
Abstract
Electrochemical capacitors are primarily limited by the low electric double-layer storage capacity and narrow operating window that avoids the formation of solid electrolyte interface layers. Herein, we demonstrate that electric double-layer adsorption of solvated Na+ in carbon nanopores is achievable under a large offset potential of −2.95 V vs. potential of zero charge in diethylene-glycol-dimethyl-ether electrolyte, even accompanying with the as-formed solid electrolyte interface layers. The largely enlarged offset potential in situ drives the continued partial desolvations in carbon nanopores, which largely reduces the average solvation numbers from 2.1 to 0.6, leading to a high electric double-layer capacitance of 172 F g−1, a high capacity of 508 C g−1 and high initial coulombic efficiency of 92.2% at 0.1 A g−1 (0.5 mA cm−2), together with high-rate capability and long-term cycling stability. Thereby, such increased electric double-layer charge storage enables the redesign and assembly of sodium-ion capacitor pouch cells that display a high specific density of 40 Wh kg−1 (on cell level) and 30,000 cycles at a fast (dis)charging rate of 51 C (20 mA cm−2). Such sodium-ion capacitors are assembled without any pretreatments that are beneficial for scale-up fabrications in industry.
Similar content being viewed by others


Introduction
Electrochemical capacitors deliver high power density and tens of thousands of cycles, which are suitable for power smoothing and phase regulation in grid storage, energy harvesting and recovery in transportation, and other applications require fast charging or high-power delivery in minutes or seconds1,2,3. Commercial electric double-layer capacitors (EDLCs) use activated carbon as both positive and negative electrodes in organic electrolytes through electric double-layer (EDL) adsorption/desorption of ions in nanopores4,5,6,7,8. Despite their attractive properties, EDLCs face limitations in terms of low gravimetric/volumetric energy densities owing to their limited EDL capacitance and capacity9,10.
The gravimetric EDL capacitance (\(C\), F g−1) is determined as follows:
where, \({\varepsilon }_{0}\) is the dielectric constant of vacuum, \({\varepsilon }_{r}\) is the electrolyte dielectric constant, \({SA}\) is the accessible surface area (m2 g−1, usually measured by gas adsorption) of the porous carbon, and \(d\) is the approaching distance between the adsorbed ions and carbon surface11. Route ① (Fig. 1a) indicates the enlargement of \(C\) by increasing the \({SA}\) of carbon materials. Unfortunately, such an enlargement is not as expected12. Liu et al.2 evaluated nanoporous carbons in standard tetraethylammonium tetrafluoroborate (TEABF4) in acetonitrile (ACN) and demonstrated that SA did not linearly affect the \(C\) (ref. 2). Instead of \({SA}\), disordered carbons with smaller graphene-like domains are more significant in determining \(C\), whereas stronger interactions between ions and carbon atoms lead to more efficient storage of ions. Compared with the other ordered carbons, the highly disordered carbon (ACS-PC) displayed a high capacitance of 138 F g−1, but leading to poorer cycling stability owing to the rich defects2. Route ② (Fig. 1a) represents reducing \(d\) through matching the sizes between nanopores and adsorbed ions, resulting in an increased capacitance of ~140 F g−1 13,14. In situ nuclear magnetic resonance (NMR) and electrochemical quartz crystal microbalance (EQCM) techniques demonstrated that such increased \(C\) is attributed to the partial desolvation of ions entering the nanopores (the pore size is smaller than the size of fully solvated ions), resulting in ions close to carbon wall with reduced \(d\), identified as nanoconfinement effect15,16,17. However, the fine tuning of the porous structure, disordering and SA of carbon materials is very complex and costly, and the accurate guidance is still lacking2,4. There is a strong desire to break the limited EDL capacitance and capacity through an efficient mechanism, yet this remains largely unexplored.

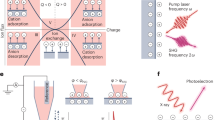
a Increasing the capacitance of EDL reactions via routes ① increasing \({SA}\) or ② reducing \(d\), and enhancing capacity via route ③ expanding ∆φ. b CV curves of the YP50F electrode with expanding ∆φ in the EC:DMC electrolyte at 1 mV s−1. c Schematic of EC-SEI layers for sieving solvated Na+ and allowing the flux of net Na+ ions, leading to the loss of EDL capacitive behavior of solvated ions in nanopores. d CV curves of the YP50F electrode in the DGDE electrolyte at 1 mV s−1. e CV curves of the YP50F electrode in the DGDE electrolyte at 0.2 mV s−1, showing a capacitive contribution of 99.1%. f Schematic of DGDE-SEI layers for allowing the flux of solvated [Na-xDGDE]+ into nanopores, resulting in efficient EDL capacitive charge storage even the potential low to 0.05 V vs. Na+/Na.
The EDL charge storage (Q, C g−1 or mAh g−1) essentially relies on the balance between surface adsorption of the opposite ions from the electrolyte and the applied potential on the electrodes4:
where Δφ = φ − φPZC represents the electrochemical polarization away from the potential of zero charge (PZC)18. Route ③ (Fig. 1a) shows the ability to increase the Q of a negative electrode by expanding Δφ to lower potentials19. The operating voltage of commercial EDLCs using TEABF4 in ACN is limited to 2.7 V (rarely up to 3 V) owing to the decomposition of solvents beyond its stable potential window20,21. However, organic solvents and slats are reduced to form solid electrolyte interface (SEI) layers on negative electrodes at low potentials22,23. One well-known function of SEI layers (formed in ethylene carbonate (EC) based electrolytes) is to sieve solvation shells and allow the intercalation of net Li+ into graphite, resulting in a theoretical capacity of 372 mAh g−1 by forming LiC6 (0.167 e− per carbon)24. This finding significantly promoted the commercialization of lithium-ion batteries since 199123. Conversely, the high-SA activated carbon anode cycling in the EC-based electrolyte displayed very low initial coulombic efficiencies (ICEs), owing to the severe decomposition of electrolyte and formation of thick EC-SEI layers25,26. This conventional view on the function of SEI layers blocks the development of unitizing the EDL adsorption of solvated ions in nanopores under a largely expanded Δφ11.
Challenging above-mentioned concepts, in this work, we demonstrate that the highly efficient EDL capacitive charge storage in nanopores is achievable even Δφ is expended to −2.95 V (corresponding to a very low potential of 0.05 V vs. Na+/Na away from the PZC) in carbon nanopores by using Na+-based diethylene glycol dimethyl ether (DGDE) electrolyte. The as-formed DGDE-SEI layers allow the flux of solvated Na+ into nanopores. Unlike tuning the pore size and disordering of carbon materials2,27,28,29 or using different sizes of cations/anions30, we find that expanding Δφ is able to electrochemically drive the continuous partial desolvation, as demonstrated by in situ EQCM and NMR techniques. The enhanced desolvation degree, but not complete, leads to an in situ reduction in d and thus a significant increase in capacitance and capacity of EDL capacitive charge storage. Under a large Δφ of −2.95 V, commercial activated carbon (YP50F) delivers a high EDL capacitance of 172 F g−1 and a high capacity of up to 508 C g−1 (refers to 0.063 e− per carbon). Moreover, it delivers a high ICE of 92.2%, a total capacitive-dominated contribution of 99.1%, high-rate capabilities, and long-term cycling stability. This finding enables us to redesign and assemble the hybrid sodium-ion capacitor (SIC) pouch cells without any pretreatments, which display improved specific energy, high power ability, and long cycle life.
Results
DGDE-SEI layers enable the EDL adsorption of solvated ions in nanopores
YP50F has a high Brunauer‒Emmett‒Teller (BET) SA of ~1700 m2 g–1 and an average pore size of 0.9 nm (Supplementary Fig. 1)2,17. In the electrolyte of 1 M NaPF6 in EC:diethylene carbonate (DMC), the YP50F electrode displays box-like cyclic voltammetry (CV) curves in 3 − 1.0 V vs. Na+/Na at 1 mV s–1 (Fig. 1b), which is typical of EDL capacitive behavior. Nevertheless, the CV curves become distorted and the box-like EDL capacitive behavior completely disappears as soon as the cut-off potential below 1.0 V vs. Na+/Na along with the formation of EC-SEI layers. When directly cycled in 3−0.05 V vs. Na+/Na, the CV curves (Supplementary Fig. 2a) and galvanostatic (dis)charge profiles at 0.1 A g–1 (0.5 mA cm–2) (Supplementary Fig. 3a) show the irreversible electrolyte decomposition at ~0.8 V vs. Na+/Na and a very low ICE of 20.9% (Supplementary Fig. 3b,c). The thick SEI layers covering on YP50F particles are observed by ex situ scanning electron microscopy (SEM) and transmission electron microscopy (TEM) (Supplementary Fig. 4), which are composed of rich Na2CO3 and R−C = O composites analysed by ex situ X-ray photoelectron spectroscopy (XPS) (Supplementary Fig. 5)31,32. After the formation of the EC-SEI layers, the characteristic microporous-type adsorption isotherms entirely disappeared, and SA decreased to only 6.5 m2 g−1 (Supplementary Fig. 6). Therefore, the YP50F electrode in EC:DMC in 3−0.05 V vs. Na+/Na displays the loss of characteristic capacitor-like CV curves (Fig. 1b and Supplementary Fig. 2a,b) and reaction kinetics (a low capacitive contribution of 47.6% at 0.2 mV s−1, Supplementary Fig. 7)33. The above results support the function of EC-based SEI layers (Fig. 1c)23,24,31, which sieve solvation shells and allow net Na+ adsorption.
Conversely, the YP50F electrode in the DGDE electrolyte continues to display butterfly-shaped CV curves during the expanding ∆φ, until the cut-off potential low to 0.05 V vs. Na+/Na (Fig. 1d). When directly cycled in 3−0.05 V vs. Na+/Na, the well-overlapping CV curves indicate the minimal electrolyte decomposition (Supplementary Fig. 8). After cycles, the YP50F particles have clean surface with the partial distributions of NaF nanograins (~3 nm, Supplementary Fig. 9)34. The ex situ XPS results (Supplementary Fig. 10) reveal a small amount of reduced products, including inorganic NaF, Na2CO3 and organic polyether (R−C=O and R−C−O), from the thin DGDE-SEI layers35. Importantly, after cycles, the YP50F electrode with DGDE-SEI layers maintains microporous adsorption isotherms and a high SA of 961 m2 g−1 (Supplementary Fig. 11), indicating that the as-formed DGDE-SEI layers do not block the micropores for the flux of solvated ions. The YP50F electrode in the DGDE electrolyte displays a total capacitive-dominant contribution of 99.1% at 0.2 mV s−1 (Fig. 1e and Supplementary Fig. 12). The functions of the EC-SEI and DGDE-SEI layers are further demonstrated by the exchange of the applied electrolyte systems (Supplementary Fig. 13). Above all, the DGDE-SEI layers on YP50F allow flux of solvation Na+ ions (Fig. 1f) and efficient EDL capacitive behavior in nanopores under Δφ = −2.95 V (in the protentional window of 3 − 0.05 V vs. Na+/Na).
Electrochemically-driven continuous partial desolvation of EDL capacitive mechanism in nanopores
In situ EQCM and NMR techniques were employed to track the fluxes of ions into nanopores in real time and the corresponding chemical environmental changes17,36. In situ EQCM was operated via an Al-coated quartz crystal resonator (Supplementary Fig. 14)17. First, CV was performed simultaneously with the in situ EQCM response in the DGDE electrolyte in 2.0 − 4.0 V vs. Na+/Na (Fig. 2a). The φPZC of ~3.02 V vs. Na+/Na is close to the open-circuit voltage (OCV) of 3.03 V. Thus, a cut-off potential of 3.0 V is applied for convenience. For the potential window of 3 − 4 V vs. Na+/Na, which is above φPZC, the fitted mass change per electron (\(\Delta m/{dq}\)) of 155 g mol−1 indicates an anion adsorption dominated mechanism (the molar mass of PF6− is 145 g mol−1)37.

CV curve of YP50F-coated quartz crystal electrodes with corresponding mass changes (∆m) via EQCM testing in a 2−4 V and b 3−0.05 V vs. Na+/Na at the 5th cycle in DGDE electrolyte at 2 mV s−1. c Linear fitting of \(\Delta m/{dq}\) during EDL adsorption in the DGDE electrolyte. d In situ 23Na NMR spectra during the 2nd discharging process, at a specific current of 7.5 mA g−1. Evolutionary trends of the fitting e content and f chemical shifts and half widths of “in-pore” 23Na peaks. g Schematic of the EDL capacitive charge storage in nanopores: region I in 3−1.5 V vs. Na+/Na, ion exchange between [Na-\(\bar{2.1}\)DGDE]+ and PF6−; region II in 1.5−0.25 V vs. Na+/Na, counter-ion adsorption of [Na-\(\bar{1.1}\)DGDE]+; and region III in 1.5−0.25 V vs. Na+/Na, counter-ion adsorption of [Na-\(\bar{0.6}\)DGDE]+. The electrochemically driven continuous partial desolvation leads to reducing d and increased capacitance.
In situ EQCM tests in the potential window of 3−0.05 V vs. Na+/Na were subsequently performed. In the DGDE electrolyte, highly reversible mass changes are exhibited (Supplementary Fig. 15), accompanied by slight electrolyte decomposition. Dissipation factors (D) of the pristine and cycled YP50F@crystal disks were further analysed (Supplementary Fig. 16), while the small ΔD/∆n values were on the order of 10−6, indicating that the stiff films were valid for the Sauerbrey equation38,39,40,41. The EQCM data at the 5th cycle (Fig. 2b) were further analysed when stable DGDE-SEI layers formed. The linear fits of \(\Delta m/{dq}\) are divided into three regions (Fig. 2c), with values increasing and then decreasing during expanding \(\Delta {\mbox{V}}\). The low \(\Delta m/{dq}\) of 80 g mol−1 in region I of 3−1.5 V vs. Na+/Na refers to an ion exchange process between [Na-\(\bar{2.1}\)DGDE]+ and PF6− (Supplementary Fig. 17a), which is further confirmed by ex situ 19F NMR spectra (Supplementary Fig. 18). The calculated solvent number of ~2.1 is consistent with the stable solvation of Na+ coordinating with two DGDE molecules42,43. In region II of 1.5−0.25 V vs. Na+/Na, the increased \(\Delta m/{dq}\) value of 174 g mol−1 suggests a transition into the counter-ion adsorption dominated process, whereas the average solvent number is ~1.1 (Supplementary Fig. 17b), corresponding to a partially desolvated [Na-\(\bar{1.1}\)DGDE]+ flux into the nanopores. Furthermore, in region III of 0.25−0.05 V vs. Na+/Na, the \(\Delta m/{dq}\) of 108 g mol−1 corresponds to an average value of 0.6, indicating a reduced partial desolvation of [Na-\(\bar{0.6}\)DGDE]+ flux into the nanopores. For the positive scan, the trends of multistep partial desolvation processes are also observed (Supplementary Fig. 19). By calculating the overall cations maintained in the nanopores during expanding ∆φ (Supplementary Fig. 20), the total average coordinated solvation number is reduced from 2.1 (at 1.5 V vs. Na+/Na) to ~1.3 (at 0.25 V vs. Na+/Na) and then to ~1.1 (at 1.5 V vs. Na+/Na), leading to dense packing of solvated [Na-xDGDE]+ ions in nanopores.
Compared to the EQCM results in DGDE electrolyte, the mass changes in the EC:DMC electrolyte are highly irreversible (Supplementary Fig. 21) because of the formation of thick EC-SEI layers. The EC-SEI layers are able to sieve the solvation shells23,24,31. The \(\Delta m/{dq}\) of 30.5 g mol−1 is slightly higher than 23 g mol−1 for net Na+ (Supplementary Fig. 22), which might be owing to the contributions of electrolyte decomposition or the adsorption of solvated ions on the surface of EC-SEI layers. However, the completely desolvated Na+ ions should cross through the thick EC-SEI layers, resulting in poor rate capability and large voltage hysteresis (Supplementary Fig. 3c−f).
Owing to “ring current effects” originating from delocalized π-electrons in the carbon rings, 23Na NMR spectra (Supplementary Fig. 23) display resonances at more negative chemical shifts for “in-pore” ions (adsorbed in nanopores) compared to “ex-pore” ions (located outside the pores)2,17. Accordingly, in situ 23Na NMR spectroscopy was employed to track the changes in the population and microstructure of “in-pore” cations2,44. An almost reversible change in the chemical shift range of 1000 to −1000 ppm indicates the EDL process during cycling in DGDE electrolyte (Supplementary Fig. 24), which differs from the formation of Na0 clusters in hard carbon anodes45,46. A clearer understanding is achieved by analysing in situ 23Na NMR spectra from the 2nd cycle (Fig. 2d), which eliminates the influence associated with SEI formation. The in situ 23Na NMR spectra (Fig. 2d) and difference spectra (Supplementary Fig. 25) revealed a notable broadening of signals at lower potentials during the expanding ∆φ. The consistent trend with upfield shifting and broadening in-pore signals was also observed by ex situ 23Na NMR spectra (Supplementary Fig. 26). This broadening indicates a stronger interaction between solvated Na+ and the carbon surface, attributed to a decrease in the d (Eq. 1). Following this, spectral deconvolution was performed on the basis of the spectra (Supplementary Fig. 27 and Supplementary Table 1). The observed two-step increases in the deconvoluted areas of the “in-pore” signals vs. capacity (Fig. 2e) correlate well with the ion exchange and counter-ion adsorption processes confirmed by the EQCM results. Notably, as the electrode discharges from 1.4 to 1.2 V, the half width of the “in-pore” signals increases appreciably from ~23 to 40 ppm (Fig. 2f). This is followed by a nearly linear increase in the half width and a corresponding linear decrease in the chemical shift to the low potential of 0.05 V vs. Na+/Na, at which point the half width of the “in-pore” signal remains constant. These enhanced solvated Na+ ion‒carbon interactions observed via in situ NMR closely align with the EQCM results. The large electrochemical polarization (Δφ = −2.95 V) electrochemically drives continuous partial desolvation from [Na-\(\bar{2.1}\)DGDE]+ to [Na-\(\bar{1.1}\)DGDE]+ and then to [Na-\(\bar{0.6}\)DGDE]+ (Fig. 2g), leading to gradually reducing d and thus the increase in capacitance. In situ 23Na NMR in EC:DMC (Supplementary Fig. 28) shows the irreversible formation of EC-SEI single during discharging to 0.05 V and recharging to 3 V, which is very different from the reversible changes of spectra cycled in DGDE electrolyte (Supplementary Fig. 24).
Boosted EDL capacitive charge storage of YP50F electrode in DGDE under large ∆φ
The YP50F electrode shows linear charge and discharge profiles during the initial 5 cycles in DGDE in 3−0.05 V vs. Na+/Na (a large ∆φ of −2.95 V), corresponding to the EDL capacitive adsorption of solvated [Na-xDGDE]+ in nanopores (Fig. 3a). Remarkably, the ICE reaches to 92.2% and subsequent CEs quickly reach to ~100% (Supplementary Fig. 29), suggesting minimal electrolyte decomposition even using high-SA carbon materials. At the low specific currents of 0.01 and 0.05 A g−1, the YP50F electrodes also deliver very high ICEs (Supplementary Fig. 30), indicating that the low current has no influence on the partial desolvation steps of [Na-xDGDE]+ in nanopores. In contrast, the YP50F electrode in EC:DMC displays a highly curved shape owing to the adsorption of net Na+ ions, which require the total stripping of solvation shells and then crossing through thick EC-SEI layers. Figure 3b shows the relationship between the capacitance and capacity with increasing ∆φ (Supplementary Fig. 31). The capacitance gradually increases from 85 to 172 F g−1 as ∆φ increases from −1.5 to −2.95 V owing to the continuous desolvation of [Na-xDGDE]+ with decreasing \(d\). Compared with the controls of pore size, surface doping or increasing the ordering of carbon materials2,27,28,29, the large increases in capacitance caused by in situ increases in ∆φ are highly effective, convenient, and cost-saving. Importantly, owing to the simultaneously increased capacitance and ∆φ, the charge storage capacity of the YP50F electrode reaches a high value of 508 C g−1 (141 mAh g−1, corresponding to 0.063 e− per carbon).

a Initial charge-discharge profiles of the YP50F electrode in DGDE in 3−0.05 V vs. Na+/Na at 0.1 A g−1. The ICE reaches to 92.2%. b Gravimetric capacitance and capacity of the YP50F electrode with increasing ∆φ. Comparison of various porous carbon materials in aqueous, organic, and ionic-liquid electrolytes, including (c) gravimetric capacity vs. capacitance profiles and (d) normalized area capacity vs. capacitance profiles. The YP50F electrode under a ∆φ of −2.95 V displays the high gravimetric and normalized area capacities among the reported values. The source of the literature data shown in this figure can be found in Supplementary Table 2. e Rate performance of the EDL and Faradaic reactions of different carbon materials, at the consistent mass loading of 5.0 ± 0.1 mg cm−2. f Cycling performance of the YP50F electrode at 10 mA cm−2.
The EDL performances of various reported porous carbons in aqueous, organic, and ionic-liquid electrolytes are collected (Supplementary Table 2). There is no apparent correlation between the capacity and SA of porous carbon or electrolyte systems (Supplementary Fig. 32)2. Nevertheless, the relationship between gravimetric capacity and capacitance is dependent on the operating potential window of different electrolytes (Fig. 3c). Owing to the EDL capacitive charge storage under a large ∆φ of −2.95 V in the DGDE electrolyte, the commercial YP50F is able to achieve the high capacity among the state-of-the-art results (Fig. 3c, d and Supplementary Table 2). The normalized capacitance of 10.1 μF cm−2 (Fig. 3d) still falls within the range of 5–20 µF cm−2 for EDL capacitive adsorption in porous carbon4. Remarkably, YP50F under the ∆φ of −2.95 V achieves the high normalized capacity of 30.0 µC cm−2.
The storage capacity and rate performance of the EDL adsorption of [Na-xDGDE]+ are further compared with those of the Faradaic reactions in carbon materials at the consistent mass loading of 5.0 ± 0.1 mg cm−2 (Fig. 3e and Supplementary Figs. 33 and 34). The capacity of 508 C g−1 for [Na-xDGDE]+ adsorption under ∆φ = −2.95 V is significantly higher than that of 135 C g−1 for commercial EDLCs (1 M TEABF4 in ACN) and getting close to 1005 C g−1 for Na+ intercalation in hard carbon and 1218 C g−1 for Li+ intercalation in graphite47,48. However, when the current density is over 5 mA cm−2 (the same charging and discharging currents), these intercalation-type materials show rapidly reduced capacity. But, importantly, the EDL capacitive adsorption of [Na-xDGDE]+ shows high rate capabilities in the current densities of 5–50 mA cm−2. The YP50F electrode displays negligible capacity loss after 10,000 cycles (Fig. 3f) in the DGDE electrolyte in 3.0−0.05 V vs. Na+/Na under half-cell tests. These improved EDL capacitive charge storages support the key concept that increased ∆φ facilitates denser ion packing in nanopores, leading to simultaneously increased capacitances and capacities.
Redesign and assembly of hybrid sodium ion capacitor pouch cells
The hybrid capacitor comprises an EDL electrode and a battery-type electrode49. Commercial hybrid LICs are composed of pre-lithiated graphite as the negative electrode and activated carbon as the positive electrode (Fig. 4a). The LICs necessarily require a pre-lithiation process for device operation, which is complex and requires extra costs in industry50. Additionally, sluggish Li+ intercalation into graphite limits the high-power capability and increases the risk of dendritic growth under fast charging22,51. Recently, hybrid SICs composed of hard carbon negative electrode and activated carbon positive electrode were proposed, accompanying the development of pre-sodiaiton methods52,53,54.

a Schematic of commercial hybrid LIC using positive activated carbon and negative pre-lithiated graphite in 1 M LiPF6 in carbonate electrolyte. b Schematic of hybrid SIC composed of NVP positive electrode and YP50F negative electrode in 1 M NaPF6 in DGDE, following the sodium-ion-transport mechanism. c Charge-discharge profiles of the assembled three-electrode cell, where the YP50F negative electrode operates until 0.05 V vs. Na+/Na. d Cycling performance of the SIC pouch cell at 20 mA cm−2, corresponding to a fast (dis)charging time of 70 s (a rate of 51 C). The inset of d is the photograph of assembled SIC pouch cell. e Charge-discharge profiles of the SIC pouch cell at current densities ranging from 0.5 to 40 mA cm−2 (IC = ID). f Ragone plots of the SIC pouch cell in comparison with state-of-the-art LICs and EDLCs. The source of the literature data shown in this figure can be found in Supplementary Tables 3 and 4.
Differently, we redesign and assemble hybrid SIC pouch cells using YP50F as the negative electrode, matching with battery-type material as positive electrode (Fig. 4b). Na3V2(PO4)3 (NVP) was selected owing to its high-rate capabilities (Supplementary Fig. 35a–d)55, which displayed well-matched reaction kinetics to the YP50F negative electrode (Supplementary Fig. 35e). The three-electrode device demonstrates that the assembled hybrid SIC follows a sodium-ion-transport mechanism (Fig. 4c), that is, the extraction of Na+ from NVP and adsorption of solvated Na+ into the YP50F. The discharging process is inverse. In particular, in this design system, the YP50F negative electrode reversibly operates low to 0.05 V vs. Na+/Na in the DGDE electrolyte (Fig. 4c), which enables the hybrid SIC to work up to 3.4 V.
The YP50F|1 M NaPF6 in DGDE|NVP hybrid SIC pouch cells (rated 545 F/515 mAh, inset of Fig. 4d and Supplementary Fig. 36) are assembled without any presodiation treatments, which delivers a high ICE of 96.1% (Supplementary Fig. 37). This assembly technology is cost-efficient and easily matched with the production processes of the battery or supercapacitor industry. The self-discharge ratio of the SIC pouch cell is ~13.0% after setting for 120 h, which is lower than that of ~38.0% for an EDLC device (Supplementary Fig. 38). The SIC pouch cell shows a capacity retention of 71.4% after 30,000 cycles at 20 mA cm−2, corresponding to a fast (dis)charging time of 70 s that is ~51 C (Fig. 4d and Supplementary Fig. 39), demonstrating the long-term cycling stability.
Moreover, the SIC pouch cell displays triangular charge-discharge curves at various current densities ranging from 0.5 to 40 mA cm−2 (Fig. 4e). At 10, 20 and 40 mA cm−2, it delivers capacities of 396, 310 and 211 mAh, corresponding to fast charging abilities with state of charge (SoC) of 76.9%, 60.6%, and 41.0% finished in 196, 70 and 26 s, respectively. The energy efficiencies of the SIC pouch cell are 93.3%, 80.4%, and 61.0% for the (dis)charge times of 5035, 196, and 26 s, respectively, which are comparable to those of the EDLC and LIB (Supplementary Fig. 40). Additionally, when the charge current was set at 4 mA cm−2, the pouch cell could output large discharge currents of up to 100 mA cm−2 with a high capacity of 196 mAh in 10 s (Supplementary Fig. 41), indicating the ability of high power delivery. The Ragone plots (Fig. 4f) show the SIC pouch cell combined with both high energy and high power densities. It delivers a high specific energy of 40 Wh \({{\mbox{kg}}}_{{\mbox{cell}}}^{-1}\) (calculated based on the total weight of pouch cell), which is comparable with state-of-the-art commercial LICs (Supplementary Table 3) and EDLCs (Supplementary Table 4)9,56,57.
Discussion
We demonstrate that efficient EDL capacitive adsorption in carbon nanopores can operate under a large Δφ of −2.95 V (from PZC (~3 V) to 0.05 V vs. Na+/Na) in Na+-based DGDE electrolyte. The DGDE-SEI layers do not block the pores and allow the flux of solvated Na+ ions, which is different from the well-known sieving function of the EC-SEI layers. The electrochemically driven continuous partial desolvation from [Na-\(\bar{2.1}\)DGDE]+ to [Na-\(\bar{1.1}\)DGDE]+ and then to [Na-\(\bar{0.6}\)DGDE]+ leads to reduced d and thus increased capacitance of 172 F g−1 under the large Δφ of −2.95 V. Beneficial from the simultaneously increased capacitance and ∆φ, the YP50F electrode reaches a high capacity of 508 C g−1 without sacrificing the features of supercapacitors, including high-rate capability and long-term cycling stability.
The efficient EDL capacitive charge storage mechanism allows the development of a 3.4 V hybrid YP50F||NVP SIC with improved specific energy compared to some commercial 2.7 V EDLCs and LICs. The SIC pouch cells maintain high power ability and cycling stability. The waiver of presodiation treatments simplifies the scale-up assembly process of the electrochemical capacitors in industry. This work opens a research direction for breaking the limited EDL charge storage capacity that has the potential to reduce the energy gap between supercapacitors and batteries. The low-cost hybrid SICs are promising for large-scale, sustainable developments in the future.
Methods
Electrode and electrolyte preparation
The slurry comprised YP50F (Kuraray Co.), ketjen black (EC-600JD, Nouryon Co.), carboxymethyl cellulose (CMC, >99.5%, Canrd New Energy Technology Co.), and styrene butadiene rubber (SBR, >99.5%, Canrd New Energy Technology Co.) at a ratio of 92:3:3:2 by weight. The slurry with deionized water as the dissolving solvent was homogeneously coated onto Al foil (16 μm in thickness, >99.5%, Shenzhen Kejing Star Technology Co., Ltd.) and dried at 100 °C for 12 h in a vacuum oven. The thick-film YP50F electrode has a single-sided thickness of 100 μm, corresponding to a mass loading of 5.0 mg cm−2. The graphite anode (mass loading of 5.0 mg cm−2) on Cu foils (25 μm in thickness, Shenzhen Kejing Star Technology Co. Ltd.) and hard carbon (mass loading of 5.0 mg cm−2) on Al foil were prepared as the same process and component as the YP50F electrode. The Na3V2(PO4)3@C (NVP@C, Hubei Energy Technology Co. Ltd.) electrode was prepared by mixing NVP@C powder, ketjen black, and polyvinylidene fluoride (PVDF, Battery grade, Shenzhen Kejing Star Technology Co. Ltd.) at a ratio of 90:5:5 by weight. The slurry with N-methylpyrrolidine (NMP, >99%, Acmec) was homogeneously coated onto Al foil and dried at 120 °C for 12 h in a vacuum oven. The single-sided mass loading of the NVP electrode was controlled at 7.5 mg cm−2. The above electrodes were cut into 14 mm in diameter for coin cell testing. The electrolytes of 1 M NaPF6 in diethylene-glycol-dimethyl-ether (DGDE, >99.8%) and 1 M NaPF6 in ethylene carbonate (EC):diethylene carbonate (DMC) (1:1 by weight, >99.8%) were obtained from DuoDuo Chemical Technology Co. The solvents of DGDE (>99.8%) and DMC (>99.8%) were obtained from DuoDuo Chemical Technology Co.
Electrochemical measurements
The half-cells were assembled in an Ar-filled glove box (H2O < 0.1 ppm and O2 < 0.1 ppm). The CR2025 coin cells were composed of a sodium foil (>99.9%, 0.6 mm in thickness, 15.8 mm in diameter, Sinopharm Chemical Reagent Co.) as the counter and reference electrode, a Celgard-2325 separator, and a YP50F electrode as the working electrode. The volume of ~60 μL electrolyte is applied for each coin cell. CV measurements were performed via potentiostats (Bio-Logic VSP). The galvanostatic charge‒discharge measurements were performed by NEWARE battery test system. Three or more cells were tested for a single electrochemical experiment. All the electrochemical results were measured at 25 °C (MSK-TE906-150P-70-5, Shenzhen Kejing Star Technology Co. Ltd.).
Characterization techniques
Powder X-ray diffraction (XRD) was performed on Bruker-Axs X-ray diffractometer (Cu Kα radiation, λ = 1.5406 Å) over a diffraction angle range of 10°−60°, at a speed size of 5° min−1. The Brunauer–Emmett–Teller (BET) specific surface area was measured via nitrogen sorption isotherms (Micromeritics Tristar 3020) at 77 K. To prepare cycled YP50F powders for nitrogen sorption isotherms, a pouch cell using YP50F electrodes and sodium foil in the DGDE (or EC:DMC) electrolyte was assembled. After 5 cycles, the cell was disassembled in a glovebox and washed with DGDE (or DMC) to remove residual electrolytes. All powders were scraped from the current collector and dried for BET analysis. Scanning electron microscopy (SEM) images were recorded by using SU-70, conducted at 20 keV. The cross-sectional SEM image of the electrodes was obtained with a Leica EM TIC 3X. Transmission electron microscopy (TEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and EDS maps were obtained through aberration-corrected transmission electron microscopy (Titan Cubed Themis G2 300). To prepare the sample for ex situ TEM analysis, the cycled YP50F electrodes were disassembled in a glovebox and washed with DGDE or DMC solvent to remove residual electrolytes. Afterwards, the powders were scraped from Al foil and then subjected to ultrasonic treatment in alcohol, and deposited onto a grid for TEM characterization58. Throughout the transfer procedure, the samples were kept in a sealed loading chamber under an Ar atmosphere to prevent oxidation. Ex situ XPS survey scans were performed on a Kratos Axis Supra system equipped with a monochromatic Al Kα X-ray source (1486.7 eV)58. Coin cells were disassembled inside the glovebox, and the YP50F electrodes were rinsed with either DGDE or DMC solvent. After drying, the anodes were introduced into the XPS chamber for measurement. To avoid air contact, the XPS chamber was directly linked to an Ar-filled glovebox for sample preparation and transfer. All spectra were calibrated for charge referencing based on the C 1s peak (at 284.8 eV).
In situ EQCM measurements and analyses
The EQCM measurements were conducted on a QCD-300 single-channel quartz crystal microbalance (Bio-Logic), lined with a potentiostat (Bio-Logic SP-200). The slurry comprising YP50F, carbon nanotubes, and CMC at a ratio of 90:5:5 by weight was dispersed in deionized water through strong ultrasonic treatment. Then, the slurry was sprayed onto an Al-coated quartz crystal disk (5 MHz). The crystal disk was placed onto a hot plate at 80 °C, while a steel mask was covered on the disk with the exposure of the Al-coated surface for the spray of slurry. Compressed air was used to control the spray pressure. The carbon deposited crystal disks were dried in a vacuum oven for 4 h at 100 °C. The mass loading was controlled at ~50 μg cm−2. The dissipation factors (D) at different overtones of the pristine YP50F@crystal disks and the electrochemically cycled disks at the fully discharged state were measured by the Q-Sense Explorer (Biolin Scientific).
Electrochemical measurements were conducted with simultaneous recording of the quartz resonance frequency. According to the Sauerbrey equation (Eq. 3), the frequency shift (Δf) of the quartz resonator is effectively converted into the corresponding mass change (Δm) of the electrodes and quartz crystal16,17:
where ρq means the density of quartz, μq means the shear modulus of quartz, \({f}_{0}\) means the fundamental resonance frequency of the quartz, and \({C}_{f}\) means the calibration constant (or sensitivity factor). The calculation of \({C}_{f}\) was based on the method provided by Griffin et al17. In detail, a sliver deposition experiment with 0.01 M silver nitrate and 0.1 M tetraethylammonium tetrafluoroborate in DGDE was applied (Supplementary Fig. 14). Thus, the calculated \({C}_{f}\) was 21.02 ng Hz−1.
In situ and ex situ NMR testing
In situ 23Na NMR measurements were performed on a AVANCE III 500 MHz spectrometer. Self-designed in situ probe and operando cells were applied (Supplementary Fig. 24a). A single pulse sequence with a π/12 angle of 1.5 μs and a recycle delay of 0.02 s was used to acquire the 23Na NMR spectra with an interval time of 30 min, and every single spectrum required ~10 min.
Ex situ NMR spectroscopy was performed on a Bruker AVANCE NEO 300 MHz NMR spectrometer. The coin cells were operated to the target potential and disassembled in an Ar-filled glove box immediately. Then materials were scraped from the Al foils and packed into 3.2 rotors for NMR tests. 23Na NMR spectra were acquired using a π/12 pulse of 0.6 μs and a recycle delay of 0.02 s with magic angele spinning (MAS) frequency of 8 kHz, referenced to NaCl powders (7.2 ppm). 19F NMR spectra were acquired using a π/4 pulse of 1.5 μs and a recycle delay of 10 s with MAS frequency of 3 kHz, referenced to LiF (−204 ppm).
Ex situ nitrogen adsorption-desorption isotherms
The Brunauer–Emmett–Teller (BET) specific surface area was measured via nitrogen adsorption-desorption isotherms (Micromeritics Tristar 3020) at 77 K. The YP50F powders (~100 mg) were placed into a glass bulb gas cell and degassed at 150 °C in vacuum for 16 h before the measurement2. To prepare the cycled YP50F electrode for nitrogen adsorption-desorption isotherms, a pouch cell using YP50F electrodes and sodium foil in the DGDE (or EC:DMC) electrolyte was assembled. After 5 cycles, the cells were disassembled in a glovebox and washed with DGDE (or DMC) to remove residual electrolytes. All powders were scraped from the current collector and dried at 120 °C in the glove box. Then, the powders (~100 mg) were placed into a glass bulb gas cell and degassed at 150 °C in vacuum for 16 h before measurement.
Fabrication of hybrid SIC pouch cells
The hybrid sodium-ion capacitor pouch cells were assembled by repeatedly stacking 16 layers of YP50F negative electrodes (double-side coating with the mass loading of 10 mg cm−2), Celgard-2325 separators, and 15 layers of NVP positive electrodes (double-side coating with the mass loading of 15 mg cm−2). The YP50F and NVP electrodes were cut into 43 mm × 56 mm in size. The pouch cells were assembled in Ar-filled glove box, injected with the electrolyte (~13 mL of 1 M NaPF6 in DGDE), and vacuum sealed at 200 °C. The pouch cells were firstly tested at 0.2 mA cm−2 for initial cycle in the voltage window of 0–3.4 V and then for other electrochemical performance measurements, under the external pressure of ~100 kPa. All the electrochemical results of pouch cells were measured at 25 °C.
The specific capacity (\({Q}_{m}\), C g−1 or mAh g−1), specific capacitance (\({C}_{m}\), F g−1), specific energy\(\,\left(\right.{E}_{m}\), Wh kg−1), and specific power (\({P}_{m}\), W kg−1) are calculated via Eq. 4−7.
where t = t2 − t1 refers to the time for a discharging process and the discharge voltage profiles are applied for the calculation of specific energy. For SIC pouch cells, the m represents total weight of cell.
Data availability
The authors declare that all data supporting the finding of this study are available within the paper and its supplementary information files. All raw data generated during the current study are available from the corresponding authors upon request. Source data are provided with this paper.
References
Simon, P. & Gogotsi, Y. Perspectives for electrochemical capacitors and related devices. Nat. Mater. 19, 1151–1163 (2020).
Liu, X. et al. Structural disorder determines capacitance in nanoporous carbons. Science 384, 321–325 (2024).
Sun, H. et al. Hierarchical 3D electrodes for electrochemical energy storage. Nat. Rev. Mater. 4, 45–60 (2019).
Shao, H., Wu, Y.-C., Lin, Z., Taberna, P.-L. & Simon, P. Nanoporous carbon for electrochemical capacitive energy storage. Chem. Soc. Rev. 49, 3005–3039 (2020).
Merlet, C. et al. On the molecular origin of supercapacitance in nanoporous carbon electrodes. Nat. Mater. 11, 306–310 (2012).
Gao, X. et al. Recent advances in carbon-based nanomaterials for multivalent-ion hybrid capacitors: a review. Energy Environ. Sci. 16, 1364–1383 (2023).
Tang, X. et al. Research advances in biomass-derived nanostructured carbons and their composite materials for electrochemical energy technologies. Prog. Mater. Sci. 118, 100770 (2021).
Li, W., Liu, J. & Zhao, D. Mesoporous materials for energy conversion and storage devices. Nat. Rev. Mater. 1, 16023 (2016).
Jeżowski, P. et al. Safe and recyclable lithium-ion capacitors using sacrificial organic lithium salt. Nat. Mater. 17, 167–173 (2018).
Li, Z. et al. Tuning the interlayer spacing of graphene laminate films for efficient pore utilization towards compact capacitive energy storage. Nat. Energy 5, 160–168 (2020).
Forse, A. C., Merlet, C., Griffin, J. M. & Grey, C. P. New perspectives on the charging mechanisms of supercapacitors. J. Am. Chem. Soc. 138, 5731–5744 (2016).
Ji, H. et al. Capacitance of carbon-based electrical double-layer capacitors. Nat. Commun. 5, 3317 (2014).
Chmiola, J. et al. Anomalous increase in carbon capacitance at pore sizes less than 1 nanometer. Science 313, 1760–1763 (2006).
Fleischmann, S. et al. Continuous transition from double-layer to Faradaic charge storage in confined electrolytes. Nat. Energy 7, 222–228 (2022).
Forse, A. lexanderC. et al. Direct observation of ion dynamics in supercapacitor electrodes using in situ diffusion NMR spectroscopy. Nat. Energy 2, 16216 (2017).
Tsai, W.-Y., Taberna, P.-L. & Simon, P. Electrochemical quartz crystal microbalance (EQCM) study of ion dynamics in nanoporous carbons. J. Am. Chem. Soc. 136, 8722–8728 (2014).
Griffin, J. M. et al. In situ NMR and electrochemical quartz crystal microbalance techniques reveal the structure of the electrical double layer in supercapacitors. Nat. Mater. 14, 812–819 (2015).
Ge, K., Shao, H., Lin, Z., Taberna, P.-L. & Simon, P. Advanced characterization of confined electrochemical interfaces in electrochemical capacitors. Nat. Nanotechnol. https://doi.org/10.1038/s41565-024-01821-z (2024).
Pandey, D., Kumar, K. S. & Thomas, J. Supercapacitor electrode energetics and mechanism of operation: uncovering the voltage window. Prog. Mater. Sci. 141, 101219 (2024).
Naoi, K., Ishimoto, S., Miyamoto, J. -i & Naoi, W. Second generation ‘nanohybrid supercapacitor’: evolution of capacitive energy storage devices. Energy Environ. Sci. 5, 9363–9373 (2012).
Noori, A., El-Kady, M. F., Rahmanifar, M. S., Kaner, R. B. & Mousavi, M. F. Towards establishing standard performance metrics for batteries, supercapacitors and beyond. Chem. Soc. Rev. 48, 1272–1341 (2019).
Cai, W. et al. A review on energy chemistry of fast-charging anodes. Chem. Soc. Rev. 49, 3806–3833 (2020).
Winter, M., Barnett, B. & Xu, K. Before Li ion batteries. Chem. Rev. 118, 11433–11456 (2018).
Liu, T. et al. In situ quantification of interphasial chemistry in Li-ion battery. Nat. Nanotechnol. 14, 50–56 (2019).
Jain, R. et al. Nanostructuring versus microstructuring in battery electrodes. Nat. Rev. Mater. 7, 736–746 (2022).
Pomerantseva, E., Bonaccorso, F., Feng, X., Cui, Y. & Gogotsi, Y. Energy storage: The future enabled by nanomaterials. Science 366, eaan8285 (2019).
Zhang, E. et al. Unraveling the capacitive charge storage mechanism of nitrogen-doped porous carbons by EQCM and ssNMR. J. Am. Chem. Soc. 144, 14217–14225 (2022).
Ge, K., Shao, H., Raymundo-Piñero, E., Taberna, P.-L. & Simon, P. Cation desolvation-induced capacitance enhancement in reduced graphene oxide (rGO). Nat. Commun. 15, 1935 (2024).
Liu, L., Raymundo-Piñero, E., Sunny, S., Taberna, P.-L. & Simon, P. Role of surface terminations for charge storage of Ti3C2T MXene electrodes in aqueous acidic electrolyte. Angew. Chem. Int. Ed. 63, e202319238 (2024).
VanAken, K. L., Beidaghi, M. & Gogotsi, Y. Formulation of ionic-liquid electrolyte to expand the voltage window of supercapacitors. Angew. Chem. Int. Ed. 54, 4806–4809 (2015).
Adenusi, H., Chass, G. A., Passerini, S., Tian, K. V. & Chen, G. Lithium batteries and the solid electrolyte interphase (SEI)—progress and outlook. Adv. Energy Mater. 13, 2203307 (2023).
Yuan, X. et al. Engineering battery corrosion films by tuning electrical double layer composition. Joule 8, 3038–3053 (2024).
Choi, C. et al. Achieving high energy density and high power density with pseudocapacitive materials. Nat. Rev. Mater. 5, 5–19 (2020).
Xia, D. et al. Self-terminating, heterogeneous solid–electrolyte interphase enables reversible Li–ether cointercalation in graphite anodes. Proc. Natl. Acad. Sci. USA 121, e2313096121 (2024).
Goktas, M. et al. Graphite as cointercalation electrode for sodium-ion batteries: electrode dynamics and the missing solid electrolyte interphase (SEI). Adv. Energy Mater. 8, 1702724 (2018).
Chubak, I. et al. Quadrupolar 23Na+ NMR relaxation as a probe of subpicosecond collective dynamics in aqueous electrolyte solutions. Nat. Commun. 14, 84 (2023).
Levi, M. D. et al. Electrochemical quartz crystal microbalance (EQCM) studies of ions and solvents insertion into highly porous activated carbons. J. Am. Chem. Soc. 132, 13220–13222 (2010).
Shpigel, N., Levi, M. D., Sigalov, S., Daikhin, L. & Aurbach, D. In situ real-time mechanical and morphological characterization of electrodes for electrochemical energy storage and conversion by electrochemical quartz crystal microbalance with dissipation monitoring. Acc. Chem. Res. 51, 69–79 (2018).
Buttry, D. A. & Ward, M. D. Measurement of interfacial processes at electrode surfaces with the electrochemical quartz crystal microbalance. Chem. Rev. 92, 1355–1379 (1992).
Lemaire, P. et al. Making advanced electrogravimetry as an affordable analytical tool for battery interface characterization. Anal. Chem. 92, 13803–13812 (2020).
Easley, A. D. et al. A practical guide to quartz crystal microbalance with dissipation monitoring of thin polymer films. J. Polym. Sci. 60, 1090–1107 (2022).
Li, Y. et al. Ether-based electrolytes for sodium ion batteries. Chem. Soc. Rev. 51, 4484–4536 (2022).
Gotoh, K. et al. Structure and dynamic behavior of sodium–diglyme complex in the graphite anode of sodium ion battery by 2H nuclear magnetic resonance. J. Phys. Chem. C. 120, 28152–28156 (2016).
Wang, H. et al. In situ NMR spectroscopy of supercapacitors: insight into the charge storage mechanism. J. Am. Chem. Soc. 135, 18968–18980 (2013).
Stratford, J. M. et al. Correlating local structure and sodium storage in hard carbon anodes: insights from pair distribution function analysis and solid-state NMR. J. Am. Chem. Soc. 143, 14274–14286 (2021).
Li, Y. et al. Origin of fast charging in hard carbon anodes. Nat. Energy 9, 134–142 (2024).
Li, Y., Lu, Y., Adelhelm, P., Titirici, M.-M. & Hu, Y.-S. Intercalation chemistry of graphite: alkali metal ions and beyond. Chem. Soc. Rev. 48, 4655–4687 (2019).
Sun, N., Qiu, J. & Xu, B. Understanding of sodium storage mechanism in hard carbons: Ongoing development under debate. Adv. Energy Mater. 12, 2200715 (2022).
Shao, Y. et al. Design and mechanisms of asymmetric supercapacitors. Chem. Rev. 118, 9233–9280 (2018).
Jin, L. et al. Progress and perspectives on pre-lithiation technologies for lithium ion capacitors. Energy Environ. Sci. 13, 2341–2362 (2020).
Li, B. et al. Electrode materials, electrolytes, and challenges in nonaqueous lithium-ion capacitors. Adv. Mater. 30, 1705670 (2018).
Ding, J., Hu, W., Paek, E. & Mitlin, D. Review of hybrid ion capacitors: from aqueous to lithium to sodium. Chem. Rev. 118, 6457–6498 (2018).
Lin, K. et al. Practical evaluation of presodiation techniques for high energy sodium-based batteries. ACS Nano 19, 17062–17086 (2025).
Chen, S. et al. Active organic salts enabling non-intrusive electrolyte presodiation strategy. Adv. Mater. 37, 2502251 (2025).
Jin, T. et al. Polyanion-type cathode materials for sodium-ion batteries. Chem. Soc. Rev. 49, 2342–2377 (2020).
Cementon, C., Ramireddy, T., Dewar, D., Brennan, M. & Glushenkov, A. M. We may be underestimating the power capabilities of lithium-ion capacitors. J. Power Sources 591, 233857 (2024).
Zhao, J. & Burke, A. F. Electrochemical capacitors: performance metrics and evaluation by testing and analysis. Adv. Energy Mater. 11, 2002192 (2021).
Wei, Q. et al. Surface-redox sodium-ion storage in anatase titanium oxide. Nat. Commun. 14, 7 (2023).
Acknowledgements
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (Grant Nos. 22479123 [Q.W.], 22179113 [Q.W.], 22272175 [G.Z.], and 52431009 [D.-L.P.]), the Fujian Provincial Natural Science Foundation of China (Grant No. 2025J010006 [Q.W.]) and Fujian Provincial Young Eagle Talents Program. G. Z. acknowledges the financial support from the Dalian Institute of Chemical Physics (Grant No. DICP I202213) and Liaoning Binhai Laboratory (Grant No. LBLF-2023-03).
Author information
Authors and Affiliations
Contributions
Q.W. and D.-L.P. conceived and supervised this work. S.F., Z.Y., B.W., and Q.W. designed the experiments and wrote the paper. S.F., Z.Y., and B.W. conducted the NMR, EQCM, the assembly of pouch cells, and electrochemistry measurements. H.L., Y.O., and G.Z. conducted the in situ NMR characterization. Y.S. collected the reported results from the literature. D.T., Z.Y., K.L., Z.P., D.-L.P., and G.Z. participated in discussions and data analysis. Q.W., G.Z., and D.-L.P. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Q.W., Z.Y., and S.F. are inventors on a patent application related to this work fled by Xiamen University. The other authors declare that they have no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fan, S., Yan, Z., Wang, B. et al. Unlocking limited electric double-layer capacity via electrochemically-driven continuous partial desolvations in carbon nanopores. Nat Commun 17, 363 (2026). https://doi.org/10.1038/s41467-025-66433-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66433-0
