
Abstract
Advances in genome engineering have improved our ability to perturb microbial metabolic networks, yet bioproduction campaigns often struggle with parsing complex metabolic datasets to efficiently enhance product titers. We address this challenge by coupling laboratory automation with machine learning to systematically optimize the production of isoprenol, a sustainable aviation fuel precursor, in Pseudomonas putida. The simultaneous downregulation through CRISPR interference of combinations of up to four gene targets, guided by machine learning, permitted us to increase isoprenol titer 5-fold in six consecutive design-build-test-learn cycles. Moreover, machine learning enabled us to swiftly explore a vast experimental design space of 800,000 possible combinations by strategically recommending approximately 400 priority constructs. High-throughput proteomics allowed us to validate CRISPRi downregulation and identify biological mechanisms driving production increases. Our work demonstrates that ML-driven automated design-build-test-learn cycles, when combined with rigorous data validation, can rapidly enhance titers without specific biological knowledge, suggesting that it can be applied to any host, product, or pathway.
Similar content being viewed by others

Introduction
Synthetic biology harnesses microbial metabolism to produce chemicals that address global challenges1. A robust array of genome engineering2,3,4,5 and modulating tools6 has provided biologists with an unprecedented capacity to perturb metabolic networks for product-specific optimization. Accordingly, synthetic biology has been used to engineer microbes to enhance crop yields7, produce pharmaceuticals that improve human health7,8, and generate sustainable aviation fuel (SAF) blendstocks that provide enhanced biofuel alternatives for the airline industry9,10,11. The production of isoprenol, a precursor of the SAF 1,4-dimethylcyclooctane (DMCO), exemplifies biomanufacturing’s capacity to generate products with improved performance. Specifically, DMCO shows increased energy density, making it suitable to extend flight ranges when blended with Jet-A or other fuels12 and is a promising component in global bioeconomy plans13.
The canonical Design-Build-Test-Learn (DBTL) paradigm has proven to be a powerful framework in synthetic biology, but its adoption in industrial biotechnology faces limitations in throughput and reproducibility14, relying heavily on manual operations15, and struggling to effectively interpret complex datasets for iterative performance improvement16. As a result, generating industrially competitive production strains has been laborious (50–300 person-years) and expensive ($10–$100 million)17,18,19.
An emerging strategy to accelerate synthetic biology is the concerted integration of precision metabolic engineering tools like CRISPR with laboratory automation and machine learning20,21. CRISPR interference (CRISPRi) can downregulate the expression of genes encoding proteins of interest using highly specific single guide RNAs (sgRNAs) to precisely perturb cellular metabolism. However, multiplexing CRISPRi sgRNA arrays to downregulate multiple genes leads to an intractably large combinatorial design space, prohibitive to conventional synthetic biology, but well suited for active learning. Active learning is a machine learning (ML) approach in which the algorithm chooses the next experiment to be performed, and a principled way to: (1) rapidly model the effects of perturbations on production (e.g., CRISPRi on titer), (2) query the possible experimental design space for new designs that improve production, and (3) integrate new data as it is collected across cumulative DBTL cycles20,21,22,23,24,25,26.
However, effective machine learning relies on large amounts of consistent and reproducible data, which has traditionally been a challenge in biological research27,28. Laboratory automation can satisfy this requirement by standardizing construct design and experimental testing, increasing reproducibility by reducing manual operations, increasing throughput, and ultimately expediting effective learning between DBTL cycles15,29. Furthermore, integration of proteomics in automation workflows facilitates the collection of high-quality data for validating successful CRISPRi. Combining CRISPRi, ML, and laboratory automation presents a unique and underexploited opportunity for advancing strain optimization.
Here, we create a laboratory automation pipeline to rapidly design, assemble, and test strains harboring multiplexed CRISPRi arrays, providing the high-quality data for machine learning to improve the production of isoprenol in Pseudomonas putida (Fig. 1a). Using active learning, we solve a problem that is intractable through traditional intuition-driven approaches: predicting the impact of multiple gene perturbations to systematically improve titers. By integrating active learning with laboratory automation, we perform six DBTL cycles, each 2–3 weeks in duration, and ultimately improve isoprenol titers by 5-fold while reducing the design space to 347 unique CRISPRi arrays (combinations of downregulated genes) from an initial pool of over 800,000 possible arrays (Fig. 1b–d). In so doing, we create and leverage new automation approaches, uncover some limitations of CRISPRi, pinpoint the metabolic mechanisms that underpin these improvements, and show that improvements from CRISPRi downregulation are difficult to recapitulate with strict gene knockouts (KOs). Because this approach can be generalized to any product, pathway, or host, it has the potential to radically improve how DBTL cycles are used in both academic and industrial environments, thereby significantly accelerating biomanufacturing workflows.

a P. putida has been engineered to generate isoprenol, a precursor to sustainable fuel for aviation, which remains a historically difficult sector to decarbonize. b Active learning was applied to explore combinations of downregulated genes through CRISPRi rather than single knockdowns. Initial gene targets were selected by both heuristic and genome-scale metabolic model (GSMM)-guided methods. Colors represent the positions of given sgRNA in the array, beginning with lone sgRNAs and building up to four-sgRNA arrays. c Pairing automation with machine learning enabled effective exploration of the massive combinatorial sgRNA design space to increase titer. The use of a high-throughput microfluidic electroporator (HTME) eliminated one of the main technical hurdles for high-throughput strain construction. This figure was partially generated using BioRender.com (https://BioRender.com/7n0g6tu). d The approach proved remarkably effective, increasing isoprenol titer by 5-fold over 6 DBTL cycles, each of which took 2–3 weeks to complete. Circles represent individual replicates from each cycle. The maximum and median performance strains (n = 3) are highlighted in orange and blue, respectively. The reproducibility of the controls (yellow) was critical to avoid biases in the machine learning recommendations for subsequent DBTL cycles. Source data are provided in the Source Data file.
Results
Iterative CRISPRi array design through laboratory automation provides large amounts of highly reproducible data for effective active learning
We developed a semi-automated pipeline for constructing and screening CRISPRi arrays to generate the large amounts of highly reproducible data needed for active learning. This pipeline provided precise titers and proteomics data from 472 unique strains (125 single perturbations and 347 combinations) in triplicate across 6 total DBTL cycles (DBTL0-6) with highly reproducible control titers (Fig. 2). Furthermore, each DBT cycle was completed in only 2–3 weeks (Supplementary Fig. 1). The CRISPRi arrays were capable of simultaneously downregulating genes situated in orthogonal metabolic pathways30 to elucidate specific genes or gene combinations favorable for isoprenol bioproduction. DBT stages were completed by a cohesive suite of automated laboratory equipment (Fig. 2a). Briefly, plasmids harboring CRISPRi arrays were built by idempotent cloning with individual sgRNA units and positionally dependent, complementary linkers (Supplementary Fig. 2). Assembled sgRNA plasmids were transformed into P. putida IY1452b in parallel using a high-throughput 384-well microfluidic electroporator (HTME)31,32, which eliminates a traditional bottleneck in high-throughput strain engineering by performing up to 384 electroporations (Supplementary Figs. 3 and4) in under 1 min31,32. Automation of plasmid construction and transformation standardized the ~3000 liquid handling steps needed in this project, consistently yielding >95% successful P. putida transformations while using 15-fold lower cell and plasmid volumes compared to conventional cuvettes (Fig. 2b). Fermentations were performed using a Beckman Coulter BioLector, which maintains excellent repeatability and strong correlations to higher scale production conditions33. The use of carefully calibrated automated equipment resulted in the excellent reproducibility we would later need to train our active learning process: our controls showed only a 5–10% coefficient of variation of isoprenol titer across all cycles (Fig. 2c). We combined isoprenol production data with high-quality, high-resolution global proteomics, which identified 1500 proteins (Top3) per replicate and offered unique insight into how gene downregulation impacts global protein abundances.

a A suite of laboratory automation equipment facilitated the construction of sgRNA arrays. sgRNA units were dispensed with positionally dependent complementary linkers using the ECHO 550, then simultaneously digested and ligated to compose linked sgRNA units. sgRNA units were magnetically purified with an Apex KingFisher, assembled into sgRNA arrays, dispensed, and transformed into competent Escherichia coli with the MANTIS, plated using a Hamilton VANTAGE, picked with a QPix 460, and ultimately sequence validated. Validated sgRNA arrays were then dispensed with electrocompetent P. putida IY1449b (ΔphaABC, ΔmvaB, ΔhbdH, ΔldhA, ΔzwfB, ΔgntZ, and ΔliuC) cells harboring pIY670 again using the ECHO 550. Electroporations were completed in parallel using the 384-HTME prior to outgrowth and plating on a selective medium using the Hamilton VANTAGE. Assembly and transformation routinely displayed a success rate above 95% with failure most often stemming from downregulation toxicity. Strains transformed with CRISPRi arrays were adapted twice in minimal medium and cultured in a microbioreactor. Cultures were harvested for proteomics and isoprenol analysis, producing a total of ~1500 samples for the full project. This figure was partially generated using BioRender.com (https://BioRender.com/ukpmliz). b Each cycle generated 60 unique sgRNA arrays in addition to a control, which were tested in four sequential 48-well BioLector fermentations, with controls included in triplicate. c Although DBTL cycles sometimes took place months apart, isoprenol titers in control strains harboring a non-target sgRNA remained remarkably precise and reproducible, varying by 5–10% CV across all cycles. In DBTL0, the control strain was cultured across plates (n = 18) while subsequent cycles had three control strains per plate (n = 12). Longer sgRNA arrays were favored in later cycles. Error bars represent standard deviations. Source data are provided in the Source Data file.
Rapid characterization of isoprenol production and gene downregulation provides a set of validated CRISPRi gene targets
Validating CRISPRi downregulation through proteomics was imperative to select functionally successful sgRNAs for iterative strain design. DBTL0 established a feasible automated workflow to perform high-throughput CRISPRi evaluation, forming a validated set of gene targets from which we would later select combinations to improve titer. We perturbed 120 unique genes across a range of metabolic functions and identified 67 genes that (1) influenced isoprenol titer and (2) were effectively downregulated by CRISPRi (i.e., exhibited a sufficient decrease in protein expression caused by dCas9 downregulation). Individual sgRNA targets were initially suggested by a combination of a priori heuristics and the GSMM-based Flux Reaction Target Prioritization Genome-Scale Modeling Technique (FluxRETAP)34 as a basis to modulate expression of genes associated with the isoprenol production pathway (Supplementary Data 1)31,34,35,36.
The proteins of interest (POIs), categorized here via Clusters of Orthologous Groups (COGs), were concentrated around protein families related to energy metabolism (oxidative phosphorylation and the TCA cycle), lipid metabolism (β-oxidation and fatty acid biosynthesis), and amino acid and carbohydrate metabolism (Fig. 3a). Downregulation of genes expressing different POIs within a COG yielded strikingly different impacts on isoprenol titer, indicating that the general protein functions captured by COG are not granular enough to predict how a specific protein will impact titer. Likewise, the level of downregulation as measured by proteomics was highly variable within the same COG, with several strains showing target expression near that of the control (Fig. 3a). The variability in isoprenol titer and apparent gene downregulation observed within COGs specifically underscores the importance of both validating POI abundance and selecting a comprehensive panel of candidate genes.

a The distribution of isoprenol titer (left) and target protein abundance (right) of strains expressing each sgRNA in our initial 125 sgRNA library. The initial library was selected by a combination of FluxRETAP and heuristics. FluxRETAP is a GSMM-based approach that uses a genome-scale model to identify which reactions must change to improve production, while heuristics involves the selection of reactions known a priori to compete for mevalonate pathway precursors. POIs are sorted by COG category and ordered by abundance in each category. Violin plots show the smoothed distribution of isoprenol or target protein abundance, while circles represent the mean values that constitute the distribution (n = 3). Vertical lines represent control values. b Example distributions of POI abundance in strains harboring a given sgRNA colored by filter status. Vertical blue lines represent the POI level in the target strain, while violins show the distribution of the POI across the whole library. c A comparison of dCas9, POI abundance, and filtering results reveals that a distinct mode of strain failure was low dCas9 expression. Joint and marginal distributions of normalized POI and dCas9 levels in each strain in the DBTL0 library. Strains are colored by their filtering result. Black contours in the joint plot represent the population density of all strains, independent of filtering. The example sgRNAs are labeled. d A similar distribution of isoprenol titers for strains passing or failing proteomics filtering suggests that some strains improve titer through mechanisms other than by downregulating the gene encoding the POI. Five sgRNA transformations failed to yield colonies, resulting in 120 unique strains. Source data are provided in the Source Data file.
Given the failures observed in downregulating certain genes, we developed a filter to avoid training the machine learning model on sgRNAs that did not effectively decrease their POI abundance, which would yield poor predictions and recommendations. Guides were filtered out from our training set if they (1) had insufficient downregulation compared to the control (e.g., PP_1023), (2) downregulated the POI nonspecifically in many other strains (e.g., PP_0582), or (3) resulted in isoprenol titers below 66.6 mg/L (Fig. 3b). PP_0814 and PP_0815 (subunits of a terminal oxidase complex PP_0812-15) were the only exceptions to these criteria as these proteins were below our minimum level of protein detection. Nonetheless, strains harboring PP_0814 and PP_0815 sgRNAs showed high levels of dCas9 protein expression and improved isoprenol titer. Overall, ~50% (67/120) of the sgRNAs in our library passed this filter (Fig. 3d).
While passing strains typically exhibited dCas9 protein levels above 50% that of the control, failing strains showed poor dCas9 levels with POI abundance comparable to the control (Fig. 3c). This failure consistently occurred across replicates, suggesting that selective pressure from the sgRNA array may have driven mutations in the dCas9 plasmid and underscoring the importance of using proteomics to validate each strain constructed. While necessary, medium adaptation inadvertently facilitated in situ adaptive evolution of CRISPRi arrays. Modes of CRISPRi failure typically fell into three categories: (1) partial deletion of dCas9, (2) single-nucleotide insertion resulting in a stop codon in dCas9, or (3) recombination across CRISPRi scaffolds (Supplementary Fig. 5).
More broadly, prior CRISPRi applications have validated downregulation using RT-qPCR37, fluorescence38,39, transcriptomics40, or not at all. Curiously, passing and failing strains exhibited similar distributions of isoprenol (Fig. 3d), suggesting that some strains improve titer through mechanisms other than POI downregulation (e.g., transient downregulation before dCas9 loss, off-target downregulation, and anomalously high pathway protein expression). Our capacity to effectively use active learning to design sgRNA combinations for iterative strain improvement relies upon proteomics validation of POI. Otherwise, we believe that the algorithm leading the active learning is unlikely to swiftly learn the titer landscape for rapid improvement. Overall, DBTL0 established a feasible automated workflow for high-throughput CRISPRi evaluation and provided a large set of validated sgRNAs that yielded a range of isoprenol titers.
Active learning identified sgRNA combinations that significantly increased isoprenol production
Having established a set of 67 validated sgRNAs, we used an active learning process to choose the combinations that would ultimately improve isoprenol titer 5-fold (Fig. 4a). We generated pseudorandom combinations of sgRNA in DBTL1, which provided training data for DBTL2. Then, in DBTL2-6, we used the Automated Recommendation Tool41 (ART) to explore CRISPRi arrays of up to four sgRNAs predicted to improve isoprenol production, based on data from previous cycles. ART uses an ensemble of machine learning models and Bayesian inference to effectively leverage small data sets to predict a target output and quantify prediction uncertainty41. In this case, we represented the gene target identity as a binary vector with length 67 for each validated target (input) and used ART to predict isoprenol titer (response, Fig. 4a). The validated sgRNAs from DBTL0 formed the building blocks of these arrays, as these were expected to exhibit more consistent target downregulation and therefore more predictable isoprenol titer. Without repetition, these 67 sgRNAs spanned a large design space of >800,000 possible combinations–a value that encompasses all possible two-, three-, and four-sgRNA arrays derived from the 67 validated sgRNAs.

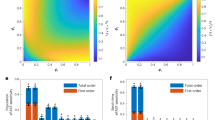
a Schematic demonstrating how ART is used to model isoprenol as a function of CRISPRi target combination identity. First, sgRNA identity is encoded as a binary vector, and ART is trained to predict titer for data from previous DBTL cycles. This produces a predictive model of titer to identify new sgRNA combinations, which are evaluated in the subsequent Design-Build-Test steps. b Distribution of isoprenol titer in passing and failing strains across all DBTL cycles. The vertical line in each distribution is the median. Passing and failing filters used in DBTL0 were different from those used in DBTL1-6 (“Methods” section). c Parity plot showing measured and predicted isoprenol for the final ART model trained on data from DBTL3-6 with mean absolute error (MAE) between measured and predicted values. d Distribution of changes in isoprenol when a sgRNA targeting a given gene is added to an array. The sgRNA targets are sorted by means of their distributions. For each box-and-whisker plot, the boxes correspond to the 25th and 75th percentiles, while the line corresponds to the median value. The whiskers extend to all values within 1.5× the interquartile range of the 25th or 75th percentile. Values outside of this range are plotted as individual circles. e Number of parent-child pairs for each sgRNA target. f Counts of all strains containing each target labeled based on whether they were below 50% of control expression (passed), above 50% of control expression (failed), or undetected. Circles represent mean values (n = 3) and error bars represent standard deviations. Source data are provided in the Source Data file.
In DBTL1, we generated data on the effect of combinations of filtered sgRNAs on isoprenol titer required by ART to inform future predictions. We generated 30 two-sgRNA and 30 three-sgRNA array combinations based on pseudorandom combinations of the successful sgRNAs from DBTL0. Target genes were sampled with a weight proportional to their isoprenol titers in DBTL0 (“Methods” section, Supplementary Data 1). Using our automation pipeline, we built and tested these multi-sgRNA strains and once again observed a wide range of isoprenol titers (Fig. 4b). The best DBTL1 strain successfully downregulated PP_0815 and PP_1317, resulting in a titer of 449 ± 22 mg/L. Following DBTL1, we refined our filtering strategy to address the observed loss of dCas9 expression (Fig. 3c). Strains were excluded if dCas9 levels were below 25% of control levels or if any constituent sgRNA target failed to be downregulated by at least 50% compared to controls. Applying these more stringent criteria, we found that 57% (17/30) of the two-sgRNA strains and 47% (14/30) of the three-sgRNA strains met the filtering requirements (Supplementary Fig. 6). In this cycle, we also observed that strains with poor target downregulation (‘Fail’) had worse isoprenol titers overall than those successfully passing the filter (Fig. 4b). Each subsequent cycle after the pseudorandom strains in DBTL1 utilized titer and sgRNA data from previous cycles (i.e., the ART model for DBTL2 was trained on validated data from both DBTL0 and DBTL1).
We measured isoprenol production and global proteomics from 347 sgRNA combinations across six DBTL cycles and generated 12 unique strains with at least 4-fold improvement over the control. In each cycle, we used our automation pipeline to construct and test the recommended strains, measure isoprenol and proteomics levels, filter strains based on target interference, and then train ART on filtered data. To get recommendations for the next cycle, we generated all possible target combinations, excluded combinations already tested, and then selected the combinations with the highest predicted titer. Combinations were subject to constraints that (1) the same sgRNA could not appear in more than a specified number of strains (between 5 and 15); and (2) that we select a specific number of combinations with two, three, or four sgRNA. We varied the number of times an sgRNA could appear in each training cycle to diversify the recommendations and better explore the experimental design space (i.e., we sampled more diverse sgRNAs if an sgRNA appeared fewer times). We found that our mean and maximum titers in passing strains increased each cycle until DBTL6 (Fig. 4b). Notably, we suffered 40% failure in DBTL2 strains owing to low dCas9 expression (Supplementary Fig. 6). Following DBTL2, we repaired the dCas9 vector to eliminate a redundant linker and reduce spurious recombination during adaptation, thereby increasing dCas9 fidelity. DBTL3 was then repeated with the same sgRNA arrays from DBTL2, with a noted 25% improvement in dCas9 fidelity. With the exception of DBTL6, subsequent cycles also maintained higher dCas9 expression (Supplementary Fig. 6). Although the repair only reduced the incidence of recombination, models after DBTL3 were trained only on data from the improved dCas9 plasmid, consolidating our design space to the 18 unique sgRNA that passed proteomics filters in DBTL3. Finally, DBTL6 exhibited an increase in strains that failed to pass the filter and exhibited poor titer, prompting the end of the campaign. The mixed performance of DBTL6 was partially due to the saturation of the two and three-sgRNA design space, which included an abundance of designs harboring the sgRNA targeting PP_1506, a vital and difficult to downregulate adenylate kinase. We also examined combinations of sgRNA for patterns of synergistic or antagonistic interactions between sgRNA, but we did not identify any obvious interactions (Supplementary Fig. 7).
Using ART to model the relationship between CRISPRi combinations and titer resulted in a predictive model that could extrapolate to new designs, enabling our active learning cycles to efficiently navigate a vast phase space. ART had very high predictive accuracy when trained on the full dataset (coefficient of determination R2 > 0.80, mean absolute error = 76 mg/L) but had lower accuracy when 5-fold cross-validation was used to test its ability to generalize (Supplementary Fig. 6). This is not surprising, given the high-dimensional input data and the relatively sparse sampling of interactions between different targets present in the training data. For example, the cross-validated R2 after DBTL1 was 0.04, indicating very limited predictive power. This low value may be due to overfitting, as the number of instances (97) was close to the number of features (67). However, as more instances were added to the training set with each new DBTL cycle, the cross-validated R2 increased to a final value of 0.65 for DBTL6 (Fig. 4c, Supplementary Fig. 8). Thus, despite low initial R2 values, ART was still able to yield productive recommendations that guided the search through the phase space to increase titer. This demonstrates that low cross-validation accuracy does not necessarily prevent the model from identifying regions of the input phase space that will yield high titers. The model may predict that these combinations will have relatively high titers, but poorly predict the numerical value, leading to low R241.
The latter three DBTL cycles were narrowed to combinations of 16 specific sgRNAs and generated strains with greatly improved titer, suggesting that these particular guides were instrumental in isoprenol biosynthesis. To quantify the relative impact of each sgRNA on titer across our dataset, we performed a parent-child analysis that examined the effect of adding a given sgRNA (child, e.g., PP_0815_PP_1317_PP_4192) to an otherwise identical array (parent, e.g., PP_1317_PP_4192) on titer. We repeated this for each of our final 16 sgRNAs (Fig. 4d). When evaluating the change in titers for all such pairs in our dataset, the majority of our most abundant final sgRNAs had a positive effect on titer. For all the most beneficial targets, our dataset included at least 10 parent-child pairs, suggesting that the effects of each sgRNA are robust to the presence or absence of other sgRNAs (Fig. 4e). Conversely, we observed that the inclusion of certain sgRNAs, PP_1506 for instance, led to a higher likelihood of failing the proteomics filter (Fig. 4f). While the PP_1506 sgRNA had a positive effect on titer in the one parent-child pair analyzed, it was predominant in strains that failed the proteomics filter.
In summary, our active learning process, enabled by automation, systematically identified numerous combinations of CRISPRi targets that contributed to high isoprenol titer, based on the data from previous DBTL cycles. This shows that ML and automation complement each other to solve a problem that is intractable through traditional intuition-driven approaches: the prediction of the impact of multiple gene perturbations to systematically improve titers. This process also provided a massive repository of high-quality proteomics data that included over 1400 entries and 472 unique CRISPRi strains from which biological insights and mechanistic hypotheses for improved isoprenol titer could then be generated.
Biological insights from CRISPRi through global proteomics
Following the approximately 5-fold increase in isoprenol titer from our reduced panel of sgRNAs, we explored how these guides enhance isoprenol production. Isoprenol biosynthesis via the isopentenyl diphosphate (IPP)-Bypass pathway utilizes ubiquitous cellular metabolites: three acetyl-CoA precursors, two ATP molecules, and one NADPH cofactor (Fig. 1a). P. putida, an obligate aerobe, maintains a branched respiratory pathway characterized by five terminal oxidases that ultimately drive ATP biosynthesis. The majority of sgRNAs associated with improved isoprenol production perturbed oxidative phosphorylation and hence ATP generation, either by directly affecting electron transport chain (ETC) complexes, the tricarboxylic acid cycle (TCA), or by influencing the supply of reducing intermediates (Fig. 5a).

a An abridged metabolic map of oxidative phosphorylation, the TCA cycle, and relevant sgRNA targets that contributed to high isoprenol production. Guides are annotated with bolded accession ID (orange) and gene name (black). b Statistically significant Log2(Fold-change) values (paired two-tailed Student’s T-test, p < 0.05) for selected sgRNAs of electron transport chain complexes and selected proteins from the TCA cycle. POI Log2FC cells are bolded. Source data are provided in the Source Data file.
Downregulation of genes involved in oxidative phosphorylation may have resulted in a cascade of regulatory changes that affected ATP generation to favor isoprenol production (Fig. 5b). To investigate the potential mechanisms by which certain sgRNAs increased isoprenol production, we examined how the downregulation of individual genes in DBTL0 affected their associated metabolic pathways (Fig. 5b). Eight sgRNAs downregulated genes encoding complexes in the ETC, four of which targeted cytochrome bo3 terminal oxidase (cyoA-D; PP_0812-15). Downregulation of this cytochrome bo3 oxidase significantly affected complementary terminal oxidases, especially increasing levels of the cyanide-resistant cytochrome oxidase CioA (Log2FC = 2.5) and cbb3-I cytochrome c oxidase (Log2FC = 0.95). This cbb3 terminal oxidase has a distinctly higher oxygen affinity compared to other terminal oxidases and is typically expressed under anaerobic conditions42. Perhaps as a consequence of this shift in terminal oxidase expression, all strains harboring sgRNAs downregulating cytochrome bo3 oxidase were observed to have significantly higher ATP synthase expression, which is consistent with previous work42. Although not directly involved in the oxidative phosphorylation, downregulation of farnesyl diphosphate synthase (ispA; PP_0528) disrupted ubiquinol biosynthesis—an essential component of the electron transport chain—and significantly increased CioA levels (Log2FC = 2.6) while downregulating native isoprenoid biosynthesis (Supplementary Fig. 9). We speculate that higher levels of the cbb3 terminal oxidase and CioA under aerobic conditions may enhance ATP generation and thereby impact isoprenol production.
Many of the most prominent sgRNAs for increased isoprenol production broadly downregulated the two-enzyme conversion of 2-ketoglutarate to succinate in the TCA cycle, previously shown to improve isoprenol titer36. In particular, these included sgRNAs targeting TCA cycle genes, namely malate oxidoreductase (mqo1; PP_0751), 2-oxoglutarate dehydrogenase (PP_4189), and succinate dehydrogenase (sdhB-C; PP_4191-92), as well as ETC complexes NADH-quinone oxidoreductase (nuoB; PP_4120) and cytochrome c reductase (petA; PP_1317). While sucB (PP_4188) was not specifically selected by ART, most high-performing sgRNAs indirectly lowered SucA-D levels (Fig. 5b), and all top-performing CRISPRi arrays lowered SucC-D levels, potentially recapitulating the previously observed high isoprenol phenotype (Supplementary Fig. 10).
Overall, titer was improved by downregulating specific genes associated with oxidative phosphorylation and the TCA cycle, with particular synergy between PP_0812-15, PP_0751, PP_0368 (an acyl-CoA dehydrogenase), and PP_0528, combinations of which saturated the best CRISPRi arrays.
Investigating off-target effects by candidate sgRNAs via gene knockout
We sought to investigate the presence of off-target gene downregulation amongst our best-performing sgRNAs by comparing isoprenol titers between pairs of knockout (KO) strains harboring either a non-target sgRNA or the “target” sgRNA previously used to downregulate the KO gene. Although we filtered strains for dCas9 expression and, where possible, POI abundance, our filter did not preclude off-target effects. Off-target effects are a well-known caveat of CRISPR. While many algorithms have been designed to predict and avoid off-target effects while maximizing sgRNA efficiency43, confident design of specific and precise sgRNAs remains challenging. CRISPRi off-target downregulation has been consistently observed with 9 nt and occasionally as few as 4 nt sequence identity 5′ of a protospacer adjacent motif44,45. We can identify whether an sgRNA affects titer through off-target effects by comparing isoprenol titers between strains harboring non-target and target sgRNA in which the targeted gene is absent.
Of our most prevalent 16 sgRNAs, 11 KOs were successful, including KOs of 6 genes that were plausibly essential based on their absence in the P. putida RB-TnSeq fitness library (Supplementary Data 1)46. However, KO strains often exhibited drastically different growth and production phenotypes than their CRISPRi counterparts. A poignant example is PP_4191: although it could be deleted, ΔPP_4191 strains showed severe growth inhibition and negligible isoprenol production, reflecting its high failure rate across sgRNA arrays (Fig. 4f) and suggesting that PP_4191 is on the boundary of essentiality. Likewise, although the PP_0751 sgRNA was abundant across high-performing strains, ΔPP_0751 strains proved difficult to culture in minimal medium. Again, this discrepancy stems from the relative burden of the deletion compared to downregulation (Fig. 6a).

a Comparison of mean isoprenol production between strains expressing either a PP_0815-targeting sgRNA or a non-targeting control sgRNA in a knockout background, demonstrating significant off-target effects of the PP_0815 sgRNA (n = 3). b Volcano plots of two KO strains, ΔPP_0812 and ΔPP_0815, showing fold-change differences in global protein expression with a target vs. non-target sgRNA. The ΔPP_0815 strain harboring a PP_0815 sgRNA shows a distinctive shift in global protein expression, whereas the ΔPP_0812 strain, a counterexample, has minimal differences in protein abundance. A panel of candidate genes that were up- and downregulated in the ΔPP_0815 strain harboring the PP_0815 sgRNA was then up- or downregulated in new strains to assess their potential for improving isoprenol production either through direct overexpression or via favorable off-target effects, respectively. c A KEGG pathway enrichment analysis of all significantly upregulated and downregulated proteins (paired two-tailed Student’s T-test, p < 0.05) in ΔPP_0815 harboring PP_0815 sgRNA indicated significant upregulation across secondary metabolism and amino acid biosynthesis with robust changes to central carbon metabolism. Despite improved strain performance, over 50 genes associated with ribosome biogenesis were downregulated. d Overall, 12 unique sgRNA arrays exhibited 4-fold improvement in titer compared to the control, all of which included the PP_0815 sgRNA (n = 3). The arrays were composed of seven sgRNAs with significant redundancy. KO strains were built to recapitulate strain performance with a mixture of KOs and sgRNA downregulating essential or unknown genes. Error bars represent standard deviations. Source data are provided in the Source Data file.
We transformed KO strains with their target sgRNA and screened against the performance of KO strains harboring non-target sgRNAs to elucidate off-target effects. As previously noted, the sgRNAs targeting PP_0814 and PP_0815 were manually passed through our proteomics filter. Although their abundances were routinely below our minimum level of detection, strains downregulating PP_0814 and PP_0815 were still observed to have lower PP_0812-encoded protein abundance with Log2FC = −3.5 and Log2FC = −1.1, respectively, while improving titer. Lower abundance of the PP_0812-encoding protein, which is encoded upstream in the cytochrome c oxidase complex operon, strongly indicated successful downregulation of their POI by association. While most KO strains showed similar titers to their CRISPRi counterparts from DBTL0 (Supplementary Fig. 11), ΔPP_0815 and ΔPP_0812-15 harboring PP_0815 sgRNA produced significantly more isoprenol compared to those with non-targeting guides, indicating that an off-target gene was driving isoprenol production level (Fig. 6a). Furthermore, both deletion strains displayed broad changes in the proteome that were not reflected in other cytochrome bo3 subunit deletions when comparing non-target and target sgRNAs (e.g., ΔPP_0812) (Fig. 6b).
In all, 636 genes spanning across many distinct metabolic pathways were significantly up- or downregulated between ΔPP_0815 harboring the non-target and target sgRNA (Fig. 6c). Differential expression between PP_0815 and the non-target sgRNA in ΔPP_0815 showed a broad reshuffling of central carbon metabolism with upregulation of amino acid biosynthesis and downregulation of ribosome biogenesis. These were accompanied by downregulation of genes associated with alcohol/aldehyde salvage47, PQQ biosynthesis, and, perhaps most peculiarly, valine tRNA-ligase (PP_0977) (Supplementary Table 1).
Although the off-target effects of the PP_0815 sgRNA led to increased isoprenol titers, identifying the responsible off-target gene proved challenging. We hypothesized that differentially expressed proteins between ΔPP_0815 strains harboring either the PP_0815 or non-target sgRNA might be driving high isoprenol titers. Therefore, we identified a set of off-target candidates derived either from this differential expression analysis or from a list of biomarkers for the PP_0815 sgRNA identified through Stabl, a sparse modeling approach for identifying biomarkers (“Methods” section) using the entire filtered DBTL proteomics dataset48. Specifically, we overexpressed two operons, PP_2791-94 (lvaA-D; levulinic acid degradation) and PP_2208-09 (phnW-X; phosphonoacetalaldehyde hydrolase) on secondary plasmids and further downregulated 14 potential off-target candidates using CRISPRi. To achieve this, IY1452b strains harboring overexpression candidates, along with IY1452b ΔPP_0815 strains expressing sgRNA for the various off-target candidates, were transformed, cultured, and characterized for isoprenol production and proteomics. Although the overexpression candidates slightly improved isoprenol titers (Supplementary Fig. 12), we were unable to either identify or, in the case of top biomarker PP_0977, successfully downregulate the off-target candidates to recapitulate PP_0815 sgRNA performance (Supplementary Fig. 13).
The noted off-target effects of PP_0815 sgRNA are particularly intriguing because they (1) significantly increased isoprenol titer across strains, (2) appeared in all top-performing CRISPRi arrays, (3) may have been overlooked in traditional heuristic methods due to PP_0812-14 redundancy, and (4) raised a difficult question of causality: whether profound shifts in global protein expression are the result or cause of increased isoprenol production. Indeed, high production outliers that failed the proteomics filter in DBTL0 (e.g., PP_5419, PP_1770, PP_1240) also had significant downregulation of genes that were downregulated in our highest production strains (Fig. 6c). Many of these downregulated genes encode proteins that are highly abundant in P. putida, broadly pertain to protein synthesis, and negatively correlate with isoprenol across our dataset (Supplementary Fig. 14). While ribosome biosynthesis proteins are essential and therefore absent from large RB-TnSeq libraries, their downregulation has not been observed in isoprenol tolerance studies49. This would suggest that a global reshuffling of expression occurs specifically due to high isoprenol production (as opposed to presence) or specifically within our IY1452b strain. In any case, these findings highlight the advantages of corresponding proteomic datasets not only to validate gene perturbations, but to parse off-target effects from broad production-induced changes in protein expression.
Overall, we found 12 unique CRISPRi arrays that yielded over 4-fold titer improvement, but that recapitulation of CRISPRi strain performance through gene KO alone proved challenging (Fig. 6d). All high-performing arrays harbored PP_0815, which had favorable off-target downregulation, and four harbored PP_0528, which downregulated an essential gene. While genomically modified strains (e.g., KO strains) are considerably more stable than those expressing CRISPRi arrays, single gene KOs only improved titer by up to 2-fold compared to the control. Combining KOs with specific sgRNAs for PP_0528 and PP_0815 further improved titer to 4-fold that of the control (651 mg/L) and 12% more isoprenol than the two-sgRNA array in a strain without KOs (580 mg/L, p < 0.02), indicating the importance of modulating rather than deleting certain genes (e.g., off-target or essential) to recapitulate production phenotypes.
Our investigation into the off-target effects of candidate sgRNAs via gene knockout revealed significant insights into the complexities of CRISPRi-mediated gene downregulation. Despite the challenges associated with off-target effects, our study successfully identified unique CRISPRi arrays that substantially improved isoprenol titers, underscoring the potential of strategic sgRNA design and application. Notably, the PP_0815 sgRNA emerged as a key component in high-performing arrays, demonstrating favorable off-target downregulation that contributed to enhanced production levels. However, the difficulty in recapitulating CRISPRi strain performance through gene knockout alone highlights the nuanced interplay between gene modulation and production phenotypes. Our findings emphasize the salience of integrating large proteomic datasets to discern off-target effects and production-induced changes, paving the way for more stable and efficient biotechnological applications.
Discussion
Here, we have demonstrated the power of synergizing active learning, CRISPRi, and laboratory automation to dramatically accelerate DBTL cycles in synthetic biology. The integration of these technologies enabled the collection of highly reproducible experimental data to guide an active learning process, which effectively identified high-titer strains in a large design space. Our pipeline enabled us to evaluate the effects of an unprecedented number of CRISPRi arrays on isoprenol titer and target protein downregulation, and our algorithm allowed us to effectively focus on the diminutive region of the design space that improves production. Collectively, we generated and evaluated 472 unique CRISPRi strains (from a total design space of ~800,000 possible strains) with 12 CRISPRi sgRNA arrays, yielding over 4-fold improvement and one with 5-fold improvement compared to the control. These results demonstrate that the combination of automation and machine learning can facilitate the prediction of the phenotypic impact of multiple gene perturbations with an efficacy that is unrivaled by traditional intuition-driven approaches.
We found that CRISPRi downregulation fails more often than expected, complicating its use and interpretation. Generally, past work has evaluated CRISPRi efficacy using a change in fluorescence, transcriptomics, or not at all, and has claimed that CRISPRi is generally effective at downregulating the measured output. However, our work, which directly measured target protein expression across a swath of metabolic pathways, challenges this paradigm. We show that CRISPRi targets across a range of cellular functions—both essential and nonessential—are frequently not downregulated, or downregulated only slightly, even with optimal sgRNA selection. Moreover, CRISPRi targets that are downregulated for a single target may not be downregulated when a combination of genes is targeted. We also found that CRISPRi, which is frequently used to prototype metabolic engineering perturbations, can have a large effect on product titer even with slight target downregulation. Finally, bioproduction performance was often distinct between CRISPRi downregulation and gene KO (e.g., PP_4191 sgRNA vs. ΔPP_4191 and PP_0751 sgRNA vs. ΔPP_0751). Such effects can result from the intrinsic complexity of metabolic pathways, in which even small changes in protein levels can have an unexpectedly large effect on titer. In practice, this observed heterogeneity in gene downregulation led us to implement a proteomics filter to provide high-quality data to train our active learning algorithm (ART).
While CRISPRi can elucidate genes whose downregulation improved titer, the recapitulation of our best-performing strains with gene KOs proved challenging due to gene essentiality and off-target effects. In particular, one gene (PP_0528) is essential, while another sgRNA (PP_0815) showed clear off-target effects. Both sgRNAs were frequent in our best-performing arrays. The noted off-target effects of PP_0815 sgRNA remain particularly intriguing because they significantly increased isoprenol titer across strains, appeared in all top-performing CRISPRi arrays, and would have been overlooked in traditional heuristic methods. Only by considering KO vs. downregulation could we discern the presence of off-target effects. This raises the troubling caveat that global -omics analysis is necessary for accurate CRISPRi characterization. More broadly, our inability to recapitulate strain performance suggests that CRISPRi is most useful to downselect to a minimum set of targets for precision genomic attenuation via engineering native regulatory elements (i.e., the associated RBS or promoter). However, such an approach would require significant screening or an accurate, predictive tool for protein expression.
Gene targets found to significantly increase titer were predominantly associated with the ETC and TCA cycles, likely affecting ATP generation, which is favorable for isoprenol production. We also found that some sgRNAs often indirectly downregulated other genes we had previously identified to improve isoprenol titer36, frequently yielding titers greater than direct downregulation of those previously identified genes. This effect showcases the importance of non-linear phenotypic effects of genetic perturbations, as well as the difficulty of predicting them from prior biological knowledge. Proteomics also revealed interesting patterns amidst high-titer production strains, including downregulation of ribosomal proteins, TCA cycle proteins, and a curious upregulation of levulinic acid degradation. These findings provide insight into how specific guides perturb cellular metabolism; however, future investigations should leverage the richness of our massive repository of global proteomics data to inform empirical models that holistically explain the patterns driving higher isoprenol production.
Collectively, our optimizations were shaped by the strategies and constraints employed in the study, namely the initial selection of sgRNA targets, native strain background, proteomics filter, and application of ART. Notably, we found that explicit, uncertainty quantification (UQ)-driven exploration of our design space was of limited utility in this case, since our binary input space resulted in ART producing very similar prediction uncertainties across many suggested designs. Furthermore, ART frequently generated degenerate recommendations in which two or three high-titer sgRNAs were combined with several other sgRNAs with no predicted effect. To address this, we adopted an ad hoc approach of limiting the number of occurrences of specific sgRNAs per round, thereby generating more exploratory designs. Consequently, our experimental approach led us to a specific maximum titer within an undoubtedly complex experimental landscape. Nonetheless, the framework we describe is broadly applicable to other host strains, other gene targets, and other filtering strategies, which may reveal additional biological mechanisms that further improve isoprenol titer.
Overall, we have shown that laboratory automation can expedite DBTL cycles and yield high-quality data for machine learning to solve a critical challenge in biomanufacturing: the prediction of titers associated with multiple genetic perturbations. While the machine learning algorithm is important (i.e., ART’s performance on small data sets and its use of uncertainty quantification to produce recommendations), the experimental design is critical. Without the systematic use of proteomics to validate reduced POI abundance, we believe that the active learning process will be more susceptible to noise due to undetected CRISPRi failure and thereby require more iterative cycles. Indeed, we can see that recommendations that did not pass the filter led to lower production increases in the later DBTL cycles (DBTL 4, 5, 6; Fig. 4b). While CRISPRi enables quick gene perturbation, off-target effects and inconsistent impact relative to knockouts indicate that it may not be ideal for developing stable production strains. Instead, CRISPRi is better suited for identifying genes that significantly influence production titers for precision engineering. Nonetheless, the advantages of combining automation and machine learning may be applied to other strategies for genetic modification50. The pipeline we developed can be adapted to various production schemes, organisms, media (e.g., lignocellulosic hydrolysate), and products, enabling the swift and efficient identification of genes for attenuation to improve bioproduction. This capability is crucial for accelerating biomanufacturing to tackle global challenges.
Methods
Pathway overview
The base pathway (Fig. 1a) is an engineered mevalonate (MVA) pathway with a promiscuous mevalonate decarboxylase (PMD*) from S. cerevisiae capable of converting mevalonate monophosphate into isopentenyl monophosphate, thereby bypassing isopentenyl diphosphate (IPP-Bypass)51,52. Pathway protein expression was monitored across all strains (Supplementary Fig. 15).
Construction of placeholder sgRNA Library
Cycle libraries were constructed via an automated Golden Gate assembly combinatorial strategy, Biopart Assembly Standard for Idempotent Cloning (BASIC)36,53. Briefly, sgRNA sequences of heuristic or FluxRETAP34 targets were generated using CRISPOR43 selected for no off-target sites, high predicted efficiency, binding to the non-target (sense) strand, and proximity to transcriptional initiation. Oligonucleotides (Supplementary Data 2) encoding sgRNAs (Integrated DNA Technologies, Redwood City, CA) with flanking type IIS endonuclease BsaI recognition sites were digested with BsaI HF v2 (New England Biolabs (NEB), Ipswich, MA), then purified with magnetic beads (AxyPrep Mag PCR Clean-Up, Axygen Inc., Union City, CA). A placeholder plasmid with sfGFP flanked by PaqCI recognition sites was likewise digested (NEB) to yield complementary cohesive ends for subsequent ligation with T4 DNA ligase (NEB) and transformation into XL-1 Blue chemically competent E. coli cells prepared by the UC Berkeley QB3 Core facility (Berkeley, CA). Transformations were plated on LB (Luria Bertani) agar (10 g/L tryptone, 5 g/L yeast extract, and 10 g/L NaCl; Millipore Sigma, Burlington, MA) with 100 mg/L carbenicillin and, following overnight growth, screened against sfGFP fluorescence. A single non-fluorescent colony was inoculated into liquid medium for plasmid extraction (QIAPrep Spin Miniprep Kit, Qiagen), then screened via Sanger sequencing (Azenta Life Sciences, Burlington, MA). Each placeholder plasmid thus maintained a “gRNA unit” composed of a J23119 constitutive Anderson promoter, a designed sgRNA, the Cas9 sgRNA handle, and the Cas9 terminator sequence again flanked by BsaI endonuclease recognition sites for future assemblies.
Automated construction of dCas9-gRNA plasmids
A 96-well plasmid extraction kit (BioBasic, Markham, ON, Canada) compatible with the Hamilton VANTAGE (Hamilton, Reno, NV) was used for purification of the plasmid libraries. Prefix and suffix linkers consist of an annealed adapter and linker oligo. In accordance with BASIC, individual oligos were first phosphorylated53 by mixing 5 μL T4 ligase buffer, 100 μM oligo, 1 μL T4 polynucleotide kinase (NEB) in a 50 μL reaction, followed by incubation at 37 °C. Phosphorylated oligos were then mixed in 400 μL of annealing buffer, heated to 95 °C for 10 min, then cooled to room temperature. Annealed prefix or suffix linkers were stored at −20 °C. Each suffix and prefix has position-dependent overhangs that facilitate the construction of multiplexed combinatorial arrays. Multiplexed sgRNA arrays demanded annealing of position-dependent linkers for each sgRNA, often with the same sgRNA unit in multiple positions on different recommendations (e.g., the PP_0812 sgRNA unit in position one as well as position two in a given cycle). A Jupyter notebook was written for rapid translation of ART recommendations to an ECHO dispense list.
The ECHO 550 acoustic liquid handler (Beckman Coulter, Brea, CA) was used to dispense 30 μL reaction mixtures for simultaneous digestion and annealing into a 384-well PCR plate as the destination plate (Beckman Coulter). Reagents for the reactions were stored in an ECHO-compatible 384-well polypropylene plate (384PP; Beckman Coulter). Each reaction mixture included 4 μL placeholder plasmid (diluted to 400 ng/μL on the plate), 0.5 μL BsaI HF v2, 0.5 μL T4 ligase, 5 μL of specified prefix and suffix linker, 3 μL ligase buffer, and 12 μL deionized (DI) water for each positional sgRNA arrangement. The dCas9-harboring vectors pIY989 or pDBTL3-Template also expressed sfGFP flanked by BsaI recognition sites, enabling fluorescent screening of successful transformants. Whole plasmid sequencing (Primordium Labs, Monrovia, CA; Plasmidsaurus, Eugene, OR) was performed on the dCas9 template vector as well as pIY670 before every cycle to ensure fidelity.
Analogous to the placeholder plasmids, the dCas9 vectors were similarly dispensed and annealed with linkers contingent upon the number of guides in the array. For context, generating the entire set of DBTL1-linked sgRNA units and destination vectors required 597 liquid handling steps. Following dispensing, the 384-well plates were sealed and transferred to a 384-well C1000 Touch Thermal Cycler (Bio-Rad, Hercules, CA). The assemblies were incubated at 37 °C for 2 min, then 20 °C for 1 min and cycled 20 times. Reactions were quenched by holding at 65 °C for 20 min before cooling to 4 °C.
Magnetic bead purification was performed using magnetic beads (Axygen Inc.) and automated using a KingFisher Apex (ThermoFisher Scientific, Waltham, MA). Purified linked sgRNA and vector units were then dispensed into another ECHO-compatible PP384 well source plate for ligation in a 384-well PCR destination plate. Owing to the designed compatible cohesive ends of the different prefixes and suffixes, ligation was accomplished simply by dispensing 0.5 μL of each DNA component with 0.5 μL cutsmart buffer (NEB) in a 5 μL reaction, subsequently incubating at 50 °C for 60 min to enable recombination. After DBTL0, each DBTL cycle consisted of 60 strains for four consecutive BioLector (Beckman Coulter) runs with on-plate controls.
Each reaction mixture was cooled and, as before, new assemblies were transformed into XL-1 Blue chemically competent E. coli cells. Here, 15 μL of chilled competent cells were dispensed into the reaction mixtures using a MANTIS liquid handler (FORMULATRIX, Dubai, United Arab Emirates). The transformation mixture was incubated on ice for 20 min, heat shocked at 42 °C for 1 min, and returned to ice for 5 min. Following heat shock, the transformations were outgrown by resuspension into a 96-well deep well plate (DWP) with 1 mL of SOC (20 g/L tryptone (Gibco Bacto, Fisher Scientific), 5 g/L yeast extract (Gibco Bacto, Fisher Scientific), 5 mM MgSO4, 20 mM glucose, 10 mM NaCl, and 2.5 mM KCl), and cultured for 1 h. Either the Biomek FX (Beckman Coulter) or the Hamilton VANTAGE was used to plate outgrowth cultures onto Q-Trays for overnight recovery. A QPix 460 colony picker (Molecular Devices, San Jose, CA) was then used to elucidate non-fluorescent colonies for overnight growth in a 96-well plate with LB and antibiotic (LB with 30 mg/L gentamicin). An aliquot of the overnight was cryostocked, with the remainder undergoing plasmid DNA extraction. Sanger sequencing confirmed a remarkably high assembly fidelity, typically surpassing 90% (55/60 transformations), though longer sgRNA arrays exhibited more frequent failure. Alignments were performed using multiple alignment using fast Fourier transform (MAFFT) in Benchling (Benchling, San Francisco, CA). In the event of sequencing failure, new non-fluorescent colonies were picked.
Automated transformation of P. putida
The selected chassis strain, P. putida IY1449b, has the in-frame deletions ΔphaABC, ΔmvaB, ΔhbdH, and 4,538,575Δ86,812 (Δzwf, ΔglZ, and ΔliuC) for improved isoprenol titers31. Each cycle, the strain was streaked onto an LB agar plate from which a single colony was inoculated into LB medium, grown overnight, and made electrocompetent for transformation with pIY670, harboring the IPP-Bypass MVA pathway, and selected (50 μg/mL kanamycin sulfate) to generate IY1452b31. From there, electrocompetent IY1452b cells were prepared for transformation with cycle-specific dCas-gRNA libraries.
A high-throughput electroporation platform was used to perform electroporation of dCas9-gRNA harboring plasmids into our P. putida production strain IY1452b32. The apparatus provides 384 individually addressable wells (0.1 mm gap width) in a format compatible with liquid handlers. The custom 384-well electroporation plates were washed with isopropyl alcohol, 70% ethanol, and MilliQ water before baking at 50 °C for 30 min. Then, plates were irradiated with 103 μJ/cm2 UV light for 30 s (UV Crosslinker, Fisher Scientific) and sealed with an AeraSeal gas-permeable membrane (Excel Scientific, Victorville, CA). The ECHO first dispensed 200 nL of each plasmid from a 384PP plasmid library plate, then dispensed 2 μL of electrocompetent P. putida IY1452 cells into individual wells. During DBTL0, transformations were completed in triplicate (Supplementary Fig. 3) while subsequent DBTL cycles of 60 sgRNA arrays were completed without replicates. Electroporations were completed with a voltage of 250 V and time constant of 5 ms, then resuspended in SOC medium with the Biomek FX (Beckman Coulter). Transformations were transferred to 96-well DWPs for recovery over 3 h at 30 °C and 1000 RPM shaking (INFORS HT Multitron, Bottmingen, Switzerland) and, finally, plated onto Q-Trays with LB medium (50 μg/mL kanamycin sulfate and 30 μg/mL gentamicin sulfate). Transformations were imaged using the QPix.
Passaging and culturing of P. putida strains
All strains were adapted to growth on minimal glucose medium. Briefly, single colonies were inoculated into 96-well DWPs with 1 mL LB medium (50 μg/mL kanamycin sulfate, 10 μg/mL gentamicin sulfate), then 40 μL was passaged twice into 1 mL of M9-NREL medium with the appropriate antibiotic. Here, 96-well DWP plates were again sealed with a gas-permeable membrane. M9-NREL medium was selected owing to its prevalence as a baseline P. putida production medium. Briefly, the medium composition included 20 g/L glucose, 0.5 g/L NaCl, 6.8 g/L Na2HPO4, 3 g/L KH2PO4, 100 μM CaCl2, 2 mM MgSO4, 10 mM (NH4)2SO4, and 500 μL of a trace metal solution (Teknova Cat no. T1001; Teknova, Hollister, CA). Gentamicin concentration was reduced during passaging and production owing to the considerable burden of dual antibiotics on growth. Following adaptation, strains were inoculated in triplicate into 1.5 mL of M9-NREL media in a 48-well BioLector flower plate without optodes and gas-permeable sealing foil (Beckman Coulter Life Sciences) to reduce evaporation. The production medium was always freshly made just prior to inoculation. A BioLector Pro (Beckman Coulter Life Sciences) was selected for strain culturing owing to its purported repeatability and scalability33,54,55. Cultures were grown at 24 °C and shaken at 1000 RPM without humidity control as batch experiments. Isoprenol pathway genes were induced after 8 h by the addition of L-arabinose to a final concentration of 2 g/L. Following 48 h of production, cultures were transferred from the 48-well BioLector plate into a 96-well DWP and pelleted by centrifugation at 3000 rpm in a benchtop centrifuge. Supernatant was extracted and transferred to a fresh deep well plate, while 10 μL of the pelleted cells were transferred to a 96-well PCR plate and frozen at −80 °C for proteomic analysis. All P. putida strains used in this study are listed in Supplementary Table 2.
Quantification of isoprenol using GC-FID
Isoprenol from BioLector experiments was detected using gas chromatography-flame ionization detection (GC-FID; Agilent Technologies, Santa Clara, CA). Here, a 400 µL aliquot of centrifuged culture supernatant was mixed with 400 µL of ethyl acetate containing 30 mg/L of 1-butanol as an internal standard. The extractions were vortexed for 10 min at 3000 RPM and then separated by centrifugation at 18,000 × g for 5 min. After separation, 200 µL of each ethyl acetate extraction was aliquoted into amber GC vials with glass vial inserts (Agilent Technologies). Samples were run on an Agilent 8890 GC System equipped with dual lines, Agilent 7693 A Autosampler, FID, and DB-Wax columns (15 m × 320 μm × 0.25 μm, Agilent J&W). The inlet and detector were held at 250 °C and 300 °C, respectively. All samples were run using splitless injection and a flow rate of 2.2 mL/min He. The oven program had an initial hold of 1 min at 40 °C, then the temperature was increased to 100 °C at a ramp rate of 15 °C/min, then increased to 230 °C at a ramp rate of 30 °C/min before holding for 1 min. All GC-FID data were quantified using OpenLab Chromatography Data Systems (Agilent Technologies).
Proteomics analysis
Protein was extracted from 10 µL cell pellets, and tryptic peptides were prepared by following an established proteomic sample preparation protocol56. Cell pellets were resuspended in Qiagen P2 Lysis Buffer (Qiagen, Germany) to promote cell lysis. Proteins were precipitated with the addition of 1 mM NaCl and 4× volume of acetone, followed by two additional washes with 80% acetone in water. The recovered protein pellet was homogenized by pipette mixing with 100 mM ammonium bicarbonate in 20% methanol. Protein concentration was determined by the DC protein assay (Bio-Rad, USA). Protein reduction was accomplished using 5 mM tris 2-(carboxyethyl)phosphine (TCEP) for 30 min at room temperature, and alkylation was performed with 10 mM iodoacetamide (IAM; final concentration) for 30 min at room temperature in the dark. Overnight digestion with trypsin was accomplished with a 1:50 trypsin:total protein ratio. The resulting peptide samples were analyzed on an Agilent 1290 UHPLC system coupled to a Thermo Scientific Orbitrap Exploris 480 mass spectrometer for discovery proteomics57. Briefly, peptide samples were loaded onto an Ascentis® ES-C18 Column (Sigma–Aldrich, USA) and were eluted from the column by using a 10 min gradient from 98% solvent A (0.1 % FA in H2O) and 2% solvent B (0.1% FA in ACN) to 65% solvent A and 35% solvent B. Eluting peptides were introduced to the mass spectrometer operating in positive-ion mode and were measured in data-independent acquisition (DIA) mode with a duty cycle of 3 survey scans from m/z 380 to m/z 985 and 45 Tandem mass spectrometry (MS2) scans with precursor isolation width of 13.5 m/z to cover the mass range. DIA raw data files were analyzed by an integrated software suite DIA-NN58. The database used in the DIA-NN search (library-free mode) included the latest P. putida KT2440 Uniprot proteome FASTA sequences in addition to the protein sequences of heterologous proteins and common proteomic contaminants. DIA-NN determines mass tolerances automatically based on first pass analysis of the samples with automated determination of optimal mass accuracies. The retention time extraction window was determined individually for all MS runs analyzed via the automated optimization procedure implemented in DIA-NN. Protein inference was enabled, and the quantification strategy was set to Robust LC = High Accuracy. The output main DIA-NN reports were filtered with a global false discovery rate set at 0.01 (FDR < = 0.01) on both the precursor level and protein group level. The Top3 method, which is the average MS signal response of the three most intense tryptic peptides of each identified protein, was used to plot the quantity of targeted proteins in the samples59,60.
Computational proteomics preprocessing
Initial CRISPRi target interference validation
In DBTL0, we used proteomics to determine whether an sgRNA downregulated a target gene. We kept the sgRNAs of strains that satisfied the following three constraints: (1) below 90% of the library mean for the target; (2) in the bottom quartile of target expression; and (3) isoprenol titer greater than 66.6 mg/L (approximately 40% of control isoprenol titer). Filtering resulted in 67 validated sgRNA targets.
Pseudorandom combinations for DBTL1
We generated pseudorandom combinations of the validated targets from DBTL0 to generate recommendations for DBTL1, biased by the target titer. We generated weights for each target using Equation 1:
Here, titer was normalized to the control mean. We generated 30 of each two-sgRNA and three-sgRNA arrays by randomly selecting from the targets based on their weighted probabilities.
Multiguide CRISPRi validation
In DBTL1-6, we used proteomics abundance measurements normalized to the control to identify CRISPRi combinations in which all individual perturbations successfully interfered with their target. We first calculated the control average for all proteins targeted in a given DBTL cycle, along with the control average for dCas9. For each individual line, we then calculated the relative abundance of all targeted proteins in the strain relative to the control. To ensure that strains used for modeling express dCas9, we discarded strains with dCas9 expression below 25% of the control in any of the replicates. Strains were discarded if any individual POI level was above 50% that of the control in any of the replicates to ensure that strains used for modeling showed target downregulation. If any target was below the proteomics limit of detection, we assumed that the specific target was successfully downregulated. This proteomics filter was applied to the data collected in each DBTL cycle.
Active learning using the automated recommendation tool
We used ART to model the relationship between sgRNA and isoprenol titer and recommend new sgRNA combinations to try in the next DBTL cycle. ART is a flexible, user-friendly active learning package that has been applied to several other metabolic engineering optimization problems, including promoter selection and media optimization21,26. We represented sgRNA inputs as binary vectors of length 67 (the number of validated sgRNAs from DBTL0), where a 1 represented the presence of a sgRNA and a 0 represented the absence of that sgRNA. We chose to use a binary representation instead of one-hot encoding based on the assumption that the position of the sgRNA in the plasmid would not affect the outcome, and in order to reduce the sparsity of the dataset.
ART is an ensemble modeling tool. For our model, we used 7 models from the scikit-learn Python package (neural regressor, random forest regressor, support vector regressor, kernel ridge regressor, k-nearest neighbor regressor, Gaussian process regressor, and gradient boosting regressor). We also included one TPOT regressor model61. Model parameters are shown in Supplementary Table 3. We first tested models using 5-fold cross-validation to evaluate predictive performance. During cross-validation, we randomly split the data, ensuring that replicates from the same strain were all included in the same split. Finally, we trained a model on the full dataset to make recommendations.
For DBTL2, we trained ART on the data from DBTL0 and DBTL1 after filtering for proteomics. For DBTL3, we did not use ART, since we remade the designs from DBTL2 with an improved dCas9 plasmid. For DBTL4, we trained an ART model only on the data from DBTL3. Since DBTL3 only included 16 different sgRNA targets, we effectively reduced our target search space in DBTL4 and onwards. In DBTL5 and DBTL6, we trained on data from DBTL3 and all subsequent runs.
To generate recommendations, we passed all possible combinations of up to 4 sgRNA into ART (816,596 combinations for 67 targets as determined via binomial coefficients, \({\sum }_{k=2}^{4}\left(\frac{67}{k}\right)\)). In later cycles (DBTL4-5), we generated recommendations based only on sgRNA that were in DBTL3, since ART did not have any information about the contribution of other sgRNA not included in DBTL3 (18 targets, 3060 total recommendations). We used the trained ART model to calculate the mean and standard deviation of the posterior predictive distribution of titers for each sgRNA combination. To select recommendations to test experimentally, we sorted the list by the mean predicted titer. Then, we selected target combinations from the top of the list, with constraints on the number of times a single target could appear in the final list of experimental combinations. Specific settings for each cycle are detailed in Supplementary Table 4.
Sparse proteomics characterization using Stabl
We used the Stabl algorithm48 to identify sparse linear regression models that could predict isoprenol titer. Briefly, Stabl is a feature selection algorithm previously validated on high-dimensional simulated and biochemical data. Stabl provides false discovery rate control by augmenting the input data with ‘knockoff’ features62, which are generated from the input data and known to be uninformative. Then, subsets of features are used to fit a sparse classification (logistic regression) or regression model (LASSO, linear regression with L1 regularization). In this way, the number of knockoff features included in each model can be calculated explicitly, and an optimal false discovery rate can be chosen. We used Stabl in two ways: (1) to identify a minimal model to predict isoprenol titer; and (2) to classify strains based on the presence or absence of a sgRNA. We used standardized proteomics measurements for all endogenous proteins present in every cycle as features. After using Stabl to identify protein features to predict isoprenol, we identified upregulation targets based on their correlation with isoprenol titer. If a single target was part of a complex or operon, we included the whole complex/operon for upregulation. For predicting the presence or absence of an sgRNA in a strain, we further removed proteins within that operon, in addition to exogenous pathway proteins.
Overexpression of candidate genes
Plasmids were constructed by amplifying native P. putida genes associated with high isoprenol titer from Stabl. These operons, PP_2208-PP_2209 and PP_2791-PP_2794, were amplified along with the DBTL3 Control Vector using Q5 DNA polymerase (NEB) and oligos with 20 bp 5′ overhangs. The vector amplicons, designed for salicylic acid induction of the inserted genes, were digested with dpnI (Thermo Fisher Scientific), assembled (NEBuilder HiFi Assembly Cloning Kit, NEB), and, as before, cloned into XL-1-blue competent cells. Plasmid sequences were verified by whole plasmid sequencing (Primordium Labs) and ultimately transformed into IY1449b with pIY670 to evaluate the impact of titrated induction on isoprenol titer. Strains harboring genes informed by Stabl were adapted to M9 medium and cultured for over 48 h in a Biolector Pro with an RFP control (JBx_266188) before GC-FID analysis.
Knockout of candidate genes
Stable gene knockouts were generated from the parent strain IY1449b via a Cpf1-mediated repair63. Briefly, plasmid pTE452 harboring an RBS-tuned recT and mutL (E36K) from Pseudomonas aeruginosa under a 3-methyl-benzoate (3MB) inducible promoter was cloned into selected P. putida competent cells63,64. Another plasmid, pTE433β, harbored cpf1 from Francisella novicida for constitutive expression of a designed sgRNA. Initially, pTE433β was digested with PmeI and AgeI (NEB), purified, and then assembled (NEBuilder HiFi Assembly Cloning Kit, NEB) with a 70-mer ssDNA oligo encoding the sgRNA and flanked by 25 bp backbone homology. A library of plasmids targeting the 16 most abundant genes in later DBTL cycles was generated. These genes included PP_1769 in addition to those in Fig. 4D.
The pTE452 plasmid was cloned into IY1449b and grown overnight. Overnight cultures were diluted 10-fold into 25 mL LB gentamicin (30 mg/L) in a baffled 250 mL flask and grown for one hour at 200 RPM and 30 °C. After growth, 1 mM of 3MB was added to induce cpf1 expression, and cultures were grown for another hour. After growth, the strains were made electrocompetent as before and transformed with 1 μL of 100 μM repair oligonucleotides as well as 50 ng of sgRNA harboring plasmid. Transformations were recovered for 4 h in SOC medium before plating on LB kanamycin (50 mg/L) plates. Colonies were typically observed after outgrowth at 30 °C for 48 h. Deletion success rate varied dramatically between KO targets (Supplementary Table 5).
Statistics & reproducibility
All strains were cultured as biological triplicates (n = 3). Experimental groups were defined based on recommendations from the machine learning algorithm and limited to 60 recommendations to facilitate four sequential BioLector runs per DBTL cycle. Each experimental group was assembled, cultured, and analyzed under identical conditions alongside experimental controls. Data used to train the active learning model was filtered according to the method above; however, no data was excluded in our analysis. Where applicable, statistical significance was determined using a paired Student’s T-test, where p < 0.05. Spearman’s rank correlation coefficients (Supplementary Fig. 14) were calculated using the SciPy package in Python. With the exception of the box-and-whisker plot in Fig. 4d, all error bars represent standard deviation.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Oligonucleotides, as well as plasmids and strains constructed and used in this work, are described in Supplementary Data 2–4 as well as Supplementary Table 2. Plasmids and strains are available in the public domain of the JBEI Registry (https://public-registry.jbei.org). Source data are provided in the Source Data file. The generated mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifiers PXD063733 (DBTL0) [https://www.ebi.ac.uk/pride/archive/projects/PXD063733], PXD063737 (DBTL1) [https://www.ebi.ac.uk/pride/archive/projects/PXD063737], PXD063738 (DBTL2) [https://www.ebi.ac.uk/pride/archive/projects/PXD063738], PXD063740 (DBTL3) [https://www.ebi.ac.uk/pride/archive/projects/PXD063740], PXD063743 (DBTL4) [https://www.ebi.ac.uk/pride/archive/projects/PXD063743], PXD063744 (DBTL5) [https://www.ebi.ac.uk/pride/archive/projects/PXD063744], and PXD063746 (DBTL6) [https://www.ebi.ac.uk/pride/archive/projects/PXD063746]65. DIA-NN is freely available for download from https://github.com/vdemichev/DiaNN. All proteomics and GC-FID data were also deposited into a publicly available DRYAD database (https://doi.org/10.5061/dryad.gtht76hzh). Source data are provided with this paper.
Code availability
ART is freely available for non-commercial use by academic institutions and for a small licensing fee for commercial use; the license can be accessed at https://github.com/JBEI/ART. All code required to reproduce this study, including ART, automation workflows, and figure generation, is provided as Jupyter Notebooks and can be accessed at https://github.com/JBEI/Isoprenol_CRISPRi or https://doi.org/10.5281/zenodo.1717868466.
References
French, K. E. Harnessing synthetic biology for sustainable development. Nat. Sustain. 2, 250–252 (2019).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Gaj, T., Gersbach, C. A. & Barbas, C. F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31, 397–405 (2013).
Wetmore, K. M. et al. Rapid quantification of mutant fitness in diverse bacteria by sequencing randomly bar-coded transposons. mBio 6, e00306–e00315 (2015).
Romero, P. A. & Arnold, F. H. Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol. 10, 866–876 (2009).
Qi, L. S. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013).
Voigt, C. A. Synthetic biology 2020-2030: six commercially-available products that are changing our world. Nat. Commun. 11, 6379 (2020).
Paddon, C. J. & Keasling, J. D. Semi-synthetic artemisinin: a model for the use of synthetic biology in pharmaceutical development. Nat. Rev. Micro 12, 355–367 (2014).
Meng, F. & Ellis, T. The second decade of synthetic biology: 2010-2020. Nat. Commun. 11, 5174 (2020).
Keasling, J. et al. Microbial production of advanced biofuels. Nat. Rev. Microbiol. 19, 701–715 (2021).
Rosales Calderon, O. et al. Sustainable Aviation Fuel (SAF) State-of-Industry Report: State of SAF Production Process. Rep. NREL/TP-5100-87802 (National Renewable Energy Laboratory, 2024).
Baral, N. R. et al. Production cost and carbon footprint of biomass-derived dimethylcyclooctane as a high-performance jet fuel blendstock. ACS Sustain. Chem. Eng. 9, 11872–11882 (2021).
Walkling, C. J., Zhang, D. D. & Harvey, B. G. Extended fuel properties of sustainable aviation fuel blends derived from linalool and isoprene. Fuel 356, 129554 (2024).
Stephenson, A. et al. Physical laboratory automation in synthetic biology. ACS Synth. Biol. 12, 3156–3169 (2023).
Gurdo, N., Volke, D. C., McCloskey, D. & Nikel, P. I. Automating the design-build-test-learn cycle towards next-generation bacterial cell factories. N. Biotechnol. 74, 1–15 (2023).
Zhang, J. et al. Accelerating strain engineering in biofuel research via build and test automation of synthetic biology. Curr. Opin. Biotechnol. 67, 88–98 (2021).
Chao, R., Mishra, S., Si, T. & Zhao, H. Engineering biological systems using automated biofoundries. Metab. Eng. 42, 98–108 (2017).
Keasling, J. D. Synthetic biology and the development of tools for metabolic engineering. Metab. Eng. 14, 189–195 (2012).
Lee, S. Y. & Kim, H. U. Systems strategies for developing industrial microbial strains. Nat. Biotechnol. 33, 1061–1072 (2015).
Pandi, A. et al. A versatile active learning workflow for optimization of genetic and metabolic networks. Nat. Commun. 13, 3876 (2022).
Zhang, J. et al. Combining mechanistic and machine learning models for predictive engineering and optimization of tryptophan metabolism. Nat. Commun. 11, 4880 (2020).
Kumar, P. et al. Active and machine learning-based approaches to rapidly enhance microbial chemical production. Metab. Eng. 67, 216–226 (2021).
Koscher, B. A. et al. Autonomous, multiproperty-driven molecular discovery: From predictions to measurements and back. Science 382, eadi1407 (2023).
Angello, N. H. et al. Closed-loop optimization of general reaction conditions for heteroaryl Suzuki-Miyaura coupling. Science 378, 399–405 (2022).
Esterhuizen, J. A., Mathur, A., Goldsmith, B. R. & Linic, S. High-performance iridium-molybdenum oxide electrocatalysts for water oxidation in acid: Bayesian optimization discovery and experimental testing. J. Am. Chem. Soc. 146, 5511–5522 (2024).
Zournas, A. et al. Machine learning-led semi-automated medium optimization reveals salt as key for flaviolin production in Pseudomonas putida. Commun. Biol. 8, 630 (2025).
Jessop-Fabre, M. M. & Sonnenschein, N. Improving reproducibility in synthetic biology. Front. Bioeng. Biotechnol. 7, 18 (2019).
Baker, M. 1,500 scientists lift the lid on reproducibility. Nature 533, 452–454 (2016).
Morrell, W. C. et al. The experiment data depot: a web-based software tool for biological experimental data storage, sharing, and visualization. ACS Synth. Biol. 6, 2248–2259 (2017).
Pandit, A. V., Srinivasan, S. & Mahadevan, R. Redesigning metabolism based on orthogonality principles. Nat. Commun. 8, 15188 (2017).
Banerjee, D. et al. Genome-scale and pathway engineering for the sustainable aviation fuel precursor isoprenol production in Pseudomonas putida. Metab. Eng. 82, 157–170 (2024).
Gaillard, W. R. et al. High-Throughput Microfluidic Electroporation (HTME): a scalable, 384-well platform for multiplexed cell engineering. Bioengineering 12, 788 (2025).
Unthan, S., Radek, A., Wiechert, W., Oldiges, M. & Noack, S. Bioprocess automation on a Mini Pilot Plant enables fast quantitative microbial phenotyping. Microb. Cell Fact. 14, 216 (2015).
Czajka, J. J. et al. FluxRETAP: a REaction TArget Prioritization genome-scale modeling technique for selecting genetic targets. Bioinformatics 41, btaf471 (2025).
Wang, X. et al. Engineering isoprenoids production in metabolically versatile microbial host Pseudomonas putida. Biotechnol. Biofuels Bioprod. 15, 137 (2022).
Yunus, I. S. et al. Predictive CRISPR-mediated gene downregulation for enhanced production of sustainable aviation fuel precursor in Pseudomonas putida. Metab. Eng. https://doi.org/10.1016/j.ymben.2025.11.007 (2025).
Reis, A. C. et al. Simultaneous repression of multiple bacterial genes using nonrepetitive extra-long sgRNA arrays. Nat. Biotechnol. 37, 1294–1301 (2019).
Peters, J. M. et al. A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell 165, 1493–1506 (2016).
Dong, C., Fontana, J., Patel, A., Carothers, J. M. & Zalatan, J. G. Synthetic CRISPR-Cas gene activators for transcriptional reprogramming in bacteria. Nat. Commun. 9, 2489 (2018).
Shin, J. et al. Genome Engineering of Eubacterium limosum Using Expanded Genetic Tools and the CRISPR-Cas9 System. ACS Synth. Biol. 8, 2059–2068 (2019).
Radivojević, T., Costello, Z., Workman, K. & Garcia Martin, H. A machine learning automated recommendation tool for synthetic biology. Nat. Commun. 11, 4879 (2020).
Morales, G., Ugidos, A. & Rojo, F. Inactivation of the Pseudomonas putida cytochrome o ubiquinol oxidase leads to a significant change in the transcriptome and to increased expression of the CIO and cbb3-1 terminal oxidases. Environ. Microbiol. 8, 1764–1774 (2006).
Concordet, J.-P. & Haeussler, M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, W242–W245 (2018).
Cui, L. et al. A CRISPRi screen in E. coli reveals sequence-specific toxicity of dCas9. Nat. Commun. 9, 1912 (2018).
Rostain, W. et al. Cas9 off-target binding to the promoter of bacterial genes leads to silencing and toxicity. Nucleic Acids Res. 51, 3485–3496 (2023).
Price, M. N. et al. Mutant phenotypes for thousands of bacterial genes of unknown function. Nature 557, 503–509 (2018).
Thompson, M. G. et al. Fatty acid and alcohol metabolism in Pseudomonas putida: functional analysis using random barcode transposon sequencing. Appl. Environ. Microbiol. 86, e01665–20 (2020).
Hédou, J. et al. Discovery of sparse, reliable omic biomarkers with Stabl. Nat. Biotechnol. 42, 1581–1593 (2024).
Lim, H. G. et al. Evolution guided tolerance engineering of Pseudomonas putida KT2440 for production of the sustainable aviation fuel precursor isoprenol. Metab. Eng. 91, 322–335 (2025).
Iwai, K. et al. Scalable and automated CRISPR-based strain engineering using droplet microfluidics. Microsyst. Nanoeng. 8, 31 (2022).
Kang, A. et al. Isopentenyl diphosphate (IPP)-bypass mevalonate pathways for isopentenol production. Metab. Eng. 34, 25–35 (2016).
Kang, A. et al. Optimization of the IPP-bypass mevalonate pathway and fed-batch fermentation for the production of isoprenol in Escherichia coli. Metab. Eng. 56, 85–96 (2019).
Storch, M. et al. BASIC: A new biopart assembly standard for idempotent cloning provides accurate, single-tier DNA assembly for synthetic biology. ACS Synth. Biol. 4, 781–787 (2015).
Funke, M. et al. Microfluidic biolector-microfluidic bioprocess control in microtiter plates. Biotechnol. Bioeng. 107, 497–505 (2010).
Rienzo, M. et al. High-throughput screening for high-efficiency small-molecule biosynthesis. Metab. Eng. 63, 102–125 (2021).
Chen, Y. et al. Alkaline-SDS cell lysis of microbes with acetone protein precipitation for proteomic sample preparation in 96-well plate format. PLoS ONE 18, e0288102 (2023).
Chen, Y., Gin, J. & Petzold, C. J. Discovery proteomic (DIA) LC-MS/MS data acquisition and analysis v2. Protocols.io https://doi.org/10.17504/protocols.io.e6nvwk1z7vmk/v2 (2022).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020).
Ahrné, E., Molzahn, L., Glatter, T. & Schmidt, A. Critical assessment of proteome-wide label-free absolute abundance estimation strategies. Proteomics 13, 2567–2578 (2013).
Silva, J. C., Gorenstein, M. V., Li, G.-Z., Vissers, J. P. C. & Geromanos, S. J. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol. Cell Proteom. 5, 144–156 (2006).
Ribeiro, P. et al. in Genetic Programming Theory and Practice XX (eds Winkler, S. et al.) 1–17 (Springer Nature Singapore, 2024).
Barber, R. F. & Candès, E. J. Controlling the false discovery rate via knockoffs. Ann. Stat. 43, 2055–2085 (2015).
Czajka, J. J. et al. Tuning a high performing multiplexed-CRISPRi Pseudomonas putida strain to further enhance indigoidine production. Metab. Eng. Commun. 15, e00206 (2022).
Wannier, T. M. et al. Improved bacterial recombineering by parallelized protein discovery. Proc. Natl. Acad. Sci. USA 117, 13689–13698 (2020).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552 (2022).
Carruthers, D. N. et al. Automation and machine learning drive rapid optimization of isoprenol production in Pseudomonas putida. JBEI/Isoprenol_CRISPRi. Zenodo https://doi.org/10.5281/zenodo.17178684 (2025).
Acknowledgements
We would like to thank Dr. Thomas Eng, who supplied protocols for gene deletion as well as several plasmids and oligonucleotides used for gene deletions in previous P. putida work. We also appreciate the expertise shared by Dr. Joonhoon Kim, who introduced us to Biocluster in R and provided insights into exploring off-target effects. This work was part of the DOE Joint BioEnergy Institute (http://www.jbei.org) supported by the U.S. Department of Energy, Office of Science, Biological and Environmental Research Program, through contract DE-AC0205CH11231 between Lawrence Berkeley National Laboratory and the U.S. Department of Energy. All authors were supported on this contract. The United States Government retains, and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The views and opinions expressed by the authors do not necessarily reflect those of the United States Government or any of its agencies. The United States Government and its employees make no warranty, expressed or implied, regarding the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, nor do they represent that the use of such information would not infringe privately owned rights.
Author information
Authors and Affiliations
Contributions
D.N.C., P.C.K., I.S.Y., H.G.M., and T.S.L. conceptualized the study. Y.C. and J.W.G. collected and curated proteomics data. P.C.K. wrote code to use ART. D.N.C., and S.T. developed the automation workflow. D.N.C. and Y.L. performed the biological experiments. W.R.G. and J.S. developed the HTME, and W.R.G. performed the electroporations. D.N.C., P.C.K., and T.R. performed data analysis. P.D.A., A.K.S., C.J.P., A.M., H.G.M., and T.S.L., secured funding and resources. All authors revised and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
A.K.S. and J.S. are founders of XLSI Bio. H.G.M. and W.R.G. are scientific advisors to XLSI Bio. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Paul Jensen and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Carruthers, D.N., Kinnunen, P.C., Li, Y. et al. Automation and machine learning drive rapid optimization of isoprenol production in Pseudomonas putida. Nat Commun 16, 11489 (2025). https://doi.org/10.1038/s41467-025-66304-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66304-8
