discovered to mediate synaptic connections by simultaneously interacting with both pre- and postsynaptic CAMs.
C1ql proteins belong to the C1q/TNF protein superfamily, characterized by the presence of a globular C1q (gC1q) domain, which typically forms a symmetric trimer with Ca2+ ions bound in the central axis16,17,18,19. C1qls have been increasingly recognized for their roles in the development and maturation of diverse neuronal synapses in the central nervous system16,20,21. In mammals, four C1ql proteins, including C1ql1, C1ql2, C1ql3, and C1ql4, have been identified, with C1ql1-3 mainly expressed in the central nervous system22,23,24,25. Among these C1ql proteins, C1ql1 serves as a core regulator for the assembly of climbing fibers (CFs)-Purkinje cells (PCs) synapses12,13,26,27,28. Importantly, improper function of the gC1q domain of C1ql1 leads to abnormalities in CF-PC synapses12.
Despite that the gC1q domain of C1qls is known to interact with various pre- and postsynaptic CAMs12,14,15,29, the underlying mechanisms remain elusive. The best-known C1ql-interacting CAMs are the brain-specific angiogenesis inhibitors (BAIs), which belong to the adhesion G protein-coupled receptor (aGPCR) subfamily B (ADGRB), with a large extracellular domain consisting of a complement C1r/C1s, Uegf, Bmp1 (CUB) domain, several thrombospondin repeats (TSR), a hormone-binding domain (HBD), a GAIN domain, and a seven-α-helical transmembrane domain (7TM) from N-terminal to C-terminal regions30,31. Three BAI proteins were identified in mammals: BAI1, BAI2, and BAI3, primarily expressed in the brain and implicated in various neurological diseases30,31,32,33. The binding of BAIs to other synaptic CAMs plays a role in synapse formation, maturation, and maintenance34,35. The C1ql1/BAI3 interaction has been established to mediate the contact between CF and PC neurons12,13, and genetic loss of C1ql1 or BAI3 causes the impairment of neural activity-dependent structural plasticity during development, in turn disrupting brain functions such as sensory omission, reward prediction errors, and motor learning deficit12,13,28.
In this work, to elucidate the molecular mechanisms governing synaptic connectivity mediated by the C1ql/BAI interaction, we carry out a range of structural, functional, and computational studies. Our crystal structure of C1ql1_gC1q shows that the trimeric gC1q domain can further assemble into a hexameric form through domain swapping in low Ca2+ conditions. Molecular dynamics (MD) simulations further confirm the conformational transition from trimer-to-hexamer in the Ca2+-modulated manner. The cryo-EM structure of the C1ql1_gC1q hexamer in complex with the extended CUB domain (eCUB) of BAI3 reveals the critical role of Ca2+ ions in regulating their interaction. Using cellular assays and C1ql1-knockout mice, we suggest that the impaired hexamer formation reduces the C1ql1/BAI3 assembly on the cell surface and diminishes the formation and maintenance of CF-PC synapses. Beyond the gC1q hexamer, the N-terminal (NT) region of C1ql1 contributes to the C1ql1/BAI3 assembly by promoting the formation of a high oligomeric form of C1ql1. Thus, we propose that C1ql1 forms a clustered structure, mediated by both the gC1q and NT regions, to effectively assemble with BAI3 at postsynaptic specializations, which is crucial for the formation and maintenance of CF-PC synapses.
Results
Domain-swapping drives the hexamer formation of the gC1q domain in C1ql1
C1ql proteins share a similar domain organization, consisting of an NT region, a middle collagen-like (CL) sequence, and a C-terminal typical gC1q domain (Fig. 1a). During the purification of the gC1q domain of C1ql1, we identified two different elution peaks using analytical size-exclusion chromatography (aSEC) (Fig. 1b), suggesting two different assembly forms of the gC1q domain in solution. Subsequently, molecular weight measurements by multiangle light scattering (MALS) indicated the hexameric assembly of C1ql1_gC1q, in addition to its classical trimer (Supplementary Fig. 1a). To understand the assembly mechanism of the C1ql1_gC1q hexamer, we collected protein fractions corresponding to the hexamer for structural analysis. By employing X-ray crystallography, we solved the crystal structure of the gC1q hexamer at a resolution of 2.22 Å (Fig. 1c and Supplementary Table 1). Interestingly, both a trimer and a hexamer of C1ql1_gC1q can be simultaneously observed in one asymmetric unit (Fig. 1c), supporting a dynamic transition between these two forms. The trimer is essentially identical to that previously reported12,19, with three chains packed around a central axis composed of four Ca2+ ions, while each chain also binds an additional Ca2+ ion on the surface (Fig. 1c and Supplementary Fig. 1b, c, and e). Interestingly, the hexamer structure is formed by the association of two gC1q trimers in a “tail-to-tail” manner, with their central Ca2+-axes positioned opposite to each other (Fig. 1c). In this hexameric arrangement, two β-hairpins, each comprising strands β4 and β5 from one protomer in the trimer, extend to the opposing side, swapping with the corresponding two β-hairpins in the second trimer. This results in a domain-swapped interface between the two trimers (Fig. 1c, d and Supplementary Fig. 1d).

a Domain organization of mouse C1ql1. SP signal peptide, NT N-terminal region, CL collagen-like region, gC1q globular C1q domain, with its boundary indicated. b aSEC and SDS-PAGE analyses of the purified C1ql1_gC1q protein. c Crystal structure of C1ql1_gC1q showing a trimer and a hexamer in an asymmetric unit. The bound calcium ions (Ca2+) and the axis across the central Ca2+ (Ca2+-axis) in each trimer are indicated by gray spheres and blue lines, respectively. d A rotated representation of the different C1ql1_gC1q protomers in the hexamer, showing domain-swapping between two trimers through the partial unfolding of the β4-β5 hairpin. e Structural superimposition of the trimeric gC1q domain in both trimer and hexamer forms of C1ql1_gC1q. The central Ca2+ ions and the Ca2+-axis across them are indicated by colored spheres and blue lines, respectively. The major structural differences in the regions surrounding the central Ca2+-axis are boxed. f Three enlarged views in parallel to display the conformational changes between the classical trimer and the domain-swapped hexamer of C1ql1_gC1q. The number of Ca2+ ions (#1–4 from bottom to top) bound along the Ca2+-axis is highlighted. Key residues D214, D208, and D210, which coordinate these central Ca2+ ions, are indicated. In the trimer, the loop between β5 and β6 (L5-6) presents with a folded back turn, while in the hexamer, the L5-6 exhibits reversed and extended directions, and the sidechain of D208 rotates outward due to the absence of the #3 Ca2+ ion. g A model to illustrate the role of the central Ca2+-axis in regulating the flipping of the β4-β5 hairpin for the transition between the trimer and hexamer forms of C1ql1_gC1q.
To elucidate the structural basis governing the hexamer formation, we compared the structures of the C1ql1_gC1q trimer and hexamer. It was found that the trimer structure is nearly superimposable in corresponding regions of the hexamer; however, significant differences exist in the number of bound Ca2+ ions in the central axis and the surrounding loops (Fig. 1e). Specifically, while the typical trimer contains four central Ca2+ ions (Fig. 1f and Supplementary Fig. 1c), the two trimers in the hexamer structure have two or three central Ca2+ ions (Fig. 1f and Supplementary Fig. 1c). The loss of Ca2+ ions is associated with the conformational change of the loop (L5-6) connecting β5 and β6 strands. In the typical trimer, L5-6 forms a sharp turn, positioning residues D208 and D210 to capture the top two central Ca2+ ions (#3 and #4 in Fig. 1f, left), with D214 bridging the bottom two Ca2+ ions (#1 and #2 in Fig. 1f, left). In contrast, loop L5-6 in the hexamer assembly extends from one trimer to the other, leading to the reorientation of D208 and D210, accompanied by the loss of the top Ca2+ ions (Fig. 1f, middle and right). Consequently, we propose that the #3 and #4 Ca2+ ions stabilize the L5-6 loop in a sharp-turn conformation through coordination with all three aspartate residues, prompting the preceding β4-β5 hairpin folded back and maintaining the trimer form (Fig. 1g). Conversely, at low calcium concentrations, the dissociation of the #3 and #4 Ca2+ ions from the central axis increases the flexibility of the L5-6 loop, allowing the β4-β5 hairpin to swing outward, enabling the formation of the domain-swapped hexamer (Fig. 1g).
The hexamer formation of C1ql1_gC1q couples with the partial rearrangement of the central Ca2+-axis
To test this hypothesis, we performed MD simulations to determine the free energy changes along the coordinates of the partial unfolding of the β4-β5 hairpin from the gC1q trimer with different numbers of Ca2+ coordination conditions. For each coordination, the MD simulation initiates from the same gC1q trimer structure. The well-equilibrated conformations were then used as the starting point for enhanced sampling, which was designed to map the free energy landscape of the protein unfolding process (Fig. 2a). For the gC1q trimer with all four Ca2+ ions bound (#1–4 Ca2+), a single dominant conformational state (S1) was identified, closely matching our crystal structure of the gC1q trimer. In this state, D208 interacts simultaneously with the #2 and #3 Ca2+ ions, maintaining a compact trimer interface (Fig. 2b, c). The free energy barrier for unfolding in the S1 state is more than 30 kcal/mol, preventing the unfolding process (Supplementary Fig. 2a). Upon the removal of the #4 Ca2+ ion (#1–3 Ca2+), the free energy map shows that the gC1q trimer can access a broader range of conformational space (Fig. 2b), exhibiting a configurational equilibration between two main basins (stable conformational microstates): the S1 state and an intermediate state (S2). Although conformation state S2 retains a similar trimeric interface, D208 only interacts with the #3 Ca2+ ion (Fig. 2b, c). Additionally, two high-energy basins (the S3 and S4 states) emerge, both featuring a loose trimeric interface and the unfolding of the β4-β5 hairpin. Notably, a structure rearrangement was observed where N212 spatially replaces D208 in the vacant coordination site of the #3 Ca2+ ion (Fig. 2b, c). The energy barriers between the S1 and S2 states and between the S3 and S4 states are lowered to ~17 kcal/mol (Supplementary Fig. 2a). This translates to a timescale of ~0.1–2.0 s for the unfolding process using transition state theory. Therefore, our simulation results indicate that it is feasible for an equilibrium transition between the folded and unfolded states of the β4-β5 hairpin upon calcium dissociation from the trimer binding pocket. Upon further reducing the number of bound Ca2+ ions to two (#1–2 Ca2+), the conformational equilibrium of the gC1q trimer is completely shifted towards the unfolded basin that samples states S3 and S4. In these states, the rearrangement of N212 attenuates the trimer contact and largely abolishes the coordination of Ca2+ (Fig. 2b, c). The transition from S3 to S4 is essentially barrierless (Supplementary Fig. 2a), thus promoting the subsequent formation of hexamers. Similar observations were further validated by parallel simulations (Supplementary Fig. 2d, e). Taken together, our MD simulations indicate that the coordination of the #3 Ca2+ ion and the conformational rearrangement of D208 and N212 acts as a switch for the unfolding mechanism.

a Illustration of the two collective variables to describe the unfolding process revealed by MD simulation. b Computed two-dimensional free energy surfaces for gC1q bound with four (left), three (middle), and two (right) Ca2+ ions along the central axis. The specific Ca2+ ions in the binding pocket are indicated at the top of each figure, and the equilibrium of four major conformational states was found in gC1q-WT (#1–3 Ca2+). c Characteristic structure features of the S1-S4 four conformational states indicated in (b). The representative structure model was extracted from MD trajectories. The spatial displacement of residue D208 by N212 was observed in the absence of the #3 Ca2+ ion. The open circles denote Ca2+ ions transiently bound from the bulk solvent during the simulation. The closed circles denote Ca2+ ions stably occupied throughout the simulation. d Computed two-dimensional free energy surface of gC1q-D208N, revealing a dominance of the unfolding states. e Computed two-dimensional free energy surface of gC1q-A209S, revealing a dominance of the folding states. f aSEC-based measurements of the trimer-to-hexamer transition for WT and mutated C1ql1_gC1q. The trimer fractions of C1ql1_gC1q proteins were collected and incubated for 0, 6, and 12 h at 37 °C. After incubation, the samples were immediately analyzed using aSEC to monitor the changes in the proportions of the trimer and hexamer.
Next, we designed two specific mutations to either promote or inhibit the conformational change during this transition and tested them with MD simulations. As residue D208 is crucial for coordinating the #3 and #4 Ca2+ ions, the charge-deficient mutation D208N disrupts this coordination, thereby favoring the formation of the domain-swapped hexamer (Fig. 2d). Indeed, our MD simulation showed that the enhanced sampling for gC1q-D208N (#1–3 Ca2+) only accesses the unfolding basin, a conformational landscape essentially the same as that of gC1q-WT (#1–2 Ca2+) (Fig. 2d). The presence of more Ca2+ ions in the simulation does not influence the unfolding basin of gC1q-D208N (Supplementary Fig. 2f). On the other hand, the A209S mutation, due to its proximity to N212 in a neighboring gC1q protomer, was expected to create an additional hydrogen bond with an adjacent chain (Supplementary Fig. 2b), locking L5-6 in the sharp-turn conformation and stabilizing the trimer form while inhibiting hexamer formation (Fig. 2e). Consistent with our design, the free energy landscape of gC1q-A209S (#1–3 Ca2+) reveals the dominance of the folding states (S1 and S2) (Fig. 2e), and the transition barrier to the unfolded states (S3 and S4) is elevated to ~33 kcal/mol (Supplementary Fig. 2c), eliminating the unfolding transition. Interestingly, the gC1q-A209S mutation appears to have no significant impact on the unfolding process when the Ca2+ ion concentration is reduced (Supplementary Fig. 2g).
To experimentally confirm these mutational effects, we collected trimer fractions of wild-type (WT) and two mutated C1ql1_gC1q proteins and analyzed the transition using aSEC assay after incubation at 37 °C to reach equilibrium (Fig. 2f). The WT protein showed the ability to generate the hexamer, although the trimer fraction remained higher than the hexamer fraction (Fig. 2f, left). However, the D208N mutant showed a clear trend towards hexamer formation even before incubation, with the hexamer fraction becoming dominant at equilibrium (Fig. 2f, middle). In contrast, the A209S mutant showed minimal hexamer formation after adequate incubation (Fig. 2f, right). Notably, the hexamer formation is reversible, as indicated by the same incubation strategy, while starting with the hexamer fraction (Supplementary Fig. 3a). Furthermore, increased Ca2+ concentrations reduce the trimer-to-hexamer transition (Supplementary Fig. 3b) while promoting the hexamer-to-trimer transition (Supplementary Fig. 3c). This suggests that low Ca2+ allows dynamic trimer-hexamer equilibria, whereas high Ca2+ stabilizes the gC1q protein in the trimeric state through Ca2+ chelation at the central axis. Collectively, we conclude that C1ql1_gC1q possesses the ability to transition between its typical trimer form and the domain-swapped hexamer form, which is coupled with a reorganization of the central Ca2+-axis.
We note that the domain-swapping and central Ca2+-binding sequences are highly conserved among the four members of the C1ql family (Supplementary Fig. 4a), suggesting that a similar hexamer formation may be shared in other C1ql proteins. In line with this, previous biochemical analyses of C1ql3_gC1q have demonstrated hexamer formation19. Nevertheless, our aSEC data reveal that C1ql3_gC1q has a significantly reduced propensity for hexamerization compared to C1ql1_gC1q, and C1ql2/4_gC1qs show minimal trimer-to-hexamer transitions under identical conditions (Supplementary Fig. 4b).
C1ql1_gC1q interacts with the C-terminal extended CUB domain of BAI3 in a Ca2+-dependent manner
BAI3 has been suggested as the specific CAM on the postsynaptic membrane of PC to interact with C1ql1 released by CF terminals, mediating the development and synaptic transmission of CF-PC synapses12,13,29. To map the minimal region in BAI3 for C1ql1_gC1q binding, we purified BAI3 extracellular fragments with different boundaries and analyzed their binding to the C1ql1_gC1q trimer by employing aSEC and ITC methods (Fig. 3a). Consistent with previous reports12, the extracellular region of BAI3 binds to C1ql1_gClq in the Ca2+-containing buffer condition with an affinity of ~1.1 μM (Fig. 3a, b). Further mapping showed that the TSR domain cannot interact with C1ql1_gC1q (Fig. 3a, b and Supplementary Fig. 5a). Instead, the C-terminal extended constructs of the CUB domain (eCUBs) show sub-μM affinities to the C1ql1_gC1q trimer (Fig. 3a, b), consistent with previous findings12. Among these, eCUB253 (aa. 25–253) demonstrates the highest binding affinity of ~0.15 μM (Fig. 3a, b). Interestingly, further truncating the extension sequence (eCUB186) renders the protein incapable of interacting with C1ql1 (Fig. 3a,b and Supplementary Fig. 5a), indicating that the sufficient CUB-extension sequence is essential for the C1ql1/BAI3 interaction.

a Domain organization of human BAI3. Six fragments with indicated boundaries in the extracellular domain (ECD) of BAI3 were used to determine their binding capacities for C1ql1_gC1q by aSEC and ITC assays in the buffer containing 2 mM Ca2+. Consequently, the C-terminal extended CUB (eCUB) domain, which contains the typical CUB domain and the following C-terminal extension, constitutes the minimal binding region required to interact with C1ql1_gC1q. b ITC-based measurements of BAI3 boundaries binding to the C1ql1_gC1q with the representation of the fitting curves for ITC titrations. 2 mM Ca2+ was added to the buffer. The calculated binding affinities have been listed in (a). c–e aSEC (c, e) and ITC (d) -based analyses of BAI3_eCUB binding to the C1ql1_gC1q trimer and hexamer at a gradient concentration of Ca2+ in the buffer. The Ca2+-dependent binding of eCUB to the gC1q trimer titration was indicated by ITC-measured binding affinities across a range of Ca2+ concentrations under buffer conditions. f Time-course aSEC analyses are used to monitor the potential transition of the C1ql1_gC1q hexamer and trimer upon BAI3_eCUB binding under high calcium conditions.
Consistent with the reported requirement of Ca2+ for the C1ql1/BAI3 interaction29, we found that this binding is abolished in the absence of Ca2+ in the buffer (Fig. 3c). To explore the specific Ca2+ concentrations required for this interaction, we measured the formation of C1ql1_gC1q and BAI3_eCUB253 complex across a range of buffer conditions with varying Ca2+ concentrations using aSEC and ITC assays. As a result, at Ca2+ concentrations exceeding 0.5 mM, the complex forms effectively (Fig. 3c). ITC-based quantification further reveals a rapid transition in binding affinity at Ca2+ concentrations around 0.1–0.5 mM (Fig. 3d and Supplementary Fig. 5b). This suggests that a sub-mM to mM level of Ca2+ may be necessary in the synaptic cleft for efficient interaction between C1ql1 and BAI3. Similarly, the C1ql1_gC1q hexamer also forms complexes with BAI3_eCUB253 in a Ca2+-dependent manner, despite requiring a higher concentration of Ca2+ for stable complex formation (Fig. 3e). Interestingly, our time-course aSEC analyses showed that BAI3 binding blocks the transition between the gC1q hexamer and trimer (Fig. 3f).
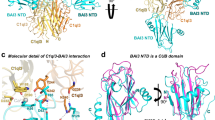
Cryo-EM structure of the C1ql1_gC1q/BAI3_eCUB complex
To understand the molecular mechanism underlying the interaction between C1ql1 and BAI3, we employed the cryo-EM technique to determine the complex structure of C1ql1_gC1q and BAI3_eCUB253. Given that protein particles with large molecular sizes are preferred for single-particle cryo-EM analysis, we focused on the complex of gC1q hexamer and eCUB253 and successfully determined the complex structure of one C1ql1_gC1q hexamer bound to four molecules of BAI3_eCUB253 at an overall resolution of ~3.5 Å (Fig. 4a, b, Supplementary Fig. 6a–d, and Supplementary Table 2). Consistent with the apo hexamer of C1ql1_gC1q observed in our crystal structure (Fig. 1c, d), the eCUB-bound hexamer is also mediated by domain-swapping from one pair of gC1q chains between two gC1q trimers (Supplementary Fig. 6e), where the two Ca2+-axes are arranged with an angle (Fig. 4b and Supplementary Fig. 7a), and each trimer in hexamer of gC1q recruits two eCUB molecules inserting into the trimer-assembling cleft formed by two adjacent gC1q chains (Fig. 4b). Upon superimposing the gC1q/eCUB interface with the two remaining eCUB-unbound gC1q molecules in the hexamer, we found that a steric hindrance precludes additional eCUB binding (Supplementary Fig. 7b), explaining why the gC1q hexamer does not form complexes with more than four molecules of eCUB. However, similar to the observation in the crystal structure of the hexamer, only the two bottom Ca2+ ions can be observed in the central Ca2+-axis of each trimer (Supplementary Fig. 6f), further confirming the coupling of domain-swapped hexamer formation and central Ca2+-axis reorganization in C1ql1_gC1q. Notably, unlike the gC1q hexamer, the gC1q trimer binds eCUB with a 3:3 stoichiometry, as indicated by our ITC data (Fig. 3b). This observation aligns with a recent structural study of the C1ql3_gC1q trimer in complex with three BAI3_eCUB molecules36, indicating the different eCUB binding stoichiometries of different gC1q oligomeric states.

a, b Cryo-EM density map (a) and atomic model (b) of the C1ql1_gC1q hexamer and BAI3_eCUB complex. In this complex structure, the C1ql1_gC1q hexamer interacts with four molecules of BAI3_eCUB. A rotational view depicting the interaction of each C1ql1_gC1q trimer and two BAI3_eCUB molecules is illustrated, further showcasing their molecular interfaces. The bound Ca2+ in the central axis and the interface, and the Ca2+-axis traversing the central calcium ions in each trimer are indicated by gray spheres and blue lines, respectively. c Cryo-EM structure of the focused subcomplex containing one gC1q trimer and one molecule of eCUB. BAI3_eCUB contains a classical CUB domain and a C-terminal CUB extension. d Detailed interface between C1ql1_gC1q and BAI3_eCUB. The interface Ca2+ ion was represented by a gray sphere. e Density-fit-model mode to show the key interface residues and the interface Ca2+ ion. f Overall structure of BAI3_eCUB. The CUB domain contains eight β-strands, while the CUB extension contains an α-helix (αC) followed by a loop region (LC). The atomic model of residues 28–203 in eCUB can be built, while the others in the two termini are flexible, indicated by dashed lines. Two and three disulfide bonds are observed in the CUB domain and CUB-extension regions, respectively. g, h ITC-based binding analyses between C1ql1_gC1q variants in their trimeric forms and BAI3_eCUB (g) and between C1ql1_gC1q trimer and BAI3_eCUB variants (h). i Multisequence alignment of the main gC1q-interacting region in the BAI family members. The key interface residues are labeled by arrowheads. The two interface residues that are not conserved in BAI1 are highlighted by blue circles. j aSEC-based analysis showing the disruptive interaction between C1ql1_gC1q and the E60R/T65R mutant of BAI3_eCUB.
In the complex structure, the four gC1q/eCUB interfaces are highly similar (Supplementary Fig. 8a). By focusing on the subcomplex of one gC1q trimer and one bound eCUB, we obtained an improved local map with 3.34-Å resolution (Fig. 4c, Supplementary Fig. 8b, c, and Supplementary Table 2), which provided sufficient quality to unambiguously assign the two protein models, including the sidechain conformations of interface residues (Fig. 4d, e). The CUB domain in BAI3_eCUB adopts a typical CUB fold with an eight-strand β-sandwich structure (Fig. 4f and Supplementary Fig. 7c). In addition to the traditional CUB domain, a C-terminal extension of the CUB domain containing an α-helix (αC) and a following loop (LC) is involved in BAI3_eCUB folding (Fig. 4f and Supplementary Fig. 7c). The αC-helix tightly packs with the β-sandwich mediated by strong hydrophobic contacts (Supplementary Fig. 7d), while the LC-loop forms three disulfide bonds with αC (Fig. 4f and Supplementary Fig. 9), coupling with the additionally surrounding hydrophobic packing, to further stabilize the overall folding (Fig. 4f and Supplementary Fig. 7e). Consistently, BAI3_eCUB186, which lacks this loop, was expressed in a more aggregate state and thereby loses its ability to bind C1ql1_gC1q (Fig. 3a, b and Supplementary Fig. 5a). Although neither the αC-helix nor the LC-loop directly contributes to the gC1q/eCUB binding interface, our results verify the importance of the extension region for maintaining the proper folding of the CUB domain, facilitating the binding with C1ql1.
Importantly, a sandwiched Ca2+ ion is observed at the gC1q/eCUB interface (Fig. 4d, e), which coordinates with several aspartate residues from both gC1q (D193 and D235) and eCUB (D63) (Fig. 4d). This Ca2+ position on the surfaces of β4 and β5 of gC1q is highly conserved in the gC1q structures of C1ql family members previously reported19 (Supplementary Fig. 7f), indicating the critical role of Ca2+ in mediating the interaction with BAI3. Consistently, mutating the Ca2+ coordination sites, including D193N and D235A in C1ql1 and D63A in BAI3, results in a significant reduction or even a complete loss of binding affinity (Fig. 4g, h). In addition to Ca2+ coordination, several residues in the L7-8 loop of gC1q, especially Y247, form hydrogen bonds with N126, E60, and D63 in eCUB, while T65 and K66 in the L3-4 loop of eCUB interact with W191 and D235 in gC1q (Fig. 4d). Consistent with our structural findings, disrupting these interactions by introducing mutations in either C1ql1 (Y247A) or BAI3 (T65A and K66A) can significantly reduce or completely abolish their interaction (Fig. 4g, h). Notably, previous studies have shown that the introduction of an artificial glycosylation site at S244, designed to create steric hindrance, blocks BAI3 binding12. This site is located between the interface residues N243 and N245 in the L7-8 loop of gC1q, which is in immediate proximity to the BAI3-binding interface (Fig. 4d), further confirming our structural findings.
The C1ql/BAI interaction likely specific to BAI2/3 but not BAI1
C1ql families have been found to interact with BAI312,13,15,29,37. To explore the potential isoform-specific binding, we analyzed the C1ql1_gC1q/BAI3_eCUB interface across different isoform sequences. The interface residues are highly conserved among C1ql1 family members (Supplementary Fig. 4a), suggesting that all C1ql proteins possess the ability to interact with BAI3. Consistently, C1ql2, C1ql3, and C1ql4 also form complexes with BAI3_eCUB in the Ca2+-containing buffer (Supplementary Fig. 4c), in agreement with previous reports15,29. Sequence alignment result shows that two interface residues, E60 and T65, in the L3-4 loop of BAI2/3 are not conserved in BAI1 (Fig. 4i and Supplementary Fig. 9). In line with this, a double mutation in BAI3 (E60R/T65R) designed to mimic BAI1 leads to a loss of interaction with C1ql1_gC1q (Fig. 4j), suggesting that BAI1 may not possess the ability to associate with C1qls. Thus, our findings indicate that the C1ql/BAI interaction is not uniformly conserved across the BAI family members, providing a potential explanation for the functional diversity of BAI isoforms.
The central Ca2+-axis allosterically modulates the binding of C1ql1_gC1q to BAI3_eCUB
By comparing the structure of eCUB-bound C1ql1_gC1q with the apo structures of the gC1q domain in C1qls, we found that while most regions maintain similar conformations (Fig. 5a), the L7-8 loop, a major component of the eCUB-binding cleft (Fig. 4d), undergoes a conformational change between gC1q structures with and without Ca2+ in the central axis (Fig. 5a). Specifically, the sidechain of N243 in L7-8 of gC1q changes its position in the absence of Ca2+ ions (Fig. 5b), consequently disrupting the hydrogen bond with E60 in eCUB that is present in the complex (Fig. 5b). In addition, the interface residues, D235 and W191, shift away from the positions for BAI3_eCUB binding (Fig. 5b), potentially losing the coordination with the interface Ca2+ ion and the interaction with T65 in eCUB. These structural changes indicate that, in addition to the interface Ca2+ ion, the central Ca2+ ions may also influence the interaction with BAI3_eCUB through allosteric regulation of the gC1q interface conformation. To verify the role of the central axis in BAI3_eCUB binding, we measured the affinities of C1ql1_gC1q with the specific mutations essential for the Ca2+-axis formation. Indeed, the triple mutant D208/210/214N, presumably disrupting the coordination with all four central Ca2+ ions, completely abolishes the binding of C1ql1_gC1q to BAI3_eCUB (Fig. 5c, d). Notably, while the single mutations D210N and D214N only cause a subtle decrease in binding affinity, the D208N mutation weakens this interaction by ~9-fold (Fig. 5c, d). This result supports the key role of D208 in coordinating Ca2+ ions, which is crucial for maintaining the central axis conformation of gC1q, indirectly impacting BAI3_eCUB binding.

a Structural comparison of the BAI3-binding surface on the gC1q domain from the eCUB-bound C1ql1_gC1q structure, the Ca2+-bound apo C1ql1_gC1q structures of both trimer and hexamer forms, and the Ca2+-free apo C1ql2_gC1q structure (PDB: 4QPY). The regions containing β4, β5, β7, and L7-8 on the gC1q domain, crucial for interacting with BAI3_eCUB and the Ca2+ ions, are highlighted in color. The conformational changes, presumably induced by Ca2+ binding, are shown in enlarged views in (b). b Detailed structural analysis of the BAI3-binding surface boxed in (a). The key residues in the gC1q domain that mediate the interaction represented in Fig. 4d are shown in stick mode. The conformational changes of interface residues D235 in β7, N243 in L7-8, and W191 in β4 are indicated, showing the differences in these residues among various gC1q structures, especially with or without Ca2+ binding. c, d ITC-based affinity measurements between BAI3_eCUB and C1ql1_gC1q variants in their trimeric forms, which have been demonstrated to alter conformational dynamics in their central Ca2+-axis (D208N, D210N, and D214N) and the hexamer formation (A209S). e Cell surface imaging analysis of the recruitment of HEK293T cell-secreted full-length (FL) C1ql1 and its mutants by overexpressed BAI3 or GFP-vector on the cell surface. The corresponding line analyses of GFP and Flag intensities along the cell surfaces are indicated by white dashed lines as shown on the bottom. f Statistical analysis of the relative intensity ratio of Flag to GFP on the cell surface derived by quantifying the fluorescent signals as shown in (e). Data were collected from 20 cells in each condition. Bars represent the means ± SEM and statistical significances were determined by one-way ANOVA with Dunnett’s multiple comparisons test compared with the WT BAI3-GFP/C1ql1-Flag group.
The gC1q trimer/hexamer transition contributes to the binding of C1ql1 to BAI3 on the cell surface
Given that the central Ca2+-axis is reorganized during the hexamer formation of C1ql1_gC1q (Fig. 1g), the gC1q trimer/hexamer transition may impact the binding of C1ql1 to BAI3_eCUB. Consistently, the A209S mutant of C1ql1_gC1q, which is primarily locked in its trimeric conformation (Fig. 2e, f and Supplementary Fig. 2b), exhibits a stronger affinity for BAI3_eCUB (Fig. 5c, d). To further analyze the role of the gC1q hexamer in the C1ql1/BAI3 interaction, we performed a cell-based recruitment assay. In this assay, a similar amount of HEK293T cell-secreted full-length C1ql1 protein or its mutants was added to the culture medium of cells overexpressing GFP-tagged BAI3 on the cell surface (Supplementary Fig. 10a). Compared to cells overexpressing GFP alone, wild-type C1ql1 was highly accumulated on the surface of cells overexpressing BAI3-GFP (Fig. 5e). As a control, the Y247A mutant of C1ql1, which was impaired in binding with BAI3_eCUB in vitro (Fig. 4g), failed to enrich on the cell surface of BAI3-overexpressing cells (Fig. 5e, f). Similarly, the D208N mutant, which weakens eCUB binding while promoting gC1q hexamer formation (Figs. 2f and 5d), shows reduced recruitment onto the surface of cells overexpressing BAI3 (Fig. 5e, f). Surprisingly, the A209S mutant of C1ql1, which was expected to have an enhanced binding due to the in vitro higher affinity (Fig. 5d), displayed relatively weakened accumulation on cells overexpressing BAI3, compared to that of wild-type C1ql1 (Fig. 5e, f). It is likely that the A209S mutant has decreased binding avidity to cell surface-accumulated BAI3, caused by its reduced oligomerization capability (Supplementary Fig. 10b), supporting a role for gC1q-mediated C1ql1 oligomerization in the assembly of C1ql1/BAI3 complexes on the cell surface. Thus, our findings suggest that the impaired transition between the gC1q trimer and hexamer may compromise the BAI3-dependent recruitment of C1ql1 to the plasma membrane.
C1ql1 forms higher-order oligomers through both NT- and gC1q-mediated hexamers
Given that the full-length C1ql1 protein has been detected to form higher-order oligomers in which an essential role of the N-terminal disulfide-linkage-mediated hexamer is identified22 (Fig. 6a), it is intriguing to explore whether the domain-swapped hexamer form of the gC1q domain indeed occurs in the full-length protein and contributes to the high oligomerization of C1ql1. Analyzing the bacteria-expressed full-length protein of C1ql1 using aSEC, we identified the hexamer (P2) and trimer fractions (P3) (Fig. 6b and Supplementary Fig. 10c, d). Employing the negatively-stained EM (NS-EM) method, we found that the trimer particles from the P3 fraction have a “one-cored” shape (Fig. 6c), akin to the trimeric structure of the gC1q domain. In contrast, the particles from P2 predominantly show a “dual-cored” shape (Fig. 6d), which can be divided into two distinct classes (Fig. 6d). The major class is featured by two closely associated “cores” (Fig. 6e), resembling the domain-swapped hexamer of C1ql1_gC1q. This configuration was further validated by subsequent cryo-EM analysis of the P2 fraction (Fig. 6e), confirming that the gC1q-mediated hexamer formation indeed exists in the full-length protein. The other class exhibits a dumbbell-like shape, with two “cores” connected by a thin linker (Fig. 6e), a configuration similar to the hexamer previously proposed to be mediated by the NT region, which includes two cysteine residues for intermolecular disulfide linkages in both C1ql and Cbln families10,22.

a Sequence alignment showing the two conserved cysteine residues indicated by orange triangles in the NT region among C1ql family members. b aSEC analyses of bacteria-expressed full-length C1ql1 proteins in the absence or presence of DTT in the buffer. Three distinct peaks were observed in the elution profile, indicating its high-oligomer, hexamer, and trimer forms, respectively. c, d Negative-staining EM analyses of the different assemblies of the full-length, wild-type C1ql1 protein samples in fractions P3 (c) and P2 (d). In the P2 fraction, two types of hexamers were observed, indicated by green (gC1q-mediated hexamers) and red arrowheads (NT-mediated hexamers). To enhance the contrast, the representative images were low-pass filtered to 10 Å. e EM analyses of protein particles from the P2 fraction using 2D-classification. f Negative-staining EM analysis of the particles in the P1 fraction. The cluster-like patterns were circled by black lines. A magnified view of one of the particle clusters observed in the P1 fraction. A serpentine structure is observed, in which two adjacent hexamers resembling gC1q- and NT-mediated hexamers are labeled by green and red arrowheads, respectively. The representative images were low-pass filtered to 10 Å. g Cell surface recruitments of HEK293T cell-secreted full-length C1ql1 and its C2S mutant by BAI3 on the cell surfaces. The corresponding line analyses of GFP and Flag intensities along the cell surfaces were indicated by white dashed lines as shown on the right. h Statistical analysis of the relative intensity ratio of Flag to GFP on the cell surface derived by quantifying the fluorescent signals as shown in (g). Data were collected from 20 cells in each condition. Bars represent the means ± SEM. The unpaired Student’s t-test two-sided analysis was used to define a statistically significant difference. i NS-EM analyses of the full-length C1ql1-C2S mutant secreted from HEK293F cells. gC1q-mediated hexamer particles were indicated by green arrowheads.
The coexistence of gC1q and NT-mediated hexamer formation raises the possibility that C1ql1 may form large oligomers through a continuous arrangement. Interestingly, aSEC analysis of C1ql1 reveals a higher-order oligomeric state corresponding to the P1 fraction (Fig. 6b and Supplementary Fig. 10c). Direct observation under NS-EM indicates that protein particles in the P1 fraction tend to aggregate, forming clusters of varying sizes (Fig. 6f). A detailed analysis of the clustered assembly of C1ql1 shows that a serpentine structure, in which adjacent hexamers are connected sequentially by gC1q and NT, can be clearly identified (Fig. 6f). The N-terminal cysteine residues from two trimers may form disulfide linkages to connect the trimers, thereby mediating C1ql1 oligomerization. Consistent with this, adding reducing reagents, which disrupt disulfide bond formation, significantly diminished the P1 fraction. (Fig. 6b). These observations collectively suggest that C1ql1 can form a higher-ordered assembly, likely arranged by the serial connection of NT- and gC1q-hexamers. Interestingly, the C2S mutation, which eliminates disulfide bond formation in the NT region22, severely reduces the ability of C1ql1 to be recruited to the cell surface by BAI3 (Fig. 6g, h and Supplementary Fig. 10a). Using full-length proteins secreted by HEK293T/F cells, we further confirmed that the C2S mutation largely disrupts C1ql1 oligomerization (Supplementary Fig. 10b), while preserving C1ql1’s capability in gC1q-mediated hexamer formation (Fig. 6i and Supplementary Fig. 10e). Our finding of the NT region in promoting C1ql1 oligomerization further implies that the higher-ordered, clustered form of C1ql1 may play a crucial role in the synaptic assembly of CF-PC synapse through multivalent interactions with BAI3 on the postsynaptic membrane.
The dynamic assemblies of C1ql1 and its interaction with BAI3 are critical for the maintenance of climbing fiber synapses
Previous works reported that C1ql proteins affect synapse density in cultured hippocampal neurons23,29, suggesting that different oligomeric forms of gC1q might have different effects on synapse properties. Thus, we directly addressed the role of the gC1q hexamer on synapses in vivo, particularly CF synapses, as C1ql1/BAI3 signaling is known to be essential for the maintenance of CF-PC synapses12,13,28. We first crossed C1ql1+/− mice to generate C1ql1+/+ (Ctrl) and C1ql1−/− mice (KO) (Fig. 7a). The impact of C1ql1 deletion on mature CFs from mice on postnatal day 21 (P21) was assessed by immunostaining vesicular glutamate transporter 2 (vGluT2) in the cerebellum, a marker for CFs (Fig. 7b). Consistent with the previous studies12,27, we observed about 30 and 60% reduction in vGluT2 density around the proximal and distal dendrites of PCs, respectively (Fig. 7c and Supplementary Fig. 11a). Moreover, the puncta size increased after C1ql1 deletion (Fig. 7d and Supplementary Fig. 11b). To check whether the gC1q trimer or hexamer can rescue the CF-PC synaptic deficits observed in KO mice, we overexpressed full-length C1ql1 or its mutants (A209S and D208N), which interfere with the gC1q trimer or hexamer formation (Fig. 2d–f); plus a control mutant Y247A, which abolishes BAI3 binding (Fig. 4g), via virus injection into the inferior olives (IOs) in C1ql1-KO mice at P21 (Fig. 7e). Comparable protein expression levels of C1ql1 variants in the IO region two to three weeks after injection were confirmed by immunohistochemistry (Fig. 7f, g and Supplementary Fig. 11c). The vGluT2 puncta along CFs with positive GFP signals in the cerebellum were selected for analysis. Quantification revealed a restoration of vGluT2 density in the wild-type C1ql1 expression group, with synaptic sizes comparable to the GFP control group (Fig. 7f, h–k). This partial rescue effect could be due to an insufficient amount of full-length C1ql1 available for the climbing fiber or insufficient time for the puncta size to recover. In contrast, both D208N and A209S mutants failed to rescue the decreased vGluT2 number, phenocopying the BAI3 binding-deficient mutant Y247A (Fig. 7f, h–k). Thus, our mouse genetic data further confirm the critical roles of both the dynamic assemblies of C1ql1, mediated by the different oligomerization of the gC1q domain, and its interaction with BAI3 in maintaining CF-PC synapses in vivo.

a Breeding strategy for generating C1ql1 wild-type (Ctrl) and C1ql1-null (KO) mice. b Immunohistochemistry of vGluT2-positive puncta onto Purkinje cells from Ctrl and KO mice. 0% and 50% are the values we set to distinguish whether the climbing fibers on the Purkinje cell reach the dendritic site or stay at the somatic site. Scale bar, 10 μm. c, d Cumulative plots of the number (c) and size (d) of vGluT2 puncta. Statistical significances were determined by the two-sided Kolmogorov–Smirnov test. e Cartoon representation of the injection of AAV viruses containing full-length, wild-type (WT) C1ql1 or its mutants into IOs of P21 C1ql1-KO mice. f Immunohistochemistry and representative images of HA-tagged C1ql1 and vGluT2 puncta along GFP-positive CFs. Scale bar, 5 μm. g Quantification of the intensity of HA-tagged C1ql1 and its mutants from (f). h, i Statistic (h) and cumulative plot (i) of vGluT2 puncta number along GFP-positive CFs from (f). The numbers of branches/mice were indicated in bars. Data are represented as means ± SEM, and statistical significances were determined by one-way ANOVA with Dunnett’s multiple comparisons test compared with the WT group. j, k Statistic (j) and cumulative plot (k) of vGluT2 puncta size along GFP-positive CFs from panel f. The numbers of branches/mice were indicated in bars. Data were represented as means ± SEM, and statistical significances were determined by one-way ANOVA with Dunnett’s multiple comparisons test compared with the WT group. l A proposed model of C1ql1 oligomerization facilitated by both NT- and gC1q-mediated hexamer formation in mediating the clustering of BAI3 GPCRs on the surface of Purkinje cells for CF-PC synapse development.
Discussion
It has been established that the CF-PC connection is mainly regulated by a specific interaction between the CF-secreted C1ql1 and the aGPCR BAI3 on the PC membrane12,13. In this study, we determined the cryo-EM structure of the C1ql1_gC1q/BAI3_eCUB complex, revealing their binding mode in a calcium-dependent manner. The hexamer formation of C1ql1_gC1q, mediated by domain swapping between two classical trimers, may enhance cell surface recruitment by BAI3 and thus contribute to CF-PC synapse organization in mice. Considering both the NT-mediated head-to-head hexamer formation and the gC1q-mediated tail-to-tail hexamer formation, we propose an assembly model for C1qls and BAI3 complexes, crucial for the CF-PC synaptic connection (Fig. 7l). In this model, secreted C1ql proteins initially form classical trimers and further connect to become hexamers through the NT-mediated disulfide bond formation, given the oxidized environment in the synaptic cleft38. Under certain conditions, such as Ca2+ fluctuation, gC1q-mediated hexamerization promotes the further assembly of C1ql proteins into higher-order oligomers in a tandem fashion. Due to the high flexibility of the linker regions between NT and gC1q, the linear C1ql oligomer forms a serpentine cluster. It is plausible that these C1ql1 clusters may efficiently bind and accumulate BAI3 on the postsynaptic surface of PCs in a Ca2+-dependent manner, thereby amplifying BAI3-mediated intracellular downstream signals, a mechanism that warrants further investigation. Nevertheless, given the partially functional disruption caused by the A209S mutation (Figs. 5e, f and 7h–k), the association of BAI3 to the trimeric form of gC1q may still contribute to the synaptic connectivity (Fig. 7l). In summary, our model provides insights into the molecular dynamics underlying the formation and maintenance of CF-PC synapses, underscoring the role of C1ql1 oligomerization in modulating synaptic connectivity. We note that the higher-order oligomeric state of C1ql1 shown in Fig. 6f was analyzed using full-length C1ql1 expressed in bacteria, lacking posttranslational modifications. Given this limitation, our model may not fully recapitulate the assembly mode of C1ql1 in vivo, where posttranslational modifications and additional cellular factors likely render the assembly process more complex.
Our findings suggest the role of the gC1q trimer/hexamer transition in C1ql1 for its binding to BAI3 on the cell surface, facilitating the establishment of CF-PC synapses. Our biochemical and MD simulation experiments suggest that the hexamer formation of the gC1q domain requires overcoming energy barriers, indicating that additional proteins or environmental factors in the synaptic cleft may modulate the dynamics of C1ql oligomerization. For example, the synaptic cleft is known to experience fluctuations in calcium ion concentration and pH levels coincident with signal transduction39. These changes could potentially influence the stability and conformation of the gC1q domain, thereby affecting C1qls’ ability to oligomerize and interact with BAI3. Specifically, the Ca2+ fluctuations in the synaptic cleft are linked with signal transmission between pre- and post-synapses, occurring within a range of millimolar concentrations40. It is important to note that Ca2+ ions exert multifaceted influences on the conformation and function of C1qls. Ca2+ ions stabilize the central axis conformation in the gC1q domain, modulate the formation of the gC1q trimer and hexamer (Fig. 2 and Supplementary Fig. 3), and directly mediate the C1ql1/BAI3 interaction (Figs. 3 and 4). It is possible that the interplay between the Ca2+-modulated gC1q hexamer of C1qls and the Ca2+-dependent interaction of C1qls with BAI3 may require a more delicate regulation of Ca2+ signal in synapses.
Notably, BAI3 binding to either the C1ql1_gC1q hexamer or trimer occludes the hexamer/trimer transition, even under high Ca2+ concentrations (Fig. 3f). Given the critical role of the gC1q β4-β5 hairpin in domain-swapped conformational transitions, its involvement in BAI3_eCUB binding (Fig. 4c, d) mechanistically explains how BAI3 binding stabilizes the hexameric state via steric hinderance of the gC1q hexamer/trimer transition. In addition to BAI3, C1ql family proteins have been reported to bind to other synaptic CAMs, including neurexin14 and GluK27, via their gC1q domains. It is tempting to speculate that these interactions may also participate in regulating the oligomeric states of gC1q, consequently modulating C1ql oligomerization and influencing the connection between the pre- and postsynaptic compartments. This regulatory mechanism may extend beyond CF-PC synapses to other synapses that are regulated by C1ql2 and C1ql3, although their propensity for gC1q hexamer formation is much less robust than that of C1ql1_gC1q (Supplementary Fig. 4b).
Considering the gC1q domain’s capacity to mediate the formation of higher-order oligomers, it is noteworthy that several secreted proteins implicated in cell adhesion also form oligomers, with the majority being members of the C1q/TNF superfamily that all contain a gC1q domain. Several higher-order oligomerization models for C1q/TNF superfamily members have been proposed10,41,42. For instance, Cbln1 is known to form hexamers via disulfide bond formation involving cysteine residues, while C1q assembles oligomers through its collagen-like sequences. Instead, we found the formation and function of gC1q-mediated hexamers through domain swapping, thereby broadening our understanding of gC1q-mediated roles in cell adhesion. We note with interest that C1q/TNF superfamily proteins like adiponectin recognize different receptors in specific tissues via different assemblies43. In addition to the C-terminal gC1q domain, the N-terminal cysteine residues and collagen-like sequences also contribute to high-molecular-weight oligomerization of C1ql proteins22,41,42. It is possible that C1ql proteins may combinatorially use these three regions to form diverse assemblies, serving various functional purposes. Notably, a cryo-EM structure of the gC1q trimer of C1ql3 in complex with BAI3_eCUB has recently been determined36, and this structure shares an essentially identical BAI3-binding interface with ours. However, based on intermolecular contacts between BAI3 and C1ql3 molecules observed in their cryo-EM sample, the authors proposed an alternative assembly mode: a hexagonal lattice network formed by the C1ql3/BAI3 complex36. Future investigations are required to validate these higher-order assembly mechanisms across different C1ql and BAI family members.
Methods
Mice
Heterozygous C1ql1 knockout mice (C57BL/6J background) were kindly provided by the laboratory of Dr. David Martinelli44. To establish a breeding colony, those mice were outcrossed to a wild-type C57BL/6J strain (Guangdong Medical Laboratory Animal Center, China). For all experiments, control (C1ql1+/+) and homozygous knockout (C1ql1−/−) littermates were produced by crossing heterozygous males and females. Mouse genotypes were determined by polymerase chain reaction (PCR) on genomic DNA extracted from tail biopsies. The PCR assay employed three primers: P1: 5’-TGAGCTACCAGTCCAAGCTG-3’; P2: 5’-CAGTGCAATTGCCCAGGATG-3’; P3: 5’-GGCTAAGGACAATTCAGCCT-3’. The wildtype allele was identified by a 314 bp band, while the knockout (KO) allele produced a 262 bp band. All mice used in this study were housed in the Peking University Shenzhen Graduate School animal facility. Mice were weaned at 21 days under a 12-h light/dark cycle and had free access to standard laboratory food and water. All animal procedures followed the guidelines approved by the Peking University Shenzhen Graduate School Animal Care and Use Committee and Shenzhen Bay Laboratory Animal Care and Use Committee.
Plasmids
The mouse C1ql1-4 genes and the human BAI3 gene were purchased from Youbio Life. For bacterial-based protein purification, C1qls’ full-length (without signal peptide sequence) or truncation genes were individually cloned into a modified pET32 vector with the thioredoxin (Trx) and His tags, where the Trx tag as an oxidoreductase may play a role in promoting proper disulfide bond formation during the folding and assembly of full-length C1ql1 protein45, followed by a 3C protease cleavage site at the N-terminus. For protein expression in insect cells, BAI3 boundaries were inserted into the pFastbac-HTB vector with a gp67 signal peptide at the N-terminus and a TEV cleavage site, and a His-Flag tag at the C-terminus. For protein production in mammalian HEK293F cells, BAI3 boundaries and full-length C1ql1-C2S were inserted into the pMC2 vector with a preprotrypsin leader signal peptide35 and a Flag tag at the N-terminus, and a 3C enzyme cleavage site and a His tag at the C-terminus. For the cell surface recruitment assay, full-length C1ql1 variants were inserted into the pCDNA3.1(+) vector with a Flag tag at the C-terminus, while full-length BAI3 was cloned into a pEGFP vector with an EGFP tag at the C-terminus. All mutations were introduced by the direct PCR method using designed mutagenesis primers.
Protein expression and purification
The full-length C1ql1, the gC1q domain of C1ql1-4, and their mutants were overexpressed using the E.coli expression system. In brief, the modified pET32 plasmids encoding the target protein fused with His and Trx tags at the N-terminus were transfected into BL21 (DE3) E. coli competent cells, which were further cultured at 37 °C for ~3.5 h until the OD600 was up to 0.8. Then, 0.2 mM isopropyl-β-D-thiogalactopyranoside (IPTG) was added to induce protein expression at 16 °C. After overnight induction, cells were collected and lysed in a buffer containing 50 mM Tris, pH 7.5, 500 mM NaCl, 5 mM imidazole, and PMSF. After centrifugation to remove precipitate, the supernatant was loaded into Ni-NTA Beads 6FF (Smart-Lifesciences), and the eluted protein was treated with 3C protease to remove the Trx-His tag. Then, the target protein without tag was subjected to size-exclusion chromatography using a HiLoad Superdex 75 pg column (Cytiva) equilibrated with a buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl. Notably, for the full-length C1ql1 protein, an additional purification by an ion exchange column (Hitrap SP HP, Cytiva) was performed before size-exclusion chromatography to remove junk proteins.
The BAI3 constructs and the C1ql1-C2S mutant were overexpressed in insect High Five cells or mammalian HEK293F cells. After 2/3-day transfection by the pre-packaged baculovirus or the plasmids, the medium containing secreted target proteins was collected, then concentrated using Amicon Stirred Cells (Merck), and incubated with Ni-NTA Beads 6FF overnight at 4 °C. The BAI3 proteins enriched on the NTA beads were digested with TEV or 3 C protease to remove the tag, and the eluted proteins were further purified by size-exclusion chromatography using a HiLoad Superdex 200 pg column (Cytiva) equilibrated with a buffer containing 50 mM Tris, pH 7.5, and 150 mM NaCl. The C1ql1-C2S protein eluted from the NTA beads was further purified by anti-Flag G1 Affinity Resin (Genscript) and finally eluted with a buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, and 0.5 mg/mL Flag-peptide.
Analytical size-exclusion chromatography (aSEC) and static light scattering
Protein samples with a volume of 100 μL were prepared at a final concentration of 50 μM and loaded onto a Superdex 200 Increase 10/300 GL column (Cytiva) equilibrated with the indicated buffer conditions, performed on an ÄKTA pure system (Cytiva). To calculate the molecular weight of target proteins, the tandem DAWN_HELEOS-II and Optilab T-rEX detectors (Wyatt Technology Corporation, Santa Barbara, CA, USA) were coupled to detect multiple-angle light scatter signals. In this assay, all the C1ql proteins, including their full-length and truncated forms, were obtained from the bacterial E.coli cell expression system, while the BAI3 truncations, including ECD, eCUB206, TSR, eCUB186, eCUB291, and eCUB253, were produced from the mammalian HEK293F cell and insect High Five cell expression systems, respectively.
Crystallization, X-ray data collection, and structure determination
For crystallization, the fractions of E.coli-expressed C1ql1_gC1q hexamer were collected in the buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, and concentrated to 15 mg/ml. Crystals were obtained by the sitting drop vapor diffusion method at 16 °C. To set up a sitting drop, 0.2 μl of protein solution was mixed with 0.2 μl of crystallization solution. The crystals of C1ql1_gC1q were grown in conditions containing 0.1 M sodium cacodylate, pH 6.0, 25% MPD, and 0.05 M calcium acetate. Before X-ray diffraction experiments, the crystals were soaked in the crystallization solutions containing an additional 30% v/v glycerol for cryoprotection. Diffraction data were collected at the BL10U2 beamline of the Shanghai Synchrotron Radiation Facility. Data were processed using XDS46 and subsequently scaled with AIMLESS47 in the autoPROC pipeline48. The initial phase was obtained by molecular replacement in PHASER49 using the C1ql1_gC1q trimer structure (PDB ID: 4QQ2) as the search model. Model building, adjustment, and refinement were carried out iteratively using COOT50 and PHENIX51. The final models were validated by MolProbity52, and the statistics are summarized in Supplementary Table 1.
Molecular simulation
Computational methods
The preparation of topology and parameters for MD simulation of the gC1q trimer complexes (WT, D208N, and A209S) were processed using CHARMM-GUI53. The protein complexes were solvated in a cubic water box extending at least 12 Å from the protein exterior, and counter ions (Ca2+ and Cl−) were added to ensure electrostatic neutrality corresponding to an ionic concentration of ~130 mM. The whole system consists of about 40 K atoms. Charmm36m force fields54 were used for the protein, along with the multisite parameters55 for Ca2+. All protein covalent H-bonds were constrained with the LINCS56 algorithm. Long-range electrostatic interactions are treated with the particle-mesh Ewald method with a real-space cutoff of 10 Å. Parallel simulations are performed simultaneously using GROMACS202257. The system was minimized using the steepest descent algorithm to remove the bad contacts, and then gradually heated to 300 K at a constant volume over 1 ns, using harmonic restraints with a force constant of 1000 kJ/(mol*Å2) on heavy atoms of both proteins and ligands. Over the following 5 ns of simulations at constant pressure (1 atm) and temperature (300 K), the restraints were gradually released. The systems were equilibrated for an additional 10 ns without positional restraints. A Parrinello-Rahman barostat58 was used to keep the pressure constant, while a V-rescale thermostat with a time step of 2 fs was used to keep the temperature constant. The system was simulated for 200 ns, with snapshots recorded every 50 ps. To mimic different calcium coordination, the third and fourth Ca2+ were removed, respectively.
Enhanced sampling using transition-temped metadynamics
To investigate the unfolding mechanism of gC1q, two collective variables (CVs) were defined for metadynamics simulation. The first CV corresponds to the contact number between one gC1q protomer and the other two protomers at the residues Q207, D210, N212, and D214, which depicts the protein-protein interaction inside the trimer. The second CV corresponds to the distance between the center of β4-β5 and the core, which describes the extent of the monomer unfolding. The Gaussian height of 1.2 kcal/mol with a bias factor of 20 was used for transition-tempered metadynamics simulation to sample the conformational change between the two basins that correspond to the folded state (0.7,110) of the trimer and the extended state (4.3, 25) of the hexamer. The deposition of bias was performed every 500 steps. The free energy landscapes were reweighted after a 1000 ns metadynamics simulation along CV1 and the new CV that defines the coordination number of Ca2+ around D208. The metadynamics simulation and analysis were performed using GROMACS2022 patched with PLUMED-2.8.359.
Trimer/hexamer transition assay
The trimer and hexamer fractions of E.coli-expressed C1qls_gC1q were individually collected from size-exclusion chromatography purification (HiLoad Superdex 75 pg column). Each sample was prepared at a final concentration of 1 mM in a volume of 50 μL and incubated at 37 °C for 0, 6, or 12 h, respectively. After incubation, each sample was immediately mixed with 100 μL of buffer and loaded onto a Superdex 200 Increase 10/300 GL column (Cytiva) performed on an ÄKTA pure system (Cytiva) at room temperature.
Isothermal titration calorimetry (ITC)
ITC measurements were performed on a PEAQ-ITC Microcal calorimeter (Malvern). The protein samples of trimeric C1ql1_gC1q and BAI3_eCUB or their variants were prepared at a concentration of 200 μM in the syringe and 20 μM in the cell, respectively. All protein samples were prepared in a buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, and the indicated concentration of CaCl2. An interval of 150 s between injections was set to ensure the curve returned to the baseline. The titration data were analyzed and fitted using a one-site binding model to determine the binding parameters. The protein source for each sample used here is identical to that in the aSEC assay described above.
Negative-staining (NS) EM
About 4 μL of E.coli-expressed or HEK293F-secreted C1ql1 proteins were applied onto a freshly glow-discharged grid. After a 1-min incubation, the excess protein solution was blotted using filter paper. Subsequently, 4 μL of 2.5% uranyl acetate solution was immediately applied to the grid for an additional 1-min staining. The stained grid was then air-dried at room temperature. All prepared grids were checked and visualized using an HT7700 (HITACHI) transmission electron microscope with 100-kV voltage. For the full-length C1ql1 protein sample, ~20–30 images were captured for further 2D-classification analysis in CryoSPARC v4.160.
Cryo-EM grid preparation and data acquisition
The freshly prepared complex of E.coli-expressed C1ql1_gC1q hexamer and HEK293F-expressed BAI3_eCUB in a buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, and 10 mM CaCl2 was concentrated to ~0.6 mg/ml. The freshly prepared hexamer fraction of E.coli-expressed full-length C1ql1 in a buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl was concentrated to ~0.5 mg/ml. About 4 μL of protein sample was injected onto a glow-discharged grid (Quantifoil Cu, 300 mesh, 1.2/1.3). After a 5-s incubation, the grid was blotted with filter paper for 3–4 s and then quickly plunged into liquid ethane. These freezing processes were performed using a Vitrobot (FEI) machine, operated under conditions of 4 °C and 100% humidity. All prepared grids were stored in liquid nitrogen for subsequent grid screening and data collection.
The screened grids were carefully transferred into a Titan Krios G3 transmission electron microscope (Thermo Fisher Scientific) operating at 300 kV and equipped with a Gatan K3 direct electron detector. A total of 18134 movies of the C1ql1_gC1q hexamer and BAI3_eCUB complex samples were collected within four separate sections using identical settings. Data collection was facilitated by SerialEM 3.7 software61. All movies were captured in the super-resolution mode with an initial pixel size of 0.536 Å and binned to 1.072 Å for data processing. Each movie is composed of 32 frames with a 2.0-s exposure at a dose rate of 1.56 e-/Å2 for each frame. The defocus range was set from −1.5 to −2.5 μm. For the full-length C1ql1 hexamer samples, ~600 movies were acquired under similar settings for further 2D-classification analysis.
Cryo-EM data processing, model binding, and refinement
To determine the overall structure of the C1ql1_gC1q and BAI3_eCUB complex, cryo-EM data processing was performed using CryoSPARC v4.160. The collected movies in each section were separately aligned with MotionCor262 or Patch Motion Correction with a B-factor of 150 to generate the micrographs, followed by CTF estimation using CTFFIND463. After manually removing junks, 16,956 micrographs were selected for structure determination. An initial particle set was picked out from 1000 micrographs in the first section by Blob-picking mode, followed by 2D-classification to quickly remove junk particles. Consequently, ~150,000 good particles were selected to generate a deep-learned particle-picking model by Topaz training64. Using this Topaz model, ~8 million particles were picked, extracted, and further cleaned by multiple rounds of 2D and 3D classifications. These iterative classifications resulted in the selection of 143,870 high-quality protein particles containing the densities of a C1ql1_gC1q hexamer and four molecules of BAI3_eCUB (referring to 6:4 complex). Finally, these selected particles were used to refine a whole complex map at an overall resolution of 3.48 Å.
To improve the density map for unambiguous visualization of the interface between C1ql1_gC1q and BAI3_eCUB, we reprocessed ~2 million particles containing the C1ql1_gC1q hexamer and different copy numbers of BAI3_eCUB that had been cleaned by 2D-classification in CryoSPARC and focused on the subcomplex containing one C1ql1_gC1q trimer and one bound BAI3_eCUB molecule (referring to 3:1 subcomplex), given the high similarity among the four binding surfaces between the C1ql1_gC1q trimer and BAI3_eCUB (Supplementary Fig. 8a). We imported these ~2 million particles into Relion4.065 for further rounds of 2D and 3D classifications, resulting in the identification of a better class containing 578,080 particles with a more complete density for one C1ql1_gC1q trimer and its bound BAI3_eCUB. Then, an additional focused 3D classification (without alignment and T = 20) was performed in Relion with a local mask created to focus on this subcomplex (Supplementary Fig. 8b). Finally, 73,947 particles with the highest quality were selected and reloaded into CryoSPARC for local refinement to generate a focused map. This resulted in a focused subcomplex map with a resolution of 3.34 Å, which provided sufficient clarity to model the sidechain orientations of the residues and pinpoint the location of Ca2+ ions at the binding interface.
The overall and focused maps were further optimized using DeepEMhancer66 with a tightTarget model. The resolution for each map was estimated using the gold-standard FSC cutoff of 0.143 in cryoSPARC. The local resolution for the two final maps was calculated using Local Resolution Estimation in cryoSPARC.
To build a complete atomic model for the complex, we rigidly fit two crystal structures of the C1ql1_gC1q trimer and four AlphaFold267-predicted models of BAI3_eCUB into the overall cryo-EM density map in UCSF-ChimeraX68. After manual adjustments in COOT, particularly to the domain-swapped regions, the modified atomic model was subjected to a real-space refinement mode against the overall map in PHENIX. Manual adjustment and automatic refinement were performed iteratively to achieve a final model. A similar approach was employed for the focused model of the subcomplex. Specifically, we extracted an initial model containing one C1ql1_gC1q trimer and one BAI3_eCUB from the overall model and used the focused map for further model adjustment and refinement. Both the overall and focused atomic models were validated against their respective maps using PHENIX. The statistical information for cryo-EM data collection, processing, model building, refinement, and validation was summarized in Supplementary Table 2. All the structural figures in this study were produced using UCSF-ChimeraX and PyMOL (https://pymol.org/).
Cell surface recruitment assay
The cell-based recruitment assay was performed as described previously29,69. Briefly, HEK293T cells transfected with different full-length C1ql1-Flag variants were individually cultured for 48 h, and the medium containing the secreted C1ql1-Flag protein was collected and further checked by Western Blotting to confirm comparable protein levels. Then, HEK293T cells transfected with BAI3-GFP were incubated with an addition of a similar amount of the harvested C1ql1-Flag secretions, respectively, supplemented with 2 mM CaCl2 and 2 mM MgCl2 in the secretion medium, for 16 h at 4 °C. Following incubation, the cells were fixed, incubated with mouse anti-Flag antibody (1:3000, Sigma-Aldrich, F4049), and visualized with secondary antibodies conjugated to Alexa Fluor™ 594 (1:1000, Invitrogen). Images were taken with a Nikon A1R confocal microscope system and analyzed using Image Pro Plus software.
Native-PAGE assay
HEK293T cells transfected with different full-length C1ql1-Flag variants and Cbln1 were individually cultured for 48 h, and then the 50 µL medium containing different secreted proteins was collected and analysed by ExpressPlus PAGE Gel (GenScript, M42015) in a SDS-free buffer containing 50 mM Tris, pH 8.0, and 50 mM MOPS. The C1ql1 variants and Cbln1 proteins were further detected by western blotting.
Antibodies
Antibodies were used with the indicated concentrations as follows. Primary antibody: rabbit anti-vGluT2 (1:1000, Synaptic Systems, 135402), guinea pig anti-vGluT2 (1:1000, Synaptic Systems, 135404), mouse anti-Calbindin (1:1000, Sigma-Aldrich, C9848), chicken anti-GFP (1:2000, Aves Labs, GFP-1020), rabbit anti-HA (1:1000, Cell Signaling Technologies, 3724), mouse anti-Flag (1:3000, Sigma-Aldrich, F4049). Secondary antibody: Goat anti-rabbit (1:10000, Sangon Biotech, D110058) and goat anti-mouse (1:10,000, ProteinFind, HS201) for western blot. Secondary antibodies conjugated with DyLight 405 (mouse, 1:500, Jackson ImmunoResearch Laboratories, 715-475-150), and Alexa 488, 546, 594, 647 (1:1000, Invitrogen) for imaging.
Immunostaining
Immunohistochemistry was performed as previously described70,71,72. Both male and female mice were collected for this experiment, and five mice were used in each group. Briefly, mice aged P21–28 were anesthetized briefly with isoflurane, followed by perfusion with cold 10 mM PBS for 2 min and 4% PFA for 10 min. The brain was dissected out and post-fixed in 4% PFA at 4 °C overnight. 30% sucrose in PBS was used to dehydrate the tissues, which were sliced on a cryostat at 30 µm. The slices were free-floating blocked for 30 min at room temperature (RT) in blocking buffer (5% BSA, 0.1% Triton X-100, 0.04% sodium azide in 10 mM PBS) and incubated with primary antibodies diluted in blocking buffer at 4 °C overnight, followed by three washes using PBS at RT. The secondary antibodies were diluted in blocking buffer and incubated with slices at RT for 2 h, followed by three washes using PBS at RT. Slices were mounted on glass slides, dried briefly, and covered with Fluoromount-G (Southern Biotech, Cat#0100-01 or 0100-20) and cover glasses.
Imaging and synapse quantification
In mouse tissues, images of cerebellar lobules IV/V in C1ql1-KO experiments or CFs with positive GFP signals in rescue experiments were collected and analyzed. Z-stacked images were required by confocal microscopies (Olympus FV3000 or IXplore SpinSR 10) with a 0.5-μm step size, 10 to 20 steps. The same parameters were used to gain images in one experiment. A custom-written macro was used in ImageJ to quantify synaptic number and size. For rescue experiments, AAVs expressing wild-type C1ql1 or its mutants were delivered in IO, with GFP-expressing AAVs serving as parallel controls in each experiment. GFP-positive branches were included for synapse analysis. To minimize batch effects during quantification, vGluT2 synaptic sizes in GFP control groups were normalized to comparable baseline levels prior to comparative analysis.
Stereotactic injections
Mice at P21 were anesthetized with ketamine/xylazine (100/10 mg/kg body weight, Jiangsu Zhongmu Beikang Pharmaceutical Co., Ltd., or HY-B0443A, MedChemExpress). Both male and female mice were collected for this experiment, and the number of mice in each group is: GFP, n = 15; WT, n = 4; A209S, n = 4; D208N, n = 6; Y247A, n = 5. The head was fixed on a stereotaxic instrument (Ruiwode Lift Technology Co., Ltd., China), followed by a surgical incision on the skin, and two small holes were made with a dental drill. The virus was delivered using a nanoliter microinjection pump with fine glass capillaries. The coordinates for bilateral IO injection were: AP: cerebellar lobule VI; ML: ±0.3 mm; DV: −6.2 to −6.8 mm from lambda, depending on the weights of mice. The virus volume was 250 nL/site with a rate of 5 nL/s, and the final volume was 500 nL. The titers were adjusted after western blot quantification to ensure similar protein expression levels among groups. The final tilters were: AAV2/9-CAG-eGFP-WPRE-pA: 0.4 × 1013 V.G./mL; AAV2/9-CAG-HA-mC1ql1-T2A-eGFP-WPRE-pA: 7×1013 V.G./mL; AAV2/9-CAG-HA-mC1ql1(A209S)-T2A-eGFP-WPRE-pA: 2.47 × 1013 V.G./mL; AAV2/9-CAG-HA-mC1ql1(D208N)-T2A-eGFP-WPRE-pA: 2.6 × 1013 V.G./mL; AAV2/9-CAG-HA-mC1ql1(Y247A)-T2A-eGFP-WPRE-pA: 3 × 1013 V.G./mL (WZ Biosciences Inc). After the suture, the mice were allowed to recover on a warm pad before returning to their home cage. Two to three weeks were taken to allow optimal viral expression.
Statistics and reproducibility
All values are displayed as the mean values with an error bar of the standard error of the mean (SEM). Significance was calculated by the indicated models in the figure legends. The protein analysis results are representative of at least three independent repeats. The number of mice and cells for statistical analyses in each condition are indicated in the figure legends or the section of Methods.
Ethical approvals
Animal experiments were performed following a protocol (No. AP20250902-01) approved by the Institutional Animal Care and Use Committee (IACUC) of Peking University Shenzhen Graduate School. No data were excluded from the study.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The crystal structure of C1ql1_gC1q has been deposited into PDB with accession code 9LKK. The overall and focused cryo-EM density maps and their corresponding models of C1ql1_gC1q and BAI3_eCUB complex have been deposited into EMDB and PDB with accession codes EMD-63181 and EMD-63182, and with PDB IDs 9LKL and 9LKM, respectively. Previously published PDB structures used for structural alignment and analysis were provided: 4QPY, 4QQ2, 4QQH, 6V55, and 3KQ4. MD simulation files, including the initial and final MD snapshots in GROMACS gro formats for C1ql1, have been deposited on Zenodo at [https://doi.org/10.5281/zenodo.17394778]. The source data underlying Figs. 1–7 and Supplementary Figs. 1, 3–5, 10–11, and AlphaFold2-predicted initial model of BAI3_eCUB are provided as a Source Data file. Source data are provided with this paper.
Code availability
The MD packages GROMACS and Plumed used in this work are publicly available at [https://www.gromacs.org] and [https://www.plumed.org].
References
Sudhof, T. C. The cell biology of synapse formation. J. Cell Biol. 220, e202103052 (2021).
Sudhof, T. C. Towards an understanding of synapse formation. Neuron 100, 276–293 (2018).
Waites, C. L., Craig, A. M. & Garner, C. C. Mechanisms of vertebrate synaptogenesis. Annu. Rev. Neurosci. 28, 251–274 (2005).
Missler, M., Sudhof, T. C. & Biederer, T. Synaptic cell adhesion. Cold Spring Harb. Perspect. Biol. 4, a005694 (2012).
Shapiro, L., Love, J. & Colman, D. R. Adhesion molecules in the nervous system: structural insights into function and diversity. Annu. Rev. Neurosci. 30, 451–474 (2007).
De Wit, J. & Ghosh, A. Specification of synaptic connectivity by cell surface interactions. Nat. Rev. Neurosci. 17, 4–4 (2016).
Dalva, M. B., McClelland, A. C. & Kayser, M. S. Cell adhesion molecules: signalling functions at the synapse. Nat. Rev. Neurosci. 8, 206–220 (2007).
Matsuda, K. et al. Cbln1 is a ligand for an orphan glutamate receptor δ2, a bidirectional synapse organizer. Science 328, 363–368 (2010).
Uemura, T. et al. Trans-synaptic interaction of GluRδ2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell 141, 1068–1079 (2010).
Elegheert, J. et al. Structural basis for integration of GluD receptors within synaptic organizer complexes. Science 353, 295–299 (2016).
Yuzaki, M. Two classes of secreted synaptic organizers in the central nervous system. Annu. Rev. Physiol. 80, 243–262 (2018).
Kakegawa, W. et al. Anterograde C1ql1 signaling is required in order to determine and maintain a single-winner climbing fiber in the mouse cerebellum. Neuron 85, 316–329 (2015).
Sigoillot, S. M. et al. The secreted protein C1QL1 and its receptor BAI3 control the synaptic connectivity of excitatory inputs converging on cerebellar purkinje cells. Cell Rep. 10, 820–832 (2015).
Matsuda, K. et al. Transsynaptic modulation of kainate receptor functions by C1q-like proteins. Neuron 90, 752–767 (2016).
Sticco, M. J. et al. C1QL3 promotes cell-cell adhesion by mediating complex formation between ADGRB3/BAI3 and neuronal pentraxins. FASEB J. 35, e21194 (2021).
Matsuda, K. Synapse organization and modulation via C1q family proteins and their receptors in the central nervous system. Neurosci. Res. 116, 46–53 (2017).
Kishore, U. et al. C1q and tumor necrosis factor superfamily: modularity and versatility. Trends Immunol. 25, 551–561 (2004).
Ghai, R. et al. C1q and its growing family. Immunobiology 212, 253–266 (2007).
Ressl, S. et al. Structures of C1q-like proteins reveal unique features among the C1q/TNF superfamily. Structure 23, 688–699 (2015).
Yuzaki, M. The C1q complement family of synaptic organizers: not just complementary. Curr. Opin. Neurobiol. 45, 9–15 (2017).
Pena Palomino, P. A., Black, K. C. & Ressl, S. Pleiotropy of C1QL proteins across physiological systems and their emerging role in synapse homeostasis. Biochem. Soc. Trans. 51, 937–947 (2023).
Iijima, T., Miura, E., Watanabe, M. & Yuzaki, M. Distinct expression of C1q-like family mRNAs in mouse brain and biochemical characterization of their encoded proteins. Eur. J. Neurosci. 31, 1606–1615 (2010).
Martinelli, D. C. et al. Expression of C1ql3 in discrete neuronal populations controls efferent synapse numbers and diverse behaviors. Neuron 91, 1034–1051 (2016).
Moghimyfiroozabad, S., Paul, M. A., Sigoillot, S. M. & Selimi, F. Mapping and targeting of C1ql1-expressing cells in the mouse. Sci. Rep. 13, 17563 (2023).
Cheung, H. W. et al. Creation of a novel CRISPR-generated allele to express HA epitope-tagged C1QL1 and improved methods for its detection at synapses. FEBS Lett. 598, 2417–2437 (2024).
Paul, M. A. et al. Stepwise molecular specification of excitatory synapse diversity onto cerebellar Purkinje cells. Nat. Neurosci. 28, 308–319 (2024).
Kakegawa, W. et al. Kainate receptors regulate synaptic integrity and plasticity by forming a complex with synaptic organizers in the cerebellum. Cell Rep. 43, 114427 (2024).
Aimi, T., Matsuda, K. & Yuzaki, M. C1ql1-Bai3 signaling is necessary for climbing fiber synapse formation in mature Purkinje cells in coordination with neuronal activity. Mol. Brain 16, 61 (2023).
Bolliger, M. F., Martinelli, D. C. & Sudhof, T. C. The cell-adhesion G protein-coupled receptor BAI3 is a high-affinity receptor for C1q-like proteins. Proc. Natl Acad. Sci. USA 108, 2534–2539 (2011).
Stephenson, J. R., Purcell, R. H. & Hall, R. A. The BAI subfamily of adhesion GPCRs: synaptic regulation and beyond. Trends Pharm. Sci. 35, 208–215 (2014).
Duman, J. G., Tu, Y. K. & Tolias, K. F. Emerging roles of BAI adhesion-GPCRs in synapse development and plasticity. Neural Plast. 2016, 8301737 (2016).
Haitina, T. et al. Expression profile of the entire family of adhesion G protein-coupled receptors in mouse and rat. BMC Neurosci. 9, 43 (2008).
Lanoue, V. et al. The adhesion-GPCR BAI3, a gene linked to psychiatric disorders, regulates dendrite morphogenesis in neurons. Mol. Psychiatry 18, 943–950 (2013).
Tu, Y. K., Duman, J. G. & Tolias, K. F. The Adhesion-GPCR BAI1 promotes excitatory synaptogenesis by coordinating bidirectional trans-synaptic signaling. J. Neurosci. 38, 8388–8406 (2018).
Wang, J. et al. RTN4/NoGo-receptor binding to BAI adhesion-GPCRs regulates neuronal development. Cell 184, 5869–5885.e5825 (2021).
Miao, Y. et al. Structure of the complex of C1q-like 3 protein with adhesion-GPCR BAI3. Commun. Biol. 8, 693 (2025).
Hamoud, N. et al. Spatiotemporal regulation of the GPCR activity of BAI3 by C1qL4 and Stabilin-2 controls myoblast fusion. Nat. Commun. 9, 4470 (2018).
Tönnies, E. & Trushina, E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimers Dis. 57, 1105–1121 (2017).
Stawarski, M. et al. Neuronal glutamatergic synaptic clefts alkalinize rather than acidify during neurotransmission. J. Neurosci. 40, 1611–1624 (2020).
Forsberg, M. et al. Ionized calcium in human cerebrospinal fluid and its influence on intrinsic and synaptic excitability of hippocampal pyramidal neurons in the rat. J. Neurochem. 149, 452–470 (2019).
Radjainia, M., Wang, Y. & Mitra, A. K. Structural polymorphism of oligomeric adiponectin visualized by electron microscopy. J. Mol. Biol. 381, 419–430 (2008).
Mortensen, S. A. et al. Structure and activation of C1, the complex initiating the classical pathway of the complement cascade. Proc. Natl Acad. Sci. USA 114, 986–991 (2017).
da Silva Rosa, S. C., Liu, M. & Sweeney, G. Adiponectin synthesis, secretion and extravasation from circulation to interstitial space. Physiology 36, 134–149 (2021).
Biswas, J. et al. C1ql1 is expressed in adult outer hair cells of the cochlea in a tonotopic gradient. PLoS ONE 16, e0251412 (2021).
LaVallie, E. R. et al. A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cytoplasm. Biotechnology 11, 187–193 (1993).
Kabsch, W. XDS. Acta Crystallogr. D. Biol. Crystallogr 66, 125–132 (2010).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution?. Acta Crystallogr. D. Biol. Crystallogr. 69, 1204–1214 (2013).
Vonrhein, C. et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D. Biol. Crystallogr. 67, 293–302 (2011).
Storoni, L. C., McCoy, A. J. & Read, R. J. Likelihood-enhanced fast rotation functions. Acta Crystallogr. D. Biol. Crystallogr. 60, 432–438 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D. Struct. Biol. 75, 861–877 (2019).
Davis, I. W. et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 (2007).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Zhang, A., Yu, H., Liu, C. & Song, C. The Ca2+ permeation mechanism of the ryanodine receptor revealed by a multi-site ion model. Nat. Commun. 11, 922 (2020).
Hess, B., Bekker, H., Berendsen, H. J. & Fraaije, J. G. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Hess, B., Kutzner, C., Van Der Spoel, D. & Lindahl, E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 (2008).
Parrinello, M. & Rahman, A. Crystal structure and pair potentials: a molecular-dynamics study. Phys. Rev. Lett. 45, 1196 (1980).
Tribello, G. A., Bonomi, M., Branduardi, D., Camilloni, C. & Bussi, G. PLUMED 2: new feathers for an old bird. Comput. Phys. Commun. 185, 604–613 (2014).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Bepler, T., Kelley, K., Noble, A. J. & Berger, B. Topaz-Denoise: general deep denoising models for cryoEM and cryoET. Nat. Commun. 11, 5208 (2020).
Kimanius, D., Dong, L., Sharov, G., Nakane, T. & Scheres, S. H. W. New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem. J. 478, 4169–4185 (2021).
Sanchez-Garcia, R. et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol. 4, 874 (2021).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Boucard, A. A., Chubykin, A. A., Comoletti, D., Taylor, P. & Sudhof, T. C. A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron 48, 229–236 (2005).
Zhang, B. et al. Neuroligins sculpt cerebellar Purkinje-cell circuits by differential control of distinct classes of synapses. Neuron 87, 781–796 (2015).
Zhang, B. & Südhof, T. C. Neuroligins are selectively essential for NMDAR signaling in cerebellar stellate interneurons. J. Neurosci. 36, 9070–9083 (2016).
Han, Y. et al. Neuroligin-3 confines AMPA receptors into nanoclusters, thereby controlling synaptic strength at the calyx of Held synapses. Sci. Adv. 8, eabo4173 (2022).
Acknowledgements
We thank Qiming Ou for his kind assistance during the revision process. We thank the assistance of the Southern University of Science and Technology (SUSTech) Cryo-EM Center and Core Research Facilities, and the synchrotron beamline BL10U2 of Shanghai Synchrotron Radiation Facility (SSRF). This work was supported by National Key R&D Program of China (Grant No. 2025YFA1308800 to Z.W.), the Key-Area Research and Development Program of Guangdong Province (Grant No. 2023B0303010001 to Z.W.), the National Natural Science Foundation of China (Grant No. 32471250 to Z.W., 82022018, 32070958, and 82161138025 to B.Z., 32471262 to F.N., and 92269102 to Y.W.), Guangdong Basic and Applied Basic Research Foundation (Grant No. 2024A1515010742 to Z.W., 2023A1515030232 and 2024A1515012651 to F.N.), Guangdong Pearl River Talent Program (2021QN02Y618 to Y.W.), Guangdong Pearl River Funding to B.Z., Shenzhen Science and Technology Program (Grant No. RCJC20210609104333007 to Z.W., and JCYJ20250604144232041 to F.N.), Shenzhen Key Laboratory of Biomolecular Assembling and Regulation (Grant No. ZDSYS20220402111000001 to Z.W.), Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions (2024SHIBS0004), Major Program of Shenzhen Bay Laboratory (S241101002 to B.Z.). All computations were performed using the central computing facilities at Shenzhen Bay Laboratory. Z.W. is an investigator at the SUSTech Institute for Biological Electron Microscopy.
Author information
Authors and Affiliations
Contributions
Z.W. and B.Z. conceived the study and supervised the project. L.L., Y.H., F.N., Y.W., Y.L., S.X., H.Z., J.X. and H.I.T. designed and performed experiments. L.L., Y.H., F.N., Y.W., J.G., B.Z., and Z.W. analyzed the data. F.N., Y.W., B.Z. and Z.W. wrote the manuscript with inputs from other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jinglei Hu, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liao, L., Han, Y., Niu, F. et al. Structural basis of calcium-dependent C1ql1/BAI3 assemblies in synaptic connectivity. Nat Commun 16, 11444 (2025). https://doi.org/10.1038/s41467-025-66254-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66254-1
