
Abstract
Three-dimensional multi-fold twin architectures are valuable for tailoring material properties but constructing them in man-made crystals remains challenging due to angular misfits from incomplete 360° space filling in large volumes, particularly lacking in-situ observations of crystal growth. Here, we successfully create three-dimensional architectures of plural multi-fold twins in a growing FeCoNiCuZn and the other multi-principal element alloy. The inherent chemical composition inhomogeneity entails twin fault energy variation across different {111} twinning planes, activating multiple twinning systems simultaneously. Importantly, we discover the commonly existed geometrically necessary amorphous zones in such three-dimensional multi-fold twin architectures. Combining in-situ transmission electron microscopy heating experiments and atomic electron tomography, we find that these geometrically necessary amorphous zones, typically several nanometers in size, fill the twin junctions and accommodate large geometric misfits, preventing crystallization even near the melting point.
Similar content being viewed by others

Introduction
Multi-fold twins in natural minerals and crystalline materials offer important opportunities for tuning their physical (such as the transport of electrons, phonons, and photons1,2) and chemical (such as the increase of active catalysis sites and chemical selectivity3,4,5) properties. In metals and alloys, multi-fold twin-induced polyhedral configurations and symmetrical variations in crystalline orientations can enhance the activation of multi-slip dislocations6,7,8,9,10. Also, the resultant changes in precipitate morphology and the addition of interfacial boundaries strongly influence strengthening11,12. However, constructing three-dimensional (3D) twin architectures in man-made materials remains challenging, as it normally requires high external stresses or special annealing conditions (e.g., to activate the slip of partial dislocations or decompose high-energy grain boundaries)13,14,15,16. The primary difficulty arises from the large closure gap due to the incomplete 360° space-filling14,17. Specifically, the accommodation of the intrinsically large angular misfit requires the introduction of a high density of lattice defects18,19,20. The associated elastic/plastic distortion and excess energy18,20,21 make the construction of 3D multi-fold twin architectures much harder to achieve15, compared to that of single-variant parallel twins with coherent twin boundaries (CTBs).
In nanoparticles (NPs) or nanograins with a very large surface-to-volume ratio, the elastic strain associated with multi-fold twin structure is relatively small and can be accommodated at nearby surfaces or interfaces22,23. However, in crystals with a larger volume, the excess energy in the angular gap is much higher due to the severe lattice distortion and a high density of stored defects. Specifically, filling the closure gap would become significantly more challenging in the presence of plural multi-fold twins. As a result, sporadic multi-fold twins, such as five-fold twins, are typically observed only in small NPs and bulk nanocrystalline metals with grain sizes below tens of nanometers18,22,24,25, where each NP or grain generally contains only a single multi-fold twin architecture. This raises the compelling question of whether plural multi-fold twin architectures can be simultaneously formed during crystal growth, and what mechanisms might enable the accommodation of angular misfit among the intersecting twin variants.
For this challenging quest, we first need to select a suitable material as our model. In general, the formation of parallel twins is favored over that of multi-fold twins, even in low stacking fault energy (SFE) materials that readily twins26,27. To construct 3D multi-fold twin architectures, it is necessary to promote the nucleation of twins via different twinning systems at separate locations, while suppressing the rapid growth of any given twin. The pronounced variations in element distribution can lead to significant differences in the energy barriers for twin nucleation and growth across different atomic planes28,29,30. Such compositional inhomogeneity is commonly observed in concentrated solid solution alloys31,32. Motivated by this, we selected multi-principal element alloy (MPEA) systems as an ideal platform. The second critical question is, even if the multi-fold twin architectures can form, how can their formation be directly observed in situ to uncover the underlying growth mechanisms? We recognized that elemental diffusion at elevated temperatures in complex alloy systems could potentially lead to new phase formation33,34,35,36. Therefore, we used an arc-discharge plasma method to synthesize MPEA NPs. Due to the non-equilibrium nature of the arc-discharge plasma process, the resulting NP compositions can deviate significantly from thermodynamic equilibrium. Upon heating, constituent atoms diffuse outward from the original NPs and nucleate new, thermodynamically stable crystals. This approach enables direct in-situ observation of microstructural formation and evolution during alloy crystal growth, with the as-synthesized NPs effectively serving as elemental reservoirs.
In this work, we chose FeCoNiCuZn MPEA as the studied system since previous studies have reported variations in twinning fault energy across different {111} planes due to chemical composition inhomogeneity in similar concentrated alloys28,29. A small amount of Zn was added to potentially increase the concentration variation, as its atomic size and diffusivity differ significantly from those of the other elements. Nevertheless, Fe40Co30Ni27Cu2.6Zn0.4 (atomic percent, at%) was first synthesized as particles in the size range from 20 to 800 nm via the arc-discharging method (Supplementary Fig. 1). The SFE corresponding to the average composition is 100.4 mJ/m2 (at 0 K). To directly monitor the construction of 3D multi-fold nanotwins, these original NPs were dispersed onto amorphous silicon nitride membranes supported by microelectromechanical systems (MEMS)-based heating chips, serving as the elemental sources for the subsequent crystal growth. Figure 1a presents a schematic illustration showing the experimental design. We then resorted to a combination of in-situ transmission electron microscopy (TEM) heating experiments at the atomic scale and atomic electron tomography (AET). Interestingly, atomic-resolution observations truly led to the discovery of the construction process of multi-fold twin architecture. Moreover, it was found that amorphous zones commonly exist at the multi-fold twin junctions and the intersections of different sets of multi-fold twins, which we term geometrically necessary amorphous zones (GNAZs). These GNAZs are needed to effectively accommodate the large geometrical misfit due to incomplete 360° space-filling, and, as such, are resistant to crystallization even at temperatures near the melting point. Additionally, a similar MPEA system FeCoNiCuNb (Supplementary Fig. 2) is also successfully constructed with multi-fold twins, showing the generality of the twinning mechanism.

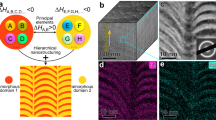
a Schematic illustration of conceptual framework of the experimental design. Scale bar, 200 nm, 10 nm, 2 nm, 10 nm. b–g The HAADF-STEM image and EDS maps of FeCoNiCuZn MPEA-NPs. Scale bar, 100 nm. h The HRTEM images acquired from the in-situ heating experiment at 650 °C, showing the nucleation of crystal embryos. Red dashed lines highlight the crystal nucleation sites. Scale bar, 2 nm. i The HRTEM images acquired from the in-situ heating experiment at 700 °C, showing the subsequent crystal growth. Scale bar, 5 nm. j The HRTEM images acquired from the in-situ heating experiment at 850 °C, showing the twin formation during crystal growth. Scale bar, 5 nm. k–p The HRTEM images from the in-situ heating experiment at 850 °C and FFT patterns acquired from the area indicated by the yellow rectangles of HRTEM images. The white dashed lines mark the sites of CTBs, and the white arrows denote the direction of twin growth. The orange overlays in the HRTEM images were generated by adjusting hue and saturation based on the original micrographs to highlight structural features. Scale bar, 2 nm (k, n) and 2 nm−1 (l, m, o, p).
Results
Multi-fold twins induced by inherent chemical composition inhomogeneity
The high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) image and corresponding energy-dispersive X-ray spectroscopy (EDS) maps of the original NPs reveal a uniform distribution of elements, with an overall composition of Fe40Co30Ni27Cu2.6Zn0.4 (“Methods”, Fig. 1b–g, Supplementary Fig. 3). With a double-tilt in-situ heating TEM holder, the as-prepared particles were gradually heated to 10 different temperature plateaus ranging from 100 °C to 850 °C at a rate of 10 °C/s, then the temperature will remain constant for 20 min at each plateau (“Methods”, Supplementary Fig. 4). At ~650 °C, Fe, Co, Ni, Cu, and Zn atoms began to diffuse outward from the original metastable FeCoNiCuZn solid-solution phase, initiating nucleation either at their periphery or independently on the amorphous silicon nitride substrate (Fig. 1h). As the temperature increased further, these newly nucleated crystals continued to grow and subsequently developed well-defined twin structures (Fig. 1i, j). When heating to approximately 850 °C, the twin growth accelerated and crossed the other to form a multi-fold twin structure (Fig. 1k–p, Supplementary Movie 1). The two parallel Σ3 CTBs formed at the beginning (named CTB-1 and CTB-2, Fig. 1k–m). As the temperature remains 850 °C at the plateau, both the matrix and top twin grain (named Twin-1) grew in the direction of \({(1\bar{1}1)}_{{{{\rm{matrix}}}}}\) and \({(1\bar{1}1)}_{{{{\rm{Twin}}}}-1}\) (Fig. 1k) which are parallel to both CTBs. After 4 min at 850 °C plateau, the growth direction of the matrix abruptly shifted, resulting in the formation of a new twin named Twin-2 (Fig. 1n). Fast Fourier transform (FFT) patterns present a three-fold feature that the angles between two twins and matrix are 141.36° and 140.77° (Fig. 1p). A stacking fault (SF) was observed along the boundary between Matrix and Twin-2 (named CTB-3), which suggests that the formation of multi-fold twins could be caused by the activation of partial dislocations via different slip systems. The formation of such kind of multi-fold twins has been rarely reported, since in the growth of pure metals and conventional alloys, twins usually grow or thicken along the original direction due to a lower energy barrier rather than forming a new CTB37,38. While this was observed during the twin growth of our FeCoNiCuZn MPEA crystals.
Consequently, at the junction of (Twin-1, Twin-2, Matrix), a stress field arises due to geometric incompatibilities and lattice mismatch among the junctions. The collision of Twin-1 and Twin-2 inevitably resulted in the formation of a high-energy ∑9 grain boundary (GB) at the multi-fold twin junction (Fig. 2a, b). However, this boundary exhibited limited mobility. While during in-situ heating, similar junctions displayed dynamic behavior. As shown in Fig. 2c, ∑9 GB was revealed to be a metastable configuration that subsequently decomposed. Meanwhile, a series of partial dislocations were observed to be emitted from CTB-3 near the junction, simultaneously migrating and forming arrays of SFs in both the Matrix and Twin-1 (Fig. 2d–f, Supplementary Fig. 5 and Supplementary Movie 2). The variable configurations of the partial dislocation arrays correspond to various widths of the SFs ribbons (Supplementary Fig. 5b). The Burgers vector of the partial dislocations is identified to be \({{{\bf{b}}}}=\frac{[112]}{6}{{{\rm{a}}}}\) (a is the lattice constant of FeCoNiCuZn MPEA), whereas the summation of the Burgers vectors of the series of partial dislocations can be \({{{{\bf{b}}}}}_{6}=\frac{[101]}{2}{{{\rm{a}}}}\) or \({{{{\bf{b}}}}}_{7}=\frac{[0\bar{1}1]}{2}{{{\rm{a}}}}\)39 (Supplementary Fig. 5c). The emission of partial dislocations facilitated the movement of CTB-3, allowing the further growth of both CTB-1 and CTB-2. As a result, Twin-1 gradually propagated during the crystal growth process, indicating that the growth of a CTB is still favored despite the involvement of complex dislocation activities.

a Atomic structure of ∑9 GB at the multi-fold twin junction. Scale bar, 2 nm. b FFT pattern corresponding to the region in a. Scale bar, 2 nm−1. c–f The HRTEM images viewed from [011]matrix presenting the coupled expansion-migration of two TBs meeting at a multi-fold twin junction. Burgers circuit analyses in (f) show Frank partial dislocations at the TB. The orange overlays in the HRTEM images were generated by adjusting hue and saturation based on the original micrographs to highlight structural features. Scale bar, 2 nm. g–l The HAADF-STEM image of atomic structure, taken along [110] zone axis, and corresponding EDS maps for individual elements of Fe, Co, Ni, Cu and Zn, show how the five elements are distributed on an atomic scale at the multi-fold twin junction. Scale bar, 1 nm. m Line profiles of the atomic fractions of each element at the junction, extracted along the orange arrow in the HAADF-STEM image.
Nonetheless, the formation of a distinct twin structure was achieved. EDS analysis reveals that a relative decrease in the average Fe concentration from ~39 at.% in the matrix to ~27 at.% at the junction, accompanied by a corresponding increase in average Ni concentration from ~29 at.% to ~39 at.%. Meanwhile, the concentrations of Co, Cu, and Zn fluctuate around their average values near the twin boundary (TB, Fig. 2g–m). Notably, the compositional values were obtained from EDS line profiles with a finite width spanning approximately three atomic columns. Elemental concentrations for the twin domains and the junction were averaged separately to minimize the effects of channeling and local signal variation40,41,42,43. Moreover, low-magnification EDS measurements under non-zone-axis conditions near the multi-fold twin junctions were performed to further exclude zone-axis-related channeling effects. The results consistently show a reduction in Fe concentration and an increase in Ni concentration at the junctions (Supplementary Fig. 6, Supplementary Table 1). And the measured elemental concentrations within the twin domains are consistent with the composition of the original Fe40Co30Ni27Cu2.6Zn0.4 NPs prior to annealing. Compared to the composition in defect-free regions and at single TBs (Supplementary Figs. 3, 7), these results indicate a significant local chemical redistribution at the twin-twin junction, which can also lead to variations in charge density distribution among neighboring elements and lattice distortion. Both contribute to changes in the local SFE and twin formation energy28,29. Consequently, the resistance to Shockley partial dislocation motion varies locally, leading to a broad distribution of energy barriers for twin thickening that often overlaps with those for twin nucleation28,29. This facilitates the nucleation of alternative/multiple twin variants rather than the continued thickening of existing twins via boundary migration. And our first-principle calculations suggest that the SFE corresponding to the average composition at the junction is 50.2 mJ/m2 at 0K, which is lower than the SFE (~100.4 mJ/m2) of the original composition (“Methods”, Supplementary Fig. 8).
Network of multi-fold twins
These observations demonstrate that local compositional fluctuations can promote the nucleation of new twins on different crystal planes before the original twin grows. To explore this further, we adjusted the in-situ heating protocols to amplify elemental diffusion differences. We found that a faster heating rate of 15 °C/s (compared to previously used 10 °C/s) and starting the first plateau at 650 °C (compared to starting at 100 °C previously) helped promote the activation of plural multi-fold twin systems (Supplementary Fig. 9). During crystal growth, the advancing front is influenced by local compositional undercooling, thermal gradients, and distribution of diffusing species. Consequently, multiple sets of multi-fold twins with distinct orientations form at different locations (Fig. 3a–e, Supplementary Movie 3). As shown in Fig. 3a, after the formation of Twin-2 upon heating, the Matrix, Twin-1 and Twin-2 collided and formed a junction (named Junction-1). Initially, Junction-1 exhibited significant lattice distortion to minimize interface mismatches, while Σ3{111} CTB and Σ3{112} incoherent TB (ITB) formed near the boundary of Junction-1 at Twin-1 and Twin-2 sides, respectively. Then they gradually aligned with the adjacent matrix after several seconds. Within each set of multi-fold twins, incoherent twin boundaries (ITBs) accommodate the orientation mismatches at twin-twin junctions, as demonstrated by the representative HAADF-STEM images and corresponding FFT patterns in Supplementary Fig. 10.

a–e The HRTEM images viewed from [011]matrix, presenting the formation of plural multi-fold twins. Scale bar, 2 nm. Different colors were overlaid on the original HRTEM images to indicate grains with different twin orientations. Grains with different twin orientations are denoted as Matrix, Twin-1, Twin-2, Twin-3, Twin-1′, Twin-3′ and Twin-4. The yellow and blue squares highlight Junction-1 and Junction-2, respectively. The inset images are the corresponding FFT patterns, where different-colored lines highlight diffraction spots from different twin variants. Scale bar, 2 nm−1. f The HAADF-STEM image of a multi-fold twin junction. The red dashed outline highlights the region with local lattice disorder, yellow dashed lines denote the TBs, and the white dashed line marks the grain boundary. Scale bar, 1 nm.
As the crystal continued to grow, Twin-3 and Twin-4 gradually formed, and a new junction (named Junction-2) emerged to accommodate the crystal growth since the crystal growth rate at the junction of multi-fold twins was relatively slower (Fig. 3b–d). Different from single multi-fold twins observed in previous studies6,16,22, the 3D architectures of plural multi-fold twins were constructed successfully in FeCoNiCuZn MPEA. The orientation of individual twin or crystal regions is shown in Supplementary Fig. 11. Figure 3f shows atomic-scale characterization of the multi-fold twin junction structure. Interestingly, a locally blurred and structurally disordered atomic region was observed at the junction of multi-fold twins and the intersection of different sets of multi-fold twins (highlighted by the red dashed outline in Fig. 3f). These disordered regions appear to play a crucial role in accommodating the geometric misfit due to incomplete 360° space-filling. Additionally, EDS analysis conducted at a five-fold twin junction similarly revealed significant compositional heterogeneity, with a distinct trend of Co enrichment and Fe depletion (Supplementary Fig. 12). The grown crystals with such 3D multi-fold twin architectures can reach sizes up to several hundred nanometers (Supplementary Fig. 13). In the other FeCoNiCuNb MPEA system, we also observed that the multi-fold twin architecture was activated by nanoscale compositional heterogeneity with the formation of planar defects (Supplementary Fig. 14, Supplementary Movie 4).
Revealing a five-fold twin architecture in 3D atomic level by atomic electron tomography (AET)
To identify the local structures of 3D multi-fold twins and further investigate the disordered atoms at the junctions, AET experiments were conducted. The AET results enable the reconstruction of a 3D atomic model, particularly the 3D disordered structure, which is then used to simulate the projected HAADF-STEM images. Using a STEM in annular dark-field mode (ADF, “Methods”), tomographic tilt series were acquired from two MPEA-NPs, named MPEA-1 (Supplementary Fig. 15) and MPEA-2 (Supplementary Fig. 16), respectively. The NPs remained stable under the electron beam, as confirmed by the consistency of zero-degree images taken before, during, and after data acquisition. After image pre-processing, each tilt series was reconstructed using an advanced tomographic algorithm suitable for particles larger than 15 nm in size (“Methods”). The average diameter of MPEA-1 was determined to be 54.77 nm, and the reconstructed volume, with sub-nanometer resolution, revealed an intersecting TB structure identified by a machine-learning method (“Methods”, Supplementary Fig. 17). A total of 24 TBs were identified, including 16 oriented along the \((1\bar{1}1)\) direction and 8 along the \((\bar{1}\bar{1}1)\) direction. In each crystallographic plane, the TBs are parallel to each other, with an overlap region of approximately 59.30 nm² (“Methods”).
Actually, the complete MPEA-2 contains two distinct yet interconnected multi-fold twin sets (Fig. 4a, Supplementary Fig. 18): a clearly identifiable five-fold twin structure on the right and a more complex, higher-order twinning system on the left. The average diameter of analyzed MPEA-2 is 22.36 nm, and the reconstruction volume achieved atomic resolution (Fig. 4a, Supplementary Figs. 19, 20 and Supplementary Movie 5, 6). To quantitatively characterize the 3D atomic structures around the multiple twins, the 3D atomic coordinates of MPEA-2 were traced, classified, and refined to produce an experimental atomic model (“Methods”). Although the atomic numbers of the elements inside MPEA-NPs differ by only 1, making them indistinguishable using HAADF-based AET, we were able to identify structural information by precisely locating the atom positions in 3D44. As shown in Fig. 4a, due to the limited field of view, the left region of MPEA-2 cannot be fully reconstructed. In order to simplify the reconstruction model and enhance the accuracy of the structural reconstruction, we focus on the five-fold twin on the right, highlighted in orange in Fig. 4a. Figure 4b shows the experimental 3D atomic model of MPEA-2, consisting of 136,314 atoms, where the five-fold twin geometry is characterized and presented45. Supplementary Fig. 20a to f shows the image slices with a thickness of 3.4 Å at different heights in the reconstructed volume of MPEA-2. All five TBs, named as TB-1 through TB-5, exhibit anomalously large atomic steps compared to traditional alloys, with changes in atomic distances corresponding to height from the bottom to the top. The largest step difference from a single TB can be as much as 7 atomic layers (Supplementary Fig. 21). The edges of the TB-3 and TB-5 did not extend to the particle boundary but terminated at 16.2 Å and 14.5 Å from the boundary, respectively. Due to the extension of the multiple twins on the other side, the edge parts of the TB-3 and TB-5 (shown by the light-yellow part in Fig. 4b) exhibit greater disorder.

a Reconstructed image slice of MPEA-2 at a height of 0 nm, with a thickness of 3.4 Å. The orange-highlighted region indicates the main area analyzed in AET. Scale bar, 5 nm. b 3D positions of individual atoms are illustrated, showing five TBs (light blue), extension regions (light yellow), and the central core (light red), as revealed by AET. Scale bar, 5 nm. c The migration of five TBs and the area of GNAZ at different heights. d The angles between five TBs at different heights, the dashed lines in each represent 70.53° and 72° respectively. e The area size of the GNAZ inversely correlates with the standard error of the twin angles and randomly varies with the height.
The center region of the five-fold twin in MPEA-2 is significantly more disordered as illustrated by the bond orientation order (BOO) parameters (Supplementary Fig. 22, “Methods”), exhibiting a non-linear trajectory along the z-axis (Fig. 4b, Supplementary Fig. 20g). Since these disordered atoms/areas appear to be needed for accommodating the angular deficiency created by the construction of multi-fold twins, we have termed them GNAZs. Figure 4c shows the distribution of GNAZ of the multiple twins in MPEA-2, with the area at the center measuring 5.97 nm2 and containing 1,686 atoms. The formation of atomic steps along the TBs is accompanied by notable changes in the size of the GNAZ (Fig. 4c), suggesting that these zones facilitate the accommodation of mismatches between adjacent twin variants. In contrast, decahedral multiple-twinned traditional alloys typically exhibit uniform shear strain with defects that compensate for the 7.35° solid-angle deficiency. Here, the angles between the five TBs in MPEA-2 are much more anisotropic (Fig. 4d). Specifically, two angles (∠12, ∠45) are, on average, larger than 72°, while ∠23 and ∠51 fluctuate around 72°, and ∠34 is significantly smaller, fluctuating around 69°. The two valleys in Fig. 4c correspond to the layers with the smallest GNAZ and the largest fluctuations in twin angles.
To quantify the degree of fluctuation in twin angles, we calculated the standard error of the twin angles at each {110} atomic layer and found that the area of the GNAZ is negatively correlated with the standard error of twin angles within the plane (Fig. 4e). When the GNAZ is larger, the uniformity of the twins is higher. This suggests that the GNAZ can relax strain and reduce the energy required for twin migration6. The correlation can be defined by the following equation:
Where A represents the GNAZ area, B represents the standard error of twin angle, and α and β are two constants. Fitting the data points with Eq. (1) gives α = 8.22 and β = 0.98 (“Methods”), suggesting a negative quadratic decay of the twin-formation energy from the amorphous matrix. This correlation suggests that larger angular deviations (i.e., greater standard error) are associated with smaller GNAZ areas, implying reduced local strain energy under conditions of enhanced TB mobility. Therefore, the GNAZ size governs the degree of strain accommodation and modulates the energy barrier for TB migration. The spatial variation in the area size of GNAZ at the five-fold twin junction makes the angles between the five TBs much more anisotropic. GNAZ provides an alternative and effective mechanism for accommodating angular misfit in the formation of multi-fold twin architectures, in addition to conventional approaches such as interfacial defects and lattice distortion. And the EDS maps of the disordering junction indicate that the GNAZ shows noticeable concentration fluctuations (Supplementary Fig. 23). By linearly projecting the 3D atomic coordinates along the viewing direction, we generated the simulated STEM projection (“Methods”, Supplementary Fig. 24d). It shows excellent agreement with the experimental HAADF-STEM image (Supplementary Fig. 24a).
Discussion
Formation mechanism of geometrically necessary amorphous zones (GNAZs)
We now discuss the formation mechanism of these GNAZs. When multiple twins/crystals of different orientations meet at their junction, an inherent geometrical gap is inevitable, see the five-fold twin cases in Figs. 3, 4. The interfacial regions between colliding twins are expected to be rugged with terraces and grain-boundary-like excess space. For these regions to connect the adjoining twins seamlessly, one could imagine that the crystal planes there would be riddled with dislocations and large lattice distortions to accommodate the geometric misfit so as to maintain the growth continuity without damage (voids). These crystal regions would thus be stored with rather high elastic strain energy, due to a high density of dislocations and atoms displaced from their comfortable lattice sites. Such zones would then rather collapse into an amorphous state, which is more lenient in accommodating all the strains. In other words, the GNAZ is a more favorable alternative/outlet, relative to other options (such as defect-ridden crystal or even voids) that would demand an even higher energy penalty. Usually, crystallization is hard to be kinetically suppressed at the elevated temperature of crystal growth46,47,48,49,50. However, being a must-have to accommodate the construction of the 3D multi-fold twin architectures, the GNAZ is resistant to crystallization. It is just that transitioning to crystalline packing would actually cause more energy elevation, because no low-defect-density ordered packing can accommodate the extensive geometric misfit associated with the extended five-fold twinned structure. We therefore conclude that GNAZ is a crystallization-defying metastable structure that necessarily accompanies complex multi-fold twin structures. Beyond its structural role, the GNAZs may contribute to strain relaxation during deformation. Geometric phase analysis (GPA) reveals that the strain field at the junction exhibits no significant stress concentration compared to the matrix and adjacent twin segments (Supplementary Fig. 25a). Instead, a nanoscale alternating tensile-compressive strain distribution is observed, suggesting effective local strain accommodation. Additionally, SFs are frequently observed around the junctions (Supplementary Fig. 25b), indicating that the multi-fold twin junctions may act as sources or sinks for dislocations. Specifically, they may emit partial dislocations into the surrounding twins or absorb gliding dislocations from the crystal interior to relieve accumulated stress.
This study provides new insights into the behavior of twins, in particular, the construction of multi-fold twin architectures during crystal growth. In the present MPEA, atomic-scale compositional fluctuations in three dimensions result in variations in the nucleation barriers of twins on different {111} planes. These fluctuations lead to a high propensity for heterogeneous twin nucleation on different {111} planes while suppressing the rate of twin growth. The significant elastic strain induced by interfacial incoherency and intrinsic geometrical misfit leads to the formation of structurally disordered GNAZ at the junctions. Once this complex multi-fold twin architecture, including the associated GNAZ, is established, it remains stable even upon subsequent heating to rather high temperatures, which was thought to be unlikely before, underscoring its structural resilience. This finding offers a new perspective on the formation mechanism of complex multi-fold twin architectures, emphasizing the critical role of compositional heterogeneity and GNAZs. Our results indicate that by tuning the interplay between heterogeneous twin nucleation and twin growth, it is possible to design novel 3D multi-fold twin architectures in alloys.
Methods
Alloy preparation
The FeCoNiCuZn MPEA-NPs were synthesized using an arc-discharge plasma method. First, cylindrical targets were prepared by mechanically mixing and compressing high-purity metal powders of each element, then placed in the anode region of a direct current arc discharge chamber. The chamber was evacuated to a vacuum level of 10−3 Pa. Next, it was filled with reaction gas (H₂) and protective gas (Ar), reaching a pressure of 0.3 × 105 Pa. The arc discharge process was sustained for 5–10 min. Finally, the NPs were collected after a 6-h passivation period in the chamber.
TEM Characterizations and in-situ heating experiments
The FeCoNiCuZn MPEA-NPs used for characterization were ultrasonically dispersed in ethyl alcohol for 10 min and then deposited onto a molybdenum grid. The microstructure of the MPEA-NPs was analyzed using TEM (Tecnai G2 F20 S-TWIN, FEI) and spherical-aberration-corrected TEM (Titan G2 60–300, FEI, operated at 200 kV, and Spectra 300-30 operated at 300 kV). EDS analysis was performed with the Spectra 300–30 microscope equipped with a Super-X EDS detector.
The FeCoNiCuZn MPEA-NPs used for in-situ heating experiments were dispersed onto MEMS-based heating chips (Nanochip, DENS solutions). In-situ TEM heating experiments were carried out using a double-tilt heating holder (Wildfire D6, DENS solutions). These experiments and characterizations were performed using both conventional TEM instruments (Tecnai G2 F20 S-TWIN, FEI) and spherical-aberration-corrected TEM instruments (Titan G2 60–300, FEI, and Spectra 300–30).
Calculation of SFE at 0 K
The FeCoNiCuZn samples, containing 12 (111) layers (12 atoms per plane) with orthogonal axes [112], \([1\bar{1}0]\) and \([1\bar{1}\bar{1}]\), are constructed, which was generated using a special quasirandom structure (SQS)51. For the 0 K SFE (stacking fault energy) calculations, a non-periodic boundary condition was applied along the normal direction of the stacking fault plane. A vacuum region of 10 Å is inserted on the top and bottom of the simulation cell in order to eliminate the interplay between the two surfaces. Energy calculations and structure optimization were performed using the Vienna ab initio simulation package (VASP 5.4.4)52 with a plane wave cutoff energy of 350 eV. Spin polarization is turned on for all calculations. Brillouin zone integrations were performed using 4 × 3 × 1 Monkhorst–Pack meshes. The generalized gradient approximation with the Perdew-Burke-Ernzerhof form was employed to describe the exchange-correlation function, and the valence electronic states of elements were handled by the projector-augmented wave method53,54. Ionic forces of initial and final configurations are relaxed to <3 meV/Å, and the energy criterion in each self-consistent field (SCF) iteration is set to 1 × 10−4 eV. The SFE was calculated by formula54:
where A is the area of SF, ESF, and E0 is the energy of configuration with and without SF, respectively. A total of 13 independent samples were used to obtain the accurate SFE.
Calculation of temperature-dependent SFEs
Based on the first-order approximation of the axial interaction model55, the temperature-dependent SFE can be estimated using:
where \(\Delta {E}_{0}\), \(\Delta {E}_{{{{\rm{ph}}}}}\) and \(\Delta {E}_{{{{\rm{ele}}}}}\) denote the differences in zero-temperature energy, phonon free energy and electronic free energy between the hexagonal close-pack (hcp) and face-centered cubic (fcc) structures, respectively. The electronic contribution is neglected due to the very small value56. Specifically, the phonon free energy was calculated using the supercell method with the force constants obtained by the finite difference approach, as implemented in the PHONOPY code56 within the harmonic approximation. Spin-polarized density functional theory approaches were performed to calculate the zero-temperature energy in the VASP code. Brillouin zone integrations were performed using 4 × 4 × 3 Monkhorst–Pack meshes with an energy cutoff of 350 eV in force calculations. Projector augmented wave potentials were adopted with the Perdew–Burke–Ernzerhof generalized-gradient approximation for the exchange-correlation functional. The energy criterion in each iteration is set to 1 × 10−5 eV. 3 independent supercells, containing 72 atoms each, were generated as special quasi-random structures (SQS). For the finite-temperature SFE calculations, both the fcc and hcp configurations were computed under periodic boundary conditions.
Electron tomography data acquisition
The FeCoNiCuZn MPEA-NPs were deposited onto 10-nm-thick carbon membranes (Electron Microscopy Sciences Inc., chip size 3 mm, window size 74 μm × 74 μm). Two tomographic tilt series were acquired from two MPEA NPs with different sizes (50 nm and 20 nm, named MPEA-1 and MPEA-2) by Thermo-fisher Spectra Ultra aberration-corrected STEM. The tilt ranges and angular increments are from −72.4° to 45.3° and 2° for MPEA-1, and from −68.5° to 67.4° and 2.5° for MPEA-2. The α rotation axis and fast scanning direction were pre-aligned for accurate Euler angle determination. At each tilt angle, high-resolution images were collected at 300 kV (MPEA-1) and 200 kV (MPEA-2) in annular dark field (ADF)-STEM mode with convergence angle 21.4 mrad and 25.1 mrad. To minimize sample drift and improve the signal-to-noise ratio, 3 images per angle were measured with 5 μs and 3 μs dwell time for two NPs. The electron current for each acquired frame is 30 pA, and the total electron doses of each tilt series for all the NPs were estimated to be \(1.60 \times {10}^{6}\,{{{{\rm{e}}}}}^{-1}{{{{\text{\AA }}}}}^{-2}\) and \(9.57 \times {10}^{5}\,{{{{\rm{e}}}}}^{-1}{{{{\text{\AA }}}}}^{-2}\).
Image pre-processing
The 3 ADF-STEM images acquired at each tilt angle were used to estimate and correct the specimen drift57. The spatial correspondences of different frames acquired at the same angle were matched by sub-pixel cross-correlation. The relative spatial shifts were used to correct the scanning drift during the ADF-STEM images acquisition due to the instability of the sample holder. The optimized ADF-STEM images were denoised by a generalized denoising algorithm, termed block-matching and 3D filtering (BM3D)58,59. The denoised images from the two tilt series were aligned to a common tilt axis using two complementary methods. Along the tilt axis, sub-pixel alignment was achieved with the common-line method, while perpendicular alignment was refined using the center-of-mass (CoM) approach, in which each image was shifted until its CoM coincided with the origin. This combined alignment ensured precise registration of all projections, enabling high-quality tomographic reconstruction60,61,62,63. For MPEA-1, since the field of view (80.1 × 80.1 nm2) can cover the whole NPs, center of mass at both directions were used to align the tilt series. While for MPEA-2, the field of view (35.2 × 35.2 nm2) cannot cover the whole NP, so the center of mass at x direction and common line of y direction were applied to align the images.
3D volume reconstruction and determination of 3D atomic coordinates
The 3D volumes of two MPEA NPs were reconstructed by an analytical gradient-descent algorithm, termed Real Space Iterative Reconstruction (RESIRE)64. The robustness and accuracy of RESIRE have been demonstrated by a few papers62. The refinement of Euler angles and spatial correspondences of tilt series were performed by optimizing the cross-correlation and L1-norm between the measured projections and simulated projections of 3D reconstructions with brutal-force input Euler angle and projection sets. After these refinement procedures, a final 3D reconstruction was obtained by running 200 iterations of RESIRE. Atomic positions within the reconstructed MPEA-2 volume were identified through a 3D polynomial fitting procedure weighted by a Gaussian function of spatial distance, enabling robust localization of atomic coordinates in regions with intensity variations65. The integer coordinates of local maxima were identified in the upsampled 3D reconstruction by a factor of three using spline interpolation. Starting from the highest intensity, we fitted each local maximum of a 2226 × 2226 × 2226 voxel volume (258.0 × 258.0 × 258.0 Å3) by a 3D polynomial method to locate its center position63. The fitted local maxima were considered as potential atoms, and the one with the minimum distance to potential atoms with higher intensity smaller than 2 Å will be removed. For every potential atom in the list, the integrated intensity of the 2226 × 2226 × 2226 voxel volume around the center position was calculated, and a K-means clustering method was used to classify the non-atoms and atoms66. Since the atomic numbers of Fe (26), Co (27), Ni (28), Cu (29) and Zn (30) are close to each other, the atom types of them cannot be differentiated from an ADF-STEM-based 3D reconstruction and all atoms are classified to a single type. After excluding the non-atoms and manually checking all the atoms, we obtained a 3D atomic model for the 3D reconstruction of MPEA-2. The 3D visualization of the atomic model was rendered using the OVITO package (Ver. 3.14.1)67.
Identifying the grain boundary
The twin grain boundaries with different orientations in MPEA-1 were segmented automatically using the machine-learning Trainable Weka Segmentation plugin in FIJI (ImageJ, Ver. 1.54 f)68. 50 representative slices from the obtained 3D reconstruction (450 slices in total) were annotated and used to train the classifier. For MPEA-2, the atomic TBs were manually traced using the reconstructed 3D atomic model. The atomic model was manually processed via slice by slice, and each slice with a thickness of 30 pixels (10.03 Å). The TBs can be well identified within the atom slice of such thickness.
The twin order parameter
The BOO method was employed to demonstrate its capability in distinguishing structurally ordered and disordered atomic configurations62,69,70. The twin order parameter is defined by two normalized BOO parameters:
where two normalized BOO parameters are respectively given by:
Firstly, the normalized BOO parameter BOOfcc of each atom (Supplementary Fig. 22a) of the central region of Fig. 4b was calculated. Applying a cutoff value of 0.5 to distinguish crystalline and amorphous phases62, it was found that the majority of atoms in this region exhibit BOOfcc values below the threshold. This confirms the findings on the GNAZ. This conclusion is further supported by the visibly disordered atomic arrangement observed in Supplementary Fig. 22b.
Analysis of the normalized twin order parameters (ΔBOO) revealed distinct structural characteristics across different regions (Supplementary Fig. 22c, d). The grain interiors showed an average ΔBOO of 0.26, while TBs exhibited a significantly higher value of 0.63. Most notably, the junction region of multi-fold TBs demonstrated the highest ΔBOO value of 0.84, substantially exceeding those of other regions. This clear gradation in ΔBOO values effectively differentiates these three structurally distinct regions.
The correlation of GNAZ size and TB angle changes
The angle induced by each TB migration can be estimated6:
where h is the step height of each TB (equal to the {111} interatomic distance) and R is the radius of the sample (TB length). On the other hand, the central amorphous core of a five-fold twin can also be estimated by assuming that it is a general grain boundary region:
where s is the area of the core region, and γ0 is the energy density in the core region.
The correlation of GNAZ size and the TB angle changes via the equation based on our experimental results:
where A represents the GNAZ area, B represents the standard error of twin angle, and α and β are two constants. Fitting the data points with Eq. (9) gives α = 8.22 and β = 0.98.
Simulation of HAADF-STEM image
The plane coordinates (x, y) of the atomic model in Supplementary Fig. 24a were accurately traced by a 2D Gaussian-fitting method57. The atomic positions along the z-direction were arranged based on the A-B stacking of the {110} plane. The central amorphous region was incorporated using coordinates cropped from the MPEA-2 particle determined by AET. Then the HAADF-STEM image in Supplementary Fig. 24d was simulated using a linear projection assumption, where the 3D intensities of all atoms have the same Gaussian distribution. The projection is calculated by the summation of the intensities of all atoms along a certain direction.
Data availability
Source data are provided with this paper. The data that support the findings of this study are available from the Zenodo link https://doi.org/10.5281/zenodo.17310133. Source data are provided with this paper.
References
Pastoriza–Santos, I., Sánchez–Iglesias, A., García de Abajo, F. J. & Liz–Marzán, L. M. Environmental optical sensitivity of gold nanodecahedra. Adv. Funct. Mater. 17, 1443–1450 (2007).
Li, H., Hu, H. & Xu, D. Silver decahedral nanoparticles-enhanced fluorescence resonance energy transfer sensor for specific cell imaging. Anal. Chem. 87, 3826–3833 (2015).
Zhang, J. et al. Cyclic penta-twinned rhodium nanobranches as superior catalysts for ethanol electro-oxidation. J. Am. Chem. Soc. 140, 11232–11240 (2018).
Lu, N. et al. 5-fold twinned nanowires and single twinned right bipyramids of Pd: utilizing small organic molecules to tune the etching degree of O2/halides. Chem. Mat. 26, 2453–2459 (2014).
Bian, T. et al. Epitaxial growth of twinned Au–Pt core–shell star-shaped decahedra as highly durable electrocatalysts. Nano Lett. 15, 7808–7815 (2015).
Chen, Y., Huang, Q., Zhao, S., Zhou, H. & Wang, J. Penta-twin destruction by coordinated twin boundary deformation. Nano Lett. 21, 8378–8384 (2021).
Narayanan, S., Cheng, G., Zeng, Z., Zhu, Y. & Zhu, T. Strain hardening and size effect in five-fold twinned Ag nanowires. Nano Lett. 15, 4037–4044 (2015).
Jin, Z. H. et al. Interactions between non-screw lattice dislocations and coherent twin boundaries in face-centered cubic metals. Acta Mater. 56, 1126–1135 (2008).
Kacher, J. & Robertson, I. M. Quasi-four-dimensional analysis of dislocation interactions with grain boundaries in 304 stainless steel. Acta Mater. 60, 6657–6672 (2012).
Zhu, Y. et al. Ultrastrong nanotwinned titanium alloys through additive manufacturing. Nat. Mater. 21, 1258–1262 (2022).
Lu, Q. et al. Synergy of multiple precipitate/matrix interface structures for a heat-resistant high-strength Al alloy. Nat. Commun. 14, 2959 (2023).
Yang, Y. et al. Bifunctional nanoprecipitates strengthen and ductilize a medium-entropy alloy. Nature 595, 245–249 (2021).
Cao, Z. H. et al. Size dependence and associated formation mechanism of multiple-fold annealing twins in nanocrystalline Cu. Acta Mater. 95, 312–323 (2015).
Zhu, Y. T., Liao, X. Z. & Valiev, R. Z. Formation mechanism of fivefold deformation twins in nanocrystalline face-centered-cubic metals. Appl. Phys. Lett. 86, 103112 (2005).
An, X. H. et al. Formation of fivefold deformation twins in an ultrafine-grained copper alloy processed by high-pressure torsion. Scr. Mater. 64, 249–252 (2011).
Wang, X. et al. In situ atomic-scale observation of 5-fold twin formation in a nanoscale crystal under mechanical loading. Nano Lett. 23, 514–522 (2023).
Hofmeister H. Fivefold twinned nanoparticles. In: Encyclopedia of Nanoscience and Nanotechnology. (American Scientific Publishers, 2004).
Johnson, C. L. et al. Effects of elastic anisotropy on strain distributions in decahedral gold nanoparticles. Nat. Mater. 7, 120–124 (2008).
Marks, L. D. & Howie, A. Multiply-twinned particles in silver catalysts. Nature 282, 196–198 (1979).
Wu, J. Y., Nagao, S., He, J. Y. & Zhang, Z. L. Role of five-fold twin boundary on the enhanced mechanical properties of fcc fe nanowires. Nano Lett. 11, 5264–5273 (2011).
Bringa, E. M. et al. Fivefold twin formation during annealing of nanocrystalline Cu. Scr. Mater. 59, 1267–1270 (2008).
Song, M. et al. Oriented attachment induces fivefold twins by forming and decomposing high-energy grain boundaries. Science 367, 40–45 (2020).
Song, M. et al. Uneven strain distribution induces consecutive dislocation slipping, plane gliding, and subsequent detwinning of penta-twinned nanoparticles. Nano Lett. 24, 1153–1159 (2024).
Deng, Z., Luo, J., Yuan, W. & Xi, W. Formation and disintegration investigation of fivefold annealing twins in copper nanoparticles. Scr. Mater. 169, 42–45 (2019).
Wang, Y., Zhou, X., Lu, Y. & Li, X. Enhanced mechanical properties in bulk nanograined ni with high-density fivefold twins. Small 21, 2410202 (2025).
Lu, L., Chen, X., Huang, X. & Lu, K. Revealing the maximum strength in nanotwinned copper. Science 323, 607–610 (2009).
Lu, L., Shen, Y., Chen, X., Qian, L. & Lu, K. Ultrahigh strength and high electrical conductivity in copper. Science 304, 422–426 (2004).
Yang, C. et al. Chemical inhomogeneity–induced profuse nanotwinning and phase transformation in AuCu nanowires. Nat. Commun. 14, 5705 (2023).
Liu, N.-J. et al. Origin of the high propensity for nanoscale deformation twins in CrCoNi medium-entropy alloy. J. Mater. Sci. Technol. 183, 63–71 (2024).
Moniri, S. et al. Three-dimensional atomic structure and local chemical order of medium- and high-entropy nanoalloys. Nature 624, 564–569 (2023).
Ma, E. & Liu, C. Chemical inhomogeneities in high-entropy alloys help mitigate the strength-ductility trade-off. Prog. Mater. Sci. 143, 101252 (2024).
Ding, Q. et al. Tuning element distribution, structure and properties by composition in high-entropy alloys. Nature 574, 223–227 (2019).
Han, X. et al. In-situ atomic tracking of intermetallic compound formation during thermal annealing. Nat. Commun. 15, 7200 (2024).
Zhang, W. et al. The role of surface substitution in the atomic disorder-to-order phase transition in multi-component core–shell structures. Nat. Commun. 15, 9762 (2024).
Rao, J. C. et al. Secondary phases in AlxCoCrFeNi high-entropy alloys: an in-situ TEM heating study and thermodynamic appraisal. Acta Mater. 131, 206–220 (2017).
Liu, C. et al. In-situ STEM imaging of growth and phase change of individual CuAlX precipitates in Al alloy. Sci. Rep. 7, 2184 (2017).
Zhong, L., Zhang, Y., Wang, X., Zhu, T. & Mao, S. X. Atomic-scale observation of nucleation- and growth-controlled deformation twinning in body-centered cubic nanocrystals. Nat. Commun. 15, 560 (2024).
Lee, S. et al. Reversible cyclic deformation mechanism of gold nanowires by twinning–detwinning transition evidenced from in situ TEM. Nat. Commun. 5, 3033 (2014).
Li, Q. et al. Migration kinetics of twinning disconnections in nanotwinned Cu: An in situ HRTEM deformation study. Scr. Mater. 194, 113621 (2021).
MacArthur, K. E., Brown, H. G., Findlay, S. D. & Allen, L. J. Probing the effect of electron channelling on atomic resolution energy dispersive X-ray quantification. Ultramicroscopy 182, 264–275 (2017).
Lugg, N. R., Kothleitner, G., Shibata, N. & Ikuhara, Y. On the quantitativeness of EDS STEM. Ultramicroscopy 151, 150–159 (2015).
Kothleitner, G. et al. Quantitative elemental mapping at atomic resolution using x-ray spectroscopy. Phys. Rev. Lett. 112, 085501 (2014).
MacArthur, K. E. et al. Optimizing experimental conditions for accurate quantitative energy-dispersive X-ray analysis of interfaces at the atomic scale. Microsc. Microanal. 27, 528–542 (2021).
Yalisove, R., Sung, S. H., Ercius, P. & Hovden, R. Limits of three-dimensional resolution and dose for aberration-corrected electron tomography. Phys. Rev. Appl. 15, 014003 (2021).
Zahiri, A. H., Chakraborty, P., Wang, Y. & Cao, L. Strong strain hardening in ultrafast melt-quenched nanocrystalline Cu: the role of fivefold twins. J. Appl. Phys. 126, 075103 (2019).
Chen, Y., Dai, Z.-W. & Jiang, J.-Z. High entropy metallic glasses: glass formation, crystallization and properties. J. Alloy. Compd. 866, 158852 (2021).
Duarte, M. J. et al. Kinetics and crystallization path of a Fe-based metallic glass alloy. Acta Mater. 127, 341–350 (2017).
Zhan, X. & Ernst, F. Crystallization micro-mechanism of near-eutectic amorphous Ni–P. Acta Mater. 104, 274–282 (2016).
Shen, Y. & Perepezko, J. H. Al-based amorphous alloys: glass-forming ability, crystallization behavior and effects of minor alloying additions. J. Alloy. Compd. 707, 3–11 (2017).
Zhang, Y. & Jiang, S. Atomistic mechanisms for temperature-induced crystallization of amorphous copper based on molecular dynamics simulation. Comput. Mater. Sci. 151, 25–33 (2018).
Zunger, A., Wei, S. H., Ferreira, L. G. & Bernard, J. E. Special quasirandom structures. Phys. Rev. Lett. 65, 353–356 (1990).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Denteneer, P. & Haeringen, W. Stacking-fault energies in semiconductors from first-principles calculations. J. Phys. C: Solid State Phys. 20, L883 (2000).
Zhao, S., Stocks, G. M. & Zhang, Y. Stacking fault energies of face-centered cubic concentrated solid solution alloys. Acta Mater. 134, 334–345 (2017).
Xu, R. et al. Three-dimensional coordinates of individual atoms in materials revealed by electron tomography. Nat. Mater. 14, 1099–1103 (2015).
Makitalo, M. & Foi, A. Optimal inversion of the generalized anscombe transformation for poisson-gaussian noise. IEEE Trans. Image Process. 22, 91–103 (2013).
Dabov, K., Foi, A., Katkovnik, V. & Egiazarian, K. Image denoising by sparse 3-D transform-domain collaborative filtering. IEEE Trans. Image Process. 16, 2080–2095 (2007).
Yang, Y. et al. Deciphering chemical order/disorder and material properties at the single-atom level. Nature 542, 75–79 (2017).
Tian, X. et al. Correlating the three-dimensional atomic defects and electronic properties of two-dimensional transition metal dichalcogenides. Nat. Mater. 19, 867–873 (2020).
Yang, Y. et al. Determining the three-dimensional atomic structure of an amorphous solid. Nature 592, 60–64 (2021).
Zhou, J. et al. Observing crystal nucleation in four dimensions using atomic electron tomography. Nature 570, 500–503 (2019).
Pham, M., Yuan, Y., Rana, A., Osher, S. & Miao, J. Accurate real space iterative reconstruction (RESIRE) algorithm for tomography. Sci. Rep. 13, 5624 (2023).
Rogers, S. S., Waigh, T. A., Zhao, X. & Lu, J. R. Precise particle tracking against a complicated background: polynomial fitting with Gaussian weight. Phys. Biol. 4, 220 (2007).
Lloyd, S. Least squares quantization in PCM. IEEE Trans. Inf. Theory 28, 129–137 (1982).
Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 18, 015012 (2010).
Arganda-Carreras, I. et al. Trainable Weka Segmentation: a machine learning tool for microscopy pixel classification. Bioinformatics 33, 2424–2426 (2017).
Yang, Y. et al. Atomic-scale identification of active sites of oxygen reduction nanocatalysts. Nat. Catal. 7, 796–806 (2024).
Yuan, Y. et al. Crystal nucleation and growth in high-entropy alloys revealed by atomic electron tomography. arXiv Preprint at https://doi.org/10.48550/arXiv (2025).
Acknowledgments
Q.Y. was supported by the National Key R&D Program of China (Grant No. 2023YFB2405802), the National Science Fund for Distinguished Young Scholars of China (Grant No. 52325102), and the Natural Science Foundation of Zhejiang Province, China (Grant No. LZ22E010001). Y.Y. was supported by the National Natural Science Foundation of China (Grant No. 52473240). The electron tomography experiments were carried out at the Instrumentation and Service Center for Physical Sciences (ISCPS) supported by the Research Center for industries of the Future (RCIF) at Westlake University and Westlake Education Foundation. We thank Y. H. Zhu from Zhejiang University of Technology and Y. X. Li from Northeastern University for providing technical support.
Author information
Authors and Affiliations
Contributions
Q.Y. proposed and supervised the research. Y.S. performed the microstructure characterization and corresponding data analysis, including in-situ electron microscopy observations and STEM. S.H. and Y.Y. carried out electron tomography experiments and corresponding data analysis. B.Z. and J.D. performed the stacking-fault energies calculations. Y.S., S.H., B.Z., Z.G., F.Z., X.Q., Y.C., J.Z., J.D., Y.Y., Q.Y., and E.M. analyzed the data and interpreted the results. Q.Y., E.M. and Y.Y. led the writing of the paper, with the initial draft from Y.S. and S.H. All the authors contributed to data analysis and scientific discussions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Wentao Hu and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Song, Y., Huang, S., Zhang, B. et al. Three-dimensional network of multi-fold twins enabled by geometrically necessary amorphous zones. Nat Commun 16, 11535 (2025). https://doi.org/10.1038/s41467-025-66378-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66378-4
