
Abstract
The domestication of camelids played a significant role in the development of Andean societies. However, distinguishing wild from domesticated forms remains difficult due to overlapping morphology. We use palaeogenomics to reveal the sex and ancestry of camelids from the Tulán ravine, a key region for early pastoral development in the South-Central Andes. We find evidence for both Vicugna and Lama based on 49 individuals with >0.001x coverage. Investigations into 26 individuals >0.01x show all individuals, except one male Lama, represent ancestry not in modern individuals. Similar male-to-female sex ratios suggest hunting or herding with selective culling. We find limited evidence for intergeneric admixture common in modern domesticates. Here, we show that early Formative communities (3360–2370 cal. yr BP) in the Tulán ravine primarily exploited wild or now-extinct early domesticate lineages, indicating that present-day domestic camelids are not direct descendants of those managed by early pastoralists.
Similar content being viewed by others


Introduction
One of the most significant cultural developments of humanity was the domestication of plants and animals1. Domestication refers to the process by which humans selectively breed wild animals and plants for specific traits that serve human needs, leading to genetic, morphological, and behavioural changes over generations2. As domesticated animals became central to human societies, pastoralism emerged as a subsistence strategy, where humans raise and manage these animals for resources such as food, fibre, and transport, often incorporating varying degrees of mobility to access pastures and water3. However, despite its remarkable significance, domestication processes (e.g., wild ancestors, timing, location, and underlying genetic mechanisms) remain unsolved for many species4. This is especially true for the only large herd animals domesticated in the Americas, South American camelids1. The domestication of South American camelids played a significant role in the development of Andean societies. Their unique status as the continent’s sole large domesticates and ability to occupy broad domesticate roles (transport, fibre, food) exemplifies the pivotal role these animals played in shaping ancient South American societies. Camelid fibres and meat served both practical and ritual purposes, while pack llama (Lama glama) acted to connect distant communities5,6,7. Even today, South American camelids remain the most important resource for many communities8.
South American camelids comprise four extant species; two domesticated (llama [Lama glama], alpaca [Vicugna pacos]) and two wild (guanaco [L.guanaco], vicuña [V. vicugna]). Within the wild species, there are two subspecies each: L.g.cacsilensis, L.g.guanicoe, V.v.mensalis, and V.v.vicugna. While multiple regions show evidence of early camelid management, such as hunting, herd protection, and the early stages of captive care, the most likely centres of domestication, where the controlled breeding of camelids began, are currently considered to be the Central Andes, Southern Bolivia, and the Circumpuna region spanning Northern Chile and NW Argentina9,10,11,12,13,14,15. However, the timing and number of domestication centres remain debated16. Archaeological evidence suggests South American camelid domestication was a complex process and not synchronous across the Andes that began with hunting, herd protection, and finally captivity/selective breeding17. However, although South American camelids can be osteometrically classified as large (putatively Lama), or small (putatively Vicugna), it is difficult to designate an individual as wild or domesticated18,19. This is primarily due to the significant overlap in size between wild and domesticated forms, as well as the lack of clear, consistent morphological differences between them. These factors complicate efforts to definitively classify individuals as wild or domesticated, limiting insights into the presence of early domesticated individuals.
Genetics is a useful tool that can complement the archaeological record to provide further insights into domestication20. However, despite attempts to address theories of South American camelid domestication, the Spanish conquest of South America beginning ~ 1500 A.D and uncontrolled post-conquest intergeneric hybridisation between domesticated species makes it difficult to identify genetic affinities prior to ~ 1500 A.D from contemporary individuals21. This intergeneric hybridisation, which is thought to have become widespread following the Spanish arrival, has significantly shaped the genetic makeup of modern domesticated llama and alpaca populations21. As a result, modern populations are highly admixed, complicating efforts to trace pure genetic lineages that could shed light on the early domestication process. Ancient DNA (aDNA) from pre-conquest individuals is therefore a useful approach to reconstructing the uncertain past of South American camelid domestication and husbandry. However, although previous studies have used pre-conquest individuals, these have been limited to mitochondrial DNA18,22,23,24. Mitogenomes are maternally inherited, only represent a single locus, and are confounded by hybridisation and stochastic events25. This can be especially problematic in the case of South American camelids, as modern individuals are known to contain high levels of interspecific nuclear ancestry resulting from post-conquest hybridisation21, limiting the power of mitochondrial reference panels for taxonomic identifications.
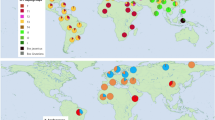
Here, we use palaeogenomic data to investigate the use of camelids by ancient South American communities. We analyse the sex and ancestry from pre-conquest South American camelids recovered from archaeological sites from the Tulán ravine (Tulán-52, Tulán-54, Tulán-85, and Tulán-94) located in the eastern border of the Salar de Atacama that descends into Northern Chile (Fig. 1). The Tulán ravine is a key area for the transition from hunter-gatherer societies, which are characterised by subsistence through hunting, and foraging of wild plants, to initial pastoral societies, where domesticated animals are herded for food, labour, and other resources, in the South-Central Andes26. The sites studied here belong to the Late Archaic period (Tulán-52; 5290-4840/4430-4090 cal. yBP) and to the Early Formative period (Tulán-54, Tulán-85, and Tulán-94; 3360–2370 cal. yr BP) when domesticated camelids are thought to have been incorporated into the economic activities of humans of this area. Specifically, archaeological remains from ritual (sites associated with ceremonial or religious practices) and domestic (sites used for daily living and subsistence activities) contexts provide evidence of a significant cultural and economic transformation from in the highlands of the Southern Andes27.

Results
Mapping results
Our study used genomic data from a total of 75 ancient camelid individuals, sourced from four archaeological sites: Tulán-52, Tulán-54, Tulán-85, and Tulán-94. These individuals had previously undergone mitochondrial and morphological analysis (Supplementary Data 1 and 2) but have yet to be analysed from a nuclear genomic perspective. After mapping to the alpaca reference genome, genome-wide coverages ranged between ~ 0 and 0.28 x or 4500 and 587 million mapped base pairs (Supplementary Data 2). The ancient samples showed typical ancient DNA damage patterns with elevated levels of A-G and C-T transitions towards the read ends (Supplementary Fig. 1).
To build a comparative reference panel, we also incorporated previously published genomic data from 80 modern individuals representing all extant Lama and Vicugna subspecies, which served as a comparative basis for the ancient samples. After mapping to the alpaca reference genome, all modern individuals had genome-wide coverages ranging from 8.34 x to 41.11 x (Supplementary Data 1).
Principal component analysis
To evaluate the broad relationships between the modern and ancient camelids included in this study, we performed principle component analyses (PCAs) using a pseudohaploid base call with ANGSD. When analysing the entire dataset (Lama sp. and Vicugna sp.), the modern individuals fell into five distinct clusters, in most cases pertaining to their subspecies classification (Fig. 2). However, we found L.g.cacsilensis and llamas clustering together, apart from two L.g.cacsilensis which clustered with L.g.guanicoe (Cacsilensis1 and Cacsilensis2). These two individuals are from outside of the known range of L.g.cacsilensis and were therefore likely misidentified21.

A PCA performed on all modern individuals and ancient individuals > 0.01 x using 5,163,744 transversion sites. B PCA performed on modern Lama individuals and ancient Lama individuals > 0.01 x using 3,668,867 transversion sites. C Ancestry states of eight ancient individuals. Ancestry states are derived from a single individual; L.g.cacsilensis - Cacsilensis4, L.g.guanicoe - Guanicoe1, V.v.mensalis - Mensalis4, and V.v.vicugna. Ancestral is defined as having combined ancestry between both Lama and Vicugna (C) or mixed ancestry between Lama (sub)species (D).
When considering the ancient samples > 0.01x, we see three groupings; One closest to V.v.vicugna and mostly containing osteometrically small size individuals, one mostly containing osteometrically large size individuals somewhat in the middle of the two modern Lama sp. clusters, and a single large ancient individual (C72) within the llama/L.g.cacsilensis cluster (Fig. 2A). When reducing the dataset to only include Lama sp. individuals, we again see C72 within the llama/L.g.cacsilensis cluster, with the remaining individuals still forming their own cluster, but sitting more closely to the llama/L.g.cacsilensis cluster (Fig. 2B). Results are similar when considering ancient individuals >0.001x; the cluster closest to V.v.vicugna remains but the structuring within Lama sp. becomes less clear (Supplementary Fig. 2). Individuals where the osteometric size did not match with their PCA placement included a single big individual (C38), and two small individuals (C69 and C70).
Phylogenetic analysis
To further investigate the broad relationships between the modern and ancient camelids included in this study, we computed a neighbour-joining tree. Our neighbour joining tree showed similar results as the PCA (Supplementary Fig. 3). There is a split in the ancient individuals, with individuals being most closely related to V.v.vicugna or the llama/L.g.cacsilensis clade. Only individual C72 was found to be within the llama/L.g.cacsilensis clade.
Ancestry proportions
To obtain high-resolution ancestry profiles, clarify the origins of each specimen, and assess possible interspecific gene flow, we calculated ancestry proportions for eight ancient individuals using admixfrog28. Investigations using simulated aDNA damaged modern individuals showed a good ability to discern subspecies designation in the wild individuals using this approach as the highest proportion of single subspecies ancestry in each individual was as expected (Supplementary Fig. 4). In the modern llama we saw a relatively high proportion of ancestral camelid DNA (0.11 vs. 0.01 in the wild species) but negligible levels of Vicugna ancestry (0.01). However, in the modern alpaca, we saw a much higher proportion of ancestral camelid DNA (0.44) as well as relatively high levels of Lama ancestry (0.13).
The archaeological specimens’ ancestry proportion results were in line with the PCA and phylogenetic tree results (Fig. 2C, D). C72 had more L.g.cacsilensis ancestry relative to L.g.guanicoe compared to the other ancient Lama sp. individuals (Fig. 2C). When looking into Lama sp. specific ancestry, C72 contained relatively higher levels of shared ancestry with modern llama than the other three investigated individuals (Fig. 2D). Overall, the genetically identified Lama sp. individuals had more L.g.cacsilensis ancestry than L.g.guanicoe and Vicugna sp (Fig. 2C). Putative Vicugna individuals have more V.v.vicugna ancestry than V.v.mensalis (Fig. 2C). The small sized C69 contained mostly L.guanicoe ancestry, and the big size C38 contained mostly V.vicugna ancestry. Only a very small proportion of each individual contained ancestral South American camelid ancestry < 0.04 (Fig. 2C).
When using the wild species reference panel, the vast majority of the oldest individual’s, C46 from Tulán-52, ancestry could not be determined beyond ancestral Lama, with the highest proportion of single subspecies ancestry (2%) coming from L.g.cacsilensis, the next closest being only 0.1% from L.g.guanicoe. When using the Lama only reference panel, we found 48.9% of its ancestry as ancestral Lama, 50.1% as ancestral L.g.cacsilensis/llama, 0.58% as L.g.cacsilensis only, 0.25% as llama only, and 0.13% as L.g.guanicoe only.
D-statistics
To further test the taxonomic placement of the archaeological specimens and to identify any signatures of interspecific gene flow, we used D-statistics. However, very low-coverage ancient DNA data has been shown to introduce biases in D-statistics analyses, especially when mapping to a divergent reference genome29. To address potential biases in our data, we conducted a simulated dataset analysis. Placing an individual with simulated ancient DNA damage into the H3 position produced comparable results to when placing a high-quality version of the same individual in the H3 position (Supplementary Fig. 5). Therefore, we could proceed with confidence knowing ancient DNA damage would not bias our topology/population structure results.
D-statistics results using the topology H1 = Lama sp. H2 = Vicugna sp. H3 = Ancient individuals returned results consistent with the PCA, phylogenetic tree, and ancestry proportion analysis; individuals C75, C72, C89, C21 were more closely related to Lama sp. and individuals C50, C39, C38, and C20 were more closely related to Vicugna sp. (Fig. 3A). Further investigations into the Lama sp. individuals showed that most had an overall closer relation to L.g.cacsilensis relative to llama or L.g.guanicoe (Fig. 3B). However, C72 had similar levels of L.g.cacsilensis and L.glama ancestry, as seen by few significant results when calculating D-scores using L.g.cacsilensis and L.glama in the H1 and H2 position respectively. Further investigations into the Vicugna sp. individuals showed an overall closer relation to V.v.vicugna relative to alpaca or V.v.mensalis (Fig. 3C).

A Comparisons between Lama and Vicugna. B Comparisons within Lama. C Comparisons within Vicugna. Non-significant values | Z | < 3 not shown.
Comparing results placing a llama individual with simulated ancient DNA damage into the H2 position, a Lama sp. in the H1 position, and a Vicugna sp. individual in the H3 position to those when placing a high-quality version of the same llama individual in the H2 position, we saw a slight discrepancy in the D-scores. The mean deviation was 0.0322, which we used to correct for ancient DNA/reference related biases in the interpretations of the empirical data (Supplementary Fig. 6).
Investigations into the topology ((Lama sp., C21), Vicugna sp.) found that individual C21 had less gene flow with Vicugna sp. than all modern llama individuals, similar levels of gene flow with V.v.vicugna and V.v.menalisis but more with alpaca compared to L.g.guanicoe, and less gene flow with alpaca but similar levels with V.v.vicugna and V.v.menalisis compared to L.g.cacsilensis (Fig. 4). Investigations into the topologies ((Lama sp., C72), Vicugna sp.) found that Tulan individual C72 had less gene flow with Vicugna sp. than modern llama, similar levels of gene flow with V.v.vicugna and V.v.menalisis but more with alpaca compared to L.g.guanicoe, and comparable levels with all Vicugna sp. compared to L.g.cacsilensis (Fig. 4).

A negative D-score shows relatively more gene flow between H1 and H3 compared to H2 and H3, whereas positive shows relatively more gene flow between H2 and H3. Non-significant values | Z | < 3 are shown with open circles.
Genetic sex determination
To evaluate whether the sex distributions in our archaeological assemblages reflect hunting or herding strategies, we genetically sexed all individuals with >5,000 mapped reads using the SeXY pipeline30. We were able to genetically determine the sex of 32 males and 29 females (Supplementary Data 2). We were unable to determine the sex of 14 individuals due to either a low number of mapping reads or ambiguous results. Further separating into each site, we found: Tulán-52 (1 F), Tulán-54 (24 F and 26 M), Tulán-85 (6 M and 3 F), and Tulán-94 (1 F). Further splitting by taxonomic status, we found similar proportions of male and female Lama at Tulán-54, slightly more females than male Vicugna at Tulán-54, and more males than females at Tulán-85 for both genera (Table 1). The one individual that showed the highest proportion of ancestry shared with modern llama (CL72) was a male. We note that while our findings provide insights into sex ratios at these sites, the limited sample size, especially at Tulán-85, presents a challenge in making broad inferences.
Discussion
The Tulán ravine, occupied during the Late Archaic and Early Formative periods, contains both ceremonial and residential sites with archaeological evidence for camelid exploitation, fibre processing, and ritual activity. Investigating palaeogenomic data, we identify the genetic ancestry and sex of camelids from these contexts and interpret these findings in light of the archaeological record to better understand the composition of camelid populations present during this critical period of economic and social transition in the South-Central Andes.
Specifically, we investigated 49 individuals with mean genome-wide coverages > 0.001 x from the ceremonial sites of Tulán-52 (Late Archaic; n = 1) and Tulán-54 (Early Formative; n = 40), as well as the residential site of Tulán-85 (Early Formative; n = 8). We found evidence for both genera, Vicugna and Lama, in our dataset. Further filtering to 26 individuals > 0.01x showed that all individuals, except one, likely represented ancestry not found in modern individuals (Fig. 2). Moreover, while our ancient data quality undoubtedly presented some limitations, particularly due to the low coverage and poor DNA preservation typical of temperate regions31, careful evaluation of biases through simulated data demonstrated that even low-coverage palaeogenomes can yield valuable insights when these biases are accounted for. This highlights the potential of low-quality data to provide meaningful results, even from challenging archaeological contexts.
The Vicugna individuals we identified most likely belonged to the subspecies V.v.vicugna, which was not unexpected given that the local subspecies of Vicugna is V.v.vicugna32. The presence of small individuals with characteristics indicative of vicuñas in the Tulán sites may reflect the value of vicuña fibre, which has been appreciated since pre-Hispanic times8, and is still greatly appreciated today. This may be the reason that most of the small animals in Tulán correspond to vicuñas. The use of camelid fibre is supported by microscopic camelid fibre analyses33. While the overwhelming majority of fibres were light brown natural colours, a small quantity of pure black fleece suggests colour variation, hinting at early colour selection of pre-domesticated camelids34. While this could suggest a lost domesticate-like lineage filling a similar role as the alpaca, this could also be the result of dye, so should be considered with caution35.
Lama individuals were more difficult to place than the Vicugna. All individuals had a tendency towards L.g.cacsilensis but all bar one had considerable levels of L.g.guanicoe ancestry. The current distribution indicates that L.g.cascilensis are generally found further north but may slightly overlap with L.g.guanicoe in Northern Chile36,37. Therefore, the ancient Tulán individuals may represent a locally extinct L.g.cacsilensis population that hybridised with the local L.g.guanicoe individuals. The one individual more closely related to L.g. cacsilensis, with relatively high levels of llama ancestry, is particularly interesting from a domestication standpoint. When placed in the broader archaeological context of the Tulán ravine, where camelids appear to have been used in a domesticated role, these genetic findings support the idea that this individual may represent either an early domesticated llama or a wild population ancestral to contemporary llamas, with full domestication likely occurring later.
Being an early domesticated llama may have also been the case for our one sample from the Late Archaic Tulán-52 individual (C46), which contained mostly L.g.cacsilensis/llama ancestry with relatively little presence of L.g.guanicoe. There is a cultural continuity between the Late Archaic and the Early Formative, so it is to be expected that these populations continued to develop the domestication/specialisation process. However, the very low coverage (0.008 x) nature of this sample prevented us from delving deeper. Taken together, our results suggest individuals that were not the direct ancestors of contemporary domesticated species were the main source of South American camelids used in both domestic and ceremonial activities in the Tulán ravine during the Early Formative period (3360–2370 cal. yr BP).
Osteometric analyses of the excavated samples of Tulán-54 previously revealed the presence of the two wild species: guanaco and vicuña, and the domestic species, llama27. However, the large degree of overlap between domestic and wild individuals makes clear results difficult to obtain38. Taking these difficulties together with the fact that there are other lines of evidence (e.g., artefacts and fibres39,40) supporting the notion that there were domesticated animals in the Tulan ravine since the Late Archaic period41, suggest the large animals were used in a domesticated role despite containing different ancestry to contemporary llama.
As it has been hypothesised that llama domestication was a complex process and may have occurred in several different areas of the Andes17, the Tulán individuals could represent a domestic lineage that was subsequently replaced or diluted through admixture with other populations. Similar patterns of replacement through introgression have been documented in other domestic mammals, such as pigs, where Near Eastern domestic ancestry was largely replaced by European wild boar ancestry following introduction42, and horses, where ancient domestic lineages were replaced by later lineages of different origin43. While the exact mechanisms and timing may differ, these cases illustrate how domesticated lineages can be transformed or supplanted through subsequent gene flow.
A previous study using mitochondrial genomes of the same individuals suggested that individuals whose size did not match their maternal ancestry (e.g., small size but Lama ancestry or big size but Vicugna ancestry) may have been hybrid individuals, which was interpreted as indicating domesticated individuals18. Our palaeogenomic analyses find that these potential hybrid individuals do not contain clearly higher levels of shared ancestry between genera. This finding challenges the assumption that mitochondrial and nuclear signatures, as well as osteometric measurements, should align to accurately determine domestication status. As mitochondrial DNA reflects only the maternal lineage, it may not fully capture genetic complexities such as intergenerational hybridisation and introgression. Moreover, osteometric measurements alone, though valuable in identifying size variation, do not account for the possibility of mislabelling or misidentification of juveniles versus adults. As such, we propose that the apparent discrepancy between mitochondrial and nuclear data regarding hybrid status is likely due to the inherent limitations of mitochondrial analysis. Consequently, the assumption that size, mitochondrial ancestry, and nuclear signatures should consistently align may not always hold due to the complexity of domestication processes and the potential for interbreeding between different camelid species and subspecies over time. While mitochondrial introgression may have occurred, a single sporadic mitochondrial introgression event could lead to inferences of widespread hybridisation, which, as shown by our nuclear genomic analysis, is not supported.
Despite early Spanish observers acknowledging the importance of camelid pastoralism, they generally failed to distinguish between domestic species in their records. This contrasts with Lama and Vicugna having long been distinct, biologically divergent genera separated approximately 2–3 million years ago21 and culturally differentiated by pre-Hispanic communities5. The subsequent indiscriminate hybridisation between domesticates resulted in a loss of their exquisite fibre qualities and colourations that made them so prized in prehistoric Andean communities5. This uncontrolled hybridisation between domesticated species and the population decline post-conquest makes the identification of pre-hispanic genetic ancestry of modern individuals difficult.
Previous work exclusively analysing genomes from modern individuals has found both llama and alpaca to contain evidence of intergeneric gene flow tracing back to ~500 years ago21. Our results show that the putative llama in our dataset contained less Vicugna ancestry than modern llama, and similar amounts to wild L.g.cacsilensis, the likely ancestral (sub)species of modern llama. Therefore, we suggest that intermixing between Vicugna (most probably alpaca) and llama was likely not a key component of the early domestication process, and only occurred within the last ~3000 years. While the absence of evidence for early hybridisation in our samples does not exclude its sporadic occurrence, it suggests that hybridisation was not a widespread practice during the studied period. Palaeogenomic data from more domesticated individuals will undoubtedly help shed further light on this issue.
In addition to the insights gained from the ancestry analysis, we were also able to infer the sex of the individuals using the palaeogenomic data. South American camelids are sexually monomorphic44, showing sexual dimorphism when specific bones are available (e.g., pelvis and canine teeth)45. While it is possible to sex South American camelids, this can rarely be done due to the fragmentary nature of most assemblages. Therefore, the palaeogenomic data presented a unique opportunity to make inferences on sex. Phalanxes and Astragali have been used in this study, due to their high frequency, completeness, and in the case of the former, as their fusion stage indicates the presence of adult animals (over 24 months)46. Thus, our results examine only the adult sex ratio in the archaeological assemblages.
Sex distribution in archaeological assemblages reflects both hunting and herding strategies. Hunting assemblages are generally expected to be dominated by adult remains, with sex ratios balanced or male-biased, whereas herding assemblages often include an adult sex ratio skewed toward females47. However, herding practices can vary, for example, through selective culling of infertile or undesirable males, which may produce more balanced adult sex ratios48. In the Tulán assemblages, we observe roughly similar proportions of males and females across sites and genera (Table 1), although the small sample from Tulán-85 requires cautious interpretation. These patterns are consistent with either hunting or herding with selective male culling and align with our genomic findings showing that most individuals at these sites were wild individuals and not the direct ancestors of domesticated camelids.
Taken together, our results suggest that early Formative communities in the Tulán ravine managed camelids in ways consistent with either mixed hunting and/or emerging herding practices rather than clearly established pastoralism. The dominance of wild or now-extinct domestic lineages indicates that communities in the region relied on populations distinct from the direct ancestors of modern llamas and alpacas, offering insight into the complex, regionally variable pathways of camelid use and domestication in the South-Central Andes.
Methods
Permits and permissions
All research was conducted in accordance with Chilean regulations. Excavation permits were issued by the Consejo de Monumentos Nacionales de Chile (CMN) under permit N° 4409 (18.11.2013). Export of osteological samples (Camelidae) to the University of Copenhagen for ancient DNA analysis was authorised by CMN ordinances ORD. N° 03642 (21.10.2016), N° 03746 (27.10.2016), and N° 03425 (20.01.2017), within the scope of FONDECYT project 1130917. All exported samples were photographed, labelled, and transported via certified courier in accordance with CMN requirements. Although none of the camelid remains analysed here were directly radiocarbon dated, chronological placement is based on radiocarbon dates from associated archaeological contexts (e.g., stratified deposits, fireplaces, and burials)49.
Archaeological contexts
It is proposed that the location of Archaic and Formative settlements in the Tulán area reflects a combination of favourable cultural, social, and environmental conditions. These conditions supported a shift from a society primarily reliant on hunting and foraging, characterised by seasonal mobility, small group sizes, and exploitation of wild resources, to one increasingly oriented toward pastoralism, marked by the management of domesticated camelids, more permanent settlement structures, and reliance on animal products for subsistence.
The Late Archaic period (ca. 5290-4090 cal. yBP) is marked by large campsites like Tulán-52, the emergence of substantial architecture with clusters of circular structures, and a diversified, innovative lithic industry. This period also saw the appearance of rock art, long-distance interactions, and a subsistence strategy that combined hunting with early camelid domestication. Altogether, these features point to a growing sociocultural complexity50.
The Early Formative period (ca. 3360−2370 cal. yBP) is characterised by the emergence of large settlements with a complex and planned ritual architecture along with the intensification and expansion of productive practices, an emphasis on ritual expressions, the appearance of new technologies such as pottery, and three large contemporary Formative settlements (Tu-54, Tu-122 and Tu- 85) have been documented for this period. These are located within a 15 km area between the border of the Salar de Atacama and the Tulán ravine, at an altitude ranging from 2300–3200 m. These sites have different functions. At Tulán-122, located in the middle sector of the ravine, residential aspects were emphasised. In contrast, Tulán-85, is located on the border of the resource-rich salt flat. These settlements shared a common political and religious organisation centred around the Tulán 54 site, the only site that possesses monumental architecture26.
The sunken temple structure identified in Tulán-54 at the centre of a refuse mound contained 27 human infant graves accompanied by offerings, rock art, structured fireplaces, and offering pits, among other elements, suggesting that the structure had a ceremonial function. The main structure identified at the site is a large, sunken semi-oval ceremonial structure surrounded by a perimeter wall made of large, vertically positioned stone blocks. The inner space is divided into six precincts, with a single oval structure in the middle26,27. The recovered camelid anatomical units display a very similar temporal and spatial pattern across all occupations and among the different precincts of the ceremonial structure, which was filled intentionally with the remains of butchering, processing, consumption and feasting activities. The waste was likely generated outside and then ritually incorporated51. On the border of the Salar de Atacama, Tulán-85 corresponds to an extensive occupation characterised by dense domestic monticulated deposits. The site also includes marginal structures and a sector with burials of newborns52. Finally, the Tulan-94 site is composed of groups of circular and sub-circular structures, with a total of 19 living enclosures. The evidence suggests the transitional character of the site, since it has material components corresponding to the Late Archaic and Early Formative phases26.
Samples
Our study consisted of 75 ancient individuals sourced from four archaeological sites: Tulán-52, Tulán-54, Tulán-85, and Tulán-94, and have previously undergone mitochondrial and morphological analysis18 (Supplementary Data 1 and 2). In brief, samples were collected from both ceremonial and domestic contexts across different sectors of the archaeological sites to capture spatial and temporal variation. Bone elements (primarily first anterior phalanges and astragali) were selected from stratified excavation units and structures, with care taken to sample different individuals. Only adult specimens, identified by fused epiphyses or advanced ossification, were included in the morphometric analysis. Measurements were taken using Vernier calipers to the nearest 0.1 mm, following established osteometric protocols, and were recorded prior to aDNA sampling to avoid damage-related bias. Key dimensions were measured on each bone type to assess size variation, and results were compared against published morphometric data for all four modern South American camelid species to evaluate patterns of morphological differentiation. The threshold for distinguishing small and large individuals was determined based on a comparison of key osteometric measurements from the first anterior phalanx (breadth of proximal articulation [BFp] and depth of the proximal epiphysis [Dp]) and the astragalus (breadth distal [Bd] and greatest length medial [GLm]). The distinction between small and large groups was tested statistically by correlating measurements from both the phalanx and astragalus using PAST53. Published morphometric data for all four modern South American camelid species were used to establish the thresholds for size differentiation54,55.
To build a comparative reference panel, we also included published genomic data from 80 modern individuals (DNAzoo.org21,36). The modern individuals represented all extant Lama and Vicugna subspecies (Fig. 1).
Bioinformatic processing
The raw sequencing data for the ancient Tulán individuals was generated in a previous study where only the mitochondrial DNA was analysed and obtained from the authors18 while the raw sequencing reads for the modern individuals were downloaded from the European Nucleotide Archive (ENA) (Supplementary Data 1 and 2). For all individuals, we removed Illumina adaptor sequences, low-quality reads (mean q < 25), short reads (< 30 bp), and merged overlapping read pairs with Fastp v0.23.256. We mapped the processed reads (merged only for the ancient individuals) to the alpaca reference genome (Genbank accession: GCF_000164845.3) using Burrows-Wheeler Aligner (BWA) v0.7.1557 and either using the aln algorithm, with the seed disabled (-l 999) (otherwise default parameters) for the ancient individuals or the mem algorithm with default parameters for the modern individuals. We chose the alpaca genome as the reference genome due to its hybrid ancestry21, making it putatively more suitable when making genomic comparisons between genera. We parsed the alignment files and removed duplicates and reads of mapping quality score < 30 using SAMtools v1.658. We checked for ancient DNA damage patterns of the ancient individuals using mapdamage v259. As the ancient individuals had also undergone targeted enrichment for mitogenomes and some selected genes, we aligned the probes to the reference genome using BLAST v 2.15.060 using default parameters, and we removed any regions of the alpaca genome aligning to the probes from the mapped bam files using bedtools v2.29.161.
Genetic sex determination
We identified scaffolds putatively originating from the sex chromosomes in the alpaca assembly by aligning the assembly to the alpaca X (included in the reference genome) and Human Y (Genbank accession: NC_000024.10) chromosomes using satsuma synteny v2.162 with default parameters. We followed the SeXY pipeline30 to identify the sex of each of the ancient individuals. We set a minimum threshold of 5000 mapped reads to ensure the reliability of the results. If an individual had a mean X:A ratio of < 0.7 it was designated as a male. If an individual had a mean X:A ratio of > 0.8 it was designated a female. Individuals with mean ratios between 0.7 and 0.8 were deemed undetermined.
Ancient DNA simulation
To assess the impact of ancient DNA damage on our analyses, we simulated the damage patterns and read lengths of individual C21 onto representatives of each modern subspecies (seven in total) using gargammel v1.1.463. We chose C21 due to it having the highest coverage of the ancient samples. To do this, we generated consensus fasta files from the mapped bam files of the modern individuals using ANGSDv0.92164 and the following parameters -dofasta 2, minimum read depth of 5 (-setmindepthind 5), and minimum mapping and base qualities of 30 (-minq 30 -minmapq 30). The modern individuals included L.glama (Llama2 and Llama3), V.pacos (Alpaca3), L.g.cacsilensis (Cacsilensis3), L.g.guanicoe (Guanicoe3), V.v.mensalis (Mensalis2), and V.v.vicugna (Vicugna1). We mapped the simulated ancient DNA reads to the alpaca reference genome following the same approach as for the ancient Tulán individuals. We additionally downsampled them to ~ 0.01 x using SAMtools.
Principal component analysis
We evaluated the relationships between the modern and ancient camelids included in this study by performing PCAs using a pseudohaploid base call with ANGSD. We initially did this for two different sample sets using both Lama and Vicugna individuals. The first sample set used all modern and ancient individuals with genome-wide coverages > 0.01x. The second used a broader sample set that also incorporated lower-coverage individuals (> 0.001 ×). We computed consensus pseudohaploid base calls specifying the parameters: minimum mapping and base qualities of 20 and 30 respectively (-minmapQ 20 -minQ 30), calculate genotype likelihoods using the GATK algorithm (-GL 2), calculate major and minor alleles based on genotype likelihoods (-doMajorMinor 1), remove transitions (-rmtrans 1), only consider autosomal chromosomes > 10 Mb (-rf), skip triallelic sites (-skiptriallelic 1), only consider reads mapping to one region uniquely (-uniqueonly 1), make a consensus identity by state base calls (-doIBS 2), output a covariance matrix (-doCov 1) and only consider variable positions where the minor allele occurs in at least two individuals (-minminor 2).
We also repeated this analysis, but only with individuals belonging to the Lama genus. We set the minimum individual threshold to the number of modern individuals + 1, i.e., 81 for the complete dataset and 57 for the Lama only.
Phylogenetic analysis
To build a neighbour joining tree, we first constructed a distance matrix, which we output from ANGSD when performing PCA (setting the parameter -makematrix 1). We converted the distance matrix into a neighbour-joining phylogenetic tree using fastME v2.1.6.165 and default parameters.
Ancestry proportions
We calculated the ancestry proportions of eight ancient individuals using admixfrog v0.7.228. We included four big size camelids (C21, C38, C72, C75) and four small size camelids (C20, C39, C50, C69). We selected these based on their higher coverage or unexpected placement within the PCA. Individuals C69 and C38 did not correspond to what was expected based on their size, and C75 clustered differently compared to the other big sized ancient individuals. We also ran this analysis on the lone individual from Tulán-52 (C46), as it comes from an earlier time period, which may be closer to the onset of domestication. Initially, we only considered the wild species as the ancestry states, each represented by a single individual; L.g.cacsilensis - Cacsilensis4, L.g.guanicoe - Guanicoe1, V.v.mensalis - Mensalis4, and V.v.vicugna - Vicugna2. We repeated the analysis for the ancient Lama individuals using the same two Lama sp. subspecies as above, plus Llama9 as ancestry states. As input for the reference panel, we created a multi-individual variant call file using BCFtools v1.1566, specifying autosomal scaffolds >10 Mb (-f) and minimum mapping and base qualities of 20 (-q 20 -Q 20). We ran admixfrog specifying a minimum read length of 30 bp, a bin size of 50 kb, and otherwise default parameters. We only considered a bin in interpretations if it contained more than five SNPs. We pooled regions showing mixed ancestry between Lama sp. and Vicugna sp. as ancestral camelid in the first run, and pooled regions showing any mixed ancestry in the Lama sp. only run as ancestral. Furthermore, to assess the accuracy and suitability of this method on our dataset, we also performed the same analysis (all wild (sub)species reference panel) on the modern individuals with simulated ancient DNA damage.
D-statistics
D-statistics calculate shared derived alleles between non-sister branches of a given topology [[[H1, H2], H3], Outgroup]. An allele is considered as derived if it is different to the outgroup allele. Although it is most commonly implemented to investigate differential levels of gene flow between non-sister taxon, it can also be used to investigate shared ancestral polymorphisms and therefore topology/population structure67,68.
We subsampled the dataset to include four modern individuals per subspecies and only the eight ancient individuals also used in the ancestry proportions analysis above. We calculated the D-statistics using a random base call approach in ANGSD (-doabbababa 1), specifying only autosomal scaffolds > 10 Mb and the following parameters: -minmapQ 30 -minQ 30 -blocksize 1000000 -rmtrans 1 -uniqueonly 1. To infer the ancestral sequence (-anc) we used a wild bactrian camel (Camelus ferus; NCBI biosample SRR1947250) which we also mapped to the alpaca genome following the approach mentioned above. We summarised the results using a block jackknife approach with the Rscript available in the ANGSD toolsuite (ANGSD_jackknife.R).
We filtered the output into three different sets. First, to investigate which genus each ancient individual was most closely related to, we filtered for H1 = Lama sp. H2=Vicugna sp. H3=Ancient individuals. Based on these results, we then looked into which subspecies the ancient individuals were most closely related to with either H1 = Lama sp. H2 = Lama sp. H3 = Ancient Lama sp. individuals or H1 = Vicugna sp. H2 = Vicugna sp. H3 = Ancient Vicugna sp. individuals. To investigate gene flow between the ancient Lama sp and Vicugna sp., we extracted results corresponding to the topologies H1 = Lama sp., H2 = C21, H3 = Vicugna sp. and H1 = Lama sp., H2 = C72, H3 = Vicugna sp.
To investigate whether the ancient DNA damage could cause biases in the topology test results, we compared results when placing the individuals with simulated ancient DNA damage in H3 with the results generated using the high-quality version of the same individual. To evaluate the ability of our dataset to infer gene flow between the putative ancient llama and Vicugna species, we also performed D-statistics with the two 0.01 x simulated aDNA llama (Llama 2 and 3) individuals in the H2 position, Vicugna sp in the H3 position and the remaining Lama sp. individuals in the H1 position. By comparing these results to those when the high-quality versions of llama 2 and 3 were in the same position, we were able to calculate a correction factor to account for potential biases. We found a mean deviation between the two results of 0.0322 (Supplementary Fig. 6), which we subtracted from the D-score and using the standard error output with ANGSD, we recalculated the Z scores.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All newly generated raw sequencing data supporting this study are available in the NCBI BioProject database under accession PRJNA1215279. Coordinates of the capture probes in the alpaca reference genome are available from the University of Copenhagen ERDA repository [https://erda.ku.dk/archives/7d1d408b1711f5ab7e34a845bf58f9a8/published-archive.html]. Modern publicly available comparative camelid genomic data used in this study, together with associated SRA accession numbers, are listed in Supplementary Data 1. The corresponding BioProjects include: PRJNA512907, PRJNA612032, PRJNA685331, PRJNA750515, and PRJNA1043467. The archaeological camelid remains analysed in this study are curated at the University of Copenhagen, Denmark and will remain in Denmark for the long term; access to the specimens can be arranged through the corresponding author.
References
Larson, G. & Fuller, D. Q. The evolution of animal domestication. Annu. Rev. Ecol. Evol. Syst. 45, 115–136 (2014).
Zeder, M. A. The domestication of animals. J. Anthropol. Res. 68, 161–190 (2012).
Salzman, P. C. Pastoralists: Equality, Hierarchy, and the State. (Routledge, Boulder, CO, 2004).
Frantz, L. A. F., Bradley, D. G., Larson, G. & Orlando, L. Animal domestication in the era of ancient genomics. Nat. Rev. Genet. 21, 449–460 (2020).
Wheeler, J. C., Russel, A. J. F. & Redden, H. Llamas and Alpacas: Pre-conquest breeds and post-conquest hybrids. J. Archaeol. Sci. 22, 833–840 (1995).
Hesse, B. Archaeological evidence for camelid exploitation in the Chilean Andes. Säugetierkundliche Mitteilungen 30, 201–211 (1982).
Broughton, K. L. South American camelids in Central Andean religious practices. (2010).
Vilá, B. & Arzamendia, Y. South American Camelids: their values and contributions to people. Sustainability Sci. 1–18 (2020).
Goñalons, G. L. M. & Yacobaccio, H. D. The domestication of South American camelids. Documenting Domestication-New Genetic and Archaeological Paradigms 228–244 (2006).
Cartajena Fasting, I. Explorando la variabilidad morfométrica del conjunto de camélidos pequeños durante el Arcaico Tardío y el Formativo Temprano en Quebrada Tulán, norte de Chile. Rev. del. Mus. de. Antropol. 2, 199–212 (2009).
Moore, K. M. In Sustainable Lifeways: Cultural Persistence in an Ever-Changing Environment 244–272 (University of Pennsylvania Press, 2012).
Moore, K. M. The Archaeology of Andean Pastoralism (2016).
Noe, S. J., Haas, R. & Aldenderfer, M. Hunting to herding on the Andean Altiplano: Zooarchaeological insights into Archaic Period subsistence in the Lake Titicaca Basin, Peru (9.0–3.5 ka). J. Anthropol. Archaeol. 77, 101658 (2025).
Le Neün, M. et al. Holocene occupation of the Andean highlands: A new radiocarbon chronology for the Telarmachay rockshelter (Central Andes, Peru). Quat. Sci. Rev. 312, 108146 (2023).
Marsh, E. J. & Rademaker, K. Human–environment interactions in the Lake Junín basin: Fire, megafauna, deforestation, and domestication, from the peopling of the Andes to the Inca Empire. Quat. Sci. Rev. 351, 109159 (2025).
Mengoni Goñalons, G. L. Camelids in ancient Andean societies: A review of the zooarchaeological evidence. Quat. Int. 185, 59–68 (2008).
Yacobaccio, H. D. The domestication of South American camelids: a review. Anim. Front 11, 43–51 (2021).
Diaz-Maroto, P. et al. Ancient DNA reveals the lost domestication history of South American camelids in Northern Chile and across the Andes. ELife 10, e63390 (2021).
Le Neün, M. et al. Can first phalanx multivariate morphometrics help document past taxonomic diversity in South American camelids?. J. Archaeol. Sci. Rep. 47, 103708 (2023).
Zeder, M. A., Emshwiller, E., Smith, B. D. & Bradley, D. G. Documenting domestication: the intersection of genetics and archaeology. Trends Genet. 22, 139–155 (2006).
Fan, R. et al. Genomic analysis of the domestication and post-Spanish conquest evolution of the llama and alpaca. Genome Biol. 21, 159 (2020).
Abbona, C. C. et al. Were domestic camelids present on the prehispanic South American agricultural frontier? An ancient DNA study. PLoS ONE 15, e0240474 (2020).
Westbury, M. et al. First complete mitochondrial genome data from ancient South American camelids - The mystery of the chilihueques from Isla Mocha (Chile). Sci. Rep. 6, 38708 (2016).
Díaz-Lameiro, A. M. et al. Ancient DNA confirms crossbreeding of domestic South American camelids in two pre-conquest archaeological sites. J. Archaeol. Sci. 141, 105593 (2022).
Westbury, M. V. et al. Hyena paleogenomes reveal a complex evolutionary history of cross-continental gene flow between spotted and cave hyena. Sci. Adv. 6, eaay0456 (2020).
Nuñez, A. L. et al. Emergencia de comunidades pastoralistas formativas en el sureste de la Puna de Atacama. Estud. Atacameños 93, 117 (2006).
Núñez, L. et al. The temple of Tulán-54: Early Formative ceremonial architecture in the Atacama Desert. Antiquity 91, 901–915 (2017).
Peter, B. M. 100,000 years of gene flow between Neandertals and Denisovans in the Altai mountains. Preprint at https://doi.org/10.1101/2020.03.13.990523 (2020).
Yuan, J. et al. Camelus knoblochi genome reveals the complex evolutionary history of Old World camels. Curr. Biol. 34, 2502–2508 (2024).
Cabrera, A. A. et al. How low can you go? Introducing SeXY: sex identification from low-quantity sequencing data despite lacking assembled sex chromosomes. Ecol. Evol. 12, e9185 (2022).
Hofreiter, M. et al. The future of ancient DNA: Technical advances and conceptual shifts. Bioessays 37, 284–293 (2015).
González, B. A. et al. Phylogeography and population genetics of vicugna vicugna: evolution in the arid Andean high plateau. Front. Genet. 10, https://doi.org/10.3389/fgene.2019.00445 (2019).
Benavente, M. A. Análisis lanimétrico de fanéreos de los sitios Tulán-52 y 54. In Transición del Arcaico Tardío al Formativo Temprano en la Cuenca de Atacama: Emergencia de complejidad sociocultural en la subárea Circumpuneña. FONDECYT (National Fund for Scientific and Technological Development) 10120216 (2005).
Dransart, P. Llamas, herders and the exploitation of raw materials in the Atacama Desert. World Archaeol. 22, 304–319 (1991).
Dransart, P. Camelid fleece and the hidden histories of colors in the andes. https://doi.org/10.32873/unl.dc.tsasp.0079 (2020).
León, F. et al. History of diversification and adaptation from north to south revealed by genomic data: Guanacos from the desert to sub-Antarctica. Genome Biol. Evol. 16, evae085 (2024).
González, B. A., Samaniego, H., Marín, J. C. & Estades, C. F. Unveiling current Guanaco distribution in chile based upon niche structure of phylogeographic lineages: Andean puna to subpolar forests. PLoS ONE 8, e78894 (2013).
López, M. et al. The use of hunting and herding spaces: Stable isotope analysis of Late Archaic and Early Formative camelids in the Tulan transect (Puna de Atacama, Chile): Stable isotopes analyses on camelids in tulan transect (Chile). Int. J. Osteoarchaeol. 27, 1059–1069 (2017).
Arias, G., Benavente, A. & Gecele, P. Identificación y variabilidad del uso del animal a través de textiles arqueológicos: Contraste con patrones fanéreos actuales. Actas del. xii Congr. Nacional de. Arqueología Chil. 151, 162 (1993).
Cartajena, I., Benavente, M. A., Núñez, L. & Thomas, C. La utilización de los camélidos durante el Formativo Temprano: una comparación entre la cuenca del Loa medio y el Salar de Atacama. Zooarqueología y. Tafonomía en. el Confín del. Mundo 45, 58 (2009).
Loyola, R. et al. Hunting and feasting in the pre-Columbian Andes: Exploring the nature and scale of early ceremonial aggregations in Tulan Ravine (5300 to 2400 yr cal. BP) through the circulation of obsidian artefacts. J. Anthropol. Archaeol. 64, 101360 (2021).
Frantz, L. A. F. et al. Ancient pigs reveal a near-complete genomic turnover following their introduction to Europe. Proc. Natl. Acad. Sci. USA 116, 17231–17238 (2019).
Librado, P. et al. Widespread horse-based mobility arose around 2200 BCE in Eurasia. Nature 631, 819–825 (2024).
Miranda-de la Lama, G. C. & Villarroel, M. Behavioural biology of South American domestic camelids: An overview from a welfare perspective. Small Rumin. Res. 220, 106918 (2023).
Kaufmann, C. A. Estructura de Edad Y Sexo En Guanaco: Estudios Actualísticos Y Arqueológicos En Pampa Y Patagonia. (Sociedad Argentina de Antropología, 2009).
Kent, J. D. The Domestication and exploitation of the South American camelids: methods of analysis and their application to circum-lacustrine archaeological sites in Bolivia and Peru. (1982).
Yacobaccio, H. Andean camelid herding in the South Andes: ethnoarchaeological models for archaeozoological research. Anthropozoologica 42, 143–154 (2007).
Wheeler, J. C. Pre-conquest alpaca and llama breeding. Camelid Quarterly (2005).
Núñez, L. et al. Presencia de un centro ceremonial formativo en la circumpuna de Atacama. Chungará 49, 3–33 (2017).
De, S. P., Cartajena, I., Núñez, L. & Carrasco, C. Cazadores-recolectores del Arcaico Tardío y desarrollo de complejidad social en la puna de Atacama: las evidencias del sitio Tulan-52 (norte árido de Chile). Rev. Werkén 13, 91–118 (2010).
Cartajena, I., Mendoza, P. L., Santander, B. & Núñez, L. Discarding practices among early herders on the western watershed of the Puna de Atacama, Chile (ca. 3000-2300 cal BP). in Animals: Cultural Identifiers in Ancient Societies? (eds. Peters, J., McGlynn, G. & Goebel, V.) 65–84 (Documenta Archaeobiologiae 15 VML Verlag Marie Leidorf, 2019).
Lopez, P., Cartajena, I. & Nunez, L. Analyses of stable isotopes in camelids collagen bones from Tulan Ravine, Atacama Puna, early formative period (CA 3, 1000-2,400 BP). Rev. de. Antropologia Chil. 45, 237–247 (2013).
Hammer, Ø, Harper, D. A. T. & Ryan, P. D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 4, 9 (2001).
Cartajena, I. Los conjuntos arqueofaunísticos del Arcaico Temprano en la Puna de Atacama, Norte de Chile. PhD diss., Freie Universität Berlin, Berlin, (2002).
L’Heureux, G. L. Morfometría de camélidos sudamericanos modernos. La variabilidad morfológica y la diversidad taxonómica. Zooarqueología a principios del siglo XXI. Aportes teóricos, metodológicos y casos de estudio 39–50 (2010).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Jónsson, H., Ginolhac, A., Schubert, M., Johnson, P. L. F. & Orlando, L. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684 (2013).
Mount, D. W. Using the basic local alignment search tool (BLAST). CSH Protoc. 2007, db.top17 (2007).
Quinlan, A. R. BEDTools: The swiss-army tool for genome feature analysis. Curr. Protoc. Bioinform. 47, 11.12.1–34 (2014).
Grabherr, M. G. et al. Genome-wide synteny through highly sensitive sequence alignment: Satsuma. Bioinformatics 26, 1145–1151 (2010).
Renaud, G., Hanghøj, K., Willerslev, E. & Orlando, L. gargammel: a sequence simulator for ancient DNA. Bioinformatics 33, 577–579 (2017).
Korneliussen, T. S., Albrechtsen, A. & Nielsen, R. ANGSD: Analysis of next generation sequencing data. BMC Bioinform. 15, 356 (2014).
Lefort, V., Desper, R. & Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 32, 2798–2800 (2015).
Narasimhan, V. et al. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 32, 1749–1751 (2016).
Westbury, M. V. et al. Extended and continuous decline in effective population size results in low genomic diversity in the world’s rarest hyena species, the brown hyena. Mol. Biol. Evol. 35, 1225–1237 (2018).
Segawa, T., Rey-Iglesia, A., Lorenzen, E. D. & Westbury, M. V. The origins and diversification of Holarctic brown bear populations inferred from genomes of past and present populations. Proc. Biol. Sci. 291, https://doi.org/10.1098/rspb.2023.2411 (2024).
R Core Team. R: A language and environment for statistical computing. (2013).
Natural Earth. Free vector and raster map data. https://www.naturalearthdata.com.
Acknowledgements
We would like to thank Anders Hansen for support in generating the sequencing data and Alba Rey-Iglesia for help curating the data. We would also like to thank Lautaro Núñez who through numerous projects (FONDECYT 1020316, 1070040 and 1130917) recovered the analysed camelid bone remains. M.V.W. acknowledges support from the Novo Nordisk Foundation Emerging Investigator Grant (NNF24SA0093839).
Author information
Authors and Affiliations
Contributions
Conceptualisation, M.V.W.; Formal analysis, C.O.H. and M.V.W.; Investigation, P.D.M. and I.C.; Writing – Original Draft, M.V.W.; Writing – Review & Editing, All authors; Resources, P.D.M. and I.C.; Supervision, M.V.W.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
O’Hare, C., Diaz-Maroto, P., Cartajena, I. et al. Palaeogenomics suggest domesticated camelid herding and wild camelid hunting in early pastoralist societies in the Atacama Desert. Nat Commun 16, 11007 (2025). https://doi.org/10.1038/s41467-025-66343-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66343-1
